

Abstract
JAK2/STAT3 is a cardio-protective, pro-inflammation pathway, the function of which in cardiomyopathy caused by diabetic (DCM) is currently unknown. Here we explore the role of the JAK2/STAT3 pathway in DCM employing different time courses and a type 2 DM (T2DM) rat model. We examined the interactions of metformin and sitagliptin treatment with the JAK2/STAT3 pathway and cardiac remodeling. A T2DM rat model was induced by high fat diet/streptozotocin (HFD/STZ) and treated with metformin, sitagliptin (10 mg/d or 20 mg/d) or a placebo. Cell inflammation markers, cardiac remodeling and cardiomyocyte apoptosis were evaluated. We observed an activated inflammation reaction as well as activation of the JAK2/STAT3 thought-out the experiment in the simple HFD group only in the early stage of the disease (until week 9). JAK2/STAT3 activity showed a phasic peculiarity as increased inflammation was observed in prolongation of the DCM accompanied with an accelerated cardiac dysfunction but reduced phosphorylation of myocardial STAT3. Moreover, in the metformin but not the sitagliptin treated group, JAK2/STAT3 activation was associated with having better improved cardiac remolding and reduced myocardial apoptosis. In vitro studies further validated that metformin could activate JAK2/STAT3 pathway and alleviate apoptosis of NRCMs under hyperglycemia incubation. The phasic feature of JAK2/STAT3 pathway activation may participate in the pathophysiological development of DCM. The superior cardio-protective effect of metformin over sitagliptin treatment may partly account for the differences we observed in JAK2/STAT3 activation, indicating that measuring JAK2/STAT3 pathway coupled with metformin treatment may give insight into a more promising DM treatment.
Keywords: Diabetic cardiomyopathy, JAK2/STAT3 pathway, inflammation, apoptosis, metformin, sitagliptin
Introduction
Diabetes mellitus (DM) is an emerging global threat to human health and is estimated to affect 366 million people by the year 2030 [1]. Besides hypertension and coronary narrowing, DM has been confirmed to be a strong independent risk factor of acquired cardiomyopathy [2]. Diabetic cardiomyopathy (DCM) is defined as diabetes-related ventricular systolic dysfunction and/or diastolic dysfunction, regardless of any other pre-existing cardiovascular diseases a patient might have. DCM is relatively common in the diabetic community with a prevalence rate of 16.9%, and is one of the leading causes of morbidity and mortality in diabetic patients [3].
Chronic inflammation and cardiomyocyte apoptosis have been found to have key roles in the development of DCM [4]. Heart failure progression usually entails a local rise in inflammation cytokines and the activation of pro-inflammatory transcription factors. Many previous studies have focused on the adverse effects of inflammation, however, recent data demonstrate that under certain conditions, the Janus Kinase 2/signal transducer and activator of transcription 3 (JAK2/STAT3) inflammation pathway may initiate a cardioprotective effect via promoting cell survival [5]. The JAK2/STAT3 pathway is a pro-inflammatory cytokine induced pathway that is also known as the survival activating factor enhancement (SAFE) pathway. It can be activated by various cytokines including interleukins, colony-stimulating factors and interferons that all initiate the inflammation reaction. However, more recently researchers have found that the activation of JAK2/STAT3 is capable of promoting an anti-apoptosis effect in multiple diseases like ischemia heart disease [6-9]. While in both DM and DCM the role of JAK2/STAT3 has been under-characterized and what has been found has been inconsistent, it is known that activated JAK2/STAT3 can result in chronic inflammation and ultimately DCM in rats fed a high-fat diet [10]. However, rats given DM induced by STZ or leptin receptor defect demonstrated a suppressed JAK2/STAT3 pathway activation at 12 weeks of age [11]. While different animal models or different experimental durations may account for the disparities seen in the function of the JAK2/STAT3 pathway in DCM, it is still reasonable for us to believe that the JAK2/STAT3 pathway plays a role in DCM development. To verify our assumptions, we chose a DCM rat model to explore JAK2/STAT3 pathway activity using two hypoglycemic drugs metformin and sitagliptin as treatment control.
Metformin is a first-line hypoglycemic drug that also shows a promising therapeutic effect in cardiovascular disease. Previous studies have confirmed that metformin can improve left ventricular remodeling in diabetic animals, with many possible mechanisms proposed including anti-inflammation [12,13]. Metformin is known to elicit a wide range of protective effects through the activation of AMPK, (adenosine 5’-monophosphate -activated protein kinase) which promotes myocardial cell survival. In fact, the AMPK-STAT3 axis may account for several biological effects of metformin like anti-atherosclerosis and anti-tumor growth through the inhibition of STAT3 activation via AMPK [14-16]. The cardio-protective effect of metformin and its possible relationship with the JAK2/STAT3 pathway in DCM have yet to be addressed, however metformin could work through amelioration of inflammation in the diabetic heart [17,18]. Metformin has also proven to be effective in suppressing apoptosis by activating heme oxygenase-1 (HO-1), which acts upstream of STAT3 [19]. So, it is reasonable for us to assume that metformin might attenuate myocyte apoptosis via STAT3 activation. Combination of sitagliptin with metformin could additionally decrease the inflammation reaction during DM, and recent studies have shown that the inhibitor DDP4 could ameliorate apoptosis by up-regulating STAT3 in rats with myocardial infarction to play a cardio-protective role [20-22]. As a novel hypoglycemia drug, sitagliptin inhibits DPP-4, improving the global levels of glucagon-like peptide-1 (GLP-1). Several in vivo and in vitro studies suggest that sitagliptin possesses antioxidant and anti-inflammatory properties, but the impact of sitagliptin on the JAK2/STAT3 pathway in DCM is still unknown. No data have been shown drawing a comparison between the therapeutic effect of metformin and sitagliptin in DCM.
Here we use a type 2 DM rat model and neonatal rat cardiac myocytes (NRCMs) under different interventions to characterize: 1) JAK2/STAT3 pathway activity and its relationship with inflammation in the progression of DCM; 2) determine and compare the intervention effect of metformin and sitagliptin on ventricular remodeling and cardiac dysfunction in DCM, and the functional role of JAK2/STAT3 pathway involved.
Methods
In vivo study: animals and experimental protocols
Male Sprague-Dawley rats (120±20 g, 5 wk) were obtained from the Vital River Laboratory (Beijing, China). Rats were housed at 22°C with 12-hour light-dark cycles and given free access to standard laboratory diet and drinking water. After 1 week of acclimatization, a baseline intraperitoneal insulin tolerance test (IPITT) was administered to determine insulin resistance levels, then rats were randomly divided into two dietary regimens of feeding either normal chow diet (NCD group) or high-fat diet (HFD, 60 kcal% fat, 20 kcal% protein, 20 kcal% carbohydrate; Beijing HFK Bio-Technology, China). After another 4 weeks, IPITT was re-administered and type 2 diabetes was induced by intraperitoneal injection of STZ (Sigma, St. Louis, MO; 27.5 mg/kg i.p. in 0.1 mol/L citrate buffer, pH 4.5) to HF diet feeding rats with insulin resistance. We defined a diabetic rat as animals with diabetic characteristics including a fasting blood glucose (FBG) level greater than 11.1 mmol/L in two consecutive analyses as well as reduced insulin sensitivity one week after STZ administration. Age-matched NCD group and HF diet feeding-induced simple obese rats (without diabetic characters) (HFD group) were used as controls. DM and control rats were then randomly divided into different groups according to their feeding duration and/or treatment: 9, 14, 21 weeks in feeding groups (DM/NCD/HFD-9w, DM/NCD/HFD-14w, DM/NCD/HFD-21w, n=6, each) and 21 weeks feeding group further divided according to their treatment by placebo (DM-21w), metformin or sitagliptin (MET, STAG10-21w, STAG20-21w, n=8, each). The detailed experimental protocol is shown in Figure 1.
