

Abstract
Chronic kidney disease (CKD) is commonly characterized by proteinuria and leads to progressive glomerulosclerosis and tubulointerstitial fibrosis. Accumulating evidence implicates mitochondrial dysfunction including reactive oxygen species (ROS) overproduction in the pathogenesis of CKD. Mitochondrial function and ROS production are regulated by mitochondrial uncoupling. Niclosamide ethanolamine salt (NEN) is a mild mitochondrial uncoupler, which reduces urinary albumin excretion in mice with diabetic kidney disease. However, its role in nondiabetic kidney disease has not been investigated. Here we show that NEN exerts renoprotective effects in adriamycin induced nondiabetic kidney disease. It reduces urinary protein excretion, restores podocyte function, ameliorates renal pathological injury, and decreases the excretion of the urinary tubular injury biomarkers NGAL and Kim-1. Specifically, NEN uncouples isolated kidney mitochondria, and dose-dependently decreases the renal production and urinary excretion of H2O2. Moreover, NEN increases catalase and PGC-1α expression, which might accelerate H2O2 scavenging. The results of this study provide the first evidence that NEN protects kidney in nondiabetic kidney disease by regulating redox balance.
Keywords: Niclosamide ethanolamine salt, adriamycin nephropathy, mitochondria, redox balance
Introduction
The global incidence and prevalence of chronic kidney disease (CKD) and end-stage kidney disease have increased significantly [1]. CKD is commonly characterized by proteinuria and leads to progressive glomerulosclerosis and tubulointerstitial damage. Therefore, drugs that reduce proteinuria are urgently needed.
Accumulating evidence implicates mitochondrial dysfunction in the pathogenesis of CKD [2]. However, the molecular mechanisms underlying mitochondrial dysfunction in the kidney and the impact on kidney function remain elusive. Reactive oxygen species (ROS) originating mainly from mitochondria play a key role in kidney injury [3], and hydrogen peroxide (H2O2), the main component of ROS, may be responsible for proteinuria and glomerular sieving defects [4]. For example, a deficiency in catalase, which decomposes H2O2 to water and oxygen, causes oxidative stress and sensitizes kidneys to proteinuria and tubulointerstitial fibrosis [5]. Acatalasemic mice are susceptible to adriamycin-nephropathy (AN) and exhibit increased albuminuria and glomerulosclerosis [6]. In addition, catalase deficiency aggravates diabetic renal injury, and the overexpression of catalase in renal proximal tubular cells may attenuate interstitial fibrosis and tubular apoptosis [7,8].
Mitochondrial ROS production can be suppressed by mitochondrial uncoupling, which also modulates mitochondrial biogenesis [9]. Mild mitochondrial uncoupling via niclosamide ethanolamine salt (NEN) increases energy expenditure and oxygen consumption and was shown to reduce urinary albumin excretion in diabetic kidney disease [10,11]. As NEN attenuates renal injury in diabetic mice, we examined whether it also efficacious in nondiabetic kidney disease. For this, we utilized the well-established AN model of kidney disease characterized by proteinuria and progressive renal impairment [12,13] to determine the renal protective effects, and the potential mechanisms involved.
Materials and methods
Mice
Male BALB/c mice (20-25 g) were purchased from the Laboratory Animal Center of Southern Medical University (Guangzhou, China) and housed in the Central Animal Facility at the Shenzhen Graduate School of Peking University. The mice were randomly allocated into AN, AN+NEN, or control groups (n = 6 each). Mice in the AN and AN+NEN groups received an intravenous injection into the tail vein of adriamycin (10.4 mg/kg, Sigma Aldrich, St. Louis, MO, USA) dissolved in normal saline; mice in the control group were injected with equivalent volumes of normal saline. Two weeks after adriamycin injections, mice in the AN+NEN group were fed a regular diet supplemented with 2 g/kg NEN (2A PharmaChem, Lisle, IL, USA) for 2 weeks. Mice in the control and AN groups were maintained on the regular diet. Animal studies were approved by the Guangzhou University of Chinese Medicine Institutional Animal Care and Use Committee and were performed using protocols in accordance with the relevant guidelines and regulations.
Urinary protein and H2O2 assays
At the end of the experiment, urine was collected for 24 h by using metabolic cages (Tecniplast, Buguggiate, Italy). The amount of urinary protein was determined with a Bio-Rad protein assay (Bio-Rad Laboratories, Hercules, CA, USA). Urinary H2O2 was measured by using Amplex UltraRed reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions.
