

Abstract
There is a lack of well-characterized models for pancreatic ductal adenocarcinoma (PDAC). PDAC itself is unique because of its pronounced tumor microenvironment that influences tumor progression, behavior and therapeutic resistance. Here we investigated, in patient-derived tumor xenograft (PDTX) models developed from fine needle biopsies, the cancer cells behavior, Epithelial-to-Mesenchymal Transition (EMT) and drug response. For this, we studied two behaviorally distinct PDTX models. Tumor volume measurement, histology, immuno-histochemical staining, RT-qPCR, RNA sequencing and Western blotting were used to further characterize these models and investigate the effect of two classes of drugs (gemcitabine and acriflavine (HIF-inhibitor)). The models recapitulated the corresponding primary tumors. The growth-rate of the poorly differentiated tumor (PAC010) was faster than that of the moderately differentiated tumor (PAC006) (P<0.05). The PAC010 model showed increased cell proliferation (Ki-67 staining) and markers indicating survival (increased p-AKT, p-ERK and p-NF-kB65 and suppression of cleaved PARP). Gene and protein analysis showed higher expression of mesenchymal markers in PAC010 model (e.g. VIM, SNAI2). Pathway analysis demonstrated activation of processes related to EMT, tumor progression and aggressiveness in PAC010. Gemcitabine treatment resulted in shrinking of the tumor volume and reduced proliferation in both models. Importantly, gemcitabine treatment significantly enhanced the expression of mesenchymal marker supportive of metastatic behavior and of survival pathways, particularly in the non-aggressive PAC006 model. Acriflavine had little effect on tumor growth in both models. In conclusion, we observed in this unique model of PDAC, a clear link between EMT and poor tumor differentiation and found that gemcitabine can increase EMT.
Keywords: Epithelial-to-Mesenchymal Transition, pancreatic ductal adenocarcinoma, patient-derived tumor xenografts, next-generation sequencing
Introduction
Pancreatic ductal adenocarcinoma (PDAC) has a unique tumor microenvironment with more than 90% of the whole tumor tissue consisting of stromal components and only less than 10% being actual tumor cells. This tumor microenvironment is complex and composed of extracellular matrix (ECM), activated fibroblasts, myofibroblasts, inflammatory cells, blood and lymphatic vessels [1,2]. It is a growing understanding that important dynamic interactions exists in PDAC between the tumor cells and their microenvironment (stromal components). Tumor cells secrete factors that stimulate the surrounding microenvironment and in response, stromal cells secrete growth factors and cytokines that in turn increase the growth, invasion, metastasis and therapeutic resistance of the tumor cells [3,4]. Metastatic dissemination requires of the tumor cells to break away from the epithelial ductal structures and take on characteristics of mesenchymal cells through the process of Epithelial-to-Mesenchymal Transition (EMT). This transition involves extensive remodeling and an interaction of cancer cells with the stroma to acquire a more invasive, metastatic and therapy-resistant phenotype [4,5]. The precise network of the molecular interactions between the tumor cells and the stromal components supporting tumor progression remains poorly understood.
The concept of therapies that simultaneously target both pro-tumoral stromal components and cancer cells is only beginning to emerge and there is now a need for a platform for basic understanding and pre-clinical trials. An ideal model that mimics the full PDAC tumor microenvironment and its progression as seen in patients is still lacking. Several animal models have been developed, including genetically engineered mouse models and orthotopic implantation of cancer cell lines [6]. One attractive model, that in part could address this demand, is the patient-derived tumor xenograft mice model (PDTX). These PDTX models are most often established from surgical specimen with subsequent transplantation into immune-compromised rodents such as athymic nude or NOD/SCID mice [7,8]. Nevertheless, because PDAC is usually diagnosed in a locally advanced or metastatic stage, obtaining tumor material from these patients by an invasive procedure to generate xenograft models is challenging from a clinical perspective. Moreover, xenografts of all clinical stages are urgently needed. Recently we have reported the first successful establishment by minimally invasive approaches, of PDTX models using fine needle biopsies (FNBs) obtained by endoscopic ultrasound (EUS) from patients with locally advanced disease [9]. This technique was recently confirmed in a study focused on drug response related to Kras mutations [10].
Several clinically promising therapies have emerged from in vitro studies however; there are limitations to repeat similar results in an in vivo situation. Previously, using the pancreatic adenocarcinoma cell lines (PANC-1, MiaPaca2) in vitro cell culture, we have shown that tumor microenvironmental factors (TGF-β1 or hypoxia) and drug resistance can induce EMT. In addition, we showed that a non-toxic concentration of acriflavine (ACF) was successful in reversing the mesenchymal differentiation and blocking aggressive behavior of cancer cell lines and of re-sensitize cancer cells to gemcitabine [11].
In the current study, we molecularly characterized two PDTX models and expanded our in vitro findings on EMT to PDTX models bearing two behaviorally different tumor types (a poorly differentiated and a well/moderately differentiated tumor model). Our study further exploited the differences between the models to investigate the link between EMT gene signature and therapeutic drug response (gemcitabine (GEM) -a standard of care drug for pancreatic cancer and acriflavine - proposed for EMT reversal).
Materials and methods
Establishment of patient-derived PDAC xenografts
The development and characterization of the PDTX model has been described in detail by Hermans et al., 2016 [9]. In short, female NMRI nude mice (6-8 weeks old, weighing 19-28 g, Taconic, Denmark) were housed individual in ventilated cages under specific pathogen free (SPF) conditions. Animal care and all research procedures were executed in accordance with the applicable legal guidelines and under approval of the medical ethical committee for laboratory animals of the KU Leuven (P147/2012). For transplantation, the subcutaneous interscapular fat pad of the mouse was externalized and a small pocket was created using an aseptic surgical technique. A small tumor piece of 8 mm3 was placed in the pocket. After 3-4 generation (F3 or F4), tumor tissues were stored in liquid N2 as starting material for experimental work.
Compounds used
Acriflavine (ACF) (Sigma-Aldrich, MO, USA) was dissolved in 100% dimethyl sulfoxide (DMSO) and stored at room temperature. Gemcitabine (Hospira 38 mg/ml solution for infusion, Hospira Benelux BVBA, BE390476, Belgium) was stored at 4°C.
Tumor implantation, treatment procedure, growth and sample collection
Stored tumor tissue from each type was implanted into a first group of 4 mice and after establishment of growth this tumor tissue was used for expansion in a new generation of mice (F5). After the tumor reached a volume of 100-200 mm3, these mice were randomly divided into 3 groups with 8 mice in each group that were treated intraperitoneal for up to 28 days. A) control group (treated with vehicle, 0.9% NaCl) and the experimental groups B) Gemcitabine (50 mg/kg, injected twice a week) or C) Acriflavine: schedule four doses of acriflavine injected daily with a fixed dose of 5 mg/kg Monday to Thursday and single dose on Friday for the weekend of 10 mg/kg. Prior to the actual study, a toxicity study for ACF was performed. The body weight and tumor size were measured thrice a week (Monday, Wednesday and Friday). The tumor volume was calculated using the formula: Tumor length (mm) × Tumor width (mm) × Tumor depth (mm) × (3.14/6) = Tumor volume (mm3). Tumors were harvested as soon as their volume reached 1000-1500 mm3. The tissue samples were weighed, photographed and stored for histological analysis and molecular profiling.
