

Abstract
Phospholipase C (PLC) enzymes hydrolyze the plasma membrane (PM) lipid phosphatidylinositol 4,5-bisphosphate (PI4,5P2) to generate the second messengers inositol trisphosphate (IP3) and diacylglycerol (DAG) in response to receptor activation in almost all mammalian cells. We previously found that stimulation of G protein–coupled receptors (GPCRs) in cardiac cells leads to the PLC-dependent hydrolysis of phosphatidylinositol 4-phosphate (PI4P) at the Golgi, a process required for the activation of nuclear protein kinase D (PKD) during cardiac hypertrophy. We hypothesized that GPCR-stimulated PLC activation leading to direct PI4P hydrolysis may be a general mechanism for DAG production. We measured GPCR activation–dependent changes in PM and Golgi PI4P pools in various cells using GFP-based detection of PI4P. Stimulation with various agonists caused a time-dependent reduction in PI4P-associated, but not PI4,5P2-associated, fluorescence at the Golgi and PM. Targeted depletion of PI4,5P2 from the PM before GPCR stimulation had no effect on the depletion of PM or Golgi PI4P, total inositol phosphate (IP) production, or PKD activation. In contrast, acute depletion of PI4P specifically at the PM completely blocked the GPCR-dependent production of IPs and activation of PKD but did not change the abundance of PI4,5P2. Acute depletion of Golgi PI4P had no effect on these processes. These data suggest that most of the PM PI4,5P2 pool is not involved in GPCR-stimulated phosphoinositide hydrolysis and that PI4P at the PM is responsible for the bulk of receptor-stimulated phosphoinositide hydrolysis and DAG production.
INTRODUCTION
All cells have mechanisms for responding to external stimuli involving receptors that bind to ligands, including small molecules, peptides, protein growth factors, and lipid mediators. A common mechanism that receptors use to initiate intracellular signaling events is the ligand-stimulated hydrolysis of phosphatidylinositol 4,5-bisphosphate (PI4,5P2) by phosphatidylinositol (PI)–specific phospholipase C (PLC) (1, 2), which results in the production of inositol 1,4,5-trisphosphate (IP3) and diacylglycerol (DAG). IP3 is a soluble second messenger that binds to IP3 receptors (IP3Rs) in the endoplasmic reticulum (ER) to release Ca2+ from intracellular stores, which leads to an increase in the Ca2+ concentration in the cytosol. DAG is a membrane-bound lipid that can activate protein kinases, such as protein kinase C (PKC) and PKD.
PI4,5P2 and the enzymes that generate it are predominantly found at the plasma membrane (PM) and inside the nucleus (3, 4). However, agonist-stimulated DAG production can occur on internal membranes, such as those of the ER, Golgi apparatus, and the mitochondria (5). DAG produced at the PM cannot readily diffuse between membranes, and, although it is possible that cellular machinery could transfer DAG between membranes, none of the multiple lipid exchanger proteins and pathways thus far discovered are implicated in DAG transport for mediating signal transduction.
In most cell types, IP3 is rapidly produced within the first 2 min of agonist addition and then quickly decreases in concentration, staying at slightly more than basal amounts in the sustained presence of agonist (6–9). On the other hand, DAG production follows a biphasic time course with an initial rapid production phase, which is followed by a slightly slower phase that can continue for hours (6, 7, 10). Close examination of the nature of the acyl chains on early- and late-phase DAG has shown that late-phase DAG contains acyl chains that tend to be enriched in phosphatidylcholine (PC). This suggests that the initial phase of DAG production comes from PI4,5P2 hydrolysis, whereas the second phase is from another substrate, such as PC (11–13). Two enzymes have been proposed to be responsible for the production of DAG from PC. One is a PC-specific PLC, but molecular identification of such an enzyme is lacking (10). Phospholipase D (PLD) has also been proposed to produce DAG in a two-step process, where PC is hydrolyzed to phosphatidic acid (PA) and then converted into DAG through the action of a PA phosphatase (10, 14). However, in some cells, DAG is the precursor of PA rather than the product (15). Although it is possible that PC-PLC or PC-PLD activities could contribute to a component of DAG production, they cannot account for the long-term accumulation of total inositol phosphates (IPs). The total IP assay measures the accumulation of IP1 and assumes that IP1 is derived from the metabolism of IP3 that was generated by the hydrolysis of PI4,5P2 by PLC. However, in theory, IP1 could also be generated through the hydrolysis of phosphatidylinositol 4-phosphate (PI4P) by PLC and subsequent metabolism of IP2. In many cells, total IPs continue to accumulate after agonist exposure, although IP3 is no longer produced (7, 8). This suggests that longer-term IP and DAG production could arise from a phosphoinositide other than PI4,5P2.
We previously found that PI4P, a precursor of PI4,5P2, is a substrate for PLC in neonatal rat ventricular myocytes (NRVMs) in cardiac disease (16). PI4P is found in multiple pools inside the cells, including the PM, Golgi, and internal vesicles (17). All mammalian PI-PLC isoforms can hydrolyze PI4P in vitro, but data supporting direct hydrolysis of PI4P in cells and tissues are lacking (16, 18, 19). This is, in large part, due to the precursor-product relationship of PI4P and PI4,5P2, where hydrolysis of PI4,5P2 leads to the phosphorylation of PI4P by PI4–5 kinase through mass action to replenish the PI4,5P2 pool, which leads to decreases in PI4P abundance. Studies have challenged this notion by demonstrating that the PM PI4P pool is independent of the bulk, steady-state PI4,5P2 pool based on the observation that depleting PI4P does not affect steady-state amounts of PM PI4,5P2 (20).
On the basis of our previous findings and because of the development of fluorescent probes to monitor the spatial organization and temporal regulation of phosphoinositides and of reagents to manipulate the amounts of specific phosphoinositides, we revisited the question of a potential source for the long-term generation of total IPs and DAG. Our results suggest that the PI4P pool, and not the PI4,5P2 pool, provides the major source of biologically relevant DAG production.
RESULTS
Agonist-stimulated Golgi PI4P hydrolysis occurs in multiple cell types
We previously demonstrated that endothelin-1 (ET-1) stimulates PI4P depletion in the Golgi of cardiac myocytes in a PLC-dependent manner (16). To measure Golgi PI4P hydrolysis in other cell types, the pleckstrin homology (PH) domain of four-phosphate adaptor protein (FAPP) fused to green fluorescent protein (GFP) (FAPP-PH-GFP) was transduced using a high-efficiency adenovirus delivery system into cells, and GFP fluorescence in the Golgi was visualized by confocal microscopy (Fig. 1). FAPP-PH-GFP selectively labels Golgi-associated PI4P because it binds to both PI4P and adenosine diphosphate (ADP) ribosylation factor-1 (ARF-1), which is predominantly localized to the Golgi (16, 20, 21). Stimulation of either mouse embryonic fibroblasts (MEFs) (Fig. 1A) or rat arterial smooth muscle (RASM) cells (Fig. 1B) with ET-1 reduced Golgi PI4P–associated fluorescence. Similarly, neurotensin (NT) or fetal bovine serum (FBS) stimulated the depletion of Golgi PI4P in a pancreatic ductal adenocarcinoma cell line (PANC-1) (Fig. 1C) or a human ovarian cancer cell line (HEY) (Fig. 1D), respectively. Thus, multiple stimuli cause Golgi PI4P depletion in either primary cells or cancer cell lines, suggesting that agonist-stimulated Golgi PI4P depletion is a generalizable signaling process.
Fig. 1. Receptor-stimulated Golgi PI4P hydrolysis occurs in MEFs, PANC-1 cells, RASMs, and HEY cells.

(A to D) Cells were transduced with adenovirus expressing FAPP-PH-GFP. After 16 hours, the cells were serum-starved for 4 hours and then imaged. Full confocal stacks were collected at each time point to ensure that the confocal plane was maintained during the experiment. (A) Left: Representative images of MEFs expressing FAPP-PH-GFP before and after stimulation with 100 nM ET-1. Right: Time course of the changes in fluorescence of n = 4 cells for each treatment. (B) Left: Representative images of RASM cells expressing FAPP-PH-GFP before and after stimulation with 100 nM ET-1. Right: Time course of the changes in fluorescence of n = 3 cells for each treatment. (C) Time course of PI4P hydrolysis in untreated PANC-1 cells and PANC-1 cells treated with 100 nM NT. Data are combined from n = 6 cells for each treatment. (D) Time course of PI4P hydrolysis in untreated HEY cells and HEY cells treated with 1% FBS. Data are combined from n = 4 cells for each treatment. All traces are means ± SEM. Each time point on the curves for treated cells was compared to those of the control curves and was analyzed by one-tailed unpaired t test. *P < 0.05 and **P < 0.01; ns, not significant (P > 0.05).
