

A global transition away from energy dense and cheap fossil fuels will require the commercial implementation of numerous new energy technologies, each of which must be scaled to large enough sizes to impact existing markets and substantially lower global CO2 emissions. The electrochemical conversion of CO2 to chemicals, fuels, and feedstocks has shown enough promise in the past decade to justify both academic and industrial sectors to increase efforts to better assess the potential economic and technical feasibility of the technology. Intensifying the process toward an industrial scale can be achieved by higher production rates, either by simply increasing the total area of catalyst in a reactor or by increasing the reaction rate (current density) for a given area of catalyst. Both approaches will be needed to economically produce an industrial quantity of product in a single plant (e.g., >100 tons product/day). In electrochemical CO2 reduction, however, increasing current densities to those needed for commercial operation (e.g., >200 mA/cm2) requires researchers to use cell designs that can supply enough CO2 to the catalyst layer to fuel the reaction, as opposed to traditional H-cell reactors. For these reasons, more and more researchers have begun using catalysts deposited onto gas-diffusion layers (GDLs), where high concentrations of CO2 can be maintained in close proximity to the catalyst layer even at high reaction rates. GDLs can also reduce overall cell potentials by directly improving catalytic activity, while a more system-focused testing platform can help reduce major system losses such as ohmic heating. Researchers operating these experimental devices at higher current densities, however, have discovered a number of operational intricacies that can make the direct switch away from lower current density experiments in an H-cell challenging. This Viewpoint is meant to describe some of these unique operational considerations that can impact catalytic activity and our ability to accurately collect data, while acting as a starting guide for researchers to transition to using gas-diffusion layers as a platform for benchmarking novel catalysts at commercially viable current densities.
Specific discussion points here may apply broadly across all platforms for performing high current density CO2 reduction, while others pertain specifically to our presented cell configuration consisting of three chambers and a catalyst layer deposited onto a GDL (Figure 1). Membrane electrode assembles, which show promise in the reduction of overall cell potentials in future systems, are a similar cell configuration with additional operational intricacies brought on by a stagnant or extremely small catholyte layer.1−5 For researchers new to high current density testing, however, the three-chamber configuration can provide an easy platform to rapidly test new catalysts and GDLs.6−9 This Viewpoint briefly discusses the intricacies of operating GDLs for CO2 electroreduction through three main subtopics: assembly, operation, and postanalysis of data.
Figure 1.

(a) A standard electrochemical for CO2 electrolysis for testing catalysts in a three-electrode configuration. (b) Side profile showing chambers for the CO2 (left), catholyte (center), and anolyte (right). (c) Isometric view of the exploded three-chamber cell showing individual components.
In electrochemical systems, a catalyst is typically deposited onto a substrate, with carbon paper, glassy carbon, and metal foils being common supports. In high current density experiments, a GDL10−12 commonly acts as a support for the catalyst. A catalyst is typically deposited directly onto the microporous layer side of the GDL, which is hydrophobic and helps to form a gas–liquid interface between the microporous layer and the catalyst. The creation of new GDLs specifically with mechanical and chemical properties suited for CO2 reduction applications will be an important avenue for new research going forward,13,14 but most current systems report using commercially available GDLs such as Sigracet 39BC or variations from Freudenberg or Covestro. The hydrophobic nature of bare carbon-based GDLs and an as-prepared GDL with a 100 nm layer of sputtered silver is demonstrated in Supporting Video 1. Here, we further show that after the surface has been briefly placed under reducing potentials, a separately prepared 150 nm thick Cu catalyst layer becomes fully wetted when a droplet of water is pipetted on top. The bare GDL without a catalyst layer, however, maintains its hydrophobic surface even after being placed in reductive conditions. During operation, commonly used metallic and porous catalyst layers (e.g., Ag, Au, Cu) are then assumed to be fully covered by the electrolyte, while CO2 diffuses across a gas–liquid interface provided by the hydrophobic surface of the microporous layer inside of the GDL. As we discuss in more detail in our recent perspective, we then assume that CO2 travels a short distance and reacts in dissolved form rather than a gaseous form with a three-phase interface.15
