

Abstract
To broaden our understanding of human neurodevelopment, we profiled transcriptomic and epigenomic landscapes across brain regions and/or cell types for the entire span of prenatal and postnatal development. Integrative analysis revealed temporal, regional, sex, and cell type–specific dynamics. We observed a global transcriptomic cup-shaped pattern, characterized by a late fetal transition associated with sharply decreased regional differences and changes in cellular composition and maturation, followed by a reversal in childhood-adolescence, and accompanied by epigenomic reorganizations. Analysis of gene coexpression modules revealed relationships with epigenomic regulation and neurodevelopmental processes. Genes with genetic associations to brain-based traits and neuropsychiatric disorders (including MEF2C, SATB2, SOX5, TCF4, and TSHZ3) converged in a small number of modules and distinct cell types, revealing insights into neurodevelopment and the genomic basis of neuropsychiatric risks.
Graphical Abstract
INTRODUCTION:
The brain is responsible for cognition, behavior, and much of what makes us uniquely human. The development of the brain is a highly complex process, and this process is reliant on precise regulation of molecular and cellular events grounded in the spatiotemporal regulation of the transcriptome. Disruption of this regulation can lead to neuropsychiatric disorders.
RATIONALE:
The regulatory, epigenomic, and transcriptomic features of the human brain have not been comprehensively compiled across time, regions, or cell types. Understanding the etiology of neuropsychiatric disorders requires knowledge not just of endpoint differences between healthy and diseased brains but also of the developmental and cellular contexts in which these differences arise. Moreover, an emerging body of research indicates that many aspects of the development and physiology of the human brain are not well recapitulated in model organisms, and therefore it is necessary that neuropsychiatric disorders be understood in the broader context of the developing and adult human brain.
RESULTS:
Here we describe the generation and analysis of a variety of genomic data modalities at the tissue and single-cell levels, including transcriptome, DNA methylation, and histone modifications across multiple brain regions ranging in age from embryonic development through adulthood. We observed a widespread transcriptomic transition beginning during late fetal development and consisting of sharply decreased regional differences. This reduction coincided with increases in the transcriptional signatures of mature neurons and the expression of genes associated with dendrite development, synapse development, and neuronal activity, all of which were temporally synchronous across neocortical areas, as well as myelination and oligodendrocytes, which were asynchronous. Moreover, genes including MEF2C, SATB2, and TCF4, with genetic associations to multiple brain-related traits and disorders, converged in a small number of modules exhibiting spatial or spatiotemporal specificity.
CONCLUSION:
We generated and applied our dataset to document transcriptomic and epigenetic changes across human development and then related those changes to major neuropsychiatric disorders. These data allowed us to identify genes, cell types, gene coexpression modules, and spatiotemporal loci where disease risk might converge, demonstrating the utility of the dataset and providing new insights into human development and disease.
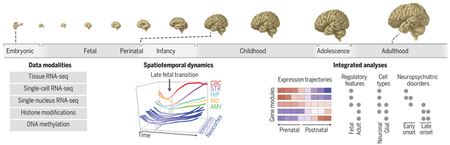
Spatiotemporal dynamics of human brain development and neuropsychiatric risks. Human brain development begins during embryonic development and continues through adulthood (top). Integrating data modalities (bottom left) revealed age- and cell type–specific properties and global patterns of transcriptional dynamics, including a late fetal transition (bottom middle).We related the variation in gene expression (brown, high; purple, low) to regulatory elements in the fetal and adult brains, cell type–specific signatures, and genetic loci associated with neuropsychiatric disorders (bottom right; gray circles indicate enrichment for corresponding features among module genes). Relationships depicted in this panel do not correspond to specific observations. CBC, cerebellar cortex; STR, striatum; HIP, hippocampus; MD, mediodorsal nucleus of thalamus; AMY, amygdala.
The development of the human central nervous system is an intricate process that unfolds over several decades, during which time numerous distinct cell types are generated and assembled into functionally distinct circuits and regions (1–4). These basic components of the brain are neither born mature nor static throughout their lifetimes; over the course of development, they undergo a variety of molecular and morphological changes. As a consequence, the characteristics of a given cell, circuit, or brain region described at a given time offer only a snapshot of that unit.
The processes guiding the development of the nervous system are reliant on the diversity and precise spatiotemporal regulation of the transcriptome (1–4). There is increasingly persuasive evidence that dysregulation of the transcriptional, regulatory, and epigenetic processes underlying the spatial architecture and temporal progression of human neurodevelopment can have dire consequences for brain function or strongly affect the risk of neuropsychiatric disorders (5–7). Indeed, many of the regulatory and epigenomic features governing the transcriptome of the developing human nervous system may be specific to particular developmental contexts in humans or closely related primate species. As such, it is difficult to identify or fully study human functional genomic elements using most common model organisms or cell culture systems (8). Assaying human cells and postmortem tissues solves some of these problems, but challenges, including the availability and quality of developmental tissue, limit the scale of such analyses. Consequently, despite ongoing efforts, our understanding of different facets of the transcriptional, regulatory, and epigenetic architecture of the human nervous system, particularly during early developmental periods, remains highly incomplete (8–21).
To begin rectifying this deficiency, the National Institutes of Health–funded PsychENCODE (http://psychencode.org) and BrainSpan Consortia (www.brainspan.org) sought to generate and analyze multidimensional genomics data from the developing and adult human brain in healthy and disease states.
Study design and data generation
Here we describe the generation and integrated analysis of multiple genomic data modalities, including transcriptomic profile, DNA methylation status, histone modifications, CTCF binding sites, and genotype generated from bulk tissue (1230 samples from 48 brains) or at the single-cell or single-nucleus level (18,288 cells or nuclei from 12 brains) from 60 de-identified postmortem brains obtained from clinically and histopathologically unremarkable donors of both sexes and multiple ancestries. Subject ages ranged from 5 postconceptional weeks (PCW) to 64 postnatal years (PY) (Fig. 1 and tables S1 to S6). Genotyping of DNA extracted from brain with a HumanOmni2.5–8 BeadChip confirmed subject ancestry and revealed no obvious genomic abnormalities (22).
Fig. 1. Overview of the data generated in this study.

(A) The developmental time span of the human brain, from embryonic ages (≤8 PCW) through fetal development, infancy, childhood, adolescence, and adulthood, with PCW and PY indicated. Below is the distribution of samples in this study across broad developmental phases (embryonic to adulthood), age [5 PCW to 64 PY (19)], and developmental windows (W1 to W9). Each circle represents a brain, and color indicates the sex [red circles (female) and blue circles (male)]. (B) Postmortem human brains sampled for different data modalities in this study are indicated.
For transcriptome analysis, tissue-level mRNA sequencing (mRNA-seq) was performed on a total of 607 histologically verified, high-quality tissue samples from 16 anatomical brain regions [11 areas of the neocortex (NCX), hippocampus (HIP), amygdala (AMY), striatum (STR), mediodorsal nucleus of thalamus (MD), and cerebellar cortex (CBC)] involved in higher-order cognition and behavior [Fig. 2A, (22)]. These regions were systematically dissected from 41 brains ranging in age from 8 PCW to 40 PY [18 females and 23 males; postmortem interval (PMI) = 12.9 ± 10.4 hours; tissue pH = 6.5 ± 0.3; RNA integrity number = 8.8 ± 1] (Fig. 1 and table S1). Because of the limited amounts of prenatal samples, small-RNA sequencing (smRNA-seq) was performed on 16 regions of 22 postnatal brains, with 278 samples passing quality control measures (Fig. 1 and table S2). These tissue-level RNA-seq analyses were complemented by single-cell RNA sequencing (scRNA-seq) data generated from 1195 cells collected from embryonic fronto-parietal neocortical wall and mid-fetal fronto-parietal neocortical plate and adjacent subplate zone of an independent set of nine brains ranging in age from 5 to 20 PCW (Fig. 1 and table S3) and single-nuclei RNA sequencing data (snRNA-seq) generated from 17,093 nuclei from the dorso-lateral prefrontal cortex (DFC, also termed DLPFC) of three adult brains (Fig. 1 and table S4). For epigenome analyses, DNA cytosine methylation was profiled with the Infinium HumanMethylation450 BeadChip in 269 postnatal samples covering the same 16 brain regions analyzed by RNA-seq (Fig. 1 and table S5). Additional epigenomic data was generated with chromatin immunoprecipitation sequencing (ChIP-seq) for histone marks H3K4me3 (trimethylated histone H3 lysine 4), H3K27me3 (trimethylated histone H3 lysine 27), and H3K27ac (acetylated histone H3 lysine 27) and the epigenetic regulatory protein CTCF, which together identify a large fraction of promoters, repressors, active enhancers, and insulators. These data were generated from DFC and CBC of a subset of samples from mid-fetal, infant, and adult brains (Fig. 1 and table S6). Stringent quality control measures (figs. S1 to S8) were applied to all datasets before in-depth analyses. We also validated some results by applying independent approaches (figs. S9, S10, and S18). Finally, to enable more powerful comparisons, we grouped specimens into nine time windows (W1 to W9) on the basis of major neurodevelopmental milestones and unsupervised transcriptome-based temporal arrangement of constituent specimens (Fig. 1A and tables S1 to S6).