Agonist-stimulated PI4P depletion occurs at the PM
We also examined PI4P depletion at the PM. Because FAPP-PH does not bind tightly to PI4P in the absence of ARF, we used a probe that does not require ARF to bind PI4P, P4M, a PI4P-binding domain of the secreted effector protein from Legionella pneumophila, SidM, fused to GFP (GFP-P4M). This probe was previously shown to bind to PI4P both in the Golgi and in the PM (17). To increase the ability to visualize PM PI4P, we used a tandem P4M domain fused to GFP (GFP-2×P4M) (22). Transduction of GFP-2×P4M into PANC-1 cells labeled both PM PI4P and what is likely Golgi PI4P as visualized by epifluorescence microscopy (Fig. 2A). In PANC-1 cells, treatment with NT activates Gq-dependent PLC signaling, which is responsible for DAG generation and the subsequent PKC and PKD activation that is required for NT-dependent cell proliferation (23). We found that NT caused a rapid decrease in PM-associated GFP-2×P4M fluorescence and accumulation at what is likely the Golgi apparatus (Fig. 2, A and B). This accumulation at the Golgi likely occurs because GFP-2×P4M freed from PM PI4P becomes available to bind to Golgi PI4P.
Fig. 2. NT stimulates PI4P depletion even after removal of the bulk of PI4,5P2.
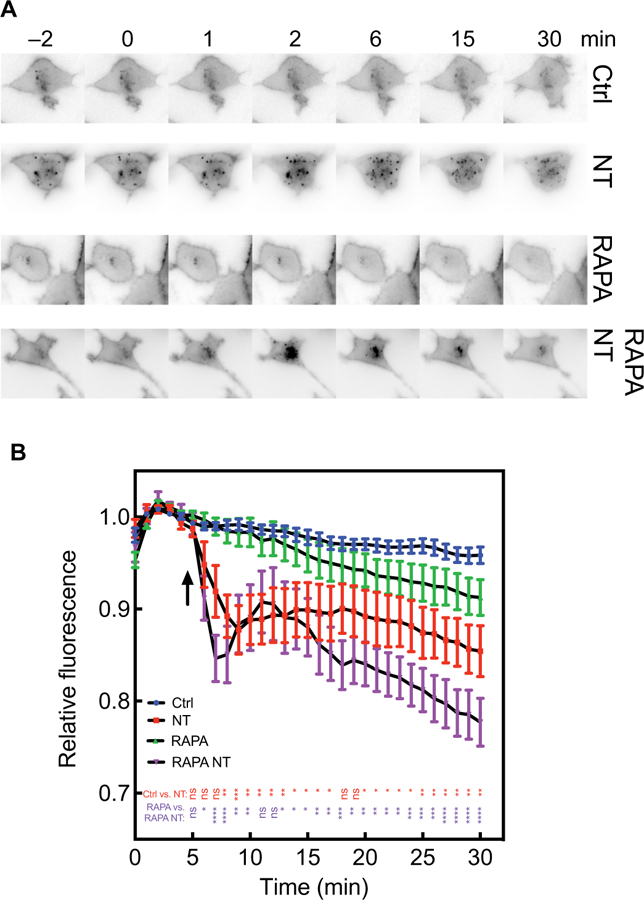
(A and B) PANC-1 cells were transduced with adenoviruses expressing the FKBP–5-phosphatase system and GFP-2×P4M and were imaged by epifluorescence microscopy. Cells were treated with dimethyl sulfoxide (DMSO) (vehicle) or RAPA for 15 min before being incubated with vehicle or 100 nM NT (at the times indicated by the arrow) for 25 min. (A) Representative image time courses were produced with the ImageJ montage setting with higher fluorescence represented in black. (B) Compiled data for at least 12 cells from at least three independent experiments. All traces are means ± SEM. All curves were compared using two-way analysis of variance (ANOVA) with multiple comparisons within each row. Comparisons between control (Ctrl) and RAPA treatments were not statistically significant at any time point. P values represent Ctrl compared to NT-treated (red) and RAPA compared to RAPA NT-treated (purple). *P < 0.05, **P < 0.01, *** P < 0.001, and ****P < 0.0001.
To confirm that the labeling at the PM corresponded to PI4P, PANC-1 cells transduced with GFP-2×P4M were treated with the PI4 kinase A (PI4KA)–specific inhibitor A1, which selectively inhibits PI4P synthesis at the PM but not at the Golgi apparatus (24). This compound causes depletion of PI4P, but not steady-state PI4,5P2, at the PM in human embryonic kidney (HEK) 293 cells (24). Treatment with A1 caused selective depletion of PM, but not Golgi-associated, GFP-2×P4M fluorescence and did not affect PI4,5P2-associated Tubby-GFP fluorescence (fig. S1A). The kinetics of A1-dependent PI4P depletion was very rapid, with a half-life (t1/2) of 2 min (fig. S1C), suggesting that the PM pool of PI4P is rapidly turned over at steady state. PI4 kinase B (PI4KB) is responsible for maintaining Golgi PI4P abundance. A selective inhibitor of PI4KB, PIK93, caused depletion of Golgi-associated fluorescence without substantially depleting PM PI4,5P2, as has been previously reported (fig. S1B). These data suggest that GFP-2×P4M labels both PM and Golgi PI4P pools in these cells, and that A1 selectively, and PIK93 somewhat selectively, acutely perturbed PM and Golgi PI4P pools, respectively. These data also confirm previous results indicating that the bulk PM pool of PI4P is not in direct equilibrium with most of the PM PI4,5P2 pool. If this were a direct equilibrium, then depletion of PI4P should lead to conversion of PI4,5P2 to PI4P by mass action and lead to substantial loss of PI4,5P2.
Receptor-dependent PI4P depletion at the PM does not depend on bulk PI4,5P2 hydrolysis
A possible explanation for the decrease in PI4P abundance is through conversion to PI4,5P2 by PI4–5 kinase, because PI4,5P2 is depleted during receptor-stimulated PLC activation. To assess the role of the bulk PI4,5P2 pool in receptor-dependent depletion of PI4P, PANC-1 cells were adenovirally transduced with a single vector expressing a phosphoinositide 5-phosphatase (5-phosphatase) fused to FK506-binding protein (FKBP) and the FKBP12-RAPA–binding subunit (FRB) fused to mRFP and targeted to the PM (PM-FRB-mRFP). Upon addition of rapamycin (RAPA), FRB and FKBP dimerize, targeting the 5-phosphatase to the PM where it can hydrolyze PI4,5P2 to PI4P (fig. S2, A and B) (16, 20, 25–27). To confirm depletion of PM PI4,5P2 in PANC-1 cells, Tubby-GFP was cotransduced with the FKBP–5-phosphatase RAPA translocation system using a high-efficiency adenoviral system, and Tubby-GFP and PM-FRB-mRFP fluorescence were visualized at the PM by confocal microscopy. Treatment of cells with RAPA resulted in a rapid loss in PM-associated Tubby-GFP fluorescence, whereas the localization of PM-FRB-mRFP fluorescence was unaffected, confirming the depletion of PM PI4,5P2 at the single-cell (fig. S2C) and population (fig. S2D) level. To determine whether the FKBP–5-phosphatase completely removed PI4,5P2, we measured the effect of FKBP–5-phosphatase treatment on ET-1–dependent Ca2+ release in populations of PANC-1 cells. The IP3R is very sensitive to low concentrations of IP3 that might be generated from a pool of PI4,5P2 that is not detected by Tubby-GFP. The RAPA-dependent recruitment of the FKBP–5-phosphatase to the PM did not substantially affect ET-1– dependent Ca2+ release (fig. S2E), indicating that a small pool of PI4,5P2 remained after FKBP–5-phosphatase treatment. Thus, most of the PI4,5P2 in the steady-state pool was not needed for receptor-dependent Ca2+ signaling, suggesting that the IP3 needed to stimulate IP3R-dependent Ca2+ release can be generated from hydrolysis of a very small pool of PI4,5P2.