Histology and immunohistochemistry
Hematoxylin and eosin (H&E) staining was performed on formalin-fixed paraffin-embedded (FFPE) tumor samples to assess the general tumor morphology including the pattern of growth, grade, and specific cyto-architectural characteristics. Immunohistochemistry (IHC) was done on formalin-fixed paraffin-embedded (FFPE) tumor to study proliferation (Ki-67 staining). Primary antibodies against human-specific Ki-67 (Abcam, Cambridge, UK) were used after antigen retrieval with citrate buffer (pH 6.0) in tissue immunohistochemically analysis.
Protein extraction and Western blot
Fifty to seventy micrograms of protein per condition, obtained by tissue lysis, were separated on a Mini-PROTEAN® TGXTM precast gel (BioRad, CA, USA). Proteins were transferred to nitrocellulose membrane and, subsequently incubated with the appropriate primary and secondary antibodies. The immunoreactive bands were visualized using the enhanced chemiluminescent Western blot detection kit (BioRad). To verify equal protein loading, the blots were reprobed with β-actin antibody (Sigma-Aldrich). Images were visualized using the ChemiDocTM MP Imaging System (BioRad).
RNA isolation and next-generation RNA sequencing (NGS)
RNA from biological tissue samples was isolated with the RNeasy Kit (Qiagen, Chatsworth, CA) according to the manufacturer’s instructions. The RNA quality and quantity were verified with a NanoDrop 1000 Spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA). VIB Nucleomics Core (www.nucleomics.be) performed RNA sequencing and processing. In short, the poly-A containing mRNA molecules were purified using poly-T oligo-attached magnetic beads. Samples were sequenced on an Illumina HiSeq 4000 full flow-cell. After preprocessing, reads were aligned to the reference genome of Homo sapiens 38 (GRCh38) and counted. With the EdgeR 3.20.8 package of Bioconductor, generalized linear model (GLM) was fitted and the resulting p-values of the Limma and EdgeR packages were corrected for multiple testing. A gene was considered differentially expressed if a 2 log fold change > + 1 or <-1 and a corrected P<0.05. RNA sequencing data have been submitted to the Gene Expression Omnibus (NCBI) accession number GSE118197.
Gene expression analysis
We applied the cut-off value was set at 2log fold change between -1 to 1 (more than 1.0: up-regulation, -1 to 1: no change, less than -1: down-regulation) and genes related to miRNA in all the xenografts were excluded from further analysis. The data were analyzed using Ingenuity Pathway Analysis (IPA) (www.ingenuity.com) and Webgestalt 2013 (http://www.webgestalt.org/webgestalt_2013/). The core analysis of IPA identifies biological functions and/or diseases and upstream regulators (genes, RNA and proteins) that are most significant to the data set. Hierarchic clustering of the individual samples was done using PermutMatrix program (Version 1.9.3 EN) [12].
Gene expression by RT-qPCR
To validate the RNA-Seq data, the expression level of selected genes was analyzed with RT-qPCR. For this, genes that have low and high expression levels were chosen. One microgram of cellular RNA was reverse transcribed into cDNA using SuperScript II reverse transcriptase and random hexamer primers (Invitrogen/Life Technologies, USA). The PCR reaction was carried out in a mixture that contained appropriate sense- and anti-sense primers and a TaqMan MGB probe in Taq-Man Universal PCR Master Mixture (Applied Biosystems, Foster City, CA, USA). Beta-2-microglobulin was used as housekeeping gene. qRT-PCR amplification and data analysis were performed using the Lightcycler 96 (Roche Applied Science, Penzberg, Germany). Each sample was assayed in duplicate. The ΔΔCq method was used to determine relative gene expression levels.
Relative EMT-score of change in gene expression in response to treatments
A relative EMT score was calculated for each sample based on the Z-score for NGS gene expression (http://www.statisticshowto.com/probability-and-statistics/z-score/). We selected 8 well-established EMT markers. As mesenchymal markers we used SNAI1, SNAI2, ZEB2, TGF-β1 and VIM, as epithelial markers we used: CDH1, CTNNB1 and AKT2. The higher this score is, the more mesenchymal characteristics the sample has. The EMT score corresponds with the hierarchic clustering (see result section).
EMT-score = sum Z - scores (+ SNAI1 + SNAI2 + ZEB2 + TGF-β1 + VIM - CDH1 - CTNNB1 - AKT2)
Statistical analysis
All statistics were performed using SPSS v23 (IBM). Statistical differences between groups was assessed with a Student’s t-test, ANOVA or the Mann-Whitney Rank Sum Test as appropriate. For differences in gene expression assessed by qRT-PCR, ANOVA test with post hoc Tukey’s procedure was used. A P value below 0.05 was considered statistically significant.
Results
Characterization of PDAC patient-derived xenograft models
Establishment and histology
We selected two cancer models with a distinct phenotype (PAC006 and PAC010) from the panel PDAC patient-derived xenograft models (PDTX) that we recently developed [9]. These PDTX lines were established from tissue that was obtained by endoscopic ultrasound (EUS)-guided fine needle biopsies (FNB). For each patient’s tumor sample a histopathological and genetic comparison of pre-graft and post-graft tumor tissues was made (Table 1).
Table 1.
Summary of the characteristics of the patient tumor and corresponding PDTX model*
| Well-moderately differentiated (PAC006) | Poorly differentiated (PAC010) | |
|---|---|---|
| Patient background | Primary tumor biopsy (EUS-FNA) from 85 years old women. | Primary tumor biopsy (EUS-FNA) from 83 years old man. |
| Patient tumor pathology | Pancreatic ductal adenocarcinoma, grows as irregular glandular structures in a desmoplastic stroma & locally, cribriform patterns. Tumor cells have enlarged, atypical and hyperchromatic nuclei. | Pancreatic ductal adenocarcinoma, metastases in liver. Tumor cells have large atypical and pleiomorphic nuclei on a background of necrosis. |
| Treatment history | None. | None. |
| PDTX tumor pathology | Pancreatic ductal adenocarcinoma, irregular glandular structures, cells with atypical nuclei and prominent nucleolus. | Large tumor cells with atypical nuclei and often prominent nucleolus. |
| Genomic profiling | RNA sequencing , Whole exome sequencing, Sequenom MassArray for KRAS, Shallow sequencing (copy number) (only PDX model, not human tumor). | RNA sequencing, Sequenom MassArray for KRAS, Shallow sequencing (copy number), Whole exome sequencing (only PDX model, not human tumor). |
| Oncogene mutation status | KRAS p.G12D, TP53 wildtype. | KRAS p.G12D, KRAS double amplified, TP53 p.R175H, TP53 homozygous deficient. |
Adapted from: Hermans et al., 2016 [9].
No major differences were observed between tumors from the same model over generations while expanding in mice with regard to genetics, cytology and tissue architecture except for a depletion of the human stroma (Figure 1).
Figure 1.
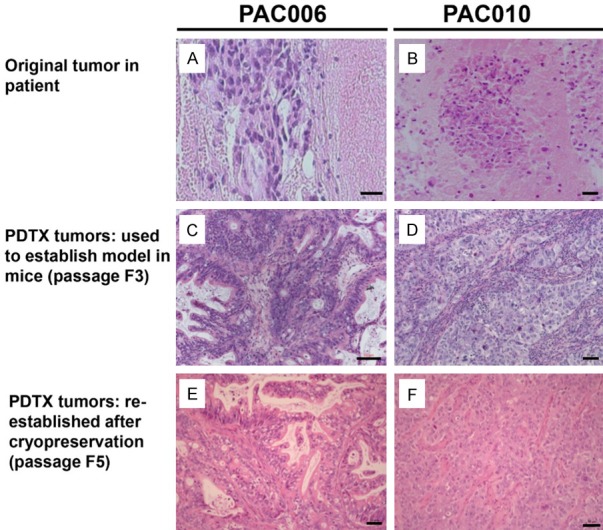
H&E staining of tumor tissue from patients, in mice before cryopreservation (passage 3) and from current experiments (passage 5). Representative H&E staining of PAC006 (A, C, & E) and PAC010 (B, D, & F) are shown here. Morphological we see a clear difference between the tumor obtained from patient PAC006 (A, well/moderately differentiated) and PAC010 (B, poorly differentiated). The characteristics seen in the original tumor tissue are preserved for each model during propagation in mice (C, D) and also after cryopreservation and re-implantation (E, F). Original magnification of histological images is × 20; scale bar 50 µm.