Figure 1.
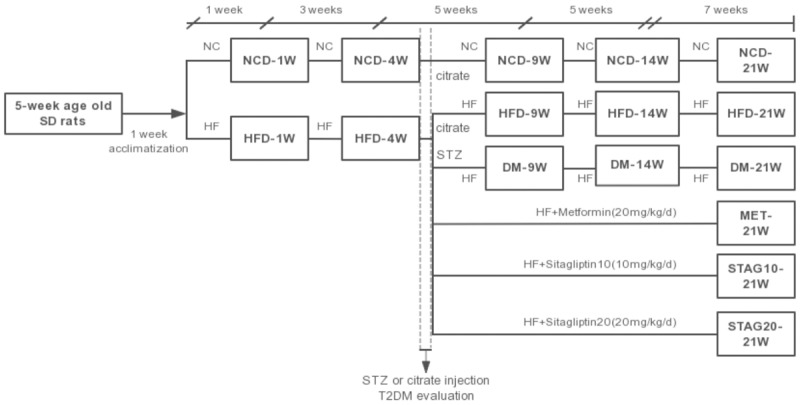
Flow chart of the animal experiment. Male Sprague-Dawley rats were first randomly divided into two dietary regimens of feeding either normal chow diet or high-fat diet (60 kcal% fat, 20 kcal% protein, 20 kcal% carbohydrate); type 2 diabetes was induced by intraperitoneal injection of STZ to HF diet feeding rats with insulin resistance. DM and control rats were randomly divided according to their feeding duration and/or treatment; 21 weeks feeding group were further divided into placebo, metformin or sitagliptin group. SD: Sprague-Dawley; HF: high fat diet; HFD: simple high-fat diet group; NCD: normal chow diet group; MET: metformin 200 mg/kg/d; STAG10: sitagliptin of 10 mg/kg/d; STAG20: sitagliptin of 20 mg/kg/d; DM-21w: 21-weeks age diabetes rats with placebo.
In vitro study: neonatal rat cardiac myocytes
Our in vitro study was designed to verify our findings in our animal experiments. NRCMs were isolated from 2-day-old neonatal SD rats (Fang-YY Laboratory Animal Co., Beijing, China) as described earlier [23] and assigned into different groups based on different glucose concentrations and experiment durations: (1) Cell apoptosis: low glucose (5.5 mmol/L), high glucose (33 mmol/L) and iso-osmotic low glucose (5.5 mmol/L glucose + 27.5 mmol/L mannitol), incubated for 0 h, 6 h, 12 h, 24 h, 36 h or 48 h. (2) JAK2/STAT3 pathway activity: low glucose (5.5 mmol/L), high glucose (33 mmol/L) and iso-osmotic low glucose (5.5 mmol/L glucose + 27.5 mmol/L mannitol), incubated for 0 h, 30 min, 60 min, 3 h, 6 h, 12 h, 18 h or 24 h. (3) Impact of Metformin on cell apoptosis and JAK2/STAT3 pathway activity: iso-osmotic low glucose group, high glucose group, high glucose + 0.1 mM Metformin group, high glucose + 0.5 mM Metformin group, high glucose + 1 mM Metformin group. (4) Possible protecting mechanism of JAK2/STAT3 in DCM: high glucose group, high glucose + AG490 group, high glucose + AG490 + Metformin group, high glucose + AICAR group (AG490 (Tyrphostin B42), a kinase inhibitor of JAK2; AICAR, a stimulator of AMPK increased Thr172 phosphorylation of AMPK).
Blood analysis
Blood samples were collected by the tail nipping method after 6-hour fasting period. Glucose levels were measured using Accucheck glucometer (Roche diagnostics) once a week throughout the entire duration of study. Insulin sensitivity was evaluated in 5 h fasted rats by IPITT by giving 1 U/kg of insulin (Lilly, Indianapolis, IN, USA) intraperitoneally. Blood glucose measurements during IPITT were done over a 2-hour period.
Abdominal aorta blood was collected at the end of the planned termination for each rat. Total cholesterol, triglyceride levels, glycosylated hemoglobin (HbAlc) and FBG were analyzed by the Bayer 1650 blood chemistry analyzer (Bayer, Tarrytown, NY). Fasting insulin levels were measured using insulin ELISA kit (ALPCO, USA) to the manufacturer’s instruction. Homeostasis model assessment of insulin resistance (HOMA-IR) was employed to assess the status of insulin action.
Inflammation markers IL-6 and TNF-α were measured in serum of related rats using ELISA kits (R&D, USA). Serum BNP level was determined using ELISA kit (Sigma-Aldrich, USA).
Echocardiographic measurements
Transthoracic echocardiography was performed using Vevo 2100 High Resolution Imaging System (Visual Sonics Inc, Canada) as previously described. The rats were anaesthetized by isoflurane inhalation. Then the chests of the rats were shaved and rats were placed in the recumbent position. The following parameters were measured and calculated from M-mode tracing and Doppler echocardiographic imaging: diameters of left ventricular end-diastole (LVEDd), ejection fraction (EF), and the ratio of the early (E) to late (A) ventricular filling velocities (E/A). Tissue Doppler imaging (TDI) was used to evaluate early diastolic (E’) and late (A’) velocity of the mitral annulus (arithmetic average travel speeds of the lateral and septal walls), E’/A’ and E/E’ ratios. Wall thickness of two segments [diameters of interventricular septum (IVSd) and diameters of end-diastole left ventricular posterior wall (LVPWd)] and fractional shortening (FS) were evaluated on short axis 2D images. Left ventricular end-diastolic volume/end-systolic volume (LV_d/LV_s) and left ventricular mass (LV mass) were calculated [24]. Every animal was tested in similar conditions (room temperature, breath, heart rate and blood pressure), and all scans of echocardiographic investigation were performed by two experienced sonographers blinded to the experimental group to ensure the reproducibility. Measurements are reported as the mean of at least five cardiac cycles on M-code tracings.
Tissue collection and histology
Cardiac tissues and blood were collected after sacrificing the rats according to the schedule. Left ventricles were fixed in 10% formalin and paraffin-embedded and sectioned at 5 µm. In each group, at least 10 randomly selected sections were stained with hematoxylin-eosin (H-E) and studied for histological changes.
The degree of fibrosis was determined by Masson trichrome staining of heart sections with a Masson kit (Maixin Biotech, Fuzhou). Dark green-staining was identified as collagen fibers. Interstitial and perivascular fibrosis were also evaluated by Picrosirius red staining. Collagen I was stained red.
To analysis cell apoptosis, the terminal deoxynucleotidyl transferase-mediated dUTP nick end-labeling (TUNEL) assay was used on sectioned mid-LV samples with an In Situ Apoptosis Detection Kit (Roche, MD, USA) according to the manufacturer’s instructions. The apoptotic index (AI) = number of TUNEL-positive myocytes/total number of myocytes stained with DAPI from a total of 40 fields per heart (n=4/5).
Immunohistochemistry assay
Immunohistochemistry assays were performed to determine the collagen distribution and content. The slides were incubated with 10% non-immune goat serum for 1 h at room temperature to block non-specific staining before incubation with the murine anti-collagen I antibody (ab34710, abcam, USA) or anti-collagen III antibody (ab7778, abcam, USA) overnight, in humidified chambers at 4°C. The immunohistochemistry assays for TNF-α and IL-6 were also performed to evaluate inflammation changes with anti-TNF-α antibodies (ab66579, abcam, USA) and anti-IL-6 (ab6672, abcam, USA). Anti-pStat3 (phospho Y705, abcam, USA) was used to detect pStat3 expression. All slides were incubated with biotinylated secondary antibody for 1 h at room temperature and HRP-conjugated streptavidin for 15 min at room temperature, followed by detection with a commercial kit (Boster, Wuhan). For quantitative analysis, the average score of 10-20 randomly selected areas were calculated using the Image Pro Plus 6.0 software.