Fig. 2. Global transcriptomic architecture of the developing human brain.

(A) mRNA-seq dataset includes 11 neocortical areas (NCX) and five additional regions of the brain. IPC, posterior inferior parietal cortex; A1C, primary auditory (A1) cortex; STC, superior temporal cortex; ITC, inferior temporal cortex; V1C, primary visual (V1) cortex. (B) The first two multidimensional scaling components from gene expression showed samples from late fetal ages and early infancy (W5, gray) clustered between samples from exclusively prenatal windows (W1 to W4, blue) and exclusively postnatal windows (W6 to W9, red). (C) Intraregional Pearson’s correlation analysis found that samples within exclusively prenatal (W1 to W4) or postnatal (W6 to W9) windows correlated within, but not across, those ages. (D) Interregional transcriptomic differences revealed a developmental cup-shaped pattern in brain development. The interregional difference was measured as the upper quartile of the average absolute difference in gene expression of each area compared to all other areas. (E) AC-PCA for samples from all brain regions at late mid-fetal ages (W4), late fetal ages and early infancy (W5), and early adulthood (W9) showed that interregional differences were generally greater during W4 and W9 but reduced across W5. (F) Pairwise distance across samples using the first two principal components for all regions (left) or excluding one region at a time (right) demonstrated that the reduction of variation we observed is common across multiple brain regions, once the most differentiated transcriptomic profile (the cerebellum) is excluded. The shaded bands are 95% confidence intervals of the fitted lines.
Global spatiotemporal dynamics
We found that most protein-coding genes were temporally (67.8%) or spatially (54.5%) differentially expressed (22) between at least two time windows or regions, respectively, with the majority of spatially differentially expressed genes(95.8%) also temporally differentially expressed. To gain a broad understanding of this transcriptomic variation, we analyzed the level of similarity between individual samples in the mRNA-seq dataset using multidimensional scaling applied to both gene and isoform transcript-level analyses (Fig. 2B and figs. S11 and S12). In both analyses, we found a clear divide between samples from embryonic through late mid-fetal development (W1 to W4) and samples from late infancy through adulthood (W6 to W9), with samples from the late fetal period through early infancy (W5) generally spanning this divide. To determine the relationship between these three groups, we performed unsupervised hierarchical clustering analysis and found that all samples from W5, including the late fetal samples, were more similar to early postnatal samples than to late mid-fetal samples (fig. S13). Analysis of large-scale, intraregional changes in the transcriptome across time also suggest a major transition that begins before birth. The transcriptomes of major brain regions and neocortical areas correlated well across both embryonic and early to mid-fetal (W1 to W4) and later postnatal (W6 to W9) development but displayed a sharp decrease in correlation across late fetal development and early infancy (W5) (Fig. 2C and fig. S14). This transition was also apparent at the interregional level. Pairwise comparisons of gene expression across all 16 brain regions found a reduction in the number of genes showing differential regional expression during W5 relative to all other windows (fig. S15). Taken together, our observation of high variation during embryonic and early to mid-fetal ages followed by a decrease across late fetal ages and the subsequent resumption of higher levels of inter- and intraregional variation during late childhood and adolescence revealed a cup-shaped, or hourglass-like, pattern of transcriptomic development (Fig. 2D).
To further explore how regional transcriptomic profiles change with age, we applied the adjustment for confounding principal components analysis algorithm (AC-PCA) (23), which adjusts for interindividual variations. Within any given developmental window, AC-PCA exhibited a clear separation of brain regions, but the average dissimilarity between transcription profiles of brain regions declined from W1 to W5 and then increased with age after W5 (Fig. 2, E and F, and fig. S16). Implying a relationship between transcriptional signatures and developmental origin, we found that dorsal pallium–derived structures of the cerebrum (i.e., NCX, HIP, and AMY) as well as STR became increasingly similar across prenatal development, whereas CBC and MD remained most distinct across all time windows. To confirm these observations and to evaluate the contribution of each brain region to the regional variation described by AC-PCA, we quantified the mean distance in the first two principal components across brain regions, excluding from the AC-PCA one region at a time. Because of the relative transcriptomic uniqueness of the CBC, its exclusion unmasked a qualitatively distinct and pronounced cup-shaped pattern with a transition beginning before birth and spanning the late fetal period and early infancy (Fig. 2F). CBC was again the most distinct region of the brain after multidimensional scaling analysis for expressed mature microRNAs (miRNAs), a small RNA species enriched within our smRNA-seq dataset, and the dominant contributor to miRNA expression variance (fig. S17).
The global late fetal transition and overall cup-shaped developmental dynamics we observed were also apparent when this analysis was repeated for the 11 neocortical areas included in this study (Fig. 3A and fig. S16). We observed greater dissimilarity across areas at early fetal ages (Fig. 3A), with prefrontal areas [medial prefrontal cortex (MFC), orbital prefrontal cortex (OFC), DFC, and ventrolateral prefrontal cortex (VFC)] being the most distinct. In addition, reflecting the spatial and functional topography of the NCX, both rostro-caudal and dorsal-ventral axes were evident in the transcriptome during fetal development. Areal differences were also seen at later ages, with functional considerations likely taking precedence over topographical arrangements. For example, VFC clustered closely with primary motor (M1C) and somatosensory (S1C) cortex, likely reflecting functional relationships with orofacial regions of the motor and somatosensory perisylvian cortex (fig. S16). Across the entirety of human brain development, transcriptomic variation between cortical regions also showed a pronounced decrease centered on the late fetal and early infancy samples of W5 (i.e., perinatal window), again reminiscent of a cup-shaped pattern (Fig. 3, A and B, and fig. S16).
Fig. 3. Dynamics of cellular heterogeneity in the human neocortex.

(A) AC-PCA conducted on 11 neocortical areas showed decreased interareal variation across W5, similar to our observations of interregional variation in major brain regions. (B) Pairwise distance across samples using the first two principal components identified a late fetal transition in all of the neocortical areas we assessed, similar to what we observed across other brain regions. (C) Deconvolution of tissue-level data using cell type–enriched markers identified through single-cell sequencing of primary cells from 5 to 20 PCW postmortem human brains as well as from single-nuclei sequencing of adult human brains (27). (D) Maximum interareal variance across cell types for each window. (E) Neocortical areal variation in the transcriptomic signatures of each major cell type assayed in each developmental window. Because of dissection protocols and rapid brain growth across early fetal development, progenitor cell proportions are nonreliable estimates after W2 [red dashed line in (C)]. The shaded bands are 95% (B) and 50% (C) confidence intervals of the fitted lines. NPC, neural progenitor cells; ExN, excitatory neurons; InN, interneurons; Astro, astroglial lineage; Oligo, oligodendrocytes; Endo, endothelial cells.
Similar to gene expression, global measures of alternative splicing, such as the ratio between reads including or excluding exons [i.e., the percent spliced in index (PSI)], were higher during prenatal than postnatal ages (fig. S18 and table S7). So too was the gene expression of 68 RNA-binding proteins selected because of their involvement in RNA splicing and their analysis in adulthood by the Genotype-Tissue Expression (GTEx) project (24). Hierarchical clustering of expression data for these proteins also revealed a late fetal transition (fig. S19). Coincident with these observations, we found that genes exhibiting the highest interregional variation in expression in any given window [see (22)] exhibited a higher PSI during that window than iteratively chosen control groups of genes (fig. S18). Taken together, these analyses suggest that broad phenomena in the developing human brain, including a late fetal transition in intra- and interregional transcriptomic variation, may be amplified by alternative splicing.
Cellular heterogeneity and developmental dynamics
The high interareal variation observed during embryonic and early to mid-fetal development (Fig. 3B) coincides with a crucial period in neural development and the suspected etiology of psychiatric diseases (4). To help understand the temporal dynamics underlying this variation in gene expression, we analyzed our scRNA-seq data from embryonic fronto-parietal neocortical wall and mid-fetal fronto-parietal neocortical plate and adjacent subplate zone alongside our snRNA-seq data from adult human NCX and other independent datasets from overlapping developmental time points (12, 25, 26). To do so, we first applied a clustering and classification algorithm (27, 28) to the prenatal scRNA-seq data after an initial division of the dataset on the basis of the age of the donor brain (i.e., embryonic or fetal), obtaining 24 transcriptomically distinct cell clusters (fig. S20). Reflecting the rapid developmental change occurring across embryonic and fetal development and the relative homogeneity of cell-type composition as compared to adult ages, as well as the specific distribution of samples in our dataset, a number of these clusters were comprised of cells from only a single donor brain, and vice versa. Suggesting that this resulted from spatiotemporal changes across brain development rather than artifactual changes related to data processing, we confirmed broad classifications of individual cells and general relationships between cell clusters and donor brains using an alternative clustering algorithm (fig. S21). Differential expression analysis and measurements of expression specificity recovered well-known gene markers of distinct types of neuronal and non-neuronal progenitor and postmitotic cell types (figs. S20 and S22 and table S8), as well as closely related groups of cell types (i.e., markers enriched in all prenatal excitatory neuron clusters) (fig. S22).