To summarize the characteristics of PAC010: this model originates from a solid tumor with metastasis, the histology showed a poorly differentiated adenocarcinoma with stromal components (fibroblast, myofibroblasts, inflammatory cells, vessels) representing the characteristics of an aggressive pancreatic cancer. The second model (PAC006): the original tumor was only localized in the pancreas, the histology showed glandular morphology and a good differentiation pattern; which all corresponds with a behavioral less aggressive cancer. We therefore could conclude that the models exhibiting faithful replication of the original patient’s tumor.
Tumor growth and stability in experimental animals
After cryopreservation, the PAC006 and PAC010 tumors were re-established for the current experiments by engraftment in a group of experimental animals. Histological assessment confirmed that the tumors from the current passages presented a good conservation of morphological features and differentiation level, comparable to the tumors they originated from (Figure 1).
PAC006 (untreated) has a relatively slow growth, reaching a maximum size of 1000 mm3 after around 36 days following implantation. In contrast, PAC010 tumors in most animals reached a volume greater than 1000 mm3 after approximately 27 days and increased to above the ethical limit within less than a month and therefore required early animal sacrifice. We calculated the tumor volume doubling time: for PAC006 (n=6) this was 5.87 days (SEM ± 0.55) whereas PAC010 (n=6) had a doubling time of 3.60 days (SEM ± 0.33), the difference was statistically significant (P=0.005).
Expression of EMT-related genes and regulators in PDTX models
Tumor tissue from both models were profiled using RNA-sequence (RNA-seq). Based on gene expression quantification, 8282 genes were differentially expressed between the PAC010 and the PAC006 models with 4249 up regulated and 4033 down regulated, using a cutoff of P<0.001 and ≥2-fold change (Figure S1). We focused our analysis on the expression of a panel of EMT related genes (identified by the Ingenuity consortium, see Figure 2A). Between our models, many mesenchymal markers were higher expressed in PAC010 (e.g. ZEB1, STAT3, VIM, and SNAI2) with a reduced expression of epithelial markers (e.g. CDH1 and CTNNB1) in comparison to the well-to-moderately differentiated model PAC006 as illustrated by hierarchic clustering (Figure 2A). These results were confirmed by RT-qPCR on selected EMT markers (Figure 2B). The expression of EMT-related protein markers was assessed by Western blot and we could confirm significantly higher expression of SNAI1, SNAI2 and VIM protein in the PAC010 and a lower expression of CDH1, all observation indicating increased features of increased mesenchymal characteristics in this model (Figure 2C).
Figure 2.
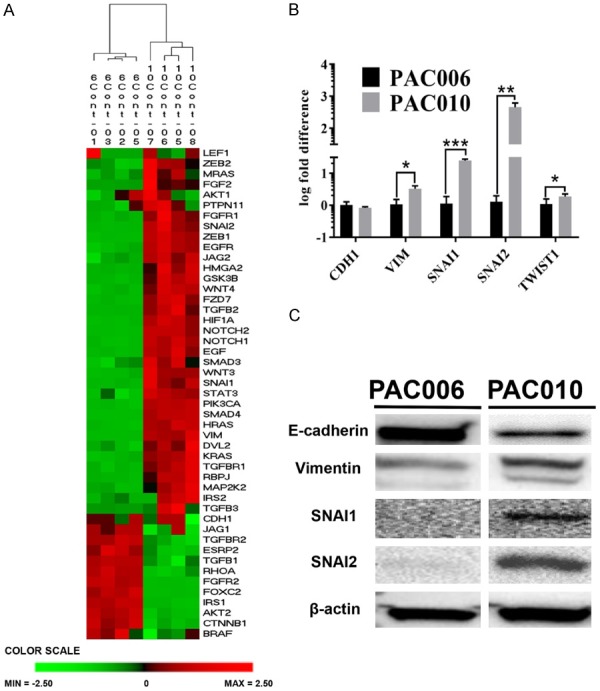
Epithelial-to-Mesenchymal transition characteristics of the PDTX models. (A) The diagram presents the result of a two-way hierarchical clustering of a panel of EMT related genes (from IPA) in the untreated PAC006 and PAC010 models. The color scale at the bottom illustrates the relative level of EMT related gene expression in our models: red, above than the mean Z-score, green, below the mean. This analysis is validated by qRT-PCR (B) and Western blot (C) on selected representative EMT related genes. All experiments were performed at least in 3 biological repeats. qRT-PCR results are given as mean 2log fold difference ± SEM. Statistical significance was assessed using parametric (one-way ANOVA) (*: P<.05, **: P<.01, and ***: P<.001).
We applied QIAGEN’s Ingenuity Pathway Analysis (IPA) on the full RNA sequence dataset to further characterize the (untreated) models (PAC010 vs PAC006) to identify upstream regulators. IPA uses the activation Z-score algorithm to make predictions of activation or inhibition. Accordingly, Table 2A showed the list of top upstream regulators activated in PAC010, which includes growth factors and cytokines that have been reported to be involve in tumor progression and EMT induction. Similarly, functional analysis listed in the top positions cellular functions that directly or indirectly are involved in the process of EMT: cell movement and invasion (Tables 2B, S1).
Table 2.
Top upstream regulators and functions identified by IPA using information from differentially expressed genes of PAC010 vs PAC006
| A. | |||
|
| |||
| Upstream Regulator | Molecule Type | Predicted Activation State* | p-value of overlap |
|
| |||
| TNF | Cytokine | Activated | 1,00E-28 |
| TGFB1 | Growth factor | Activated | 1,70E-26 |
| ESR1 | Ligand-dependent nuclear receptor | Activated | 1,83E-25 |
| ERBB2 | Kinase | Activated | 3,61E-22 |
| SMARCA4 | Transcription regulator | 4,04E-22 | |
| IL1B | Cytokine | Activated | 4,06E-20 |
| TP53 | Transcription regulator | 4,04E-19 | |
| PGR | Ligand-dependent nuclear receptor | 2,86E-18 | |
| SP1 | Transcription regulator | Activated | 3,74E-17 |
| VEGF | Group | Activated | 5,67E-17 |
| NFkB (complex) | Complex | Activated | 6,48E-17 |
| CTNNB1 | Transcription regulator | 7,20E-17 | |
| OSM | Cytokine | Activated | 1,06E-16 |
| Estrogen receptor | Group | Inhibited | 3,64E-16 |
| TP63 | Transcription regulator | Activated | 8,60E-16 |
| HNRNPA2B1 | Other | 1,42E-15 | |
|
| |||
| B. | |||
|
| |||
| Diseases or Functions Annotation | p-Value | Predicted Activation State | #Molecules |
|
| |||
| Cell movement | 2,58E-59 | Increased | 1348 |
| Migration of cells | 2,53E-54 | Increased | 1202 |
| Invasion of cells | 4,17E-35 | Increased | 591 |
| Cell movement of tumor cell lines | 1,89E-33 | Increased | 598 |
| Invasion of tumor cell lines | 2,66E-30 | Increased | 472 |
| Migration of tumor cell lines | 3,47E-29 | Increased | 496 |
| Cell movement of blood cells | 4,47E-21 | Increased | 526 |
| Migration of blood cells | 6,62E-21 | Increased | 521 |
| Leukocyte migration | 8,00E-21 | Increased | 520 |
| Cell movement of cancer cells | 2,27E-18 | Increased | 104 |
| Cell movement of leukocytes | 1,12E-17 | Increased | 456 |
blank = IPA analysis did not assign activation status (both up and down regulatory factors).