Western blot analysis
Tissue samples were lysed with RIPA lysis buffer containing protease and phosphatase inhibitors (Applygen Technologies, Beijing). The lysates were homogenized and the homogenates were centrifuged at 13000 rpm for 15 min at 4°C. The supernatants were collected and protein concentrations were determined by bicinchoninic acid assay (BCA). Equivalent amounts of protein were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred onto a polyvinylidene difluoride membrane (Millipore). The membranes were blocked with 5% milk and incubated overnight with anti-pStat3 (Tyr705, 9145, CST, USA), anti-Stat3 (12640, CST, USA), anti-pJak2 (Tyr1007/1008, 3771, CST, USA), anti-Jak2 (3230, CST, USA), anti-Cleaved (c)-Caspase3 (9664, CST, USA), anti-Bax (2772, CST, USA), anti-Bcl-2 (2870, CST, USA), anti-pERK (4370, CST, USA), anti-ERK (9102, CST, USA) and anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH; loading control). The membranes were incubated with horseradish peroxidase-coupled secondary antibodies. After immune-blotting, films were scanned and detected by the luminescence way.
Lysed NRCMs from each group were collected for protein analysis. Samples were homogenized in RIPA buffer (Roche) and centrifuged at 13000 rpm for 10 minutes, the supernatants were collected. Protein concentration was determined by bicinchoninic acid assay (BCA). Samples (30 μg/lane) were mixed with loading buffer in a 12% SDS polyacrylamide gel electrophoresis and transferred to nitrocellulose membranes. The membranes were blocked in 5% skim milk for 1 hour at room temperature and incubated overnight at 4°C with the primary antibodies as follows: anti-Bcl2 (Cell Signaling Technology, #2870), anti-Bax (Cell Signaling Technology, #2772), anti-Caspase3 (Cell Signaling Technology, #9662), anti-Akt (Cell Signaling Technology, #4691), anti-Phospho-Akt (Cell Signaling Technology, #4060) and anti-GAPDH (GSGB-BIO, Beijing, China), all diluted 1:1000. Membranes were then washed and incubated in secondary antibodies diluted at 1:5000. Protein expression levels were determined by analyzing the signals captured on the nirtocellulose membranes using a chemi-doc image analyzer (FluorChemo M FM0488, Protein Simple) and analyzed by Image J (NIH, Bethesda, MD, USA).
Hemodynamic measurements
Rats under deep anesthesia underwent hemodynamic measurement at the end of the experiment. A fluid-filled catheter was advanced from the right carotid artery into the LV, and the left ventricular end diastolic pressure (LVEDP), dp/dtmax, -dp/dtmin were measured.
Flow cytometry
NRCM apoptosis in different groups were quantified by Annexin V-FITC/PI apoptosis detection kit (BD Biosciences Clontech, USA) following the manufacturer’s instructions. Apoptotic and necrotic cells were distinguished on the basis of annexin V-FITC reactivity and PI exclusion and detected by the Accuri C 6 flow cytometer (Becton Dickinson, USA). Annexin-V-only positive cells indicated early apoptosis, whereas PI-positive cells indicated damaged cell membranes and secondary/late apoptosis. For the assay, cells were dissociated with trypsin and then re-suspended in buffer to a final concentration of 1×106 cells/L. Each cell sample was incubated with annexin-V (5 µL) and PI (4 µL) in the dark at room temperature for 20 minutes and then analyzed. The results were analyzed by FlowJo Software (TreeStar, Shenzhen, China).
Statistical analysis
All data are presented as mean ± SD. Statistical analysis were performed with Graph-Pad Prism 5.0 (GraphPad Software, San Diego, CA) or SAS 9.4 (SAS Institute, Cary, North Carolina). Parameters among the groups were compared via one-way ANOVA. Differences with P<0.05 were considered statistically significant.
Results
In vivo study
Metabolism evaluation of different groups
We characterized our diabetic model induced a by HF diet coupled with a low-dose of STZ by moderate hyperglycemia and hyperlipidemia. Insulin resistance in the animals was confirmed by IPITT and HOMA-IR. DM rats showed evident hyperglycemia (all p<0.05 vs. NCD, HFD) and insulin resistance after a 4-week period of HF diet both in the DM and HFD groups (Table 1 and Figure S1). The HOMA-IR of the DM group was significantly higher than that of the HFD group, however at the end of the experiment, the difference between the two groups did not reach statistical significance (P=0.35, Table 1). Specifically, at week 4 we observed a significant increase in fasting insulin levels along with HOMA-IR in HFD animals compared to NCD animals (77.47±5.16 vs. 55.21±1.46, P<0.01 for insulin and 2.95±5.16 vs. 1.58±0.15, P=0.04 for HOMA-IR respectively, Table 1). Serum triglyceride levels were also significantly higher in the HFD group when compared to the NCD group at 4 weeks of a HF diet (0.44±0.09 vs. 0.22±0.11, P=0.04). After STZ injection, both serum total cholesterol and total triglycerides were sustained at higher levels in the DCM group when compared to the NCD group (P<0.05) during diabetes (Table 1).
Table 1.