Tissue preparation
All of the mice were sacrificed 2 weeks after NEN treatment (4 weeks after adriamycin injections). The kidneys were dissected and rinsed in phosphate-buffered saline. Kidney tissues were fixed in 10% formalin for histopathological examination and immunohistochemical staining. Samples of renal cortices (1 mm3) were fixed in 2.5% glutaraldehyde and then post-fixed in 1% osmic acid for electron microscopy (EM). The remaining renal tissues were immediately snap-frozen in liquid nitrogen and stored at -80°C for later analysis.
Light microscopy
Paraffin-embedded kidney sections (4 μm) were stained with periodic acid-Schiff (PAS) to evaluate glomerulosclerosis and tubular injury, and with Masson’s trichrome to evaluate interstitial fibrosis according to previous methods [14,15]. In each section, 40 glomeruli were selected randomly under 40 × magnification to evaluate renal glomerulosclerosis. The glomerulosclerosis index was scored on a scale of 0 to 4 on the basis of the percentage of PAS-positive area within the glomerulus divided by the glomerular capillary area (0, < 5%; 1, 5-25%; 2, 25-50%; 3, 50-75%; 4, > 75%). The tubular injury was identified by the presence of diffuse tubular dilation, intraluminal casts, and/or tubular cell vacuolization and detachment in the cortex and medulla. Tubular injury index was assessed in 20 randomly captured fields per section under 20 × magnification, and the index was scored on a scale of 0 to 4 (0, < 5%; 1, 5-25%; 2, 25-50%; 3, 50-75%; 4, > 75%). Interstitial fibrosis was defined by the area of the interstitium with trichrome stain. At least 10 randomly chosen fields in each section under 20 × magnification were scored. The interstitial fibrosis index was estimated by evaluating the percentage of interstitial fibrosis per field and was graded on a scale of 0 to 4 (0, < 5%; 1, 5-25%; 2, 25-50%; 3, 50-75%; 4, > 75%).
EM analysis
ImageJ software (National Institutes of Health, Bethesda, MD, USA) was used to analyze the images collected by EM (JEM-1400, JEOL, Tokyo, Japan). Eight photographs for each sample were selected to measure the foot process width (FPW). The average podocyte FPW was calculated using a previously described method [16].
Immunohistochemistry
Immunohistochemical studies were performed on 4 μm thick formalin-fixed, paraffin-embedded whole tissue sections in a Bond-III automated immunostainer (Leica, Wetzlar, Germany). Antigen retrieval was performed in an EDTA-based solution (pH 9) in the instrument for 10 min. Sections were exposed to 3% H2O2 for 8 min, and then incubated with a primary antibody against WT-1 (MaiXin, Fuzhou, China) for 8 min, with labeled polymer for 4 min, with diaminobenzidine as a chromogen for 2 min, and with hematoxylin as a counterstain for 8 min. Incubations with primary antibodies against catalase and superoxide dismutase 2 (SOD2) (Cell Signaling Technology, Danvers, MA, USA) were performed overnight, followed by incubations with labeled polymer for 15 min, with diaminobenzidine for 1 min, and with hematoxylin for 2 min.
Enzyme-linked immunosorbent assays
Urinary levels of NGAL and Kim-1 were detected with enzyme-linked immunosorbent assays (R&D Systems, Minneapolis, MN, USA) according to the manufacturer’s instructions.
Immunoblotting
Renal cortical tissues were homogenized in lysis buffer and prepared in sample loading buffer (Bio-Rad, Hercules, CA, USA). The proteins were separated on SDS-PAGE gels and transferred to polyvinylidene difluoride membranes (Merck Millipore, Danvers, MA, USA). After blocking in Tris-buffered saline containing 5% nonfat dry milk for 1 h at room temperature, the membranes were incubated with primary antibodies overnight at 4°C with gentle shaking. After washing with Tris-buffered saline, the membranes were incubated with secondary antibodies for 1 h at room temperature with shaking. After washing, the protein bands were detected and analyzed by a ChemiDoc MP imaging system (Bio-Rad, Hercules, CA, USA). The results are expressed as the integrated optical density relative to that of β-actin, used as the loading control. The primary antibodies were against catalase and SOD2, peroxisome proliferator-activated receptor gamma coactivator-1α (PGC-1α; Abcam, Cambridge, UK), and β-actin (Sigma Aldrich, St. Louis, MO, USA).