A) List of top 16 upstream regulatory molecules, their predicted activation state and p-value; B) List of top 11 disease and cellular functions.
Molecules: number of genes from differentially expressed gene list linked to disease or function.
The findings indicate that our tumor models are different in gene expression and growth.
Response to therapy in PDAC models
Tumor growth during treatment
After the characterization of our models, we tested, as proof-of-principle, two different classes of drugs: gemcitabine (GEM, the standard chemotherapy for patients with PDAC) and the HIF-inhibitor and antibacterial compound acriflavine (ACF), and characterized the tumor response for growth and molecular factors.
Gemcitabine treatment resulted in a significant shrinking of the tumor volume after 10 days in the PAC006 model (P<0.05) and after 7 days in the PAC010 model (P<0.01) (Figure 3). Acriflavine, as single compound, has in the early phase of treatment little effect on tumor growth but we could observe in PAC006 a trend towards stabilization with prolonged treatment. In the PAC010, no effect of ACF on growth was observed. Important to note, as the tumors in mice bearing PAC010 increased rapidly in size, the experiment had to be stopped after 10 days for ethical/animal welfare reasons thereby limiting the statistical evaluation. For both models, during the treatment period, there was no significant effect on body weight of the mice or other side effects observed related to the treatments (Figure 3C).
Figure 3.

Tumor development over time under treatment in the different PDTX pancreatic cancer models. (A, D) Tumors volume/time growth curves, (C, F) mice body weight/time curve and (B, E) representative pictures of the tumors at the end of the experiment. Results for PAC006 are shown in (A-C) and for PAC010 in (D-F). Each of the groups consisted of eight animals. A significant shrinkage of tumor volume was observed after 10 days in PAC006 (P<0.05) and in PAC010 (P<0.01) after 7 days of GEM treatment versus their respective controls. Results are given as mean ± SEM.
Effect of treatment on tumor histology
For the present study, additional histological characterization was performed (Figure 4). In tumors treated with ACF, the H&E staining pattern is not distinct from that seen in the untreated models. In general, both PAC006 and PAC010 under GEM treatment showed highly pleomorphic nuclei and eosinophilic cytoplasm that indicates a degenerative phenotype.
Figure 4.
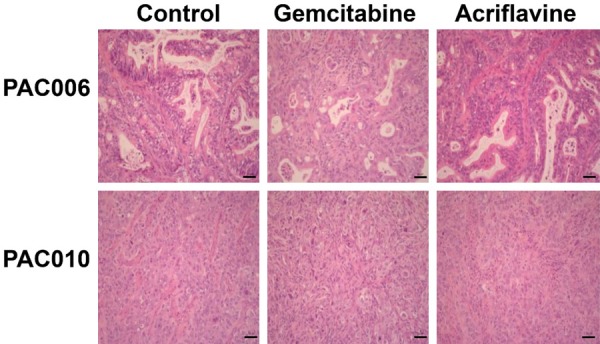
Representative H&E stain of tumor tissues at the end of the treatment. Tumor tissue was obtained from the mice at the end of the treatment period or their respective controls. Routine H&E staining was performed and histology was visualized by microscope. Original magnification of histological images is × 20; scale bar 50 µm.
Changes in the expression of EMT related genes under drug treatment
RNA-sequencing data showed that the overall number of genes of which the expression was significantly changed by drug treatment was high for GEM treatment whereas ACF had a much more limited effect (in both models ca 2000 for GEM vs 10-20 for ACF, Figure S1). Focusing on EMT related genes, we observed that hierarchic clustering could separate the GEM treated animals of PAC006 from the other samples for that model (untreated and ACF treated animals) (Figure 5B) and the same was true for PAC010 (Figure 5C). The shift to EMT was quantified by calculating the relative EMT score from a panel of EMT marker genes. What is interesting to note, and with possible clinical implications, is that GEM treatment of the moderate-well differentiated/less aggressive PAC006 cells not only killed a large fraction of the tumor (Figure 3) but also induced a shift to a more EMT-like pattern for the surviving cells as illustrated in Figure 5A. Importantly, this shift of the EMT score to a more mesenchymal phenotype after gemcitabine treatment for the PAC006 was statistically significant (P<0.05).
Figure 5.
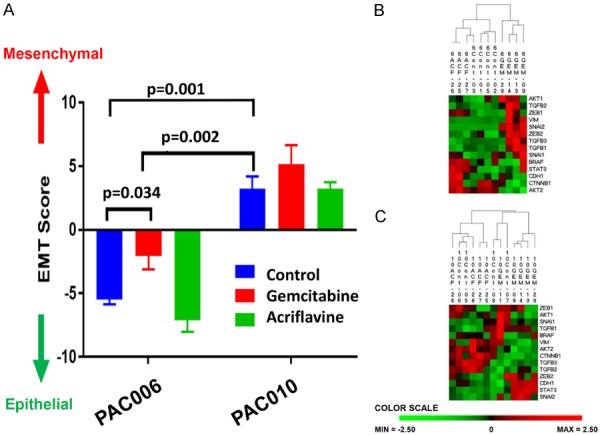
Effect of treatment on EMT gene expression. Next Gen sequencing data were analyzed in detail. (A) To quantify Epithelial vs Mesenchymal phenotype of the tumors at the end of treatment we used a representative 8-gene relative EMT score (see materials and methods). Differences within a single model were statistically assayed with one-way ANOVA-test, between untreated PAC006 and PAC010 a t-test was used. (B) Heat map presentation of two-way hierarchical clustering analysis of prototype EMT genes for PAC006. The samples that had received gemcitabine clearly separated from the untreated control and ACF treated samples. (C) In PAC010, the animals that received GEM clustered together and the most of the ACF and control samples formed the second cluster. Most of the mesenchymal markers were up-regulated. The color scale illustrates the relative level of EMT related gene expression in our models: red, above than the mean Z-score; green, below the mean.
Looking at the processes involved in this EMT-shift, IPA analysis identified for PAC006 after GEM-treatment the top regulatory molecules (Table 3A). In addition, functional annotation by IPA listed in the top a high enrichment of processes related to cellular movement (Table 3B). Both analyses, looking at full-scale RNA-expression data, underline that GEM-treatment could shift the tumor to a higher metastatic behavior. For PAC010, where the cells already have a high expression of EMT markers, no statistical significant shift to increased EMT profile was observed after GEM-treatment (Figure 5A and 5C). IPA analysis for PAC010 after GEM treatment (Table 4A and 4B) showed the same trend as seen in PAC006 but the effects were statistically less pronounced and that probably originated from the already high EMT score of the untreated PAC010 cells. An effect of ACF on EMT in either of the models was not observed.
Table 3.