Having confirmed that the FKBP–5-phosphatase system removed the bulk of the PI4,5P2 at the PM, we tested whether agonist-dependent PI4P depletion was affected by this treatment. Our hypothesis was that if PI4P depletion was the result of hydrolysis of PI4,5P2 in the bulk pool, then removal of the bulk pool would substantially decrease the agonist-dependent depletion of PI4P. PANC-1 cells were transduced with viruses expressing the FKBP–5-phosphatase system and GFP-2×P4M. The cells were then treated with DMSO (vehicle) or RAPA for 20 min to eliminate PI4,5P2 and then were stimulated with NT. PM and Golgi PI4P–associated GFP fluorescence were monitored by confocal microscopy. RAPA-dependent PI4,5P2 depletion alone did not substantially alter PM PI4P abundance, as assessed by measurement of GFP-2×P4M fluorescence (Fig. 2, A and B). Surprisingly, the rate of NT-dependent depletion of PM PI4P was enhanced after pretreatment with RAPA (Fig. 2, A and B).
To confirm these results in MEFs, we used total internal reflection fluorescence (TIRF) microscopy to visualize PI4P-associated GFP-P4M fluorescence. In MEFs, a PM edge is not readily visualized by either epifluorescence or confocal microscopy, perhaps because the cells are very flat. TIRF has the advantage that it visualizes only fluorescence within 100 nm of the coverslip, and thus, most of the fluorescence observed in the field is PM fluorescence. In cells transduced with GFP-2×P4M, the entire visualized cell was fluorescent (Fig. 3A). Treatment with ET-1 stimulated a time-dependent loss of PM fluorescence, suggesting that PM PI4P was depleted (Fig. 3, A and B), consistent with what was observed in PANC-1 cells. Treatment with RAPA to eliminate detectable PI4,5P2 from the PM substantially enhanced the rate of ET-1–dependent PM PI4P depletion (Fig. 3, A and B), as was seen for NT in PANC-1 cells. This indicates that the bulk of the PM PI4,5P2 pool was not required for ET-1–dependent PI4P depletion. The reason for the increase in the rate of PI4P depletion in these experiments is unclear but could be due to the enhanced availability of PI4P after the cleavage of PI4,5P2 by the 5-phosphatase.
Fig. 3. ET-1 stimulates PI4P depletion even after removal of the bulk of PI4,5P2.
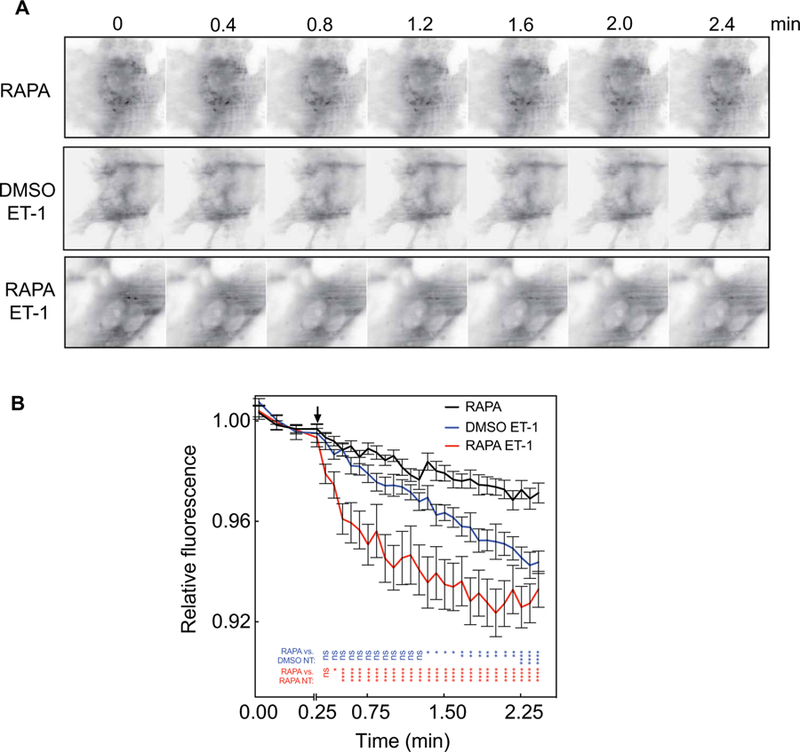
(A and B) MEFs were transduced with adenoviruses expressing the FKBP–5-phosphatase system and GFP-P4M and then were imaged by TIRF microscopy. Cells were treated with 1 μM RAPA or DMSO (vehicle) for 20 min, which was followed by the addition of 100 nM ET-1 or vehicle at the time indicated by the arrow. (A) Representative images of MEFs before and after treatment with 100 nM ET-1 with DMSO vehicle or 1 μM RAPA. (B) Time courses of PI4P depletion for combined experiments with six or seven cells analyzed for each condition in each of three independent experiments. All traces are means ± SEM. All curves are compared by two-way ANOVA with multiple comparisons within each row. P values represent RAPA, Ctrl compared to DMSO, ET-1–treated (blue) and RAPA compared to RAPA, ET-1–treated (red). *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
IP3 is transiently produced but PI4,5P2 depletion is not detected in response to NT in PANC-1 cells
We examined receptor-dependent PI4,5P2 hydrolysis at the PM in PANC-1 cells by monitoring either Tubby-GFP or GFP-PLCδ-PH domain localization to the PM. Upon PI4,5P2 hydrolysis by PLC, Tubby-GFP is released from the PM due to the loss of PM PI4,5P2. GFP-PLCδ-PH domain translocation can occur as a result of either loss of PI4,5P2 at the PM or accumulation of soluble IP3 that competes for PI4,5P2 binding to GFP-PLCδ-PH. Stimulation of endogenous receptors with NT in PANC-1 cells caused a transient decrease in the PM association of GFP-PLCδ-PH fluorescence (Fig. 4, A and B) but had no statistically significant effect on PM-associated Tubby-GFP fluorescence (Fig. 4C). This indicates that NT caused the transient hydrolysis of PI4,5P2 to generate a transient increase in IP3 but that this did not substantially deplete the PI4,5P2 pool. Together, these data suggest that most of the PM PI4,5P2 pool is insensitive to receptor-dependent PLC activation and only results in transient IP3 production. This transient increase in IP3 concentration is consistent with observations from multiple other studies (6–9). Thus, activation of endogenous NT-1 receptors can markedly deplete PM PI4P without substantial hydrolysis of PM PI4,5P2.
Fig. 4. NT-1 does not affect the amount of steady-state PM PI4,5P2 and causes only transient IP3 production.
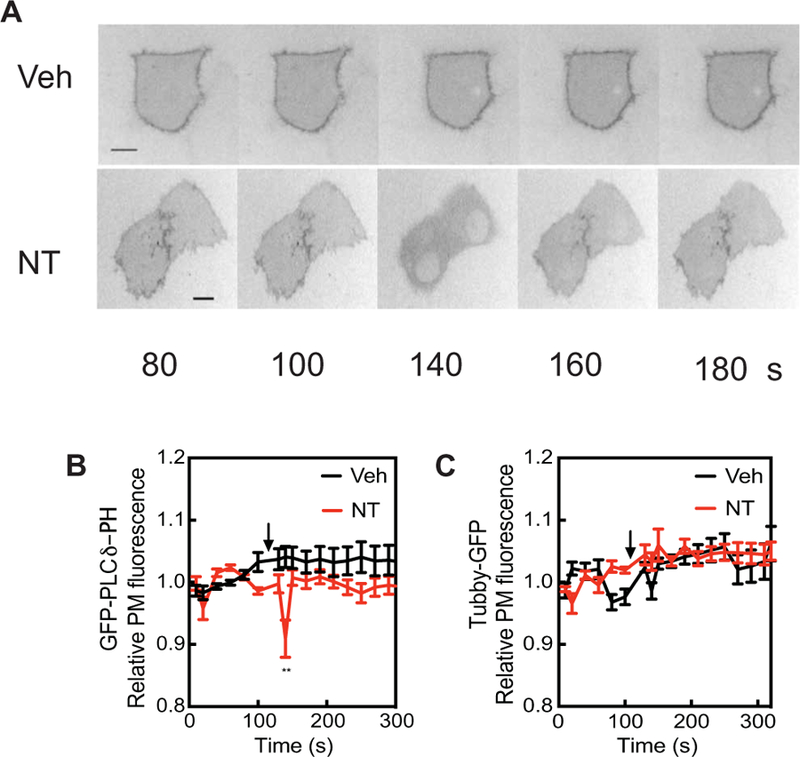
(A) PANC-1 cells were transfected with plasmid encoding the GFP-PLCδ-PH domain, treated with vehicle or 100 nM NT, and visualized by epifluorescence microscopy. Representative GFP images are shown. Time is seconds after beginning imaging. NT was added after 100 s. (B) Regions of interest containing GFP-PLCδ-PH fluorescence were identified and quantitated over time with ImageJ software. (C) Regions of interest containing Tubby-GFP fluorescence were identified and quantitated over time with ImageJ. Data in (B) and (C) are compiled from 10 cells in at least three independent experiments. All traces are means ± SEM. All curves were compared by two-way ANOVA with multiple comparisons within each row. **P < 0.01; values at all other time points were not statistically significant.