Comparisons of metabolic and inflammation markers among different groups
| FBG (mmol/L) | TG (mmol/L) | TC (mmol/L) | HDL-c (mmol/L) | LDL-c (mmol/L) | Fasting insulin (pmol/L) | HOMA-IR | IL-6 (pmol/mL) | TNF-α (pmol/mL) | |
|---|---|---|---|---|---|---|---|---|---|
| 1 week | 0.83 | 0.16 | 0.89 | 0.37 | 0.83 | 0.76 | 0.65 | 0.33 | 0.71 |
| NCD | 5.70±0.49 | 0.14±0.04 | 1.65±0.22 | 1.14±0.25 | 0.73±0.07 | 52.93±1.40 | 1.56±0.12 | 34.01±0.86 | 3.96±1.19 |
| HFD | 5.76±0.52 | 0.21±0.12 | 1.67±0.42 | 1.29±0.37 | 0.72±0.11 | 55.62±2.68 | 1.91±0.15 | 33.36±0.88 | 4.03±1.30 |
| 4 weeks | 0.07 | 0.04 | 0.13 | 0.07 | 0.83 | <0.01 | 0.04 | <0.0001 | <0.0001 |
| NCD | 5.53±0.59 | 0.22±0.11 | 1.74±0.17 | 0.80±0.13 | 0.54±0.05 | 55.21±1.46 | 1.58±0.15 | 34.82±0.79 | 4.00±1.16 |
| HFD | 6.33±0.75 | 0.44±0.09 | 1.94±0.20 | 1.01±0.20 | 0.53±0.12 | 77.47±5.16* | 2.95±0.31* | 184.29±7.80*** | 6.64±1.20*** |
| 9 weeks | <.0001 | 0.05 | 0.04 | 0.02 | 0.03 | 0.08 | <.01 | <.0001 | <.0001 |
| NCD | 5.40±0.58 | 0.26±0.17 | 1.78±0.08 | 1.06±0.16 | 0.48±0.07 | 54.67±3.00 | 1.62±0.14 | 36.36±0.86 | 4.01±1.23 |
| HFD | 6.53±0.96 | 0.63±0.13* | 2.31±0.18* | 1.57±0.20* | 0.58±0.09 | 82.27±9.72** | 2.93±0.46* | 289.50±11.61*** | 12.14±0.80*** |
| DM | 17.30±3.09***,### | 0.57±0.20* | 1.80±0.37# | 0.94±0.33## | 0.45±0.04# | 72.37±21.25* | 6.28±1.81***,## | 445.43±6.76***,### | 17.76±1.90***,### |
| 14 weeks | <.0001 | 0.02 | 0.11 | 0.21 | <.01 | 0.27 | <.001 | <.0001 | <.0001 |
| NCD | 5.2±0.80 | 0.43±0.19 | 1.67±0.19 | 1.01±0.25 | 0.49±0.07 | 55.60±2.31 | 1.72±0.09 | 36.21±0.73 | 4.08±1.20 |
| HFD | 8.20±2.17 | 1.59±0.28* | 2.30±0.49* | 1.14±0.23 | 0.39±0.07* | 65.35±8.57 | 3.24±0.29* | 418.41±14.66*** | 17.32±0.66*** |
| DM | 15.83±3.45***,### | 1.68±1.08* | 2.07±0.06 | 0.80±0.40 | 0.30±0.08*** | 61.64±7.87 | 5.32±1.25*** | 638.48±15.25***,### | 23.44±0.67***,### |
| 21 weeks | <.0001 | <.01 | 0.02 | 0.53 | 0.16 | <.0001 | <.0001 | <.0001 | <.0001 |
| NCD | 6.27±1.00 | 0.29±0.19 | 1.70±0.10 | 1.47±0.33 | 0.66±0.11 | 54.68±1.26 | 1.78±0.21 | 36.45±1.00 | 4.10±1.11 |
| HFD | 8.70±4.18 | 2.11±0.16** | 2.20±0.12** | 1.12±0.25 | 0.40±0.10 | 106.28±32.13*** | 4.45±0.27*** | 580.02±10.56*** | 25.38±0.50*** |
| DM | 18.98±1.85*** | 2.78±1.56*** | 2.33±0.71** | 1.21±0.47 | 0.68±0.49 | 55.97±2.10 | 5.92±0.73*** | 850.36±9.27*** | 32.97±1.07*** |
| MET | 16.15±5.65*** | 2.41±0.34*** | 1.87±0.22 | 1.12±0.23 | 0.49±0.10 | 61.95±11.55 | 5.53±1.99*** | 634.49±7.06‡‡‡ | 24.77±1.50‡‡‡ |
| STAG10 | 17.17±3.81*** | 2.32±1.00*** | 2.26±0.28** | 1.27±0.27 | 0.55±0.17 | 56.64±1.62 | 5.30±1.13*** | 630.91±10.17‡‡‡ | 24.81±0.36‡‡‡ |
| STAG20 | 17.24±3.86*** | 1.56±0.51*,‡ | 2.16±0.37** | 1.21±0.31 | 0.40±0.10*,‡ | 56.13±2.61 | 5.93±0.63*** | 633.08±14.29‡‡‡ | 24.94±0.69‡‡‡ |
HOMA-IR = fasting insulin (μU/mL)*fasting glucose (mmol/L)/22.5.
P<0.05, other groups VS. NCD group;
P<0.01, other groups VS. NCD group;
P<0.001, other groups VS. NCD group.
P<0.05, other groups VS. HFD group;
P<0.01, other groups VS. HFD group;
P<0.001, other groups VS. HFD group.
P<0.05, drug groups VS. DM group;
‡‡P<0.01, drug groups VS. DM group;
P<0.001, drug groups VS. DM group.
n=5 in each group. P value: comparison among NCD, HFD, DM, MET, STAG10 and STAG20 groups.
Treatment with both metformin and sitagliptin significantly lowered FBG as well as ameliorate insulin resistance in DM rats with no significant difference between the two drugs. High dosage STAG (STAG20 group) could reduce triglycerides, while metformin regulated total cholesterol levels (Table 1).
Pathology remodeling and LV function of DCM rats
All baseline data including body weight (BW), heart rate (HR), systolic and diastolic blood pressure showed no significant differences among all experimental groups (Table 2). Structural remodeling of left ventricle appeared starting in week 4 in DM rats and developed into diabetic cardiomyopathy by week 9. H-E staining revealed DCM hearts patterned in small, diffuse, non-uniform patches, as well as destroyed and disorganized collagen network structures in the interstitial and perivascular areas of animals with DCM (Figure 2A). The HW/BW ratio decreased after metformin or sitagliptin treatment to different extents when compared with the DM group (MET 2.77±0.18, P<0.001; STAG-10 3.10±0.32, P=0.10; STAG-20 3.06±0.11, P=0.09 vs. DM 3.29±0.18). We surveyed interstitial fibrosis with Masson trichrome (Figure 2B1, 2B2) and Picrosirius red staining (Figure 2C1, 2C2). We found a prominent reduction of interstitial fibrosis in treatment groups with a significant loss of interstitial fibrosis in groups treated with MET compared to animals treated with STAG (P<0.001). In addition, total collagen content was much lower in the MET group in comparison to the DM and STAG group (P<0.001, Figure 2D, 2E).
Table 2.
Comparisons of cardiac function index among different groups
| BW (g) | HW (mg) | HW/BW (mg/g) | HR (bpm) | SBP (mmHg) | DBP (mmHg) | BNP | |
|---|---|---|---|---|---|---|---|
| 1 week | 0.13 | 0.51 | 0.4 | 0.67 | 0.24 | 0.13 | 0.25 |
| NCD | 283±22.05 | 463.77±207.44 | 1.95±0.06 | 356.51±71.16 | 96.57±8.53 | 38.91±13.19 | 575.35±20.86 |
| HFD | 300.78±21.96 | 533.78±207.33 | 1.99±0.08 | 377.23±57.85 | 104.70±8.96 | 52.31±7.54 | 603.47±38.47 |
| 4 weeks | 0.02 | <0.01 | 0.41 | 0.31 | 0.01 | 0.63 | 0.19 |
| NCD | 317.83±17.99 | 657.38±35.78 | 2.07±0.06 | 365.41±46.36 | 108.60±14.44 | 83.25±22.56 | 566.73±30.69 |
| HFD | 351.33±21.94* | 772.07±9.24** | 2.10±0.03 | 391.96±40.05 | 128.48±8.03 | 88.93±16.56 | 608.30±22.17 |
| 9 weeks | 0.02 | 0.17 | 0.06 | 0.29 | 0.37 | 0.26 | <.0001 |
| NCD | 440.00±54.86 | 904.33±182.26 | 2.04±0.17 | 367.59±14.43 | 114.37±9.45 | 84.53±11.23 | 583.23±23.62 |
| HFD | 537.00±14.73* | 1262.67±222.63 | 2.35±0.38 | 379.47±44.69 | 127.10±11.50 | 87.61±17.04 | 1038.68±48.36*** |
| DM | 443.00±25.00# | 1119.00±190.89 | 2.56±0.24* | 400.25±5.73 | 139.36±30.87 | 108.02±23.54 | 1357.42±63.60***,### |
| 14 weeks | 0.02 | <.01 | <.001 | 0.63 | 0.08 | 0.03 | <.0001 |
| NCD | 492.00±39.57 | 1006.50±220.91 | 2.03±0.34 | 399.36±34.31 | 132.96±9.52 | 102.79±10.89 | 590.87±15.81 |
| HFD | 573.33±20.82* | 1675.32±144.85*** | 2.85±0.24** | 384.36±5.76 | 135.10±1.08 | 95.41±4.27 | 2400.45±129.52*** |
| DM | 485.67±35.84# | 1219.60±169.32# | 2.69±0.25*** | 384.13±29.16 | 147.39±13.14* | 115.07±9.71*,# | 3055.79±200.22***,### |
| 21 weeks | <.001 | 0.0001 | <.001 | 0.04 | 0.15 | 0.65 | <.0001 |
| NCD | 517.83±30.68 | 1121.48±84.37 | 2.19±0.10 | 400.89±42.60 | 135.82±20.16 | 107.83±27.94 | 594.45±20.11 |
| HFD | 615.67±39.80** | 1709.49±147.37*** | 2.84±0.13*** | 394.63±15.16 | 145.12±2.30 | 114.27±4.03 | 2859.26±131.22*** |
| DM | 513.00±6.24 | 1511.33±131.88*** | 3.29±0.18*** | 329.86±26.44* | 121.06±10.18 | 99.68±10.67 | 3545.06±202.36*** |
| MET | 480.67±5.03 | 1299.67±58.50‡‡‡ | 2.77±0.18***,‡‡‡ | 303.75±86.98** | 140.95±6.40 | 95.30±22.40 | 2437.61±53.08‡‡‡ |
| STAG10 | 455.67±31.89*,‡ | 1400.40±154.34**,‡‡ | 3.10±0.32*** | 329.65±47.19* | 123.85±14.94 | 101.38±13.43 | 2788.85±131.25‡‡‡ |
| STAG20 | 467.33±60.27* | 1451.50±194.43** | 3.06±0.11*** | 331.92±28.50 | 132.20±11.65 | 88.84±29.99 | 2677.14±131.41‡‡‡ |
P<0.05, other groups VS. NCD group;
P<0.01, other groups VS. NCD group;
P<0.001, other groups VS. NCD group.