Top upstream regulators and functions identified by IPA using information from differentially expressed genes of PAC006 in response to GEM treatment
| A. | |||
|
| |||
| Upstream Regulator | Molecule Type | Predicted Activation State | p-value of overlap |
|
| |||
| IFNG | Cytokine | Activated | 3,20E-38 |
| TNF | Cytokine | Activated | 1,72E-37 |
| IL1B | Cytokine | Activated | 9,87E-35 |
| NFkB (complex) | Complex | Activated | 2,89E-29 |
| RELA | Transcription regulator | Activated | 4,27E-28 |
| IL1A | Cytokine | Activated | 5,36E-25 |
| TGFB1 | Growth factor | Activated | 1,55E-24 |
| IL6 | Cytokine | Activated | 2,23E-24 |
| IL1 | Group | Activated | 2,46E-21 |
| IL27 | Cytokine | Activated | 6,55E-21 |
|
| |||
| B. | |||
|
| |||
| Diseases or Functions Annotation | p-Value | Predicted Activation State | #Molecules |
|
| |||
| Cell movement of blood cells | 1,14E-35 | Increased | 128 |
| Leukocyte migration | 1,57E-35 | Increased | 127 |
| Cell movement of phagocytes | 1,05E-30 | Increased | 92 |
| Cell movement of myeloid cells | 1,79E-29 | Increased | 90 |
| Migration of phagocytes | 1,91E-29 | Increased | 60 |
| Cell movement of leukocytes | 1,29E-28 | Increased | 108 |
| Recruitment of cells | 5,53E-27 | Increased | 66 |
| Cell movement of granulocytes | 6,48E-26 | Increased | 67 |
| Migration of cells | 2,57E-25 | Increased | 182 |
| Cell movement | 2,69E-25 | Increased | 197 |
A) List of top 10 upstream regulatory molecules, their predicted activation state and p-value; B) List of top 10 diseases and cellular functions detected
Molecules: number of genes from differentially expressed gene list linked to disease or function.
Table 4.
Top upstream regulators and functions identified by IPA using information from differentially expressed genes of PAC010 in response to GEM treatment
| A. | |||
|
| |||
| Upstream Regulator | Molecule Type | Predicted Activation State* | p-value of overlap |
|
| |||
| CBX5 | Transcription regulator | Inhibited | 2,43E-13 |
| STAT5A | Transcription regulator | 5,04E-11 | |
| VEGF | Group | 3,04E-08 | |
| S100A9 | Other | Activated | 6,06E-08 |
| IL1B | Cytokine | 4,25E-07 | |
| SMARCA4 | Transcription regulator | 5,12E-07 | |
| TNF | Cytokine | Activated | 7,52E-07 |
| CHUK | Kinase | Activated | 1,25E-06 |
| JUN | Transcription regulator | Activated | 2,00E-06 |
| TREM1 | Transmembrane receptor | Activated | 2,89E-06 |
|
| |||
| B. | |||
|
| |||
| Diseases or Functions Annotation | p-Value | Predicted Activation State* | #Molecules |
|
| |||
| Cell movement of phagocytes | 1,27E-05 | 32 | |
| Leukocyte migration | 1,85E-05 | 44 | |
| Cellular infiltration by phagocytes | 2,66E-05 | 19 | |
| Cell movement of myeloid cells | 2,91E-05 | 31 | |
| Cellular infiltration by granulocytes | 3,63E-05 | 16 | |
| Attraction of blood cells | 3,88E-05 | 8 | |
| Cellular infiltration by myeloid cells | 4,40E-05 | 20 | |
| Cell movement of leukocytes | 5,29E-05 | 39 | |
| Attraction of granulocytes | 8,51E-05 | 5 | |
| Transmigration of leukocytes | 8,67E-05 | 11 | |
blank = IPA analysis did not assign activation status (both up and down regulatory factors).
Molecules: number of genes from differentially expressed gene list linked to disease or function.
A) List of top 10 upstream regulatory molecules, their predicted activation state and p-value; B) List of top 10 diseases and cellular functions detected # Molecules: number of genes from differentially expressed gene list linked to disease or function.
Effect of drug treatment on cell proliferation and survival pathways
The effects of treatments on cell proliferation and survival was investigated with key molecular markers. Cellular proliferation in the models and the effect of treatments was evaluated by Ki-67 staining. In the poorly differentiated tumor model (PAC010), the mean number of Ki-67 positive stained cells was relatively higher than in the moderate-to-well differentiated tumor type (PAC006). Within both models, no significant difference in labeling was found between untreated and acriflavine treated groups. In contrast, the number of Ki-67 stained cells was significantly reduced (P<0.001) in GEM treated groups (Figure 6A, 6B).
Figure 6.
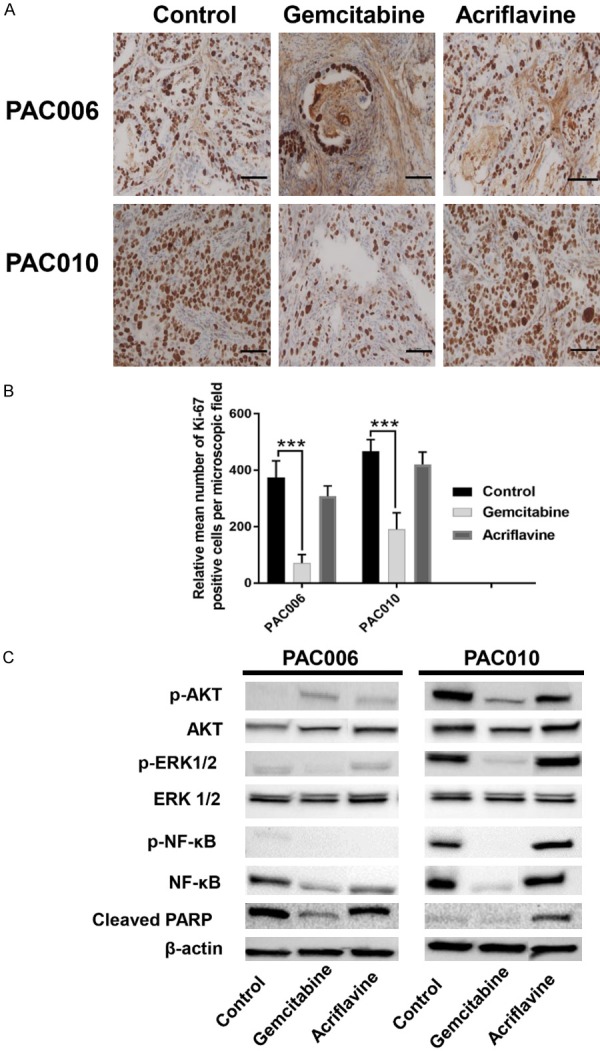
Effect of drug treatment on cell proliferation and survival pathways. (A) Immunohistochemistry staining for Ki-67 shows nuclear positivity in representative sections of control and drug treated PDTX mice, bearing PAC006 or PAC010 models. Original magnification of images: × 20; scale bar 50 µm. (B) The quantification of nuclear Ki-67 immunostaining of tumor cells was done manually on sections of four different animals per condition. The number of Ki-67 stained cells is given as mean ± SEM (*: P<.05, **: P<.01, and ***: P<.001). Statistical significance was assessed using parametric (one-way ANOVA). (C) Representative Western blots of the two tumor models showing expression of phosphorylated and total Akt, phosphorylated and total ERK (42/44), phosphorylated and total NF-kB65 and cleaved PARP with β-actin as reference.