Most of the IP produced in cells is derived from the PI4P pool
A standard method for measuring receptor-stimulated PLC activation in cells is to measure the production and accumulation of inositol monophosphate (IP), a product of IP3 metabolism (8), in the presence of LiCl to block inositol monophosphate dephosphorylation. This assay is based on the assumption that the only PLC-dependent reaction in cells is the hydrolysis of PI4,5P2 to generate IP3, which is subsequently metabolized to generate IP1. On the other hand, agonist-stimulated, PLC-dependent hydrolysis of PI4P could also contribute to the pool of total IP (Fig. 5A), given that statistically significant PI4,5P2 hydrolysis was not observed in Tubby-GFP–expressing cells. First, we fractionated the total IP pool by ion exchange chromatography into IP1, IP2, and IP3 from MEFs stimulated with ET-1 for 30 min. ET-1 produced substantial increases in the amounts of IP1 and IP2, but an increase in IP3 abundance was not detectable (Fig. 5, B and C). We then examined the time course for the production of each of the IP species after ET-1 treatment (Fig. 5D). We found that IP2 was rapidly produced in the first minute and that its abundance was sustained for at least 10 min before decreasing at 30 min. The increases in IP3 abundance were statistically significantly smaller, and IP3 decayed rapidly in the first 5 min, consistent with our earlier findings (Fig. 4B). The abundance of IP1 began to increase at 5 min after stimulation because of the conversion of other IP species to IP1 and then accumulated over the entire time course as IP1 degradation was prevented by LiCl. The predominance of IP2 production over IP3 production supports the idea that PI4P is a substrate for receptor-stimulated IP production.
Fig. 5. NT-1 stimulates in the rapid accumulation of IP2 and the generation of very low amounts of IP3.

(A) Scheme showing the potential paths to total IP production. The reagents listed in blue were used to manipulate PI4P abundance throughout the study. (B) A representative chromatogram from an experiment in which MEFs were labeled with [3H]myo-inositol before being stimulated with 100 nM ET-1 for 30 min, and then IP species were extracted and fractionated by ion exchange chromatography as described in Materials and Methods. CPM, counts per minute. (C) Quantitation of the relative contributions of each of the indicated IP species to the total IPs produced by MEFs in response to stimulation with ET-1. Data are compiled from three separate experiments performed as described for (B). Data are means ± SEM. (D) Time course of the production of the indicated IP species after stimulation with 100 nM ET-1. Data are from duplicate measurements and are representative of three independent experiments.
As another approach to examine the contribution of PI4P to the total IPs produced, we depleted PI4P with phenylarsine oxide (PAO), a broad specificity inhibitor of both PM and Golgi PI4Ks, that we and others have previously shown does not substantially affect PM PI4,5P2 abundance (16). MEFs or PANC-1 cells were treated with DMSO or PAO for 15 min and then stimulated for 30 min with ET-1 or NT before total IPs were measured. Despite an apparent lack of hydrolysis of the bulk PI4,5P2 pool, and only transient IP3 production (Fig. 4, B and C), statistically significant total IP production was observed and pretreatment with PAO greatly reduced agonist-stimulated IP production in both cell types (Fig. 6A). To determine whether the source of total IPs was from the Golgi or PM, cells were treated with A1 to deplete PM PI4P or with PIK93 to deplete Golgi PI4P, which was followed by stimulation of MEFs with ET-1 or of PANC-1 cells with NT. Depletion of PM PI4P abolished both ET-1–stimulated IP production in MEFs and NT-stimulated IP production in PANC-1 cells (Fig. 6B). Depletion of Golgi PI4P did not statistically significantly affect total IP production in either cell type (Fig. 6C). These data suggest that, in primary cells and in a cancer cell line, PM PI4P is a major source of the total IPs produced upon PLC stimulation by activation of endogenously expressed G protein (heterotrimeric guanine nucleotide–binding protein)–coupled receptors (GPCRs) and that Golgi PI4P may only contribute a small fraction of the total.
Fig. 6. PI4P depletion at the PM blocks total IP production.
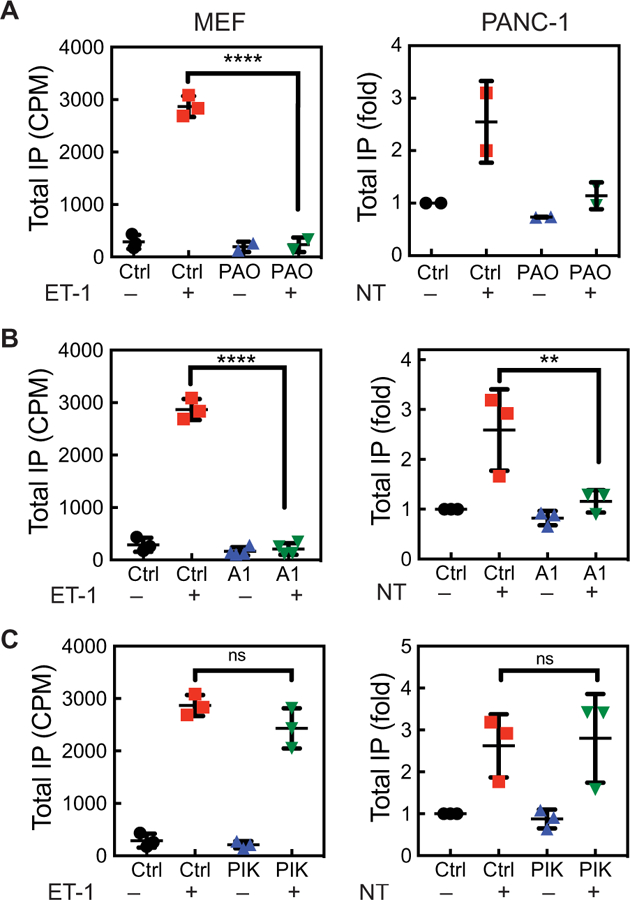
(A to C) Analysis of total IP production in vehicle- or ET-1–treated MEFs (left) and in vehicle- or NT-treated PANC-1 cells (right) after global PI4P depletion with PAO (A), after PM PI4P depletion with A1 (B), and after Golgi PI4P depletion with PIK93. The indicated cells were serum-starved and labeled with [3H]inositol for 24 hours and then were pretreated for 15 min with 10 μM PAO, 100 nM A1, or 300 nM PIK93 before being treated for 30 min with vehicle, 100 nM ET-1, or 100 nM NT in the presence of LiCl. The results are expressed in counts per minute (left) or fold over unstimulated cells (right). Data are means ± SD and were analyzed by one-way ANOVA with Tukey’s post-test. **P < 0.01 and ****P < 0.0001.
Activation of PKD depends on DAG derived from PI4P
We previously showed that, in NRVMs, PI4P depletion with Sac1(K2A) or PAO inhibited the activation of a nuclear pool of PKD (16, 28). Because PI4P hydrolysis produces IP2, which is biologically inert with respect to Ca2+ mobilization, we hypothesized that the role of PI4P hydrolysis is to produce DAG for the activation of PKC and PKD. We used a similar strategy as that described earlier (Fig. 6), except that the cells were collected and lysed to measure PKD activation by Western blotting with phosphospecific antibodies for the activated form of PKD, which is phosphorylated at Ser916 (Fig. 7). We found that the depletion of PI4P with PAO before agonist stimulation reduced PKD activation to basal levels in both NRVMs and MEFs (Fig. 7A). In PANC-1 cells, treatment with PAO did not inhibit agonist-stimulated PKD activation; instead, basal PKD activation was increased, which was further increased upon agonist stimulation (Fig. 7A). Depletion of PM PI4P with A1 also statistically significantly inhibited PKD activation in MEFs and PANC-1 cells in response to agonist (Fig. 7, B and C), whereas depletion of Golgi PI4P with PIK93 had a less substantial effect on global PKD activation (Fig. 7, B and C). PAO is an effective inhibitor of PI4Ks but has multiple off-target effects, whereas A1 is a much more specific inhibitor of PI4KB. Thus, blockade of PKD activation by A1 in PANC-1 cells is likely correct and suggests that the increase in PKD activation in PAO-treated cells is likely the result of off-target effects. Together, these data suggest that PM PI4P is critical for DAG production and global PKD activation.