We complemented these data with snRNA-seq from adult human DFC (fig. S20), from which we identified 29 transcriptomically distinct cell clusters representing various populations of glutamatergic excitatory projection neurons, GABAergic interneurons, oligodendrocyte progenitor cells, oligodendrocytes, astrocytes, microglia, endothelial cells, and mural cells (i.e., pericytes and vascular smooth muscle cells) (fig. S21). Alignment of our prenatal data with adult snRNA-seq data revealed hierarchical relationships and similarities between major cell classes, reflecting their developmental origins and functional properties (fig. S23). Notably, putative embryonic and fetal excitatory neurons clustered near, but did not wholly overlap with, their adult counterparts. We also observed transient transcriptomic entities, such as fetal cells in the oligodendrocyte lineage that clustered separately from their adult counterparts. Similarly, nascent excitatory neurons generally did not cluster with progenitor cells nor with fetal or adult excitatory neurons, indicating their maturationally distinct status. Confirming the validity of our prenatal scRNA-seq and adult snRNA-seq data, alignment of our prenatal data with cells from a previously published dataset (9) consisting of mid-fetal and adult human neocortical cells yielded similar relationships between prenatal and adult cell types (fig. S23). Comparison of neuronal transcriptomes from our prenatal single cells with both our adult single-nucleus data and independently generated adult single-nucleus data (27) also confirmed key differences between embryonic, mid-fetal, and adult populations. We observed limited transcriptional diversity in embryonic and mid-fetal excitatory and inhibitory neuron populations in the NCX as compared to the adult counterparts. The clusters identified in our prenatal dataset did not express specific combinations of marker genes described for the adult excitatory (fig. S24) and inhibitory (fig. S25) neurons. For example, the embryonic and mid-fetal neocortical excitatory neurons expressed combinations of genes known to be selectively enriched in different layers in adult human or mouse NCX (29–31), as previously shown in the prenatal human and mouse NCX (12, 31). Notably, genes enriched in adult excitatory projection neuron subtypes located in layer (L) 5 and L6, such as BCL11B (CTIP2) and FEZF2 (FEZL, ZFP312, or ZNF312), were coexpressed with L2 to L4 intracerebral excitatory projection neuron markers, such as CUX2, in certain embryonic and mid-fetal excitatory cell types (figs. S24 and S26). We also observed temporal changes in the coexpression patterns of cell type–specific marker genes in other cell types. For example, single-cell data from mid-fetal NCX revealed frequent coexpression of RELN, a marker for L1 Cajal-Retzius neurons (32), and PCP4 [75.9% of 133 PCP4 -expressing cells; reads per kilobase of exon model per million mapped reads (RPKM) ≥ 1], a marker previously shown to be expressed by deep-layer excitatory neurons (33). By contrast, analysis of snRNA-seq data suggested only sporadic coexpression of these genes [10.8% of 6084 PCP4-expressing cells; unique molecular identifier (UMI) ≥ 1] in the adult human DFC. Subsequent immunohistochemistry on independent specimens confirmed the robust coexpression of these genes in L1 of the prenatal cortex, but not in L1 or in other cortical layers of the adult cortex (fig. S26). These data imply that the molecular identities of many neuronal cell types are not fully resolved before the end of mid-fetal development and are likely malleable during early postmitotic differentiation.
Next, we utilized our single-cell and single-nucleus datasets to deconvolve bulk tissue mRNA-seq samples and estimate temporal changes in the relative proportions of major cell types in the NCX. The combined analysis revealed the cellular architecture of distinct neocortical areas and their variations across development. We observed temporal changes in cellular composition and maturational states, including the most dramatic changes during a late fetal transition (Fig. 3, C to E). For example, transcriptomic signatures for fetal excitatory neurons and fetal interneurons were generally inversely correlated with progenitor cell signatures during embryonic and early fetal development, but fetal neuron signatures nonetheless decreased across mid-fetal to late fetal development despite a concomitant reduction in the progenitor cell signature, an observation that was likely affected by our dissection strategy [Fig. 3C, (22)]. Similarly, signatures for adult excitatory neurons increased rapidly across the late fetal period and early infancy, coincident with the decrease in signatures of fetal excitatory neurons and interneurons (Fig. 3C). As expected, the molecular signatures for early born, deep-layer excitatory neurons preceded those for late born, upper-layer excitatory neurons (fig. S27). Transcriptomic signatures for prenatal oligodendrocytes and prenatal astrocytes also began to emerge during mid-fetal periods and increased rapidly across the late fetal transition and early infancy (Fig. 3C). Demonstrating the robustness of these observations, independent deconvolution using two alternate fetal single-cell datasets (12, 26) yielded similar results (figs. S27 and S30).
Given the increase in adult cell-type signatures during W5, we next reasoned that the observed decrease in interregional transcriptomic divergence during late fetal periods and infancy may reflect a synchronized transition from fetal to more mature features of neural cells. Consequently, we analyzed the variance in cell type–specific signatures across neocortical areas, which varies in accordance with their relative proportion, and found that the maximum cell type interareal variation through time recapitulated the developmental cup-shaped pattern (Fig. 3D), with large variation in the proportion of neural progenitor cells and fetal excitatory neurons (figs. S28 and S29). Beginning during early postnatal periods, we observed increased proportions and variance in the signatures of astrocytes and, by adulthood, mature excitatory neurons (Fig. 3E). These observed temporal differences in the magnitudes and variances of the relative proportions of certain cell types and the global heterogeneity of the cell type composition at each window at least partially explain the observed pattern of interareal differences across development. Gene Ontology (GO) enrichment analysis using the top variant genes in each window, with all genes expressed in each window as background, provided further support for these changes in cell composition across areas and time. Commensurate with the changes we observed in discrete cell populations, biological processes—including neurogenesis in early developmental windows (W1 to W4), myelination in the perinatal window (W5), and sensory and ion activity calcium-related biological processes in later postnatal windows (W7 to W9), among others—exhibited regional variation in the global brain transcriptome (fig. S31 and table S9). Similar patterns of interregional variation involving discrete cell types were also observed in the macaque neocortical transcriptome (34), indicating that these are conserved and consistent features of prenatal primate NCX.
Other lines of evidence also suggested pronounced and qualitatively distinct regional differences in myelination, synaptic function, and neuronal activity. For example, although we observed differences in the expression of genes associated with these processes (10) across the NCX (fig. S31 and table S9), TempShift, a Gaussian-based model that allows the quantification of temporal shifts in the trajectories of groups of genes represented by their first principal components (34), indicated that of these processes, only genes associated with myelination displayed such a shift (Fig. 4A). Conversely, perhaps reflecting functional or areal diversity in cell subtypes, we observed no similar temporal shift in the expression of genes associated with synaptogenesis or neuronal activity, confirming these results through reference to published posttranslational analyses of myelinated fiber density (35) and synaptic density (36) conducted across multiple neocortical areas (Fig. 4B). Crucially, although genes associated with these processes were expressed across the late fetal transition (Fig. 4C), of the processes analyzed, only myelination contributed to the increased interareal differences we observed during this period (Fig. 4D). Suggesting that these differences are a conserved feature of primate development, we also observed similar areal differences in the transcriptional signatures of oligodendrocytes in the macaque NCX.
Fig. 4. Timing and temporal variation of gene expression associated with key neurodevelopmental processes.

(A) Temporal variation, as determined by the TempShift algorithm (34), in the expression of genes associated with myelination showed a broad gradient across the NCX and other brain regions, whereas synaptogenesis showed only a shift between brain regions (but not neocortical areas) and neuronal activity indicated the distinct nature of the cerebellum. (B and C) Application of the TempShift algorithm to previously published posttranslational analyses of myelinated fiber density (35) (B) and synaptic density (36) (C) in multiple neocortical areas yielded relationships between areas similar to those observed in the transcriptome. (D) Expression of genes associated with assorted biological processes highlights pronounced change during the late fetal period and W5. (E) Variation in myelination-associated genes peaks during W5, as evidenced by the standard deviation of the fitted regional mean, driving interregional variation during this and neighboring (W4 and W6) windows.
Overall, these observations indicate that higher levels of divergence during early prenatal and later postnatal development reflect regional variations in cell type composition, likely arising from topographical variation in progenitor populations and neuron development during prenatal ages and cell type and functional diversification during later postnatal ages.