Kidney mitochondria isolation and oxygen consumption rate assay
Mitochondrial oxygen consumption was measured with an oxygen electrode (Hansatech Instrument, Norfolk, UK) as previously described [17]. Briefly, mitochondria (1 mg/ml) isolated from normal fresh kidney tissues and the following substrates and inhibitors were added to the respiration system (final concentrations): glutamate/malate, 2.5 mM/2.5 mM; ADP, 200 µM; oligomycin, 5 µg/ml (all from Sigma Aldrich); and NEN, 10 µM.
Measurement of kidney mitochondrial H2O2 release
Release of H2O2 from kidney mitochondria was measured using Amplex UltraRed reagent according to the manufacturer’s instructions. Mitochondria (0.2 mg/ml) isolated from kidneys were incubated in mitochondrial assay medium (sodium citrate buffer, 50 mM, pH 6.0) as previously described [18], Different concentrations of NEN dissolved in 1 μl dimethyl sulfoxide (DMSO) were pre-added to the assay medium to final concentrations of 0.5 μg/μl, 0.2 μg/μl, 0.1 μg/μl, 0.02 μg/μl, 0.01 μg/μl, and 0 μg/μl (only DMSO). The Amplex UltraRed/HRP working solution was then added to each microplate well to initiate the reaction. The fluorescence was measured at excitation/emission of 490/585 nm using a Synergy H1 microplate reader (BioTek Instruments, Winooski, VT, USA).
Mitochondrial DNA (mtDNA) copy number measurement and mRNA analysis
Genomic DNA (gDNA) was extracted from renal cortices by using a gDNA purification kit (Magen, Guangzhou, China) according to the manufacturer’s protocol. Total DNA concentrations were determined using a Synergy H1 microplate reader and adjusted to equal concentrations. Total RNA was isolated from renal cortices by using the TRIzol Plus RNA purification kit (Invitrogen, Carlsbad, CA). First-strand cDNA was synthesized with oligo(dT) [12-18] primers and M-MLV reverse transcriptase (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions.
Real-time quantitative PCR was performed using the Stratagene M × 3000P real-time PCR system (Agilent Technologies, Santa Clara, CA). PCR reactions were carried out using cDNA or 150 ng of gDNA, 1 mM of each primer, and SYBR green PCR master mix (Applied Biosystems, Foster City, CA). The amplification conditions were 95°C for 5 min followed by 45 cycles of 95°C for 15 s, 55°C for 15 s, and 72°C for 20 s. The relative mtDNA copy number or mRNA expression was calculated by 2-ΔΔCT and normalized against the housekeeping gene β-actin. Primers were synthesized by Sangon Biotechnology Company (Shanghai, China) and designed to detect cytochrome b (forward, GCCACCTTGACCCGATTCTTCGC; reverse; TGAACGATTGCTAGGGCCGCG) and NADH dehydrogenase, subunit 1 (ND1) (forward, CTAGCAGAAACAAACCGGGC; reverse, CCGGCTGCGTATTCTACGTT) for mtDNA, and β-actin (forward, GGACTCCTATGTGGGTGACG; reverse, AGGTGTGGTGCCAGATCTTC), catalase (forward, GGACGCTCAGCTTTTCATTC; reverse, TTGTCCAGAAGAGCCTGGAT), and SOD2 (forward, CCCAGACCTGCCTTACGACTAT; reverse, GGTGGCGTTGAGATTGTTGA) for nuclear DNA.
Statistical analysis
Data are expressed as the mean ± SD. Data analysis was performed using SPSS software (IBM software). Comparisons between two groups were performed using unpaired Student’s t tests. Differences among multiple groups were analyzed by using one-way ANOVA. Significance was defined as P < 0.05.
Results
NEN reduces protein excretion and recovers podocyte dysfunction in AN mice
Compared to that in control mice, urinary protein excretion increased significantly in mice in the AN group 4 weeks after adriamycin injection (Figure 1A). NEN treatment for 2 weeks reduced proteinuria significantly (Figure 1A). As effacement of the podocyte process and a progressive loss of podocytes are the most common pathological changes accounting for proteinuria. We measured the FPW and evaluated the number of podocytes (WT-1 positive cells). As shown in Figure 1B, the average FPW was significantly wider in the mice in the AN group than in the control group. NEN treatment reduced the FPW significantly. The EM images in Figure 1D show the podocyte lesions in various groups. Immunostaining for WT-1 revealed that the number of podocytes was reduced significantly in AN mice and NEN ameliorated the podocyte loss (Figure 1C, 1E).
Figure 1.