Fig. 7. PI4P depletion at the PM inhibits PKD activation.

(A) The indicated cells were treated with 10 μM PAO for 15 min to deplete global PI4P, which was followed by the addition of agonist for 30 min (100 nM ET-1 or 100 nM NT). The cells were then analyzed by Western blotting with antibodies against total PKD and pPKD (Ser916). (B) MEFs were treated with 100 nM A1 or 300 nM PIK93 for 15 min, which was followed by the addition of 100 nM ET-1 for 30 min. The cells were then analyzed by Western blotting with antibodies against total PKD and pPKD (Ser916). (C) PANC-1 cells were treated with A1 or PIK93 for 15 min, which was followed by the addition of 100 nM NT for 30 min. The cells were then analyzed by Western blotting with antibodies against total PKD and pPKD (Ser916). Western blots in all panels are representative of three independent experiments. Graphs in each panel show the abundance of pPKD relative to that of total PKD from three independent experiments. Data are means ± SEM and were analyzed by one-way ANOVA with Tukey’s post-test. *P < 0.05, **P < 0.01, and ***P < 0.001.
Agonist-stimulated PI4,5P2 hydrolysis does not contribute substantially to total IP production or global PKD activation
The data so far indicate that GPCR stimulation does not affect the PM PI4,5P2 pool or produce substantial amounts of IP3 relative to IP2 yet decreases PM PI4P abundance and stimulates total IP production and PKD activation in two different cell types in response to two different agonists. As another approach to determine the involvement of PI4,5P2 hydrolysis in PKD activation and total IP accumulation, we depleted most of the PM PI4,5P2 with the FKBP– 5-phosphatase system described earlier. For these cell population– based experiments to be successful, the FKBP–5-phosphatase system has to be successfully delivered, and PI4,5P2 needs to be depleted with high efficiency in the cell population. We first analyzed a field of cells adenovirally transduced with the FKBP–5-phosphatase system and Tubby-GFP (fig. S2D). In this field, >90% of the cells expressed Tubby-GFP and PM-FRB-mRFP. Treatment with RAPA for 10 s eliminated PM-associated Tubby-GFP in almost all of the cells. Thus, this approach markedly decreased PM PI4,5P2 abundance in the overall population. However, depletion of PI4,5P2 did not alter agonist-stimulated total IP production in MEFs, PANC-1 cells, or HEK 293 cells (Fig. 8A). Similarly, depletion of PI4,5P2 did not inhibit agonist-dependent PKD activation in NRVMs, MEFs, or PANC-1 cells (Fig. 8, B and C). Together, these results suggest that, in at least two different cell types representing primary and immortalized cells, direct PI4P hydrolysis at the PM is likely the major source of IPs, as well as being the primary source for DAG production leading to PKD activation.
Fig. 8. PI4,5P2 depletion inhibits neither total IP production nor PKD activation.

Cells were transfected (HEK) or transduced (PANC-1, MEF, and NRVM) with an adenovirus expressing the FKBP–5-phosphatase (5P) system as described in fig. S2 or yellow fluorescent protein (YFP) (YF). (A) Cells were treated with 1 μM RAPA for 20 min, which was followed by stimulation with the indicated agonists [100 nM NT, 100 nM ET-1, or 2 μM adenosine triphosphate (ATP)]. Total IPs were analyzed as described in Fig. 6. Data are means ± SEM of three independent experiments. (B) Cells were treated with 1 μM RAPA for 20 min, which was followed by stimulation with the indicated agonists (100 nM ET-1 and 100 nM NT). PKD activation was analyzed by Western blotting as described in Fig. 7. Western blots are representative of three independent experiments. (C) Grouped data from MEFs from three experiments are shown as means ± SEM and were analyzed by one-way ANOVA with Tukey’s post-test. *P < 0.05.
DISCUSSION
It is well known that the stimulation of GPCRs that activate PLC causes PI4P abundance to transiently decrease. Determining the mechanism for the receptor-stimulated reduction in PI4P abundance has been confounding, because the precursor-product relationship between PI4P and PI4,5P2 would lead to PI4P depletion as PI4P is converted to PI4,5P2 to replenish the PI4,5P2 hydrolyzed by PLC. We present several lines of evidence to show that simple replenishment of the bulk PI4,5P2 pool does not underlie receptor-stimulated PM PI4P depletion. First, receptor stimulation led to substantial decreases in the abundance of PM PI4P but not the PI4,5P2 pool. Second, GPCR stimulation led to only transient IP3 production. Third, depletion of the PM PI4P pool did not substantially affect the PM PI4,5P2 pool but markedly inhibited total IP production and PKD activation. Fourth, depletion of the bulk PI4,5P2 pool with a PI5 phosphatase did not substantially affect total IP production or PKD activation. These data all indicate that the bulk PI4,5P2 pool does not contribute substantially to longer-term total IP production.
The simplest model to explain these observations is one in which most of the total IPs accumulated over time are derived from direct hydrolysis of PI4P. Most PLC isoforms have the capacity to directly hydrolyze PI4P in vitro at a rate comparable to that of PI4,5P2 hydrolysis. Thus, there is no intrinsic biochemical reason why PLC would not be able to access PI4P in the cell. In addition, our group previously identified PI4P hydrolysis by PLCε in the Golgi of cardiac myocytes, where no detectable PI4,5P2 is present (16, 21). The finding that PM PI4P constitutes an independent phosphoinositide pool that may not be in equilibrium with the bulk PM PI4,5P2 pool provides a framework that enables PI4,5P2-independent, receptor-stimulated hydrolysis of PI4P (20).
An alternative model that could account for receptor-stimulated PI4P depletion in the absence of detectable depletion of the bulk PI4,5P2 pool posits that a very small, rapidly turning over pool of PI4,5P2 participates in a cycle of hydrolysis by PLC and is rapidly replenished from PI4P by a highly active PI4–5 kinase. In this model, it is the constant cycle of hydrolysis and replenishment that ultimately leads to PI4P depletion. Three lines of evidence that we presented argue against this possibility. The first is based on data from experiments with the GFP-PLCδ-PH domain (Fig. 4B), where only a transient decrease in PM-associated fluorescence was observed upon treatment with NT. This decrease must be due to IP3 generation, which led to competition for binding of the PH domain to membrane PI4,5P2, because Tubby-GFP, which does not bind to IP3 and is only sensitive to depletion of PI4,5P2, was unaffected by NT (Fig. 4C). A possible cave-at to this is the previous demonstration that the GFP-PLCδ-PH domain more sensitively detects PIP2 hydrolysis than does Tubby-GFP. This could be because IP3 competes for the binding of PIP2 to PLCδ-PH but not Tubby, or because Tubby could have a higher intrinsic affinity for PIP2 than for PLCδ-PH (29). If Tubby does bind PIP2 more tightly, this supports our contention that treatment with PI4,5 phosphatase markedly depletes PM PIP2. Nevertheless, sustained IP3 release would be expected to result in sustained displacement of the GFP-PLCδ-PH probe, and a transient increase in IP3 is not consistent with a model in which a pool of PI4,5P2 is rapidly turning over to deplete PI4P in the long term. This supports the idea that only a minor fraction of the PIP2 pool is hydrolyzed during agonist stimulation in PANC-1 cells and that this is not responsible for the PI4P depletion that is readily observable. Finally, the stimulation of MEFs with ET-1 stimulated marked increases in IP1 and IP2 abundance, but IP3 production was barely detectable. The ready detection of IP2 without a concomitant detection of substantial amounts of IP3 supports the idea that PI4P is a direct substrate for receptor-stimulated PLC activity.