Spatiotemporal and multimodal integration
We next sought to assess temporal variation in epigenetic signatures and their relationships to gene expression, development, and biological processes. Global DNA methylation profiling revealed that most CpG loci were either hypermethylated [37.5%; beta value (β) ≥ 0.8] or hypomethylated (31.8%; β ≤ 0.2) in at least one sample (fig. S32), but only about 10% of the tested methylation sites were progressively hyper-or hypomethylated through prenatal windows, postnatal windows, or both. Similarly, most methylation sites also exhibited regional variation, with 64% of tested sites differentially methylated between at least two brain regions at postnatal ages. Additionally, 16% of tested sites were differentially methylated between at least two neocortical areas. Conversely, most putative promoters (66%) and a substantial proportion of putative enhancers (43%) were not differentially enriched between DFC and CBC at either fetal or adult ages. However, a greater proportion of putative enhancers [H3K27ac-enriched regions not overlapping H3K4me3-enriched regions or proximal to a transcription start site (TSS)] were regionally (15%), temporally (17%), or spatio-temporally (24%) enriched than putative promoters (8, 14, and 12%, respectively). These differences, which suggest a greater role for enhancers relative to promoters in contributing to differential spatiotemporal gene expression, were selectively validated using quantitative droplet digital polymerase chain reaction (ddPCR) (fig. S10). We next explored correlations between methylation, histone modifications, and gene expression (figs. S32 to S34). In the adult, we found that TSSs that were more highly methylated were associated with genes that were expressed at low levels at the corresponding age, and vice versa. These relationships were not strongly indicated for methylation at other locations in the gene body (fig. S32). The presence of CBC-enriched H3K4me3 and H3K27ac marks in the adult human brain also correlated strongly with increased gene expression in CBC relative to DFC (fig. S33), and vice versa. Similarly, putative fetal-active and adult-active enhancers were associated with higher fetal or adult gene expression, respectively.
In addition to epigenetic effects on gene expression, we observed discrete relationships between specific enhancers, methylation sites, and cell type–specific signatures. For example, enhancers identified during the fetal period were enriched for methylation sites that were progressively more methylated across postnatal ages (post-up), whereas adult-active enhancers were enriched for methylation sites that were progressively less methylated across postnatal ages (post-down) (P < 0.05, Fisher’s exact test) [Fig. 5A and fig. S35, (22)]. Both post-up and post-down sites were themselves depleted at TSSs and enriched for sites undermethylated in neurons [neuron undermethylated (NUM) sites] and undermethylated in non-neurons (non-NUM sites) (fig. S35). They were also enriched for fetal and adult enhancers, respectively (Fig. 5B). Post-up sites were also enriched in both neuron- and glia-enriched-genes, whereas post-down sites were enriched only in glial genes (Fig. 5B) (P < 0.05, Fisher’s exact test). Further suggesting a relationship between enhancer activity, methylation, and cell type, genes associated with fetal-active enhancers, as well as those associated with differentially methylated regions (DMRs) composed of post-up sites (22), were enriched for GO terms related to early events in neural development—such as neurogenesis, cell differentiation, and synaptic transmission—but generally not for processes occurring later in development (Fig. 5B and fig. S35). By contrast, genes near adult-active enhancers and post-down DMRs exhibited enrichment for postnatal or adult processes including myelination and axon ensheathment (P < 0.01, Fisher’s exact test) (Fig. 5B and fig. S35). Taken together, these data demonstrate relationships between gene expression and epigenetic modifications, including methylation status and putative regulatory elements, as well as signatures of specific cell types and developmental programs.
Fig. 5. Integration of gene expression and epigenetic regulation with cell types and biological processes.

(A) Fetal-active enhancers (top left) were generally enriched for sites where methylation progressively increased across postnatal ages and associated with genes whose expression was higher during fetal development than adulthood and whose expression was enriched in neurons as compared to glia. Conversely, adult-active enhancers were enriched for sites exhibiting progressively lower methylation across postnatal ages and depleted for associations with higher fetal gene expression and expression in neurons. These enhancers were also enriched for gene ontology terms generally involving neurons and glia, respectively. OR, odds ratio. (B) Sites where methylation progressively increased across postnatal ages and where methylation progressively decreased across postnatal ages were generally enriched for fetal enhancers and genes whose expression was enriched in neurons, or adult enhancers and genes whose expression was enriched in glia, respectively, as well as related gene ontology terms. (C) Modules identified through WGCNA were segregated by regulation across brain regions, prenatal and postnatal gene expression in the NCX, both, or neither. Spatiotemporal modules (right) were enriched for modules that are themselves enriched for genes associated with enhancers active in the fetal DFC, associated with sites undermethylated in NeuN-positive (neuronal) cells, and/or enriched in neurons (N-type associations). Temporal, nonspatial modules (second from left) were enriched for modules that are themselves enriched for genes associated with enhancers active in the adult DFC, associated with sites undermethylated in non-NeuN-positive (non-neuronal) cells, and/or genes enriched in glia (G-type associations). Modules exhibiting no spatial or temporal specificity (left) were enriched for genes exhibiting sex-biased gene expression across neocortical development. Full circles (gray) indicate the proportion of modules in each category of modules exhibiting their greatest rate of change in W1 through W9.
We next sought further evidence that cellular dynamics contributed to the late fetal transition through the analysis of cell type– and spatiotemporal-specific patterns of gene expression and epigenetic regulation. We curated 73 gene coexpression modules resulting from weighted gene correlation network analysis (WGCNA) according to spatial relationships between brain regions and the temporal relationships of gene expression in the NCX across the late fetal transition (fig. S36 and tables S10 and S11). We found 44 modules that showed expression differences among regions in the brain (spatial), 40 modules that showed expression differences between prenatal and postnatal neocortical areas (temporal), 16 modules that were neither spatially nor temporally dynamic, and 27 modules that exhibited both spatial and temporal differences (Fig. 5C). A significantly greater than expected number of these spatiotemporally dynamic modules (including modules 2, 10, 32, and 37) exhibited their greatest change in neocortical expression from W2 through W5 (P < 0.0118, hypergeometric test) (Fig. 5C, fig. S37, and table S12). Genes whose expression was enriched in excitatory neurons, genes associated with putative fetal-active enhancers, and/or genes associated with NUM sites—a selection of characteristics we refer to collectively as neuronal (N)–type associations—were also enriched in spatiotemporal dynamic modules (P < 0.0029, hypergeometric test) (Fig. 5C, fig. S37, and table S12). Conversely, genes associated with adult-active enhancers, methylation sites hypomethylated in non-NUM sites, and glial genes [glial (G)–type modules or associations in Fig. 5C, fig. S37 and table S12] were enriched among the 13 modules where temporal (P < 0.0002, hypergeometric test), but not spatial, specificity was observed. These observations indicate increased spatial diversity of neuronal cell types relative to glial cell populations.
Analyses by sex revealed that modules enriched for the 783 genes exhibiting sex-differential expression (sex-DEX) in at least two consecutive windows in at least one brain region were enriched among modules with no spatial or temporal differential expression in the NCX (P < 0.0029, hypergeometric test) and depleted among spatiotemporal modules (P < 0.0021, hypergeometric test) (Fig. 5C and fig. S37). There were four modules exhibiting temporal expression differences in the NCX that were also enriched for sex-biased genes, as well as glial and other cell type–enriched markers, but these did not represent a significant enrichment in sex-DEX enriched modules among strictly temporal modules (P < 0.132, hypergeometric test). In addition, no module comprised of autosomal genes exhibited persistent male or female dimorphism across both prenatal development and later postnatal ages such as adolescence or adulthood (figs. S38 and S39); in cases in which an autosomal module was sex-DEX throughout development, the sex exhibiting higher expression reversed between early and late postnatal development (fig. S39). This observation was upheld when multiple thresholds were used for the identification of sexual dimorphism (fig. S40). Similarly, we identified no autosomal genes that exhibited sexual dimorphism throughout development in all brain regions or neocortical areas (figs. S38 and S39).
Cellular and temporal convergence of neuropsychiatric disease risks
Loci implicated in several neuropsychiatric disorders have been identified through genome-wide association studies (GWAS) and are enriched in putative noncoding regulatory elements (29–31). We sought to determine whether the proportion of phenotypic variance explained by common single-nucleotide polymorphisms (SNPs) in large neuropsychiatric GWAS (i.e., SNP heritability) was enriched in the cis-regulatory elements we identified at W1, W4, W5, and W9 in DFC and CBC. Toward this end, we collected GWAS data concerning neuropsychiatric disorders or personality traits including schizophrenia (SCZ) from CLOZUK (37), Alzheimer’s disease (AD) from IGAP (38), Parkinson’s disease (PD) (39), autism spectrum disorder (ASD) (40), attention deficit hyperactivity disorder (ADHD) from iPSYCH (41), major depressive disorder (MDD) (42), bipolar disorder (BD) (43), intelligence quotient (IQ) (44), and neuroticism (45), as well as non-neural traits such as height from GIANT (46), inflammatory bowel disease (IBD) (47), total cholesterol levels (48), and an endophenotype associated with diabetes (HBA1C) (49). Using partitioned linkage disequilibrium (LD) score regression analysis, we found that SNP heritability in SCZ, IQ, and neuroticism were exclusively enriched in DFC-specific, but not CBC-specific, regulatory elements as identified by peak regions of H3K27ac activity. By contrast, SNP heritability in AD or PD rendered no significant associations, and the analysis on ASD, ADHD, BD, and MDD was only nominally enriched or not enriched in putative region-specific fetal enhancers [Fig. 6 and fig. S41, (22)]. Non-neural traits (such as height and HBA1C) were also not enriched in either DFC- or CBC-specific regulatory elements but were instead enriched in regulatory elements active in the two brain regions (fig. S41), indicating a general enrichment of many of our tested GWASs in H3K27ac regions when considering a set of more ubiquitous regulatory regions.