Similarly, the cancer cell survival in our models and the effects of treatments were evaluated by selected pro-survival factors (p-ERK1/2, p-NF-kB65 and p-AKT) and pro-apoptotic factor (cleaved PARP) [13,14]. Our result indicated a clear association between activation of pro-survival pathways and suppression of pro-apoptotic molecules with increased tumor proliferation and de-differentiation. In the PAC010 model, there is higher activation of pro-survival molecules and suppression of pro-apoptotic factors compared to the PAC006 model. Upon treatment with GEM, we found the activation of the main pro-survival molecule (p-AKT) and suppression of pro-apoptotic molecule (cleaved PARP) in the PAC006 model (Figure 6C). In addition, hierarchic clustering evaluation of the apoptosis pathway genes (KEGG hsa:04210, Figure S2) also indicates that GEM treatment shifts the gene expression pattern significantly in both models.
Discussion
From the clinical perspective, PDAC offers many challenges mainly related to late diagnosis, rapid disease progression and drug resistance, all leading to a very high mortality. Central to this are not only the characteristics of the cancer cells but also that of the tumor microenvironment (stromal components) and their specific interplay that determines PDAC growth and progression. Hence, the use of appropriate in vivo models, that can mimic the tumor microenvironment as is found in patients, remains fundamental. What makes our PDTX models [9] unique is that they were developed from tissue obtained by EUS, a technique applied for tumors that are not eligible for surgical resection, which is the big majority of up to 85%. Studies in this group of patients are therefore presently limited [10,15] and using our technique we could select untreated tumors and develop them into two behavioral different models in contrast to genetically engineered mouse or cell line models.
As we previously reported, during expansion the tumors showed no major changes in histopathological characterization or mutational status, except for the depletion of human stromal content. After storage, all tumor characteristics were in agreement with the initial observations in the patients. This agreement was also reported in other studies on PDAC-PDTX [7,10,16] but this confirmation of stability remains essential before any further use of the models. The differences in gene expression we found between the PAC006 and PAC010 model indicates that the PAC010 resembles a highly metastatic tumor, with a mesenchymal phenotype and high expression of human vimentin protein, one of the main EMT markers. Our models can be classified into two distinct molecular subtypes using the PDAssign gene set: PAC006 resembles the classical subclass and PAC010 the quasi-mesenchymal subclass (with reduced disease free and overall survival) [17], which is in agreement with their original behavior.
Until now, full transcriptome analysis by RNA-sequencing following drug treatment has not been reported for EUS-derived PDAC-PDTX. In the present study we characterized at the molecular level our models and we investigated specifically what we had observed previously in vitro on Epithelial-to-Mesenchymal Transition (EMT), tumor microenvironment and tumor aggression [11]. We find in our models that treatment with gemcitabine resulted in a significant reduction of tumor size and the cell proliferation. Morphological we see highly pleomorphic nuclei and eosinophilic cytoplasm. This coincided with the reduction of Ki-67 staining, fitting with the degenerative status of a large fraction of the cells under treatment. This is similar to findings following neoadjuvant therapy in PDAC [18] or in rectal cancer [19].
There is a lack of in vivo studies on EMT and tumor behavior. The majority of these studies are cell culture based induction of EMT and characterizing the cells by their loss of epithelial markers (E-cadherin, claudin, occludin) and gain of mesenchymal markers (increased expression of N-cadherin, vimentin, and transcription factors such as SMAD, TWIST, SNAI1 and ZEB1) [11,20,21]. We found that treating PAC010 and in particular PAC006 with gemcitabine could upregulate mesenchymal markers (SNAI2, TWIST, VIM etc). Furthermore, even though the majority of the tumor mass had shrunk after gemcitabine treatment, our observations show that survival pathways were activated particularly in well-to-moderately differentiated model (PAC006) and that the residual tumor cells are shifted to a more EMT-like (Figure 5), more aggressive phenotype (migration and drug resistance, Table 3). In this process, the interplay between the natural tumor with its microenvironment must have been involved. This could indicate, in the patient setting, a therapy escape after a period of initial response. Apoptotic evasion is considered as one of the main causes of chemotherapeutic and radiotherapeutic resistance that characterizes most aggressive tumors [22]. We found that GEM has an effect on several survival pathways as well as on the apoptotic process (with a reduction of PARP-cleaving), this combination argues in favor of treatment evasion. The complexity of the apoptosis process with respect to GEM response requires additional studies to determine how this can be exploited in PDAC treatment. Many studies also have associated drug resistance cells with the acquisition of an aggressive EMT features [11,23-25]. Even though, abundant evidence supports that EMT is actively involved in tumor metastasis, invasion and resistance to treatment, recently contradicting ideas have emerged that tumor cells may disseminate without switching to a mesenchymal phenotype [26,27]. Other studies indicate that signals coming from the stroma dominate EMT development, impact cancer cell progression and can as well mediates drug resistance [28,29]. Our results are in agreement with previous report on the relevance of EMT [30,31] and tumor stromal factors like IL-6, TGF-β1, TNF [32-34] that are potential involved in pancreatic cancer progression. To understand the contribution of the microenvironment on EMT and cancer progression, the PDTX models offer good insight into important aspects. In different studies it was shown that, the human cancer stroma, included in the implanted tumor pieces, is rapidly replaced by murine stroma and that after three to five passages this human stroma is almost depleted. In a PDAC tumor as it develops in PDTX, the original well-matured human stromal that has developed over months or years is then replaced with immature mice stroma and that will not have the same physical properties/shielding efficiency. Also: -the formation of new murine stroma probably results in changes in paracrine regulation of the tumor that may limit the effect of agents directed against the tumor mass [35,36]. Not with standing, PDTX is the model to study the interaction of the stroma and the tumor, signaling and the innate immune system.
It was previously shown by us in vitro that acriflavine (ACF) is a potent EMT inhibitor and also can restore drug sensitivity in cell models of mesenchymal differentiation upon acquired gemcitabine resistance [11]. In the present study, we have found that ACF alone did not show a big impact on tumor growth in both PDTX models. This can be related to the short duration of treatment and the way the drug was administered considering its chemical properties. Previously, multiple in vivo mice experiment on colorectal, lung, hepatocellular, prostate, and glioblastoma cancer revealed ACF reduced tumor growth, macrophage infiltration, intratumoral expression of angiogenic factor and tumor vasculature [37-40]. We still think further studies in different mice models, tumor types and different ways of drug administration need to be conducted to explore the ability of ACF to inhibit tumor growth and progression as a single agent and/or in combination with other drugs.
In general: we need to be determined how representative our models are for the different clinical presentation of PDAC. If confirmed that a mild disease can become, under treatment, more aggressive (like we have seen under gemcitabine) then this response needs to be taken into account in therapy.
Conclusions
We have characterized two behavioral different human pancreatic ductal adenocarcinoma PDTX models from EUS-obtained untreated tumor tissues. We investigated EMT gene and protein expression of the tumor in a setting of tumor microenvironment and demonstrated that EMT was most pronounced in the poorly differentiated model. Drug treatment with gemcitabine shifts the gene expression to a more aggressive phenotype, which is of translational and clinical relevance. We propose that PDTX is the model to study the interaction of the stroma and the tumor, cell signaling and the innate immune system all to improve treatment options.