The data presented here are supported by various results from other laboratories. Dickson et al. reported that activation of endogenous P2Y2 receptors in tsa-201 cells causes Ca2+ release without substantial PI4,5P2 depletion or measurable IP3 generation. To observe substantial PI4,5P2 depletion, high-density overexpression of a Gq-coupled GPCR (M1 muscarinic acetylcholine receptor) was required. Depletion of PM PI4,5P2 in these studies did not substantially affect uridine triphosphate (UTP)–dependent Ca2+ release (30). These results are similar to our observations with endogenous ET-1 and NT receptors. In that work (30), it was postulated that, under strong stimulation of high-density receptors, PI4P-5 kinase supplies the PI4,5P2 required for continued IP production. However, it was also suggested that direct PI4P hydrolysis could account for some of these results. It has also been shown in other studies that inhibition of PI4K with wortmannin inhibits the production of total IPs (31). Here, we combined imaging analysis with PI-specific fluorescent probes, tools for manipulating the amounts of PI4P and PI4,5P2, and biochemical analyses to examine the roles of various pools of PI species in long-term DAG generation, and we conclude that most of the total IPs and DAG that are generated in the cell come from PI4P.
It has also been known for many years that, in many cell types, the production of IP3 after receptor activation is transient, whereas DAG production proceeds on a longer time course. Our data suggest that the PI4P pool may be the source of this DAG and that this would be consistent with the observed long-term IP production that also occurs in some cells. Several reports indicate that PI4P is present at an amount similar to that of PI4,5P2 in cells (32, 33), so how can this serve as a source for extended DAG production downstream of PLC? One possibility is that PI4P is rapidly synthesized from relatively abundant PI by PI4KA as PI4P is depleted because of PLC activity. Treatment of PANC-1 cells with A1 caused the rapid depletion of PI4P with a t1/2 of 2 min (fig. S1). This indicates that the PI4P pool at steady state is in rapid flux and that constant replenishment is needed to maintain steady-state PI4P abundance. Our experiments did not reveal the absolute rate of PI4P hydrolysis, because the observed decreases in PI4P fluorescence reflected the sum of the rates of PI4P degradation by PLC and resynthesis by PI4KA. To observe a decrease in PI4P abundance, the rate of hydrolysis must outpace the rate of synthesis.
As was mentioned earlier, PC has been considered as a probable source for long-term DAG production in some cell types because of the acyl chain composition of DAG. These data are hard to reconcile with the idea of PI4P as a source of DAG, because the acyl chain composition of PI4P is very similar to that of PI4,5P2 but different from that of PC (32). One possibility is that PC may be a substantial source of DAG mass but that the relevant DAG pool for PKD activation may be derived from PI4P. Another possibility is that the source of DAG is cell type– and receptor-specific. The development of fluorescence-based methods for the subcellular detection of lipid pools, the identification of enzymes that generate PI4P in different compartments, the development of tools to manipulate amounts of specific PI species, and the finding that the PI4P pool at the PM is not in direct equilibrium with the PM PI4,5P2 pool have facilitated an approach that has revealed processes that were not accessible in earlier experimentation. With the advent of advanced lipid mass spectrometry methods, it will be important to determine the lipid composition of the receptor-generated DAG pools in the context of manipulation of PI4P and PI4,5P2 to address the apparent discrepancy.
Our study in cardiac myocytes identified PLCε as the PLC enzyme responsible for PI4P hydrolysis at the Golgi–nuclear envelope interface (16). Previous work indicated that in RAT-1 fibroblasts, PLCε is responsible for late-stage IP production (3 to 30 min after stimulation), whereas PLCβ is responsible for early-stage IP production (0 to 3 min after stimulation) (8). On the basis of this result, one might conclude that PLCε is the enzyme responsible for PI4P hydrolysis. PLCε can be found at many intracellular locations and likely at the PM, so it is possible that PLCε may be involved in PI4P hydrolysis at some locations, but all mammalian PLC isoforms are capable of using PI4P as a substrate in vitro. It remains to be determined whether other PLC isoforms hydrolyze PI4P in response to GPCR stimulation.
In summary, we found that PI4P hydrolysis occurs in multiple cell types and substantially contributes to the production of biologically relevant amounts of DAG and total IPs. On the basis of these data, we propose a model in which PI4,5P2 hydrolysis serves as the primary source of IP3 for Ca2+ signaling, whereas PI4P hydrolysis serves as the primary source of DAG. An additional function of direct PI4P hydrolysis may be to provide a PLC substrate for the subcellular, PLC-dependent generation of DAG.
MATERIALS AND METHODS
Chemicals
ATP (A3377), ET-1 (E7764), NT (N6383), PIK93 (SML0546), and RAPA (R8781) were obtained from Sigma-Aldrich Co. [3H]Myo-inositol (NET116800) was purchased from PerkinElmer Inc. The PI4KA inhibitor A1 was provided by T. Balla (National Institutes of Health).
Antibodies
Rabbit polyclonal antibodies against total PKD/PKCμ (2052) and Ser916-phosphorylated PKD/PKCμ (2051) were obtained from Cell Signaling Technology Inc.
Isolation, culture, and transfection or adenoviral infection of cultured cells
The isolation and culture of NRVMs were performed as previously described (11). NRVMs were transduced with adenovirus at a multiplicity of infection (MOI) of 50. To prepare MEFs, day 13 to 14.5 C57/B6 mouse embryos were dissected into a 10-cm dish with phosphate-buffered saline (PBS). The top portion of the head, limbs, and organs was discarded, and the carcasses were minced and digested in 0.05% trypsin/EDTA for 5 min at 37°C. Tissue was dissociated further with a 20-gauge needle and further digested for an additional 10 min in 0.05% trypsin/EDTA at 37°C. MEFs were washed and plated in Dulbecco’s modified Eagle’s medium (DMEM) [with glucose (4.5 g/liter), 4 mM L-glutamine, and sodium pyruvate (110 mg/liter) containing penicillin (100 μg/ml) and streptomycin (100 U/ml)] and 10% FBS for 24 hours. Plates were further expanded into a second passage, and cells were allowed to grow to confluency before they were frozen at −140°C. MEFs were thawed into DMEM–10% FBS and used for experiments between passages 2 and 7. MEFs were transduced with adenoviruses at an MOI of 50. PANC-1 cells (CRL-1469) were purchased from the American Type Culture Collection, transduced with adenoviruses at an MOI of 30, and used from passages 2 to 15. HEY cells were provided by I.-m. Kim (Augusta University), transduced with adenoviruses at an MOI of 10, and used from passages 5 to 15. Mixed-population rat aortic smooth muscle cells were provided by C. Yen (University of Rochester) and transduced with adenoviruses at an MOI of 30. These cells were subcultured with DMEM with glucose (4.5 g/liter), 4 mM L-glutamine, sodium pyruvate (110 mg/liter), and 10% FBS. HEK 293 cells were subcultured with DMEM with glucose (4.5 g/liter), 4 mM L-glutamine, sodium pyruvate (110 mg/liter), and 5% FBS and transfected with 500 ng of the appropriate plasmids. All subcultured cells were passaged twice weekly according to a normal passaging protocol, briefly washed with PBS, treated with 0.25% trypsin to dislodge, and resuspended in culture medium. Transfections were performed using a standard protocol of Lipofectamine 2000, with 3 μl of Lipofectamine 2000 to 1 μg of DNA.
Plasmid and adenoviral constructs
FAPP1-PH-GFP, GFP-P4M, PLCδ-PH-GFP (Addgene plasmid #51407), Tubby-GFP, and GFP-2×P4M (Addgene plasmid #51472) were all provided by T. Balla and G. Hammond (University of Pittsburgh). PM–FRB–mRFP–T2A–FKBP–5-phosphatase was a gift from P. Varnai (Addgene plasmid #40896). All reporters were incorporated into adenoviral constructs by standard methods. Polymerase chain reaction– amplified fragments of the appropriate complementary DNAs were subcloned into an adenoviral shuttle vector under the control of the mouse cytomegalovirus promoter. The shuttle vector was recombined with the parent vector in HEK 293 cells to generate recombinant adenovirus. Recombined virus was amplified in HEK 293 cells and purified by CsCl gradient centrifugation. Viral titers were determined by an agarose overlay plaque assay.