Fig. 6. Enrichment analysis for GWAS loci among putative regulatory elements.

Putative promoters and enhancers (H3K27ac peaks) specific for DFC or CBC in the fetal, infant, or adult were enriched for SNP heritability identified through partitioned LD score regression analysis from GWASs for autism spectrum disorder [ASD, (40)], attention-deficit hyperactive disorder [ADHD, (41)], schizophrenia [SCZ, (37)], major depressive disorder [MDD, (42)], bipolar disorder [BD, (43)], Alzheimer’s disease [AD, (38)], Parkinson’s disease [PD, (39)], IQ, (44), or neuroticism [Neurot, (45)] but not for non-neural disorders or traits such as height [HGT, (46)] or diabetes [HBA1C, (49)]. Solid color indicates significance for Bonferroni adjusted P value, and faint color indicates nominal significance at LD score regression P < 0.05.
After aggregating GWAS SNPs and identifying candidate associated regions on the basis of their P values and LD patterns in individuals of northwest European ancestry (50), we next leveraged partially overlapping Hi-C datasets, derived from mid-fetal and adult NCX and processed by two independent research groups (51–53), as well as H3K27ac activity in the brain, to develop two lists of genes putatively associated with those GWAS-associated regions. To do so, we initially populated both lists of disease-associated genes by identifying TSSs overlapping H3K27ac peaks that themselves overlapped a GWAS significant region, as well as genes directly affected by GWAS significant variants within the LD region, as predicted by EnsemblV78. We next expanded these lists of disease-associated genes by identifying TSSs that interact with H3K27ac peaks overlapping GWAS significant regions, excluding interactions that did not overlap with at least one H3K27ac peak at each end or where peak-to-peak interactions were not concordant in time and brain region. In the first, less stringent list (list 1), a single interaction from either of the two Hi-C datasets was sufficient to associate a gene to a GWAS locus (table S13). For the second, more stringent list (list 2), we excluded those genes whose only association to a GWAS locus was via Hi-C interactions identified in only one of the two Hi-C datasets (table S14).
We next sought to determine the cell types enriched for the expression of the high-stringency genes implicated in neuropsychiatric disorders or brain-based traits, using our prenatal scRNA-seq and adult snRNA-seq datasets and matching prenatal and adult datasets generated from the macaque (34). We found numerous cell types enriched for disease-associated loci in both human and macaque (fig. S42). For example, neocortical excitatory neurons were enriched for the expression of genes we associated with IQ in both the fetal and adult human as well as the fetal and adult macaque. However, we found no other excitatory neuron populations in the macaque AMY, STR, HIP, or thalamus enriched for genes associated with IQ. Similarly, neural progenitors in the prenatal macaque AMY, but not progenitors in the prenatal macaque HIP, thalamus, NCX, or STR, were enriched for the expression of genes associated with MDD, a finding especially intriguing given the variable or potentially increased size of some amygdalar nuclei in MDD patients (54, 55). Similarly confirmatory was the enrichment of SCZ risk genes in cortical excitatory neurons (56), with enrichment also observed in embryonic and/or fetal progenitor cells and adult cortical interneurons.
Analysis of gene coexpression modules found that genes in the more-stringent early-onset disease (ADHD, SCZ, and MDD) risk lists converged on 7 of 73 coexpression modules, whereas adult-onset disease (AD and PD) risk-gene lists converged on five partially overlapping modules (fig. S37 and table S12). Eight of these 10 total disease-associated modules (Fig. 7A) exhibited spatiotemporal or temporal specificity, and all modules exhibited their greatest spatiotemporal change during either W2 or W5 (fig. S37). A significant number of modules associated with adult-onset disorders were enriched for signatures of glial gene expression (P < 0.0266, hypergeometric test, table S12), and of particular interest were modules ME3 and ME7, which, in addition to glial signatures, were enriched for non-NUM sites, adult-active enhancers, sex-DEX genes, and AD-associated risk genes (Fig. 7A).
Fig. 7. Convergence of risk for brain-based traits and disorders on discrete coexpression modules and cell types.

(A) Genes associated with disease risk (right; light yellow indicates neuropsychiatric disorder or brain-based trait, and dark yellow indicates adult-onset disorder) were identified by integrating GWAS, Hi-C, and H3K27ac data and converged on 10 WGCNA modules. Many of these modules exhibited dynamic expression across time; the bold rectangles in the left panel indicate the windows with greatest rate of change. Many were also enriched for gene expression associated with distinct cell types (orange), putative active enhancers (green), and/or sites undermethylated in NeuN-positive (NUM) or NeuN-negative cells (blue, non-NUM). (B) Schematic highlighting genes in ME37 that were implicated by our study in multiple neuropsychiatric disorders (ADHD, SCZ, MDD, or BD) and neurological traits (IQ or Neurot) (list 1, light blue; list 2, dark blue), as well as neurodevelopmental disorder (NDD) risk genes, including two independent lists of high-confidence risk genes associated with ASD through de novo mutations or copy number variants [dark blue, (66)] as well as ASD risk genes identified from the SFARI dataset (light blue, http://gene.sfari.org) or for developmental delay (67). Genes implicated in only a single disorder or trait are not shown in this panel. (C) Network representation of ME37 showing connectivity between genes based on Pearson correlation. Genes linked to NDDs or neurological characteristics in our study are indicated using either dark blue–shaded or light blue–shaded hexagons, as in (B). The size of a given hexagon (or circle, indicating no association in this study) is proportional to the degree of each gene under a minimum correlation value of 0.7. (D) Enrichment for genes in ME37 or two lists of ASD risk genes among the fetal and adult cell types we identified from human NCX and multiple regions of the macaque (34) brain. For graphical representation, log10 P values are capped at 25. *Adult macaque cells were classified into human adult clusters using Random Forest. NEP/RGC, neural epithelial progenitor/radial glial lineage; MSN, medium spiny neurons; NasN, nascent neurons; GraN, granule neurons; PurkN, Purkinje neurons; IPC, intermediate progenitor cells; OPC, oligodendrocyte progenitor cells.
Another module of interest was ME37, a module of 145 genes enriched for NUM sites and fetal enhancers and whose expression was enriched specifically in neurons as opposed to neural progenitors or glia. ME37 was also exceptional for its disease association, as it was enriched for genes associated with SCZ, IQ, and neuroticism but not for non-neurological characteristics such as height or a HBA1C-related trait (Fig. 7A). Complementary module-based association analysis with Multi-marker Analysis of GenoMic Annotation (MAGMA), which tested for an enrichment in association to disease specifically around genes in any given module, confirmed enrichment for SCZ, IQ, and neuroticism in ME37 [MAGMA P values < 0.01; the false discovery rate (FDR) for all traits and modules was <0.3] (table S11). At the gene level, multiple genes in ME37 identified using our less stringent criteria for interaction were associated with up to four or more different traits and disorders, including MEF2C, ZNF184, TCF4, and SATB2, all genes critical for neurodevelopment and/or implicated in neurodevelopmental disorders (57–65) (Fig. 7, B and C). We also found that ME37 was specifically enriched in clusters of excitatory neurons in the fetal and adult NCX (Fig. 7D), and further analysis of adult excitatory neuron populations identified in this study and an independent database of adult single nucleus data (27) suggested that this enrichment was selective for deep-layer neocortical neurons (fig. S43).
As the ASD GWAS resulted in only 13 significant genes, eight of which were non-protein coding, and because de novo germline mutations are known to contribute to ASD risk (66), we next developed two nonoverlapping lists of neurodevelopmental disorders (NDDs) [ASD, intellectual disability (ID), and developmental delay (DD)]. The first list was comprised of 65 high-confidence ASD risk genes (hcASD) associated with de novo mutations (66). The second list included all ASD genes documented in the SFARI database (http://gene.sfari.org) under categories “syndromic” or with scores from 1 to 4, as well as an independent list of genes associated with DD (67), with genes overlapping the hcASD list removed. We found that these genes were also significantly enriched in ME37 (FDR < 0.0001, Fisher’s exact test), and, commensurate with the cell-type enrichment found in ME37, the expression of genes in both of these lists was also enriched in several clusters of fetal and adult excitatory neurons identified in our single-cell dataset (Fig. 7D). Medium spiny neurons in the STR, a population that has also been previously linked to ASD (68), were also enriched for the expression of ASD risk genes in the prenatal macaque (Fig. 7D).
We finally studied the overlap between WGCNA modules and modules significantly enriched in differentially expressed genes in postmortem brains from patients of SCZ, BD, and ASD (69). Interestingly, we found little overlap between modules enriched in genes exhibiting postmortem differences in expression between SCZ, BD, or ASD, as compared with neurotypical controls, and modules enriched in GWAS risk genes for these same disorders (P > 0.05, hypergeometric test) (fig. S37). Emphasizing the necessity of studying neurotypical brain development, these observations may suggest a decoupling between the primary genetic causes of some neurological or psychiatric disorders and second-order effects manifesting as changes in gene expression months or years after disease onset.