NEN reduces urinary protein, FPW, and loss of podocytes. Quantification and statistical analyses of urinary protein excretion (n = 6/group) (A), FPW (n = 3/group) (B), and the number of podocytes (n = 6/group) (C) in control mice, those with AN, and those with AN treated for 2 weeks with NEN. **P < 0.01 and ***P < 0.001 vs. control; ##P < 0.01 and ###P < 0.001 vs. AN. (D) Representative EM images of the podocyte processes in each group. The podocytes in the AN group exhibited diffuse fusion of foot processes, which was attenuated by NEN treatment. Scale bar, 500 nm. (E) Representative images of WT-1 immunostaining in the three groups. Scale bar, 20 µm.
NEN ameliorates renal pathological injury
Compared to that in control mice, AN mice displayed focal segmental glomerulosclerosis (Figure 2A, 2E), tubular injury (Figure 2B, 2F), and interstitial fibrosis (Figure 2C, 2G). NEN treatment significantly attenuated these pathological alterations (Figure 2A-C, 2E-G). The number of nuclei per glomerulus also decreased significantly in the AN mice and was increased by NEN treatment (Figure 2D).
Figure 2.
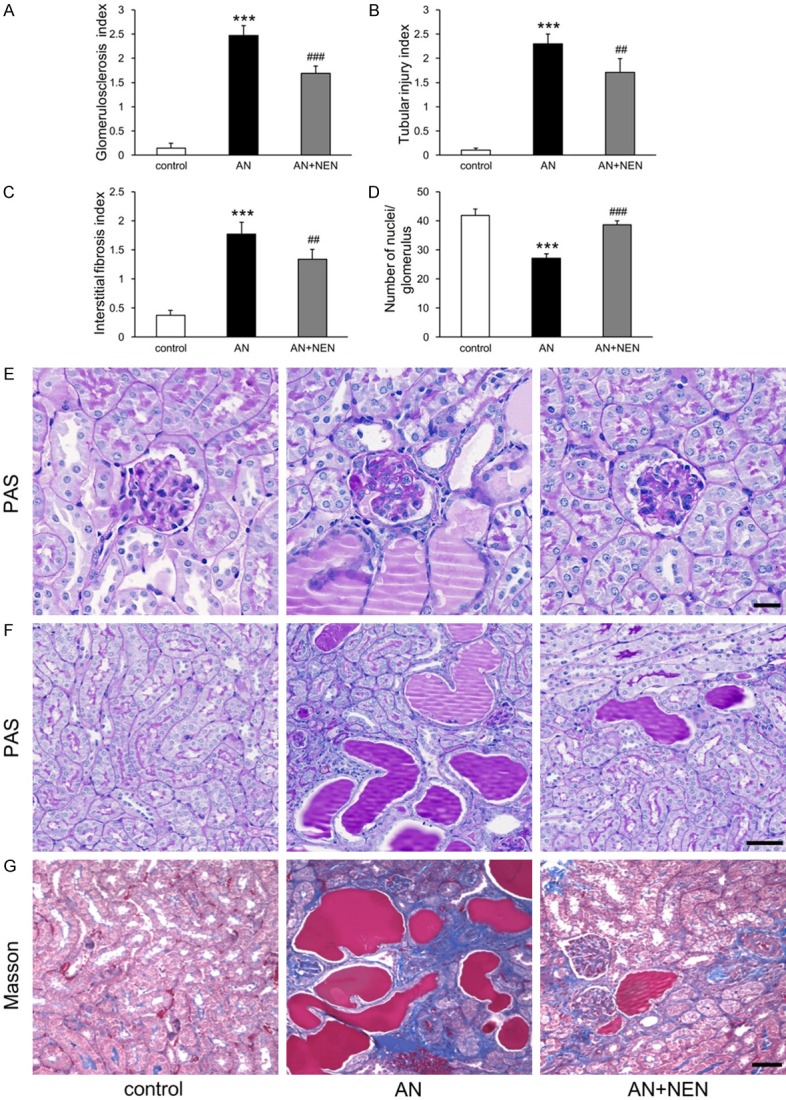
Effects of NEN on renal pathology. Quantification of glomerulosclerosis (A), tubular injury (B), interstitial fibrosis (C), and the number of nuclei/glomerulus (D) in control mice, those with AN, and those with AN treated for 2 weeks with NEN (n = 6/group). ***P < 0.001 vs. control; ##P < 0.01 and ###P < 0.001 vs. AN. Representative images of PAS staining for glomeruli (scale bar, 20 µm) (E) and for tubules (scale bar, 50 µm) (F). (G) Representative images of Masson’s trichrome staining for renal tubulointerstitium. Scale bar, 50 µm.