Acknowledgements
The authors wish to thank Dr E Hermans and Profs S van der Merwe and F Amant and the members of TRACE (TRACE: a patient-derived tumor xenograft platform (PDX) in translational cancer research at KU Leuven, Belgium) for their excellent work in the development of the EUS-PDTX models. We also want to thank the VIB Nucleomics Core facility (VIB institutional service facility, a life sciences research institute in Flanders, at KU Leuven, Belgium (Drs R. Janky, S. Plaisance) for bioinformatics support. CV holds a mandate as Senior Clinical Investigator of the Research Foundation-Flanders (Belgium) (FWO). This study was partly supported by a research grant from “Kom op tegen Kanker” Belgium and VUYLSTEKE-FLIPTS FONDS LEVERKANKER.
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Neesse A, Patrick MP, Frese KK, Feig C, Cook N, Jacobetz MA, Lolkema MP, Buchholz M, Olive KP, Gress TM, Tuveson DA. Stromal biology and therapy in pancreatic cancer. Gut. 2011;60:861–868. doi: 10.1136/gut.2010.226092. [DOI] [PubMed] [Google Scholar]
- 2.Olive KP. Stroma, stroma everywhere (Far More Than You Think) Clin Cancer Res. 2015;21:3366–3368. doi: 10.1158/1078-0432.CCR-15-0416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Heinemann V, Reni M, Ychou M, Richel DJ, Macarulla T, Ducreux M. Tumour-stroma interactions in pancreatic ductal adenocarcinoma: rationale and current evidence for new therapeutic strategies. Cancer Treat Rev. 2014;40:118–128. doi: 10.1016/j.ctrv.2013.04.004. [DOI] [PubMed] [Google Scholar]
- 4.Nielsen MF, Mortensen MB, Detlefsen S. Key players in pancreatic cancer-stroma interaction: cancer-associated fibroblasts, endothelial and inflammatory cells. World J Gastroenterol. 2016;22:2678–2700. doi: 10.3748/wjg.v22.i9.2678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maupin KA, Sinha A, Eugster E, Miller J, Ross J. Glycogen expression alterations associated with pancreatic cancer Epithelial-Mesenchymal Transition in complementary model systems. PLoS One. 2010;5:e13002. doi: 10.1371/journal.pone.0013002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Logsdon CD, Arumugam T, Ramachandran V. Animal models of gastrointestinal and liver diseases. The difficulty of animal modeling of pancreatic cancer for preclinical evaluation of therapeutics. Am J Physiol Gastrointest Liver Physiol. 2015;309:283–291. doi: 10.1152/ajpgi.00169.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rubio-Viqueira B, Jimeno A, Cusatis G, Zhang X, Iacobuzio-Donahue C, Karikari C, Shi C, Danenberg K, Danenberg PV, Kuramochi H, Tanaka K, Singh S, Salimi-Moosavi H, Bouraoud N, Amador ML, Altiok S, Kulesza P, Yeo C, Messersmith WA, Eshleman J, Hruban RH, Maitra A, Hidalgo M. An in vivo platform for translational drug development in pancreatic cancer. Clin Cancer Res. 2006;12:4652–4661. doi: 10.1158/1078-0432.CCR-06-0113. [DOI] [PubMed] [Google Scholar]
- 8.Tentler JJ, Tan AC, Weekes CD, Jimeno A, Leong S, Pitts TM, Arcaroli JJ, Messersmith WA, Eckhardt SG. Patient-derived tumour xenografts as models for oncology drug development. Nat Rev Clin Oncol. 2012;9:338–350. doi: 10.1038/nrclinonc.2012.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hermans E, Van der Merwe SW, Depreeuw J, Dekervel J, Radaelli E, Roskams T, van Pelt J, Topal B, Verslype C, Prenen H, Van Steenbergen WV, Nevens F, Lambrechts D, Amant F. Successful application of endoscopic ultrasound-guided fine needle biopsy to establish pancreatic patient-derived tumor xenografts: a pilot study. Endoscopy. 2016;48:1016–1022. doi: 10.1055/s-0042-113597. [DOI] [PubMed] [Google Scholar]
- 10.Berry W, Algar E, Kumar B, Desmond C, Swan M, Jenkins BJ, Croagh D. Endoscopic ultrasound-guided fine-needle aspirate-derived preclinical pancreatic cancer models reveal panitumumab sensitivity in KRAS wild-type tumors. Int J Cancer. 2017;140:2331–2343. doi: 10.1002/ijc.30648. [DOI] [PubMed] [Google Scholar]
- 11.Dekervel J, Bulle A, Windmolders P, Lambrechts D, Van Cutsem E, Verslype C, van Pelt J. Acriflavine inhibits acquired drug resistance by blocking the Epithelial-to-Mesenchymal Transition and the unfolded protein response. Transl Oncol. 2017;10:59–69. doi: 10.1016/j.tranon.2016.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Caraux G, Pinloche S. PermutMatrix: a graphical environment to arrange gene expression profiles in optimal linear order. Bioinformatics. 2005;21:1280–1281. doi: 10.1093/bioinformatics/bti141. [DOI] [PubMed] [Google Scholar]
- 13.Hoesel B, Schmid JA. The complexity of NF-κB signaling in inflammation and cancer. Mol Cancer. 2013;12:86. doi: 10.1186/1476-4598-12-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yamamoto S, Tomita Y, Hoshida Y, Morooka T, Nagano H, Dono K, Umeshita K, Sakon M, Ishikawa O, Ohigashi H, Nakamori S, Monden M, Aozasa K. Prognostic significance of activated Akt expression in pancreatic ductal adenocarcinoma. Clin Cancer Res. 2014;10:2846–2850. doi: 10.1158/1078-0432.ccr-02-1441. [DOI] [PubMed] [Google Scholar]
- 15.Bian B, Bigonnet M, Gayet O, Loncle C, Maignan A, Gilabert M, Moutardier V, Garcia S, Turrini O, Delpero JR, Giovannini M, Grandval P, Gasmi M, Ouaissi M, Secq V, Poizat F, Nicolle R, Blum Y, Marisa L, Rubis M, Raoul JL, Bradner JE, Qi J, Lomberk G, Urrutia R, Saul A, Dusetti N, Iovanna J. Gene expression profiling of patient-derived pancreatic cancer xenografts predicts sensitivity to the BET bromodomain inhibitor JQ1: implications for individualized medicine efforts. EMBO Mol Med. 2017;9:482–497. doi: 10.15252/emmm.201606975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pergolini I, Morales-Oyarvide V, Mino-Kenudson M, Honselmann KC, Rosenbaum MW, Nahar S, Kem M, Ferrone CR, Lillemoe KD, Bardeesy N, Ryan DP, Thayer SP, Warshaw AL, Fernández-Del Castillo C, Liss AS. Tumor engraftment in patient derived xenografts of pancreatic ductal adenocarcinoma is associated with adverse clinicopathological features and poor survival. PLoS One. 2017;12:e0182855. doi: 10.1371/journal.pone.0182855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Janky R, Binda MM, Allemeersch J, Van den Broeck A, Govaere O, Swinnen JV, Roskams T, Aerts S, Topal B. Prognostic relevance of molecular subtypes and master regulators in pancreatic ductal adenocarcinoma. BMC Cancer. 2016;16:632. doi: 10.1186/s12885-016-2540-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pai RK, Pai RK. Pathologic assessment of gastrointestinal tract and pancreatic carcinoma after neoadjuvant therapy. Modern Pathol. 2018;31:4–23. doi: 10.1038/modpathol.2017.87. [DOI] [PubMed] [Google Scholar]