P<0.05, other groups VS. HFD group;
##P<0.01, other groups VS. HFD group;
P<0.001, other groups VS. HFD group.
P<0.05, drug groups VS. DM group;
P<0.01, drug groups VS. DM group;
P<0.001, drug groups VS. DM group.
n=5 in each group. P value: comparison among NCD, HFD, DM, MET, STAG10 and STAG20 groups.
Figure 2.

Variation of pathology remodeling, cardiomyocyte apoptosis and JAK2/STAT3 pathway activity in different groups. A-F: A. H&E staining; B. Masson trichrome staining, C. Picrosirius red staining, D, E. Immunohistochemical staining of collagen I& collagen III, F. TUNEL staining (1) and their quantitative analysis (2) the order of each picture band represents NCD, HFD, DM, MET, STAG10, STAG20 group from left to right, respectively. G1&G2: Western blot and quantitative analysis of p-STAT3/STAT3, p-ERK/ERK, C-caspase3. Data are mean ± SEM; n=4-5 per group, *P<0.05 vs. DM; #P<0.05 vs. MET.
Furthermore, hemodynamic and echocardiography measurements showed decreased LVEDd (Figure 3A, 3E) and increased LVEF, E’/A’ in hypoglycemic treated rats, indicating improved LV function (Figure 3G). Particularly, Metformin treated animals showed the best improvement in LV diastolic dysfunction versus sitagliptin treated animals. LV dysfunction was drastically improved in the MET group with a higher E’/A’ compared to STAG groups (Figure 3C, 3F). To further confirm the LV diastolic dysfunction, hemodynamic measurements were performed to confirm LVEDP through cardiac catheterization (Figure 3D, 3H), with the results being consistent with UCG. Both dosages of STAG (STAG10 and STAG20) alleviated cardiac pathology remodeling as compared with the DM group, but the improvement was inferior to that of the group treated with metformin.
Figure 3.

Echocardiography and hemodynamics measurement images with quantitative analysis at week 21 of different groups. A-C, E-G: Typical image of echocardiography and the quantitative analysis results of LEVDd, E/A and E’/A’, D, H: hemodynamics measurement and analysis of LVEDP. Picture represents NCD, HFD, DM, MET, STAG10, STAG20 group respectively from left to right. *P<0.05, **P<0.01 and ***P<0.005 between compared groups.
Inflammation reaction and JAK2/STAT3 pathway during DCM development
Coincident with cardiac interstitial fibrosis and apoptosis, immunohistochemistry analysis revealed an abundant inflammatory response in DM rats (Figure 4A). The expression levels of TNF-α (Figure 4A1) and IL-6 (Figure 4A2) were significantly aggravated in the DM group when compared with the NCD group. Likewise, the TNF-α and IL-6 expression levels were also increased in the HFD group when compared with the NCD group, but less so than the DM group. The MET group also displayed a significant decrease of TNF-α and IL-6 content compared with those of the DM group. No significant differences were found in the levels of TNF-α and IL-6 among the MET and STAG groups (Table 1). As a pro-inflammatory cytokine induced pathway, JAK2/STAT3 showed disease course-dependent changes during DCM development. As shown in Figure 4B1-B3, total expression of JAK2 and STAT3 did not significantly differ among groups at different time points. Phosphorylation of JAK2 and STAT3 were higher in HFD rats compared to NCD rats at week 4 as assessed by antibody specific for pJAK2 and pSTAT3 in the tyrosine set. After STZ administration and DM was induced, the STAT3 phosphorylation levels in the HFD and DM rats were still significantly higher than the NCD rats at week 9. Phosphorylation of JAK2 and STAT3 continued to increase in HFD rats at week 14 and 21. Interestingly, phosphorylation levels of JAK2 and STAT3 were dramatically down-regulated in the DM group compared to the HFD group starting at week 14, with a further decrease at week 21. This is in accordance with the variation of rats’ heart function and myocardial inflammation reaction after DM was induced.
Figure 4.

Inflammation activity, cell apoptosis and time-dependent changes of JAK2/STAT3 pathway in different groups of DM rats. A: Immunohistochemical staining of TNF-α (A1) and IL-6 (A2) (brown staining considered positive staining; scale bar: 200 μm) and their quantitative analysis (A3, A4). *P<0.05 vs. NCD; #P<0.05 vs. DM; B: Representative western blot of JAK2/STAT3, BAX/BCL-2 and C-caspase3 in different groups (B1) and their quantitative analysis (B2~B5). JAK2/STAT3 pathway activity changes in a time-dependent pattern. *P<0.05 vs. NCD; #P<0.05 vs. HFD Data are mean ± SEM; n=4-5 per group.
Improved cardiomyocyte apoptosis in DCM accompanied with JAK2/STAT3 pathway activation
To explore the possible mechanisms underlying DCM treatment, we quantified apoptosis levels. Both the ratio of BAX/BCL-2 and the level of cleaved Caspase3 increased gradually starting at week 9 in the DM group in comparison to other groups. Similarly, BAX/BCL-2 and cleaved Caspase3 levels were also significantly increased in the HFD group when compared with the NCD group (Figure 4B1, 4B4, 4B5). TUNEL-staining was strongest in the DM group. Increased cardiomyocyte apoptosis levels in DM rats were significantly improved by MET administration, as evidenced by cleaved-Caspase 3 expression and TUNEL-positive cells showing decreased levels in the MET group but not in STAG groups (both STAG10 and STAG20).This indicates that metformin, but not STAG, could inhibit cell apoptosis in DM rats (Figure 2F1, 2F2). For further analysis, we measured pERK/ERK levels in MET and STAG group at week 21. Results showed that metformin treatment significantly induced ERK phosphorylation in DM rats while sitagliptin made no difference (Figure 2G1, 2G2). To further understand the role of the JAK2/STAT3 pathway in hypoglycemic therapy during DCM, we analyzed JAK2/STAT3 pathway activity in both MET and STAG groups. We found that metformin significantly increased the phosphorylation level of STAT3 at week 21 while sitagliptin treated groups showed no difference in phosphorylation levels (Figure 2G1, 2G2).