Imaging
Cells were imaged at room temperature 24 to 48 hours after transduction with the appropriate adenoviruses. Culture medium was exchanged for serum-free DMEM culture medium at least 4 hours before imaging, and the cells were stimulated with agonist. FAPP1-PH-GFP imaging was performed on an Olympus FV1000MP microscope in confocal mode with a 40× 0.8-NA (numerical aperture) water immersion (Olympus) lens. Enhanced GFP (EGFP) was excited at 488 nm, and emission was monitored at 510 nm. For each cell, the entire confocal stack was collected to ensure that changes in fluorescence were not due to changes in the confocal plane. Data collection and analysis were performed with Olympus Fluoview software. Single-cell imaging of Tubby-GFP with FRB-RFP (red fluorescent protein) was performed in a Nikon C1 confocal microscope with a 60× 1.3-NA oil immersion (Nikon) lens. EGFP was excited at 488 nm with an argon laser and emission was monitored at 510 nm, whereas RFP was excited at 560 nm with a HeNe laser and emission was monitored at >600 nm. Imaging was performed in PBS. Imaging of Tubby-GFP and GFP-2×P4M was performed on a Leica DMi8 epifluorescence microscope in standard epifluorescence mode with autofocus and a 100× 1.4-NA oil immersion (Leica) lens, whereas imaging of GFP-PLCδ-PH was performed with a 63× 1.4-NA oil immersion (Leica) lens. EGFP was excited at 488 nm and emission was monitored at 510 nm, whereas RFP was excited at 560 nm with an X-Cite XLED1 light source. Emission monitored at >600 nm was imaged on a backlit CMOS Photometrics Prime 95B camera. To measure PM GFP–associated fluorescence, regions of PM GFP fluorescence were identified and circled as regions of interest and quantified in each frame of the time course of the experiment with ImageJ software unless otherwise stated. All data were normalized to the initial fluorescence before agonist addition. For TIRF microscopy imaging, we used Sutter Instrument Lambda SC SmartShutter controllers that enable rapid selection of two shutter openings sequentially, a 488-nm excitation (for GFP-P4M) and a 561-nm excitation (for FRB-mRFP), one at a time. The common beam path was focused through a custom side port to a side-facing filter cube placed below the objective turret of an Olympus IX81 inverted microscope to produce objective-based TIRF illumination. The cube with the dichroic mirror and emission filter used for the experiments was a Chroma Technology z488/561. For directing the incident beam at 70° from the normal on the coverslip, the beam was focused on the periphery of the back focal plane of a 60× 1.49-NA oil immersion (Olympus) lens, thereby giving a decay constant for the evanescent field of 110 nm. Images were collected with a charge-coupled device camera (iXon3, Andor Technology) controlled by MetaMorph software. The acquisition of the digital images was in synchrony with the opening of the smart shutters specific for the individual beams. The images were acquired at 5 Hz with 50-ms exposures and 300 gain (EM setting).
Ca2+ measurements
MEF cells were plated at 10,000 cells per well in 96-well plates. One day later, the cells were infected with the FKBP–5-phosphatase system adenovirus. On the next day, the medium was removed from plates and replaced with 50 μl of medium containing 2 μM Fluo-4 in Ca2+- and Mg2+-containing Hanks’ balanced salt solution (HBSS). Plates were incubated at room temperature for 30 min and washed twice (at 100 μl per well) with Ca2+-free HBSS. Ca2+-free HBSS (80 μl) containing RAPA or DMSO was then added to each well, and the plates were incubated at room temperature for 10 min. The plates were then inserted into the plate reader (Hamamatsu FDSS7000EX Functional Drug Screening System), and ET-1 was added to each well to a final concentration of 100 nM by the addition of a total volume of 40 μl of 300 nM ET-1 at 14 s after the initiation of imaging. Plates were imaged for a total of 2.5 min. Each point in the graph is the average of six wells, and the data are representative of five separate experiments.
Measurement of IP production
Cells were plated in either uncoated or, for FKBP–5-phosphatase experiments, poly-D-lysine–coated 12-well plates. MEFs were plated at a density of 4 × 104 cells/ml, whereas PANC-1 cells were plated at 1 × 105 cells/ml. NRVMs were treated as previously described (34). Twenty-four hours after adenovirus infection, the cells were labeled for 16 hours with [3H]inositol (3 μCi/ml) in serum-free, low-inositol Gibco F-10 medium. LiCl was added to a final concentration of 10 mM for 10 min at 37°C, which was followed by the appropriate treatments and agonist stimulation for 30 min. At each end point, the cells were lysed, and the soluble fraction was extracted and separated by anionic exchange chromatography. [3H]Total IPs were measured by liquid scintillation counting. For fractionation of different IP species, MEFs were cultured as described earlier after seeding at 1.5 × 105 cells per well in a six-well culture plate. After 1 day in culture, the medium was exchanged for F-10/Ham’s containing [3H] inositol (6 μCi/ml) at 1.25 ml per well. One day later, LiCl was added to a final concentration of 10 mM for 10 min before the cells were stimulated with 100 nM human recombinant ET-1 for 30 min before extraction. The cells were then extracted into 750 μl of ice-cold formic acid per well on ice for 30 min. Extracts were then mixed with 250 μl of ice-cold 150 mM ammonia and 4 ml of ice-cold water. This mixture was then applied to 1 ml of Dowex AG 1-X8 columns (Bio-Rad) that were prepared by running 9 ml of 2 M ammonium formate, 100 mM formic acid, 2 × 9 ml of water, and 9 ml of 10 mM tris-carbonate (pH 8.5) sequentially over the columns. After sample loading, the columns were washed with 3 ml of 10 mM tris-CO3 (pH 8.5), and IP1 was eluted, together with residual inositol, in the same buffer (~20 ml). After a 12-ml wash with 50 mM ammonium sulfate in the same buffer, IP2, IP3, and IP4 were eluted in buffer containing 75, 150, and 190 mM ammonium sulfate in ~12-, 15-, and 9-ml samples, respectively. The position of IP3 elution was determined with [3H]IP3 (PerkinElmer). Fractions of 6 to 9 ml were mixed with 10 ml of high salt–compatible scintillation cocktail (EcoLume ES, MP Biomedicals) for scintillation counting. Cell residue after formic acid extraction was dissolved in 750 μl per well of 0.5 N NaOH and counted, and the values were used for normalization.
Western blotting
Cells were lysed in 2× SDS sample buffer, boiled, and loaded onto a 7.5 or 10% (w/v) SDS–polyacrylamide gel electrophoresis (PAGE). After SDS-PAGE, proteins were transferred to nitrocellulose for 16 hours at 25 V, which was followed by incubation with an antibody against either total or activated (phosphorylated at Ser916) form of PKD (1:1000 each). Goat anti-rabbit DyLight 800 (Thermo Fisher Scientific) secondary antibody (1:10,000) was added. Western blots were imaged and quantified with a LI-COR Odyssey imaging system.
Statistical analysis
All analyses were performed using GraphPad Prism statistical analysis software. Time courses were analyzed by two-way ANOVA with multiple comparisons, except for Fig. 1. For Fig. 1, each time point in the treated curves compared to control curves was analyzed by a one-tailed, unpaired t test. All other data were analyzed with a one-way ANOVA and Tukey’s post analysis. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. All data are presented as means ± SEM or ± SD as indicated.
Supplementary Material
www.sciencesignaling.org/cgi/content/full/11/547/eaan1210/DC1
Fig. S1. Effects of A1 and PIK93 on PM PI4,5P2 abundance.
FFig. S2. PI4,5P2 depletion in PANC-1 cells by RAPA-induced targeting of PI5 phosphatase to the PM.
Acknowledgments:
We would like to thank T. Balla (NIH) for providing A1 and the FAPP-PH-GFP, PLCδ-PH-GFP, and Tubby-GFP plasmids; G. Hammond (University of Pittsburgh) for providing the P4M-SidM-GFP and 2×P4M-SidM-GFP plasmids; and P. Varnai (Semmelweis University, Budapest, Hungary) for the PM–FRB–mRFP–T2A–FKBP–5-phosphatase plasmid.
Funding: This work was supported by NIH grants RO1GM053536 (to A.V.S.), RO1GM111735 (to A.V.S.), F31GM116557 (to R.G.d.R.), DE014756 (to D.I.Y.), and GM111997 (to A.A.).
Footnotes
Competing interests: The authors declare that they have no competing interests.
Data and materials availability: All data needed to evaluate the conclusions of this study are available in the paper or the Supplementary Materials.