NEN reduces urinary excretion of tubular injury biomarkers NGAL and Kim-1
As shown in Figure 3A, 3B, the urinary excretion levels of tubular injury biomarkers NGAL and Kim-1 were significantly higher in the AN group than in the control group. NEN treatment reduced these biomarkers significantly.
Figure 3.
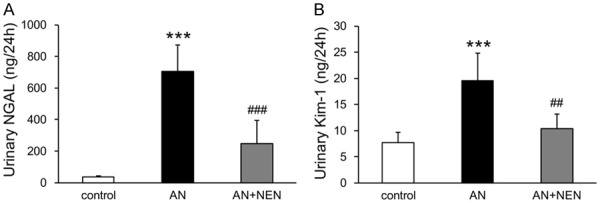
NEN reduces urinary NGAL and Kim-1 excretion. Quantification of urinary NGAL (A) and Kim-1 (B) excretion in control mice, those with AN, and those with AN treated for 2 weeks with NEN (n = 6/group). ***P < 0.001 vs. control; ##P < 0.01 and ###P < 0.001 vs. AN.
NEN uncouples isolated kidney mitochondria, dose-dependently decreases renal mitochondrial H2O2 production, reduces urinary H2O2 excretion
Compared to that in control mice, urinary H2O2 excretion increased significantly in AN mice. NEN treatment reduced it significantly (Figure 4A). As shown in Figure 4B, NEN dose-dependently decreased renal mitochondrial H2O2 production. Moreover, NEN increased oxygen consumption in the presence or absence of oligomycin in mitochondria isolated from the kidneys of control mice, demonstrating direct uncoupling of mitochondria (Figure 4C, 4D).
Figure 4.

NEN uncouples mitochondria isolated from kidneys, dose-dependently decreases renal mitochondrial H2O2 production, and reduces urinary H2O2 excretion. (A) Urinary H2O2 excretion in control mice, those with AN, and those with AN treated for 2 weeks with NEN (n = 6/group). **P < 0.01 vs. control; ###P < 0.001 vs. AN. (B) H2O2 release rates in isolated kidney mitochondria (n = 4/group). ***P < 0.001 vs. Mito (mitochondria) group; ##P < 0.01 and ###P < 0.001 vs. DMSO. Uncoupling in isolated kidney mitochondria (measured as oxygen consumption) from normal control mice in the absence (C) or presence (D) of oligomycin.
NEN regulates catalase and SOD2 expression
Immunohistochemical staining revealed that catalase and SOD2 were mainly expressed in tubules (Figure 5A, 5B). The protein and mRNA levels of catalase in renal cortical tissues from the AN group decreased significantly and were increased significantly by NEN treatment (Figure 5C, 5D, 5F). Although the mRNA expression of SOD2 was significantly decreased in the AN group compared to that in controls (Figure 5G), the protein levels were not significantly different (Figure 5E).
Figure 5.
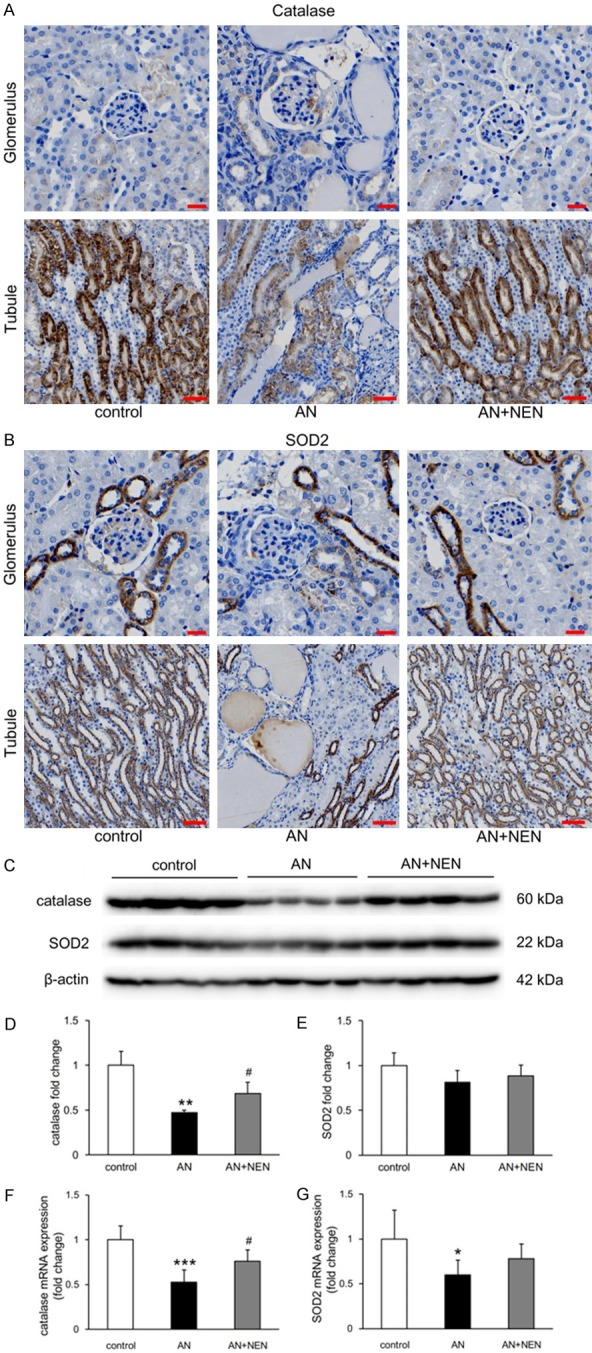
Effects of NEN on catalase and SOD2 expression in renal cortex. (A) Immunohistochemical staining of catalase (A) and SOD2 (B) in glomeruli (scale bars, 20 µm) and tubules (scale bars, 50 µm) in the renal cortices from control mice, those with AN, and those with AN treated for 2 weeks with NEN. (C) Representative Western blots for catalase and SOD2 expression. Quantification of catalase (D and F) and SOD2 (E and G) protein and mRNA levels, respectively, normalized to β-actin (n = 4/group). *P < 0.05, **P < 0.01 and ***P < 0.001 vs. control; #P < 0.05 vs. AN.