Discussion
In this study, we have presented a comprehensive dataset and a multiplatform functional genomic analysis of the developing and adult human brain. The presence of these multiple data modalities in a unified resource, and largely from the same tissue samples, allows the integration of information spanning prenatal and postnatal human brain development. Resource description and access are available at development.psychencode.org and www.brainspan.org.
Although transcriptomic differences between distinct brain regions remain across time, they are developmentally specified and exhibit an overall cup-shaped pattern centered on a late fetal transition after a period of high intra- and interregional variation during embryonic and early or mid-fetal development. Multiple analyses of distinct transcriptomic features all confirm this transition begins well before birth. Our complementary transcriptomic study of the developing rhesus macaque brain (34) also revealed a similar global developmental pattern, with a first transition beginning before birth, indicating that this is a conserved feature of catarrhine primate neurodevelopment and not due to an artifact resulting from difficulties acquiring samples from late fetal and early postnatal development. Such a phenomenon is consistent with previously observed differences in transcriptomic and methylomic profiles of mid-fetal and postnatal human NCX (17–20) and coincident with processes involved in region-specific cell type generation, differentiation, and maturation (2). Crucially, this transition is notably distinct from previously reported phylogenetic hourglass-like patterns that occur during the embryonic organogenetic period in several invertebrate and vertebrate species (70, 71). Moreover, the developmental (ontogenetic) cup-shaped pattern we observe coincides with an “evolutionary” (phylogenetic) cup-shaped pattern, in which developmental periods exhibiting high levels of interregional differences (for example, early to mid-fetal periods) also exhibit less conservation in gene expression patterns between human and macaque (34).
Among the processes that become prominent during the late fetal period are astrogliogenesis, synaptogenesis, dendritogenesis, and neuronal activity. In contrast to a previous report of robust areal differences in the progression of synaptogenesis during the same time period in humans (36), this and an accompanying study (34) found that genes associated with these processes exhibit largely synchronous expression trajectories across the developing NCX in both humans and macaque. However, myelination—which sharply increases during late fetal development, peaks after birth, and extends through childhood and adolescence (72)—is temporally asynchronous. This asynchronicity in oligodendrocyte development and myelination is not apparent at the level of oligodendrocyte progenitor cells (OPCs), which suggests that the maturation of OPCs into myelinating oligodendrocytes is a process with a variable onset and pace across areas. Similar observations were made in macaque (34), indicating that this may be another conserved catarrhine feature.
Transcriptomic variation may reflect several distinct cellular and maturational reorganizational events. For example, as first described by Brodmann (73), an ontogenetic six-layered Grundtypus foreshadows the adult NCX and transiently transforms the entirety of the neocortical plate beginning in the late fetal period, or in our W5. Furthermore, consistent with the extensive changes we observed in the cerebellar transcriptome during late fetal development and early postnatal ages, cerebellar granule cells, a cell type that represents about two-thirds of all neurons in the brain, are also generated predominately during this period (74). The late fetal transition may therefore follow an inflection point after which developmental and spatiotemporal transcriptomic variations are transiently consolidated in advance of the emergence of cellular and functional differences between adult brain regions.
The mid-fetal period of high intra- and interregional divergence that immediately precedes the late fetal transition also coincides with a key developmental period previously associated with the etiology of ASD and SCZ (63, 65, 75). Consequently, understanding the developmental and evolutionary history of this period may be essential for understanding neuropsychiatric disease. Integrating our multiple data modalities with gene coexpression modules allowed us to organize and characterize the whole-brain developmental transcriptome and identify modules with dynamic spatiotemporal trajectories, many of them showing a sharp late fetal transition, and enrichment in specific cell types, epigenetic activity, and disease-associated genes. Of particular interest is ME37, a module displaying the greatest rate of change in the NCX within the late fetal transition and in which putative risk genes for ASD, NDD, SCZ, IQ, and neuroticism converged. Several of the genes in ME37 were implicated by our study in multiple disorders and traits and have been linked previously to neurodevelopment and human disease. For example, MEF2C controls activity-dependent expression of neuronal genes, including those linked to synapse function and ASD (61, 63), and Mef2c-mutant mice display numerous behaviors reminiscent of ASD, ID, and SCZ (58). Similarly, TCF4 regulates key neurodevelopmental processes, such as neurogenesis and synaptic plasticity, DNA methylation, and memory function processes (62, 64). Moreover, mutations in both MEF2C and TCF4 result in intellectual disability in humans (57, 59, 60). Numerous other genes in this module are similarly involved in neurodevelopment, have been implicated in human brain disease, and are highly plausible disease-risk genes and potentially therapeutic candidates. For example, NR4A2, a gene encoding another transcription factor in ME37 that we linked to neuroticism and IQ, has been linked to ASD and SCZ, among other disorders. Our study also links the gene for the transcription factor TSHZ3 to neuroticism and IQ, and previous efforts have linked murine Tshz3 to ASD and the fetal development of cortical excitatory projection neurons (76), a cell type and developmental period also implicated in ASD (63, 65). Other genes in ME37, such as SATB2, FEZF2, SOX5, and TBR1, play critical roles in the development of cortical excitatory projection neurons and are mutated in NDDs (29–31, 65, 77, 78). Similarly, the population of genes included in ME37, as well as genes linked to ASD and NDD, also exhibit regional and cell type–specific convergence in neocortical excitatory neurons. Moreover, the identification of ME37 and the overlap of genes in this module with those implicated in ASD and NDD illustrates how disease-association signals from common variants unveiled by GWAS for any given neuropsychiatric disorder can identify genes that have also been associated with the etiology of a different disease through the study of de novo mutations in patient populations (76). Although not every gene in ME37 is likely to contribute to neuropsychiatric disease etiology, the coincident enrichment within this module of genes associated with multiple disorders or neurological traits, along with the multitude of genes in this module that are associated directly, suggests that neuropsychiatric disease might be considered through a broader lens encompassing additional aspects of brain dysfunction.
Interestingly, there is little overlap between the risk gene–associated modules we identified and modules enriched in genes that are differentially expressed in postmortem brains of SCZ, ASD, and BD, as compared to controls (69). This comparison may help discriminate gene networks that are primary causes from those that are secondary or reactive in these neuropsychiatric disorders while emphasizing the importance of studying disease in the context of neurotypical development.
Taken together, these observations demonstrate the utility of this resource to perform integrated analysis for the understanding of brain development and function and for the rapid interpretation of findings from neuropsychiatric genomics.
Materials and methods summary
A full description of the materials and methods is available in the supplementary materials. Briefly, we precisely dissected multiple brain regions (HIP, STR, AMY, cerebellum, thalamus, and 11 neocortical areas) in more than 60 postmortem human brains ranging in age from 5 PCW to 64 PY. We then applied bulk tissue RNA-seq, scRNA-seq and snRNA-seq, smRNA-seq, DNA methylation assay, or ChIP-seq to generate multimodal datasets, often from the same brain. After applying stringent quality control checks and independent analysis of each dataset, we performed integrated analyses to gain insights into human brain development, function, and disease.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to the many individuals in our laboratories and at our institutions who have provided support and contributed to these projects. In addition, we would like to thank the National Institute of Mental Health (NIMH), and, in particular, A. Beckel-Mitchener, M. Freund, T. Lerner, and G. Senthil, for providing invaluable institutional support and guidance for these projects.
Funding: Data was generated as part of the PsychENCODE Consortium, supported by NIMH grants U01MH103392, U01MH103365, U01MH103346, U01MH103340, U01MH103339, R21MH109956, R21MH105881, R21MH105853, R21MH103877, R21MH102791, R01MH111721, R01MH110928, R01MH110927, R01MH110926, R01MH110921, R01MH110920, R01MH110905, R01MH109715, R01MH109677, R01MH105898, R01MH105898, R01MH094714, R01MH109901, P50MH106934, and 5R24HD000836 awarded to S. Akbarian (Icahn School of Medicine at Mount Sinai), G. Crawford (Duke University), S. Dracheva (Icahn School of Medicine at Mount Sinai), P. Farnham (University of Southern California), M.B.G. (Yale University), D.H.G. (University of California, Los Angeles), F. Goes (Johns Hopkins University), T. M. Hyde (Lieber Institute for Brain Development), A. Jaffe (Lieber Institute for Brain Development), J.A.K. (University of Southern California), C. Liu (SUNY Upstate Medical University), D. Pinto (Icahn School of Medicine at Mount Sinai), P. Roussos (Icahn School of Medicine at Mount Sinai), S.J.S. (University of California, San Francisco), N.S. (Yale University), P. Sklar (Icahn School of Medicine at Mount Sinai), M.W.S. (University of California, San Francisco), P.F.S. (University of North Carolina), F. Vaccarino (Yale University), D.R.W. (Lieber Institute for Brain Development), S. Weissman (Yale University), K. White (University of Chicago), J. Willsey (University of California, San Francisco), and P. Zandi (Johns Hopkins University). The BrainSpan Project Consortium was supported by grants MH089929, MH090047, and MH089921 from NIMH. Additional support was provided by the Kavli Foundation, the James S. McDonnell Foundation, the Beatriu de Pinós program (BP-DGR 2014 to B.L.-G.), and the Eunice Kennedy Shriver National Institute of Child Health and Human Development (5R24HD000836). S.J.S. and D.M.W. were supported by a grant from the Simons Foundation (SFARI #307705) and the Autism Science Foundation (Postdoctoral Fellowship #16–009), respectively. The human embryonic and some of the fetal material was provided by the Joint MRC–Wellcome Trust (MR/R006237/1) Human Developmental Biology Resource (www.hdbr.org). This work also received funding from The Netherlands Organization for Scientific Research (NWO VICI 435-14-005), Sophia Foundation for Scientific Research. Part of the analyses were carried out on the Genetic Cluster Computer, which is financed by the Netherlands Scientific Organization (NWO, 480-05-003), by the VU University, Amsterdam, Netherlands, and by the Dutch Brain Foundation, and is hosted by the Dutch National Computing and Networking Services SurfSARA.