We proposed that the superior effect associated with the better improved pathology remodeling and LV function in metformin treated DCM rats, might be from JAK2/STAT3 pathway activation and we therefore performed the experiment in vitro to test this possibility as reported below.
In vitro study
Hyperglycemia induced cardiomyocyte apoptosis and optimal concentration
Our in vivo experiments show that cell apoptosis levels were significantly decreased accompanied with a lower fasting blood glucose in the HFD group in comparison to the DM group. We theorized that hyperglycemia may account for this difference, which agrees with findings in previous studies [4]. To test this idea, we explored the effect of different hyperglycemia conditions on cardiomyocyte apoptosis in vitro to determine the optimum glucose concentration and incubation duration. We found hyperglycemia with 33 mM for up to 48 hours increased the BAX/BCL-2 ratio most prominently. Flow cytometry was also performed to further investigate cardiomyocyte apoptosis. Quantitative analysis of flow cytometry showed a 53.8±4.73% apoptotic rate in high-glucose (33 mM) treated cells, which indicate that high-glucose (33 mM) could induce cardiac myocytes apoptosis regardless of osmotic pressure (P<0.001, Figure 5A, 5D).
Figure 5.

Results of in vitro study. NRCMs are assigned into different groups based on different glucose concentration and incubation durations to verify the interaction of metformin treatment and apoptosis induced by hyperglycemia. A-C: Expression of pSTA3/STAT3, BAX/BCL-2 tested by western blot in different treatment groups and time phase including HG, OC, MET, AG490 and AICAR. D: Detection of pro-survival effect of metformin by flow cytometry. Data are mean ± SEM; n=4-5 per group. A, B: *P<0.05 vs. 0h group, C: #P<0.05 vs. HG; *P<0.05 vs. OC, D: *P<0.05 vs. HG; #P<0.05 vs. MET + HG. OC: osmotic control, HG: high glucose, AICAR: 5-aminoimidazole-4-carboxamide ribonucleoside, a stimulator of AMPK. AG490: Tyrphostin B42, a kinase inhibitor of JAK2. Other abbreviations are same as Figure 1.
Alteration of JAK2/STAT3 pathway under high-glucose
Next we explored the activity of the JAK2/STAT3 pathway in cardiomyocytes under a high-glucose condition. The results show that cardiomyocytes incubated in 33 mM glucose demonstrated a significant inhibition of JAK2/STAT3 pathway activity, as well as STAT3 phosphorylation at tyrosine 705 compared to low osmotic glucose control after 48 h of incubation without altering total STAT3 levels (P<0.001, Figure 5A).
Effects of metformin on cell apoptosis
To verify the protective effect we saw from metformin treatment in regulating cardiac myocyte survival/death in vivo, we searched for the lowest therapeutic dose of metformin (0.1 mM, 0.5 mM, 1 mM). The results showed that expression of Bax was significantly reduced in the heart following 24 h of treatment with metformin (1 mM) as compared with DMSO, while Bcl-2 was significantly increased. Consequently, the ratio of Bcl-2 to Bax was increased with metformin treatment as compared to DMSO (Figure 5B, n=5, P<0.05). The benefit of metformin was also indicated by flow cytometry (the apoptosis rate was 32.03% vs. 53.08%, P<0.01), similar to 5-aminoimidazole-4-carboxamide ribonucleoside (AICAR), a stimulator of AMPK which could increase Thr172 phosphorylation of AMPK (35.77% vs. 32.03%, P>0.05, Figure 5D).
Inhibition of JAK2/STAT3 abolishes metformin-induced cardio-protection
To examine the role of the JAK/STAT3 pathway in mediating metformin-induced cardio-protection in vitro, JAK2 blockade AG490 (100 µmol/L) was used. We found that AG490 can block the cardio-protective effect induced by metformin treatment (1 mM) with an increased rate of apoptosis as indicated by Bax/Bcl2 ration and flow cytometry (the apoptosis rate in HG vs. MET + AG490 + HG group was 53.80% vs. 45.99, P>0.05, Figure 5C, 5D). These results suggest that JAK2/STAT3 activation is involved in metformin-induced cardiomyocyte apoptosis and cell survival.
Discussion
There is increasing evidence suggested that chronic low-grade inflammation can promote a pro-survival signaling pathway termed SAFE (survivor activating factor enhancement), of which STAT3 is the central component. The JAK2/STAT3 pathway plays a critical role in cardio-protection following many cardiac disorders, however its role in diabetic cardiomyopathy is still debated. To the best of our knowledge, this is the first study to demonstrate a time-dependent change in the JAK2/STAT3 pathway activity during the development of DCM. Our results indicate that STAT3 activation might be a novel mechanism taking part in the cardiovascular protection effect of metformin treatment in diabetic patients.
Our study also reveals that the JAK2/STAT3 pathway is continually activated in rat models fed a simple HFD, with rats demonstrating chronic inflammatory activation and left ventricular remodeling. However, the myocardial injury was significantly less than that in T2DM rats. Long-term simple HFD in rats could induce obesity and myocardial hypertrophy, which can develop into heart failure while the mechanisms mediating this development are still unclear [24]. These results also indicate that the JAK2/STAT3 pathway might play a compensatory protective role in simple HFD.
STAT3 signaling had been identified to be essential for maintaining glucose homoeostasis, and disturbance of this signaling pathway could contribute to the onset and progression of diabetes. However, the reported regulation of STAT3 activity in T2DM have been previously inconsistent. The knock-down of hepatic SirT1 can increase STAT3 acetylation and STAT3 phosphorylation (Y705), leading to decreased amounts of endogenous and insulin-stimulated glucose production and reduced fasting hyperglycemia in a T2DM rat model [25]. Treatment of high concentration insulin results in a reduction of both total and phosphorylated STAT3 protein levels, suggesting that STAT3 may demonstrate the same changes in the condition of hyperinsulinemia [26]. Chowdhury et al said that the JAK2/STAT3 pathway is inhibited in the axons of neurons derived from STZ-diabetic rats, and that enhancement of JAK2/STAT3 signaling can normalize mitochondrial polarization status and promote neurite growth [27]. As described by numerous studies, STAT3 activation is implicated in modulating the activity of downstream mediators such as p27kip1, p16ink4a and p21kip1, hence playing a key role in cell survival, proliferation, and differentiation [28-30]. However, very few previous studies have subcategorized a diabetes model into different stages like early, middle and late stages of diabetes, which might account for the inconsistent results reported. In our experiment, we used T2DM rats with different time courses and found that the JAK2/STAT3 pathway is regulated in a time-dependent manner, with pathway activity showing significant up-regulation in the early stage of diabetes and down-regulation in the middle to late stages.
Specifically, by comparing the different diabetes durations in the present study, we show that the JAK2/STAT3 pathway is significantly activated in the early stage of T2DM heart, but is then gradually suppressed. Contrarily, the inflammatory state was continually activated, and the relationship between the classical pathway and inflammatory did not match. This may partly explain the inconsistency reported between the previous findings, which may be related to the complicated regulation factors of the pathway. IL-6 paradox and IL-6 resistance might also partially explain the differences reported. Recently, some studies have highlighted that IL-6 resistance develops in the liver and changes sensitivity to IL-6 signals in peripheral tissues, along with insulin and leptin resistance [31]. A recent study also suggested that the resistance of cytokines could limit the biological efficiency of the inflammatory factors. For example, some patients who received interferon (IFN) treatment did not produce a response, known as IFN resistance [32]. These previous studies make us believe the inflammation cytokines resistance may also exist in diabetic cardiomyopathy. Comparing a simple HFD model with a type 2 diabetes rat model, we found similar degrees of inflammation in cardiomyodium but differences in JAK2/STAT3 signaling pathway. We therefore speculated hyperglycemia might be the main factor inducing JAK2/STAT3 pathway inhibition in the middle and late stages of diabetes. Our in vitro study further verified that indeed hyperglycemia alone was able to inhibit STAT3 activation and induce cardiomyocyte apoptosis.