Effects of NEN on PGC-1α expression and mtDNA copy number
Compared to that in control mice, PGC-1α expression in renal cortex was not significantly decreased (Figure 6A, 6B). However, expression levels in the AN+NEN group were significantly higher than in the AN group (Figure 6A, 6B). As PGC-1α is the master regulator of mitochondrial biogenesis, we measured the mtDNA copy number in kidney tissues. The results in Figure 6C show that mtDNA copy numbers (determined by cytochrome b and ND1 levels) decreased significantly in AN mice but were not significantly different in the AN+NEN group.
Figure 6.

Effects of NEN on PGC-1α expression and mtDNA copy number. (A) Representative Western blots of PGC-1α in the renal cortices from control mice, those with AN, and those with AN treated for 2 weeks with NEN. Quantification of PGC-1α levels (B) and mitochondrial DNA copy number (determined by real-time quantitative PCR for cytochrome b and ND1) (C) normalized to β-actin (n = 6/group). *P < 0.05 vs. control; ##P < 0.01 vs. AN.
Discussion
In the present study, we report for the first time that NEN exerts renoprotective effects in the AN model of nondiabetic kidney disease via a mechanism that likely involves mitochondrial uncoupling in the kidney. The results show that treatment with NEN attenuates the loss of podocytes, which are highly specialized and terminally differentiated cells that form intercellular junctions with each other, and reversed the widening of podocyte foot processes induced by AN. Notably, the loss of slit diaphragms and foot effacement of podocytes are key steps leading to proteinuria [19], which contributes to CKD. The recovery of podocyte injury by NEN may well explain the reduced excretion of urinary proteins.
NEN treatment ameliorated pathological renal injury, including glomerulosclerosis, tubular injury, and interstitial fibrosis, and reversed the reduction in the number of nuclei per glomerulus induced by AN. We speculate that this result is associated with the protection of podocytes. Whether other cells were involved needs further study. In addition, the excretion of tubular injury biomarkers NGAL and Kim-1 was reduced by NEN treatment. These results indicate that NEN is protective not only for glomeruli but also for renal tubules.
The kidney is an energy-demanding organ rich in mitochondria. Mitochondrial dysfunction mediated by the overproduction of ROS contributes to CKD progression [20]. Although ROS stimulate the mild uncoupling of mitochondria via uncoupling proteins, the resulting increase in proton conductance may negatively feedback on ROS production [21], and mitochondrial uncouplers can decrease ROS production [9]. We found that NEN uncoupled mitochondria isolated from kidneys and dose-dependently decreased renal mitochondrial H2O2 production. Together with the observed decrease in urinary protein excretion, we speculate that the reduction of proteinuria by NEN is related to the mitochondrial uncoupling effects in the kidney.