- 19.Debucquoy A, Libbrecht L, Roobrouck V, Goethals L, McBridee W, Haustermans K. Morphological features and molecular markers in rectal cancer from 95 patients included in the european organization for research and treatment of cancer 22921 trial: prognostic value and effects of preoperative radio (chemo) therapy. Eur J Cancer. 2008;44:791–797. doi: 10.1016/j.ejca.2008.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hotz B, Arndt M, Dullat S, Bhargava S, Buhr HJ, Hotz HG. Epithelial to mesenchymal transition: expression of the regulators snail, slug, and twist in pancreatic cancer. Clin Cancer Res. 2007;13:4769–4776. doi: 10.1158/1078-0432.CCR-06-2926. [DOI] [PubMed] [Google Scholar]
- 21.Chen S, Chen J, Zhang J, Chen H, Yan M, Huang L, Tian Y, Chen Y, Wang Y. Hypoxia induces TWIST-activated epithelial-mesenchymal transition and proliferation of pancreatic cancer cells in vitro and in nude mice. Cancer Lett. 2016;383:73–84. doi: 10.1016/j.canlet.2016.09.027. [DOI] [PubMed] [Google Scholar]
- 22.Garajová I, Le Large TY, Frampton AE, Rolfo C, Voortman J, Giovannetti E. Molecular mechanisms underlying the role of microRNAs in the chemoresistance of pancreatic cancer. Biomed Res Int. 2014;2014:678401. doi: 10.1155/2014/678401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang Z, Li Y, Kong D, Banerjee S, Ahmad A, Azmi AS, Ali S, Abbruzzese JL, Gallick GE, Sarkar FH. Acquisition of epithelial-mesenchymal transition phenotype of gemcitabine-resistant pancreatic cancer cells is linked with activation of the notch-signaling pathway. Cancer Res. 2009;69:2400–2407. doi: 10.1158/0008-5472.CAN-08-4312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tsukasa K, Ding Q, Yoshimitsu M, Miyazaki Y, Matsubara S, Takao S. Slug contributes to gemcitabine resistance through epithelial mesenchymal transition in CD133+ pancreatic cancer cells. Human Cell. 2015;28:167–174. doi: 10.1007/s13577-015-0117-3. [DOI] [PubMed] [Google Scholar]
- 25.Samulitis BK, Pond KW, Pond E, Cress AE, Patel H, Wisner L, Patel C, Dorr RT, Landowski TH. Gemcitabine resistant pancreatic cancer cell lines acquire an invasive phenotype with collateral hypersensitivity to histone deacetylase inhibitors. Cancer Biol Ther. 2015;16:43–51. doi: 10.4161/15384047.2014.986967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Banyard JL, Bielenberg DR. The role of EMT and MET in cancer dissemination. Connect Tissue Res. 2015;56:403–413. doi: 10.3109/03008207.2015.1060970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zheng X, Carstens JL, Kim J, Scheible M, Kaye J, Sugimoto H, Wu CC, LeBleu VS, Kalluri R. EMT program is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature. 2015;527:525–530. doi: 10.1038/nature16064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang Z, Li Y, Ahmad A, Azmi AS, Banerjee S, Kong D, Sarkar FH. Targeting notch signaling pathway to overcome drug-resistance for cancer therapy. Biochim Biophys Acta. 2010;1806:258–267. doi: 10.1016/j.bbcan.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Khalafalla FG, Khan MW. Inflammation and epithelial-mesenchymal transition in pancreatic ductal adenocarcinoma: fighting against multiple opponents. Cancer Growth Metastasis. 2017;10:1–13. doi: 10.1177/1179064417709287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nishioka R, Itoh S, Gui T, Gai Z, Oikawa K, Kawai M, Tani M, Yamaue H, Muragaki Y. SNAIL induces epithelial-to-mesenchymal transition in a human pancreatic cancer cell line (BxPC3) and promotes distant metastasis and invasiveness in vivo. Exp Mol Pathol. 2010;89:149–157. doi: 10.1016/j.yexmp.2010.05.008. [DOI] [PubMed] [Google Scholar]
- 31.Krebs AM, Mitschke J, Losada LM, Schmalhofer O, Boerries M, Busch H, Boettcher M, Mougiakakos D, Reichardt W, Bronsert P, Brunton VG, Pilarsky C, Winkler TH, Brabletz S, Stemmler MP, Brabletz T. The EMT-activator Zeb1 is a key factor for cell plasticity and promotes metastasis in pancreatic cancer. Nat Cell Biol. 2017;19:518–529. doi: 10.1038/ncb3513. [DOI] [PubMed] [Google Scholar]
- 32.Baran B, Bechyne I, Siedlar M, Szpak K, Mytar B, Sroka J, Laczna E, Madeja Z, Zembala M, Czyz J. Blood monocytes stimulate migration of human pancreatic carcinoma cells in vitro: the role of tumour necrosis factor-alpha. Eur J Cell Biol. 2009;88:743–752. doi: 10.1016/j.ejcb.2009.08.002. [DOI] [PubMed] [Google Scholar]
- 33.Huang L, Hu B, Ni J, Wu J, Jiang W, Chen C, Yang L, Zeng Y, Wan R, Hu G, Wang X. Transcriptional repression of SOCS3 mediated by IL-6/STAT3 signaling via DNMT1 promotes pancreatic cancer growth and metastasis. J Exp Clin Cancer Res. 2016;35:27. doi: 10.1186/s13046-016-0301-7. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 34.Tjomsland V, Sandnes D, Pomianowska E, Cizmovic ST, Aasrum M, Brusevold IJ, Christoffersen T, Gladhaug IP. The TGFβ-SMAD3 pathway inhibits IL-1α induced interactions between human pancreatic stellate cells and pancreatic carcinoma cells and restricts cancer cell migration. J Exp Clin Cancer Res. 2016;35:122. doi: 10.1186/s13046-016-0400-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Erkan M, Reiser-Erkan C, Michalski CW, Kleeff J. Tumor microenvironment and progression of pancreatic cancer. Exp Oncol. 2010;32:128–131. [PubMed] [Google Scholar]
- 36.Hidalgo M, Amant F, Biankin AV, Budinská E, Byrne AT, Caldas C, Clarke RB, de Jong S, Jonkers J, Mælandsmo GM, Roman-Roman S, Seoane J, Trusolino L, Villanueva A. Patient-derived xenograft models: an emerging platform for translational cancer research. Cancer Discov. 2014;4:998–1013. doi: 10.1158/2159-8290.CD-14-0001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee K, Zhang H, Qiana DZ, Rey S, Liuc JO, Semenza GL. Acriflavine inhibits HIF-1 dimerization, tumor growth, and vascularization. PNAS. 2009;106:7910–17915. doi: 10.1073/pnas.0909353106. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 38.Lee CJ, Yue CH, Lin YJ, Lin YY, Kao SH, Liu JY, Chen YH. Antitumor activity of acriflavine in lung adenocarcinoma cell line A549. Anticancer Res. 2014;34:6467–6472. [PubMed] [Google Scholar]
- 39.Shay JE, Imtiyaz HZ, Sivanand S, Durham AC, Skuli N, Hsu S, Mucaj V, Eisinger-Mathason TS, Krock BL, Giannoukos DN, Simon MC. Inhibition of hypoxia-inducible factors limits tumor progression in a mouse model of colorectal cancer. Carcinogenesis. 2014;35:1067–1077. doi: 10.1093/carcin/bgu004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Voss DM, Spina R, Carter DL, Lim KS, Jeffery CJ, Bar EE. Disruption of the monocarboxylate transporter-4-basigin interaction inhibits the hypoxic response, proliferation, and tumor progression. Sci Rep. 2017;7:4292. doi: 10.1038/s41598-017-04612-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.