An interesting and novel observation in our study is that metformin treatment can reduce the inflammation response in diabetic rat myocardium by activating the JAK2/STAT3 pathway. Although this is the first time reporting this finding, there are several theoretical foundations supporting this result. First, Xu found metformin could increase HO-1 expression and activate STAT3 [19]. Others have also shown that metformin can regulate SOCS3, a negative feedback factor of the JAK2/STAT3 pathway, and improve pathway “dis-regulation” [33,34]. In the present study, both metformin and sitagliptin were found to alleviate the inflammation reaction in DCM animal models. However, metformin showed a stronger protective effect in improving both cardiac remodeling and heart function, suggesting there might be some protection mechanisms independent from the upstream inflammation pathway. Further, we found metformin is able to activate the JAK2/STAT3 pathway while neither a low or high dosage of sitagliptin could. NRCM incubation showed that high glucose can induce cardiomyocyte apoptosis and that STAT3 activation is inhibited independent of osmatic pressure. Metformin can also ameliorate cell apoptosis, and increase activated levels of STAT3, indicating that AMPK may be involved. As we know, metformin is an anti-diabetic drug conclusively probed to avoid cardiac complications in diabetes. Consistent with former studies, our study also finds that metformin can ameliorate diabetic myocardium injury and hyperglycemic cardiomyocytes injury significantly better than treatment with sitagliptin.
Conclusions
The abnormal regulation of the JAK/STAT3 pathway may be one of the mechanisms for the development of diabetic cardiomyopathy with the synergy or subsequent uncontrollable inflammatory response. The favorable cardio-protective effect of metformin in type 2 diabetic rats may also be partly owing to its ability to activate the JAK2/STAT3 pathway. Based on the pivotal role of metformin and the inflammation reaction in DCM, it’s reasonable for us to conjecture that the JAK/STAT3 pathway has a more potent protective effect than other pre-existing therapies which might change the therapeutic concept of DCM and application of metformin in DM.
Acknowledgements
We acknowledge the invaluable help of all facilities (Zi-He Yang, Peihe Wang, Tuo Liang, Xiaopeng Liu, Guangpu Fan, Jue Ye, et al) of State Key Laboratory of Cardiovascular Disease, Department of Cardiology, Cardiovascular Institute, Fuwai Hospital. This paper was supported by grants from the National Natural Science Foundation of China 81700405 to Zhang Erli.
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Wild S, Roglic G, Green A, Sicree R, King H. Global prevalence of diabetes: estimates for the year 2000 and projections for 2030. Diabetes Care. 2004;27:1047–1053. doi: 10.2337/diacare.27.5.1047. [DOI] [PubMed] [Google Scholar]
- 2.Rubler S, Dlugash J, Yuceoglu YZ, Kumral T, Branwood AW, Grishman A. New type of cardiomyopathy associated with diabetic glomerulosclerosis. Am J Cardiol. 1972;30:595–602. doi: 10.1016/0002-9149(72)90595-4. [DOI] [PubMed] [Google Scholar]
- 3.Dandamudi S, Slusser J, Mahoney DW, Redfield MM, Rodeheffer RJ, Chen HH. The prevalence of diabetic cardiomyopathy: a population-based study in olmsted county, minnesota. J Card Fail. 2014;20:304–309. doi: 10.1016/j.cardfail.2014.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pan Y, Wang Y, Zhao Y, Peng K, Li W, Wang Y, Zhang J, Zhou S, Liu Q, Li X, Cai L, Liang G. Inhibition of JNK phosphorylation by a novel curcumin analog prevents high glucose-induced inflammation and apoptosis in cardiomyocytes and the development of diabetic cardiomyopathy. Diabetes. 2014;63:3497–3511. doi: 10.2337/db13-1577. [DOI] [PubMed] [Google Scholar]
- 5.Lecour S, James RW. When are pro-inflammatory cytokines SAFE in heart failure? Eur Heart J. 2011;32:680–685. doi: 10.1093/eurheartj/ehq484. [DOI] [PubMed] [Google Scholar]
- 6.Fischer P, Hilfiker-Kleiner D. Survival pathways in hypertrophy and heart failure: the gp130-STAT axis. Basic Res Cardiol. 2007;102:393–411. doi: 10.1007/s00395-007-0674-z. [DOI] [PubMed] [Google Scholar]
- 7.Verma SK, Krishnamurthy P, Barefield D, Singh N, Gupta R, Lambers E, Thal M, Mackie A, Hoxha E, Ramirez V, Qin G, Sadayappan S, Ghosh AK, Kishore R. Interleukin-10 treatment attenuates pressure overload-induced hypertrophic remodeling and improves heart function via signal transducers and activators of transcription 3-dependent inhibition of nuclear factor-κB. Circulation. 2012;126:418–429. doi: 10.1161/CIRCULATIONAHA.112.112185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee JK, Won C, Yi EH, Seok SH, Kim MH, Kim SJ, Chung MH, Lee HG, Ikuta K, Ye SK. Signal transducer and activator of transcription 3 (Stat3) contributes to T-cell homeostasis by regulating pro-survival Bcl-2 family genes. Immunology. 2013;140:288–300. doi: 10.1111/imm.12133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Szczepanek K, Chen Q, Derecka M, Salloum FN, Zhang Q, Szelag M, Cichy J, Kukreja RC, Dulak J, Lesnefsky EJ, Larner AC. Mitochondrial-targeted Signal transducer and activator of transcription 3 (STAT3) protects against ischemia-induced changes in the electron transport chain and the generation of reactive oxygen species. J Biol Chem. 2011;286:29610–20. doi: 10.1074/jbc.M111.226209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Alvarez-Guardia D, Palomer X, Coll T, Serrano L, Rodríguez-Calvo R, Davidson MM, Merlos M, El Kochairi I, Michalik L, Wahli W, Vázquez-Carrera M. PPARβ/δ activation blocks lipid-induced inflammatory pathways in mouse heart and human cardiac cells. Biochim Biophys Acta. 2011;1811:59–67. doi: 10.1016/j.bbalip.2010.11.002. [DOI] [PubMed] [Google Scholar]
- 11.Katare RG, Caporali A, Oikawa A, Meloni M, Emanueli C, Madeddu P. Vitamin B1 analog benfotiamine prevents diabetes-induced diastolic dysfunction and heart failure through Akt/Pim-1-mediated survival pathway. Circ Heart Fail. 2010;3:294–305. doi: 10.1161/CIRCHEARTFAILURE.109.903450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xie Z, Lau K, Eby B, Lozano P, He C, Pennington B, Li H, Rathi S, Dong Y, Tian R, Kem D, Zou MH. Improvement of cardiac functions by chronic metformin treatment is associated with enhanced cardiac autophagy in diabetic OVE26 mice. Diabetes. 2011;60:1770–1778. doi: 10.2337/db10-0351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.He C, Zhu H, Li H, Zou MH, Xie Z. Dissociation of Bcl-2-Beclin1 complex by activated AMPK enhances cardiac autophagy and protects against cardiomyocyte apoptosis in diabetes. Diabetes. 2013;62:1270–81. doi: 10.2337/db12-0533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vasamsetti SB, Karnewar S, Kanugula AK, Thatipalli AR, Kumar JM, Kotamraju S. Metformin inhibits monocyte-to-macrophage differentiation via AMPK-mediated inhibition of STAT3 activation: potential role in atherosclerosis. Diabetes. 2015;64:2028–2041. doi: 10.2337/db14-1225. [DOI] [PubMed] [Google Scholar]