ROS are mainly produced by mitochondria and play important roles in cell signaling and homeostasis [22]. Previous studies demonstrated that the transcriptional coactivator PGC-1α, which is the master regulator of mitochondrial biogenesis [23], induces ROS-detoxifying enzymes under oxidative stress conditions and that PGC-1α-null mice are more sensitive to oxidative stress [24,25]. The results of this study show that treatment with NEN increased the expression of PGC-1α as well as that of catalase, thereby reducing urinary H2O2 excretion induced by AN. However, there was no effect on SOD2 expression. Immunohistochemical analyses revealed that catalase is mainly expressed in tubules. Thus, we inferred that the renal protective effects of NEN are associated with its regulation of tubular mitochondria. As mtDNA copy numbers were not significantly altered by NEN treatment, another mechanism may be involved in the mitochondrial biogenesis contributing to the observed effects.
In summary, the results presented here suggest that NEN protects the kidney in nondiabetic kidney disease by regulating the balance of mitochondrial H2O2 production and scavenging. Further studies are needed to detail the mechanisms involved.
Acknowledgements
This study was supported by grants from National Natural Science Foundation of China (81673794, 81202818) and Shenzhen Science and Technology Project (JCYJ20160330171116798, JCYJ20160428182542525, ZDSYS201606081515458).
Disclosure of conflict of interest
None.
References
- 1.Webster AC, Nagler EV, Morton RL, Masson P. Chronic kidney disease. Lancet. 2017;389:1238–1252. doi: 10.1016/S0140-6736(16)32064-5. [DOI] [PubMed] [Google Scholar]
- 2.Galvan DL, Green NH, Danesh FR. The hallmarks of mitochondrial dysfunction in chronic kidney disease. Kidney Int. 2017;92:1051–1057. doi: 10.1016/j.kint.2017.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sedeek M, Nasrallah R, Touyz RM, Hebert RL. NADPH oxidases, reactive oxygen species, and the kidney: friend and foe. J Am Soc Nephrol. 2013;24:1512–1518. doi: 10.1681/ASN.2012111112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yoshioka T, Ichikawa I, Fogo A. Reactive oxygen metabolites cause massive, reversible proteinuria and glomerular sieving defect without apparent ultrastructural abnormality. J Am Soc Nephrol. 1991;2:902–912. doi: 10.1681/ASN.V24902. [DOI] [PubMed] [Google Scholar]
- 5.Kobayashi M, Sugiyama H, Wang DH, Toda N, Maeshima Y, Yamasaki Y, Masuoka N, Yamada M, Kira S, Makino H. Catalase deficiency renders remnant kidneys more susceptible to oxidant tissue injury and renal fibrosis in mice. Kidney Int. 2005;68:1018–1031. doi: 10.1111/j.1523-1755.2005.00494.x. [DOI] [PubMed] [Google Scholar]
- 6.Takiue K, Sugiyama H, Inoue T, Morinaga H, Kikumoto Y, Kitagawa M, Kitamura S, Maeshima Y, Wang DH, Masuoka N, Ogino K, Makino H. Acatalasemic mice are mildly susceptible to adriamycin nephropathy and exhibit increased albuminuria and glomerulosclerosis. BMC Nephrol. 2012;13:14. doi: 10.1186/1471-2369-13-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hwang I, Lee J, Huh JY, Park J, Lee HB, Ho YS, Ha H. Catalase deficiency accelerates diabetic renal injury through peroxisomal dysfunction. Diabetes. 2012;61:728–738. doi: 10.2337/db11-0584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brezniceanu ML, Liu F, Wei CC, Chenier I, Godin N, Zhang SL, Filep JG, Ingelfinger JR, Chan JS. Attenuation of interstitial fibrosis and tubular apoptosis in db/db transgenic mice overexpressing catalase in renal proximal tubular cells. Diabetes. 2008;57:451–459. doi: 10.2337/db07-0013. [DOI] [PubMed] [Google Scholar]
- 9.Berry BJ, Trewin AJ, Amitrano AM, Kim M, Wojtovich AP. Use the protonmotive force: mitochondrial uncoupling and reactive oxygen species. J Mol Biol. 2018;430:3873–3891. doi: 10.1016/j.jmb.2018.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Han P, Shao M, Guo L, Wang W, Song G, Yu X, Zhang C, Ge N, Yi T, Li S, Du H, Sun H. Niclosamide ethanolamine improves diabetes and diabetic kidney disease in mice. Am J Transl Res. 2018;10:1071–1084. [PMC free article] [PubMed] [Google Scholar]