Footnotes
Competing interests: The authors declare no competing interests.
Data and materials availability: Scripts used in this manuscript can be found at https://github.com/GSantpere/PEC_DEV/tree/master/Code. scRNA-seq and snRNA-seq data have been deposited at http://psychencode.org. Tissue RNA-seq, ChIP-seq, DNA methylation, and SNP array data have been deposited in dbGAP under accession number phs000755. All processed files and scripts pertaining to this manuscript are available on http://development.psychencode.org.
SUPPLEMENTARY MATERIALS www.sciencemag.org/content/362/6420/eaat7615/suppl/DC1
Materials and Methods
Figs. S1 to S43
Tables S1 to S16
Consortia Authors and Affiliations References (79–124)
REFERENCES AND NOTES
- 1.Lein ES, Belgard TG, Hawrylycz M, Molnár Z, Transcriptomic perspectives on neocortical structure, development, evolution, and disease. Annu. Rev. Neurosci 40, 629–652 (2017). doi: 10.1146/annurev-neuro-070815-013858; [DOI] [PubMed] [Google Scholar]
- 2.Silbereis JC, Pochareddy S, Zhu Y, Li M, Sestan N, The cellular and molecular landscapes of the developing human central nervous system. Neuron 89, 248–268 (2016). doi: 10.1016/j.neuron.2015.12.008; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Geschwind DH, Rakic P, Cortical evolution: Judge the brain by its cover. Neuron 80, 633–647 (2013). doi: 10.1016/j.neuron.2013.10.045; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lui JH, Hansen DV, Kriegstein AR, Development and evolution of the human neocortex. Cell 146, 18–36 (2011). doi: 10.1016/j.cell.2011.06.030; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Paus T, Keshavan M, Giedd JN, Why do many psychiatric disorders emerge during adolescence? Nat. Rev. Neurosci 9, 947–957 (2008). doi: 10.1038/nrn2513; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McCarroll SA, Hyman SE, Progress in the genetics of polygenic brain disorders: Significant new challenges for neurobiology. Neuron 80, 578–587 (2013). doi: 10.1016/j.neuron.2013.10.046; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bae BI, Jayaraman D, Walsh CA, Genetic changes shaping the human brain. Dev. Cell 32, 423–434 (2015). doi: 10.1016/j.devcel.2015.01.035; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Keil JM, Qalieh A, Kwan KY, Brain transcriptome databases: A user’s guide. J. Neurosci 38, 2399–2412 (2018). doi: 10.1523/JNEUROSCI.1930-17.2018; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Darmanis S et al. , A survey of human brain transcriptome diversity at the single cell level. Proc. Natl. Acad. Sci. U.S.A 112, 7285–7290 (2015). doi: 10.1073/pnas.1507125112; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Johnson MB et al. , Functional and evolutionary insights into human brain development through global transcriptome analysis. Neuron 62, 494–509 (2009). doi: 10.1016/j.neuron.2009.03.027; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miller JA et al. , Transcriptional landscape of the prenatal human brain. Nature 508, 199–206 (2014). doi: 10.1038/nature13185; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nowakowski TJ et al. , Spatiotemporal gene expression trajectories reveal developmental hierarchies of the human cortex. Science 358, 1318–1323 (2017). doi: 10.1126/science.aap8809; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.de la Torre-Ubieta L et al. , The dynamic landscape of open chromatin during human cortical neurogenesis. Cell 172, 289–304.e18 (2018). doi: 10.1016/j.cell.2017.12.014; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Doan RN et al. , Mutations in human accelerated regions disrupt cognition and social behavior. Cell 167, 341–354.e12 (2016). doi: 10.1016/j.cell.2016.08.071; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Florio M et al. , Human-specific gene ARHGAP11B promotes basal progenitor amplification and neocortex expansion. Science 347, 1465–1470 (2015). doi: 10.1126/science.aaa1975; [DOI] [PubMed] [Google Scholar]
- 16.Johnson MB et al. , Single-cell analysis reveals transcriptional heterogeneity of neural progenitors in human cortex. Nat. Neurosci 18, 637–646 (2015). doi: 10.1038/nn.3980; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lister R et al. , Global epigenomic reconfiguration during mammalian brain development. Science 341, 1237905 (2013). doi: 10.1126/science.1237905; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Colantuoni C et al. , Temporal dynamics and genetic control of transcription in the human prefrontal cortex. Nature 478, 519–523 (2011). doi: 10.1038/nature10524; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kang HJ et al. , Spatio-temporal transcriptome of the human brain. Nature 478, 483–489 (2011). doi: 10.1038/nature10523; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pletikos M et al. , Temporal specification and bilaterality of human neocortical topographic gene expression. Neuron 81, 321–332 (2014). doi: 10.1016/j.neuron.2013.11.018; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reilly SK et al. , Evolutionary changes in promoter and enhancer activity during human corticogenesis. Science 347, 1155–1159 (2015). doi: 10.1126/science.1260943; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Materials and methods are available as supplementary materials.
- 23.Lin Z et al. , Simultaneous dimension reduction and adjustment for confounding variation. Proc. Natl. Acad. Sci. U.S.A 113, 14662–14667 (2016). doi: 10.1073/pnas.1617317113; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ardlie KG et al. , The Genotype-Tissue Expression (GTEx) pilot analysis: Multitissue gene regulation in humans. Science 348, 648–660 (2015). doi: 10.1126/science.1262110; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lake BB et al. , Integrative single-cell analysis of transcriptional and epigenetic states in the human adult brain. Nat. Biotechnol 36, 70–80 (2018). doi: 10.1038/nbt.4038; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhong S et al. , A single-cell RNA-seq survey of the developmental landscape of the human prefrontal cortex. Nature 555, 524–528 (2018). doi: 10.1038/nature25980; [DOI] [PubMed] [Google Scholar]
- 27.Lake BB et al. , Neuronal subtypes and diversity revealed by single-nucleus RNA sequencing of the human brain. Science 352, 1586–1590 (2016). doi: 10.1126/science.aaf1204; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mi D et al. , Early emergence of cortical interneuron diversity in the mouse embryo. Science 360, 81–85 (2018). doi: 10.1126/science.aar6821; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lodato S, Arlotta P, Generating neuronal diversity in the mammalian cerebral cortex. Annu. Rev. Cell Dev. Biol 31, 699–720 (2015). doi: 10.1146/annurev-cellbio-100814-125353; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nord AS, Pattabiraman K, Visel A, Rubenstein JLR, Genomic perspectives of transcriptional regulation in forebrain development. Neuron 85, 27–47 (2015). doi: 10.1016/j.neuron.2014.11.011; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shibata M, Gulden FO, Sestan N, From trans to cis: Transcriptional regulatory networks in neocortical development. Trends Genet. 31, 77–87 (2015). doi: 10.1016/j.tig.2014.12.004; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.D’Arcangelo G et al. , A protein related to extracellular matrix proteins deleted in the mouse mutant reeler. Nature 374, 719–723 (1995). doi: 10.1038/374719a0; [DOI] [PubMed] [Google Scholar]
- 33.Rowell JJ, Mallik AK, Dugas-Ford J, Ragsdale CW, Molecular analysis of neocortical layer structure in the ferret. J. Comp. Neurol 518, 3272–3289 (2010). doi: 10.1002/cne.22399; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhu Y et al. , Spatiotemporal transcriptomic divergence across human and macaque brain development. Science 362, eaat8077 (2018). doi: 10.1126/science.aat8077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Miller DJ et al. , Prolonged myelination in human neocortical evolution. Proc. Natl. Acad. Sci. U.S.A 109, 16480–16485 (2012). doi: 10.1073/pnas.1117943109; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huttenlocher PR, Dabholkar AS, Regional differences in synaptogenesis in human cerebral cortex. J. Comp. Neurol 387, 167–178 (1997). doi: 10.1002/(SICI)1096-9861(19971020)387:2<167:AID-CNE1>3.0.CO;2-Z; [DOI] [PubMed] [Google Scholar]
- 37.Pardiñas AF et al. , Common schizophrenia alleles are enriched in mutation-intolerant genes and in regions under strong background selection. Nat. Genet 50, 381–389 (2018). doi: 10.1038/s41588-018-0059-2; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lambert JC et al. , Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet 45, 1452–1458 (2013). doi: 10.1038/ng.2802; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nalls MA et al. , Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nat. Genet 46, 989–993 (2014). doi: 10.1038/ng.3043; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Grove J et al. , ASD Working Group of the PGC; BUPGEN; Major Depressive Disorder Working Group of the PGC; 23andMe Research Team, Common risk variants identified in autism spectrum disorder. bioRxiv 224774 [Preprint]. 27 November 2017. doi: 10.1101/224774 [DOI] [Google Scholar]