REFERENCES AND NOTES
- 1.Kadamur G, Ross EM, Mammalian phospholipase C. Annu. Rev. Physiol 75, 127–154 (2013). [DOI] [PubMed] [Google Scholar]
- 2.Yang YR, Follo MY, Cocco L, Suh P-G, The physiological roles of primary phospholipase C. Adv. Biol. Regul 53, 232–241 (2013). [DOI] [PubMed] [Google Scholar]
- 3.Balla T, Phosphoinositides: Tiny lipids with giant impact on cell regulation. Physiol. Rev 93, 1019–1137 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cocco L, Martelli AM, Billi AM, Cataldi A, Miscia S, Mottola MR, Manzoli L, Phospholipids as components of the nuclear matrix: Their possible biological significance. Basic Appl. Histochem 31, 413–419 (1987). [PubMed] [Google Scholar]
- 5.Díaz Añel AM, Malhotra V, PKCη is required for β1γ2/β3γ2- and PKD-mediated transport to the cell surface and the organization of the Golgi apparatus. J. Cell Biol 169, 83–91 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wright TM, Rangan LA, Shin HS, Raben DM, Kinetic analysis of 1,2-diacylglycerol mass levels in cultured fibroblasts. Comparison of stimulation by α-thrombin and epidermal growth factor. J. Biol. Chem 263, 9374–9380 (1988). [PubMed] [Google Scholar]
- 7.Griendling KK, Rittenhouse SE, Brock TA, Ekstein LS, Gimbrone MA Jr., Alexander RW, Sustained diacylglycerol formation from inositol phospholipids in angiotensin II-stimulated vascular smooth muscle cells. J. Biol. Chem 261, 5901–5906 (1986). [PubMed] [Google Scholar]
- 8.Kelley GG, Kaproth-Joslin KA, Reks SE, Smrcka AV, Wojcikiewicz RJH, G-protein-coupled receptor agonists activate endogenous phospholipase Cε and phospholipase Cβ3 in a temporally distinct manner. J. Biol. Chem 281, 2639–2648 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Truett AP III, Verghese MW, Dillon SB, Snyderman R, Calcium influx stimulates a second pathway for sustained diacylglycerol production in leukocytes activated by chemoattractants. Proc. Natl. Acad. Sci. U.S.A 85, 1549–1553 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Exton JH, Signaling through phosphatidylcholine breakdown. J. Biol. Chem 265, 1–4 (1990). [PubMed] [Google Scholar]
- 11.Leach KL, Ruff VA, Wright TM, Pessin MS, Raben DM, Dissociation of protein kinase C activation and sn-1,2-diacylglycerol formation. Comparison of phosphatidylinositol- and phosphatidylcholine-derived diglycerides in α-thrombin-stimulated fibroblasts. J. Biol. Chem 266, 3215–3221 (1991). [PubMed] [Google Scholar]
- 12.Pessin MS, Raben DM, Molecular species analysis of 1,2-diglycerides stimulated by α-thrombin in cultured fibroblasts. J. Biol. Chem 264, 8729–8738 (1989). [PubMed] [Google Scholar]
- 13.Bocckino SB, Blackmore PF, Exton JH, Stimulation of 1,2-diacylglycerol accumulation in hepatocytes by vasopressin, epinephrine, and angiotensin II. J. Biol. Chem 260, 14201–14207 (1985). [PubMed] [Google Scholar]
- 14.Kennerly DA, Diacylgyceral metabolism in Mast cells. J. Biol. Chem 262, 16305–16313 (1987). [PubMed] [Google Scholar]
- 15.Scott SA, Xiang Y, Mathews TP, Cho HP, Myers DS, Armstrong MD, Tallman KA, O’Reilly MC, Lindsley CW, Brown HA, Regulation of phospholipase D activity and phosphatidic acid production after purinergic (P2Y6) receptor stimulation. J. Biol. Chem 288, 20477–20487 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang L, Malik S, Pang J, Wang H, Park KM, Yule DI, Blaxall BC, Smrcka AV, Phospholipase Cε hydrolyzes perinuclear phosphatidylinositol 4-phosphate to regulate cardiac hypertrophy. Cell 153, 216–227 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hammond GRV, Machner MP, Balla T, A novel probe for phosphatidylinositol 4-phosphate reveals multiple pools beyond the Golgi. J. Cell Biol 205, 113–126 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smrcka AV, Hepler JR, Brown KO, Sternweis PC, Regulation of polyphosphoinositide-specific phospholipase C activity by purified Gq. Science 251, 804–807 (1991). [DOI] [PubMed] [Google Scholar]
- 19.Rhee SG, Suh PG, Ryu SH, Lee SY, Studies of inositol phospholipid-specific phospholipase C. Science 244, 546–550 (1989). [DOI] [PubMed] [Google Scholar]
- 20.Hammond GRV, Fischer MJ, Anderson KE, Holdich J, Koteci A, Balla T, Irvine RF, PI4P and PI(4,5)P2 are essential but independent lipid determinants of membrane identity. Science 337, 727–730 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Malik S, deRubio RG, Trembley M, Irannejad R, Wedegaertner PB, Smrcka AV, G protein βγ subunits regulate cardiomyocyte hypertrophy through a perinuclear Golgi phosphatidylinositol 4-phosphate hydrolysis pathway. Mol. Biol. Cell 26, 1188–1198 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Levin R, Hammond GRV, Balla T, De Camilli P, Fairn GD, Grinstein S, Multiphasic dynamics of phosphatidylinositol 4-phosphate during phagocytosis. Mol. Biol. Cell 28, 128–140 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guha S, Rey O, Rozengurt E, Neurotensin induces protein kinase C-dependent protein kinase D activation and DNA synthesis in human pancreatic carcinoma cell line PANC-1. Cancer Res 62, 1632–1640 (2002). [PubMed] [Google Scholar]
- 24.Bojjireddy N, Botyanszki J, Hammond G, Creech D, Peterson R, Kemp DC, Snead M, Brown R, Morrison A, Wilson S, Harrison S, Moore C, Balla T, Pharmacological and genetic targeting of the PI4KA enzyme reveals its important role in maintaining plasma membrane phosphatidylinositol 4-phosphate and phosphatidylinositol 4,5-bisphosphate levels. J. Biol. Chem 289, 6120–6132 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Szentpetery Z, Várnai P, Balla T, Acute manipulation of Golgi phosphoinositides to assess their importance in cellular trafficking and signaling. Proc. Natl. Acad. Sci. U.S.A 107, 8225–8230 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zoncu R, Perera RM, Sebastian R, Nakatsu F, Chen H, Balla T, Ayala G, Toomre D, De Camilli PV, Loss of endocytic clathrin-coated pits upon acute depletion of phosphatidylinositol 4,5-bisphosphate. Proc. Natl. Acad. Sci. U.S.A 104, 3793–3798 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tóth DJ, Tóth JT, Gulyás G, Balla A, Balla T, Hunyady L, Várnai P, Acute depletion of plasma membrane phosphatidylinositol 4,5-bisphosphate impairs specific steps in endocytosis of the G-protein-coupled receptor. J. Cell Sci 125, 2185–2197 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vega RB, Harrison BC, Meadows E, Roberts CR, Papst PJ, Olson EN, McKinsey TA, Protein kinases C and D mediate agonist-dependent cardiac hypertrophy through nuclear export of histone deacetylase 5. Mol. Cell. Biol 24, 8374–8385 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Szentpetery Z, Balla A, Kim YJ, Lemmon MA, Balla T, Live cell imaging with protein domains capable of recognizing phosphatidylinositol 4,5-bisphosphate; a comparative study. BMC Cell Biol 10, 67 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dickson EJ, Falkenburger BH, Hille B, Quantitative properties and receptor reserve of the IP3 and calcium branch of Gq-coupled receptor signaling. J. Gen. Physiol 141, 521–535 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nakanishi S, Catt KJ, Balla T, A wortmannin-sensitive phosphatidylinositol 4-kinase that regulates hormone-sensitive pools of inositolphospholipids. Proc. Natl. Acad. Sci. U.S.A 92, 5317–5321 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Traynor-Kaplan A, Kruse M, Dickson EJ, Dai G, Vivas O, Yu H, Whittington D, Hille B, Fatty-acyl chain profiles of cellular phosphoinositides. Biochim. Biophys. Acta 1862, 513–522 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kruse M, Vivas O, Traynor-Kaplan A, Hille B, Dynamics of phosphoinositide-dependent signaling in sympathetic neurons. J. Neurosci 36, 1386–1400 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang L, Malik S, Kelley GG, Kapiloff MS, Smrcka AV, Phospholipase Cε scaffolds to muscle-specific A kinase anchoring protein (mAKAPβ) and integrates multiple hypertrophic stimuli in cardiac myocytes. J. Biol. Chem 286, 23012–23021 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
www.sciencesignaling.org/cgi/content/full/11/547/eaan1210/DC1
Fig. S1. Effects of A1 and PIK93 on PM PI4,5P2 abundance.
FFig. S2. PI4,5P2 depletion in PANC-1 cells by RAPA-induced targeting of PI5 phosphatase to the PM.