- 11.Han P, Zhan H, Shao M, Wang W, Song G, Yu X, Zhang C, Ge N, Yi T, Li S, Sun H. Niclosamide ethanolamine improves kidney injury in db/db mice. Diabetes Res Clin Pract. 2018;144:25–33. doi: 10.1016/j.diabres.2018.08.003. [DOI] [PubMed] [Google Scholar]
- 12.Wang Y, Wang YP, Tay YC, Harris DC. Progressive adriamycin nephropathy in mice: sequence of histologic and immunohistochemical events. Kidney Int. 2000;58:1797–1804. doi: 10.1046/j.1523-1755.2000.00342.x. [DOI] [PubMed] [Google Scholar]
- 13.Wang YM, Wang Y, Harris DC, Alexander SI, Lee VW. Adriamycin nephropathy in BALB/c mice. Curr Protoc Immunol. 2015;108:15.28.1–6. doi: 10.1002/0471142735.im1528s108. [DOI] [PubMed] [Google Scholar]
- 14.Wang Y, Wang YP, Tay YC, Harris DC. Role of CD8(+) cells in the progression of murine adriamycin nephropathy. Kidney Int. 2001;59:941–949. doi: 10.1046/j.1523-1755.2001.059003941.x. [DOI] [PubMed] [Google Scholar]
- 15.Cao Q, Wang Y, Wang XM, Lu J, Lee VW, Ye Q, Nguyen H, Zheng G, Zhao Y, Alexander SI, Harris DC. Renal F4/80+ CD11c+ mononuclear phagocytes display phenotypic and functional characteristics of macrophages in health and in adriamycin nephropathy. J Am Soc Nephrol. 2015;26:349–363. doi: 10.1681/ASN.2013121336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sun H, Wang W, Han P, Shao M, Song G, Du H, Yi T, Li S. Astragaloside IV ameliorates renal injury in db/db mice. Sci Rep. 2016;6:32545. doi: 10.1038/srep32545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li Z, Graham BH. Measurement of mitochondrial oxygen consumption using a Clark electrode. Methods Mol Biol. 2012;837:63–72. doi: 10.1007/978-1-61779-504-6_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Frezza C, Cipolat S, Scorrano L. Organelle isolation: functional mitochondria from mouse liver, muscle and cultured fibroblasts. Nat Protoc. 2007;2:287–295. doi: 10.1038/nprot.2006.478. [DOI] [PubMed] [Google Scholar]
- 19.Assady S, Wanner N, Skorecki KL, Huber TB. New insights into podocyte biology in glomerular health and disease. J Am Soc Nephrol. 2017;28:1707–1715. doi: 10.1681/ASN.2017010027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Che R, Yuan Y, Huang S, Zhang A. Mitochondrial dysfunction in the pathophysiology of renal diseases. Am J Physiol Renal Physiol. 2014;306:F367–378. doi: 10.1152/ajprenal.00571.2013. [DOI] [PubMed] [Google Scholar]
- 21.Echtay KS, Roussel D, St-Pierre J, Jekabsons MB, Cadenas S, Stuart JA, Harper JA, Roebuck SJ, Morrison A, Pickering S, Clapham JC, Brand MD. Superoxide activates mitochondrial uncoupling proteins. Nature. 2002;415:96–99. doi: 10.1038/415096a. [DOI] [PubMed] [Google Scholar]
- 22.Shadel GS, Horvath TL. Mitochondrial ROS signaling in organismal homeostasis. Cell. 2015;163:560–569. doi: 10.1016/j.cell.2015.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li SY, Susztak K. The role of peroxisome proliferator-activated receptor gamma coactivator 1alpha (PGC-1alpha) in kidney disease. Semin Nephrol. 2018;38:121–126. doi: 10.1016/j.semnephrol.2018.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.St-Pierre J, Drori S, Uldry M, Silvaggi JM, Rhee J, Jager S, Handschin C, Zheng K, Lin J, Yang W, Simon DK, Bachoo R, Spiegelman BM. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell. 2006;127:397–408. doi: 10.1016/j.cell.2006.09.024. [DOI] [PubMed] [Google Scholar]
- 25.Rabinovitch RC, Samborska B, Faubert B, Ma EH, Gravel SP, Andrzejewski S, Raissi TC, Pause A, St-Pierre J, Jones RG. AMPK maintains cellular metabolic homeostasis through regulation of mitochondrial reactive oxygen species. Cell Rep. 2017;21:1–9. doi: 10.1016/j.celrep.2017.09.026. [DOI] [PubMed] [Google Scholar]