- 41.Demontis D et al. , ADHD Working Group of the Psychiatric Genomics Con; Early Lifecourse & Genetic Epidemiology (EAGLE); 23andMe Research Team, Discovery of the first genome-wide significant risk loci for ADHD. bioRxiv 145581 [Preprint]. 3 June 2017. doi: 10.1101/145581 [DOI] [Google Scholar]
- 42.Wray NR et al. , Genome-wide association analyses identify 44 risk variants and refine the genetic architecture of major depression. Nat. Genet 50, 668–681 (2018). doi: 10.1038/s41588-018-0090-3; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ruderfer DM et al. , Genomic dissection of bipolar disorder and schizophrenia, including 28 subphenotypes. Cell 173, 1705–1715.e16 (2018). doi: 10.1016/j.cell.2018.05.046; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Savage JE et al. , Genome-wide association meta-analysis in 269,867 individuals identifies new genetic and functional links to intelligence. Nat. Genet 50, 912–919 (2018). doi: 10.1038/s41588-018-0152-6; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Luciano M et al. , Association analysis in over 329,000 individuals identifies 116 independent variants influencing neuroticism. Nat. Genet 50, 6–11 (2018). doi: 10.1038/s41588-017-0013-8; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wood AR et al. , Defining the role of common variation in the genomic and biological architecture of adult human height. Nat. Genet 46, 1173–1186 (2014). doi: 10.1038/ng.3097; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu JZ et al. , Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat. Genet 47, 979–986 (2015). doi: 10.1038/ng.3359; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Willer CJ et al. , Discovery and refinement of loci associated with lipid levels. Nat. Genet 45, 1274–1283 (2013). doi: 10.1038/ng.2797; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wheeler E et al. , Impact of common genetic determinants of Hemoglobin A1c on type 2 diabetes risk and diagnosis in ancestrally diverse populations: A transethnic genome-wide meta-analysis. PLOS Med. 14, e1002383 (2017). doi: 10.1371/journal.pmed.1002383; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.1000 Genomes Project Consortium, A map of human genome variation from population-scale sequencing. Nature 467, 1061–1073 (2010). doi: 10.1038/nature09534; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Giusti-Rodriguez PM, Sullivan PF, Schizophrenia and a high-resolution map of the three-dimensional chromatin interactome of adult and fetal cortex. bioRxiv (2018). [Google Scholar]
- 52.Wang D et al. , Comprehensive functional genomic resource and integrative model for the human brain. Science 362, eaat8464 (2018). doi: 10.1126/science.aat8464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Won H et al. , Chromosome conformation elucidates regulatory relationships in developing human brain. Nature 538, 523–527 (2016). doi: 10.1038/nature19847; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hamilton JP, Siemer M, Gotlib IH, Amygdala volume in major depressive disorder: A meta-analysis of magnetic resonance imaging studies. Mol. Psychiatry 13, 993–1000 (2008). doi: 10.1038/mp.2008.57; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rubinow MJ et al. , Basolateral amygdala volume and cell numbers in major depressive disorder: A postmortem stereological study. Brain Struct. Funct 221, 171–184 (2016). doi: 10.1007/s00429-014-0900-z; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Skene NG et al. , Genetic identification of brain cell types underlying schizophrenia. Nat. Genet 50, 825–833 (2018). doi: 10.1038/s41588-018-0129-5; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Amiel J et al. , Mutations in TCF4, encoding a class I basic helix-loop-helix transcription factor, are responsible for Pitt-Hopkins syndrome, a severe epileptic encephalopathy associated with autonomic dysfunction. Am. J. Hum. Genet 80, 988–993 (2007). doi: 10.1086/515582; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Harrington AJ et al. , MEF2C regulates cortical inhibitory and excitatory synapses and behaviors relevant to neurodevelopmental disorders. eLife 5, e20059 (2016). doi: 10.7554/eLife.20059; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zweier C et al. , Haploinsufficiency of TCF4 causes syndromal mental retardation with intermittent hyperventilation (Pitt-Hopkins syndrome). Am. J. Hum. Genet 80, 994–1001 (2007). doi: 10.1086/515583; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zweier M et al. , Mutations in MEF2C from the 5q14.3q15 microdeletion syndrome region are a frequent cause of severe mental retardation and diminish MECP2 and CDKL5 expression. Hum. Mutat 31, 722–733 (2010). doi: 10.1002/humu.21253; [DOI] [PubMed] [Google Scholar]
- 61.Ebert DH, Greenberg ME, Activity-dependent neuronal signalling and autism spectrum disorder. Nature 493, 327–337 (2013). doi: 10.1038/nature11860; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kennedy AJ et al. , Tcf4 regulates synaptic plasticity, DNA methylation, and memory function. Cell Reports 16, 2666–2685 (2016). doi: 10.1016/j.celrep.2016.08.004; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Parikshak NN et al. , Integrative functional genomic analyses implicate specific molecular pathways and circuits in autism. Cell 155, 1008–1021 (2013). doi: 10.1016/j.cell.2013.10.031; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schmidt-Edelkraut U, Daniel G, Hoffmann A, Spengler D, Zac1 regulates cell cycle arrest in neuronal progenitors via Tcf4. Mol. Cell. Biol 34, 1020–1030 (2014). doi: 10.1128/MCB.01195-13; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Willsey AJ et al. , Coexpression networks implicate human midfetal deep cortical projection neurons in the pathogenesis of autism. Cell 155, 997–1007 (2013). doi: 10.1016/j.cell.2013.10.020; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sanders SJ et al. , Insights into autism spectrum disorder genomic architecture and biology from 71 risk loci. Neuron 87, 1215–1233 (2015). doi: 10.1016/j.neuron.2015.09.016; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Deciphering Developmental Disorders Study, Prevalence and architecture of de novo mutations in developmental disorders. Nature 542, 433–438 (2017). doi: 10.1038/nature21062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Xu X, Wells AB, O’Brien DR, Nehorai A, Dougherty JD, Cell type-specific expression analysis to identify putative cellular mechanisms for neurogenetic disorders. J. Neurosci 34, 1420–1431 (2014). doi: 10.1523/JNEUROSCI.4488-13.2014; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gandal MJ et al. , Shared molecular neuropathology across major psychiatric disorders parallels polygenic overlap. Science 359, 693–697 (2018). doi: 10.1126/science.aad6469; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kalinka AT et al. , Gene expression divergence recapitulates the developmental hourglass model. Nature 468, 811–814 (2010). doi: 10.1038/nature09634; [DOI] [PubMed] [Google Scholar]
- 71.Domazet-Lošo T, Tautz D, A phylogenetically based transcriptome age index mirrors ontogenetic divergence patterns. Nature 468, 815–818 (2010). doi: 10.1038/nature09632; [DOI] [PubMed] [Google Scholar]
- 72.Huttenlocher PR, Synaptic density in human frontal cortex—Developmental changes and effects of aging. Brain Res. 163, 195–205 (1979). doi: 10.1016/0006-8993(79)90349-4; [DOI] [PubMed] [Google Scholar]
- 73.Brodmann K, Vergleichende Lokalisationslehre der Grosshirnrinde in ihren Prinzipien dargestellt auf Grund des Zellenbaues (Barth, 1909). [Google Scholar]
- 74.Kiessling MC et al. , Cerebellar granule cells are generated postnatally in humans. Brain Struct. Funct 219, 1271–1286 (2014). doi: 10.1007/s00429-013-0565-z; [DOI] [PubMed] [Google Scholar]
- 75.Gulsuner S et al. , Spatial and temporal mapping of de novo mutations in schizophrenia to a fetal prefrontal cortical network. Cell 154, 518–529 (2013). doi: 10.1016/j.cell.2013.06.049; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Caubit X et al. , TSHZ3 deletion causes an autism syndrome and defects in cortical projection neurons. Nat. Genet 48, 1359–1369 (2016). doi: 10.1038/ng.3681; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kwan KY, Transcriptional dysregulation of neocortical circuit assembly in ASD. Int. Rev. Neurobiol 113, 167–205 (2013). doi: 10.1016/B978-0-12-418700-9.00006-X; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lamb AN et al. , Haploinsufficiency of SOX5 at 12p12.1 is associated with developmental delays with prominent language delay, behavior problems, and mild dysmorphic features. Hum. Mutat 33, 728–740 (2012). doi: 10.1002/humu.22037; [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.