- 15.Zheng L, Yang W, Wu F, Wang C, Yu L, Tang L, Qiu B, Li Y, Guo L, Wu M, Feng G, Zou D, Wang H. Prognostic significance of AMPK activation and therapeutic effects of metformin in hepatocellular carcinoma. Clin Cancer Res. 2013;19:5372–80. doi: 10.1158/1078-0432.CCR-13-0203. [DOI] [PubMed] [Google Scholar]
- 16.Feng Y, Ke C, Tang Q, Dong H, Zheng X, Lin W, Ke J, Huang J, Yeung SC, Zhang H. Metformin promotes autophagy and apoptosis in esophageal squamous cell carcinoma by downregulating Stat3 signaling. Cell Death Dis. 2014;5:e1088. doi: 10.1038/cddis.2014.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen X, Walther FJ, Sengers RM, Laghmani el H, Salam A, Folkerts G, Pera T, Wagenaar GT. Metformin attenuates hyperoxia-induced lung injury in neonatal rats by reducing the inflammatory response. Am J Physiol Lung Cell Mol Physiol. 2015;309:L262–70. doi: 10.1152/ajplung.00389.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cameron AR, Morrison VL, Levin D, Mohan M, Forteath C, Beall C, McNeilly AD, Balfour DJ, Savinko T, Wong AK, Viollet B, Sakamoto K, Fagerholm SC, Foretz M, Lang CC, Rena G. Anti-inflammatory effects of metformin irrespective of diabetes status. Circ Res. 2016;119:652–65. doi: 10.1161/CIRCRESAHA.116.308445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xu J, Li H, Irwin MG, Xia ZY, Mao X, Lei S, Wong GT, Hung V, Cheung CW, Fang X, Clanachan AS, Xia Z. Propofol ameliorates hyperglycemia-induced cardiac hypertrophy and dysfunction via heme oxygenase-1/signal transducer and activator of transcription 3 signaling pathway in rats. Crit Care Med. 2014;42:e583–94. doi: 10.1097/CCM.0000000000000415. [DOI] [PubMed] [Google Scholar]
- 20.Kelleni MT, Amin EF, Abdelrahman AM. Effect of metformin and sitagliptin on doxorubicin-induced cardiotoxicity in rats: Impact of oxidative stress, inflammation, and apoptosis. J Toxicol. 2015;2015:424813. doi: 10.1155/2015/424813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nagamine A, Hasegawa H, Hashimoto N, Yamada-Inagawa T, Hirose M, Kobara Y, Tadokoro H, Kobayashi Y, Takano H. The effects of DPP-4 inhibitor on hypoxia-induced apoptosis in human umbilical vein endothelial cells. J Pharmacol Sci. 2017;133:42–48. doi: 10.1016/j.jphs.2016.12.003. [DOI] [PubMed] [Google Scholar]
- 22.Kubota A, Takano H, Wang H, Hasegawa H, Tadokoro H, Hirose M, Kobara Y, Yamada-Inagawa T, Komuro I, Kobayashi Y. DPP-4 inhibition has beneficial effects on the heart after myocardial infarction. J Mol Cell Cardiol. 2016;91:72–80. doi: 10.1016/j.yjmcc.2015.12.026. [DOI] [PubMed] [Google Scholar]
- 23.Tan WQ, Wang K, Lv DY, Li PF. Foxo3a inhibits cardiomyocyte hypertrophy through transactivating catalase. J Biol Chem. 2008;283:29730–9. doi: 10.1074/jbc.M805514200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang Z, Li L, Zhao H, Peng S, Zuo Z. Chronic high fat diet induces cardiac hypertrophy and fibrosis in mice. Metabolism. 2015;64:917–25. doi: 10.1016/j.metabol.2015.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Erion DM, Yonemitsu S, Nie Y, Nagai Y, Gillum MP, Hsiao JJ, Iwasaki T, Stark R, Weismann D, Yu XX, Murray SF, Bhanot S, Monia BP, Horvath TL, Gao Q, Samuel VT, Shulman GI. SirT1 knockdown in liver decreases basal hepatic glucose production and increases hepatic insulin responsiveness in diabetic rats. Proc Natl Acad Sci U S A. 2009;106:11288–93. doi: 10.1073/pnas.0812931106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xu J, Ji S, Venable DY, Franklin JL, Messina JL. Prolonged insulin treatment inhibits GH signaling via STAT3 and STAT1. J Endocrinol. 2005;184:481–92. doi: 10.1677/joe.1.05977. [DOI] [PubMed] [Google Scholar]
- 27.Chowdhury SR, Saleh A, Akude E, Smith DR, Morrow D, Tessler L, Calcutt NA, Fernyhough P. Ciliary neurotrophic factor reverses aberrant mitochondrial bioenergetics through the JAK/STAT pathway in cultured sensory neurons derived from streptozotocin-induced diabetic rodents. Cell Mol Neurobiol. 2014;34:643–9. doi: 10.1007/s10571-014-0054-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Siggins RW, Melvan JN, Welsh DA, Bagby GJ, Nelson S, Zhang P. Alcohol suppresses the granulopoietic response to pulmonary Streptococcus pneumoniae infection with enhancement of STAT3 signaling. J Immunol. 2011;186:4306–13. doi: 10.4049/jimmunol.1002885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang S, Suvannasankha A, Crean CD, White VL, Chen CS, Farag SS. The novel histone deacetylase inhibitor, AR-42, inhibits gp130/Stat3 pathway and induces apoptosis and cell cycle arrest in multiple myeloma cells. Int J Cancer. 2011;129:204–13. doi: 10.1002/ijc.25660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sen N, Che X, Rajamani J, Zerboni L, Sung P, Ptacek J, Arvin AM. Signal transducer and activator of transcription 3 (STAT3) and survivin induction by varicella-zoster virus promote replication and skin pathogenesis. Proc Natl Acad Sci U S A. 2012;109:600–5. doi: 10.1073/pnas.1114232109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cansby E, Nerstedt A, Amrutkar M, Durán EN, Smith U, Mahlapuu M. Partial hepatic resistance to IL-6-induced inflammation develops in type 2 diabetic mice, while the anti-inflammatory effect of AMPK is maintained. Mol Cell Endocrinol. 2014;393:143–51. doi: 10.1016/j.mce.2014.06.014. [DOI] [PubMed] [Google Scholar]
- 32.Brinckmann A, Axer S, Jakschies D, Dallmann I, Grosse J, Patzelt T, Bernier T, Emmendoerffer A, Atzpodien J. Interferon-alpha resistance in renal carcinoma cells is associated with defective induction of signal transducer and activator of transcription 1 which can be restored by a supernatant of phorbol 12-myristate 13-acetate stimulated peripheral blood mononuclear cells. Br J Cancer. 2002;86:449–55. doi: 10.1038/sj.bjc.6600066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mori H, Hanada R, Hanada T, Aki D, Mashima R, Nishinakamura H, Torisu T, Chien KR, Yasukawa H, Yoshimura A. Socs3 deficiency in the brain elevates leptin sensitivity and confers resistance to diet-induced obesity. Nat Med. 2004;10:739–43. doi: 10.1038/nm1071. [DOI] [PubMed] [Google Scholar]
- 34.Sarvas JL, Khaper N, Lees SJ. The IL-6 paradox: context dependent interplay of SOCS3 and AMPK. J Diabetes Metab. 2013;(Suppl 13) doi: 10.4172/2155-6156.S13-003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.