The diffusion of CO2 from a nearby gas phase not only allows for much higher current densities than an H-cell but has consequences for the morphology, activity, and stability of the catalyst layer. In an H-cell, for example, even a nanostructured catalyst functions as a planar electrode, relying on CO2 to diffuse 40–120 μm to reach the catalyst layer. In this configuration, almost all CO2 reduction measurements are then performed under some degree of mass transport limitations, resulting in concentration polarizations and making intrinsic activity difficult to accurately determine and potentially overestimated.16 For a rough nanostructured electrode with a surface porosity of several microns, only the outermost surfaces may have access to sufficient reagent, while the base can be depleted of CO2.17 In a GDL, however, the catalyst layer must allow for access to CO2 on one side and electrolyte on the other side, resulting in a porous electrode structure compared to the planar system in an H-cell. In this configuration, a large electrochemically active surface area is then possible, with all surfaces of the catalyst having comparatively greater access to CO2 due to the short diffusion pathway compared to that in an H-cell. Thus, the greater surface area for CO2 reduction can then result in greater geometric activity at lower overpotentials than H-cell experiments. Further, a GDL can allow for gaseous products to diffuse into the gas phase prior to nucleating at the surface and blocking active sites. These structural differences between H-cells and GDLs would similarly result in different pH and concentration gradients along catalyst nanostructures, meaning that structural benefits of catalysts derived in an H-cell may not have performance that translates directly into a GDE configuration.
A challenge that results from this orientation, however, is the potential for hydroxide and carbonates to build up inside of the structure before they can be transported to the bulk electrolyte. Crystallization of salts can then be a common occurrence within the porous catalyst layer, especially at higher current densities where substantial hydroxide is generated.18 These considerations should then be factored into the design of catalyst structures for CO2 reduction, in addition to the design and composition of the material itself. Finally, in an H-cell configuration, impurities are easily deposited onto the outermost catalyst sites, which can cause fast deactivation of CO2 reduction in favor of hydrogen evolution, as these sites possess the greatest access to CO2 coming from the bulk electrolyte. Due to the opposite direction of CO2 transport within a GDL system, however, impurities are likely to deposit onto the catalyst surfaces furthest from the source of CO2.15
As illustrated in Supporting Video 2, the electrical connection and sealing of a GDL within a cell is also important. Here, conductive tape is applied on the gas side (noncatalyst side) of the GDL composed of carbon fibers to provide an electrical connection to the potentiostat but also to physically fix it to the sealing gasket during cell assembly. More importantly, conductive tape is applied around the entire electrode instead of just at the top in order to minimize the in-plane distance that electrons travel through the GDL to reach the CO2 reduction catalyst. Sufficient current collection is extremely important for higher current density operation due to the relatively high resistivity of commonly used GDLs (∼106× more resistive than pure Cu), which can cause potential/voltage variations across the GDL and catalyst layer that scale with the applied current density (via Ohm’s Law; Vdrop_GDL = IRGDL). These potential variations can then result in heterogeneous local current distributions across the catalyst layer. In terms of CO2 reduction and catalyst characterization, the nonuniform potential and current density throughout the catalyst layer could then result in location-dependent product formation and differences in the local reaction environment (e.g., pH), irrespective of the catalyst used.
This intricacy of high current density CO2 electrolysis is particularly important for CO2 reduction on copper electrodes due to the sensitivity of pH and current density on H2, CO, and C2 product formation.13,19,20 These implications can be illustrated in Figure 2a by assuming an ideal catalyst with an overall activity of 120 mV/dec (after iR and local pH correction). In this figure, we can see the differences in the sensitivity of the overpotential and the local pH as a function of the geometric current density. An applied potential difference of only 36 mV induced by resistive losses on this electrode could then result in current density variations from 100 to 200 mA/cm2, which could then result in large spatial differences in the local reaction environment across the electrode surface. The lower current density region (e.g., 0.01–10 mA/cm2), however, is less sensitive to these effects due to the exponential relationship between the overpotential and current density and the dependency of the GDL’s resistive losses on the current density. These considerations are then unique to only high current density catalyst testing and applications. It is thus important to emphasize that catalytic activity at higher current densities can be strongly influenced by noncatalytic electrode variations, such as the resistivity of the GDL, how current is collected, and the overall electrode size.
Figure 2.
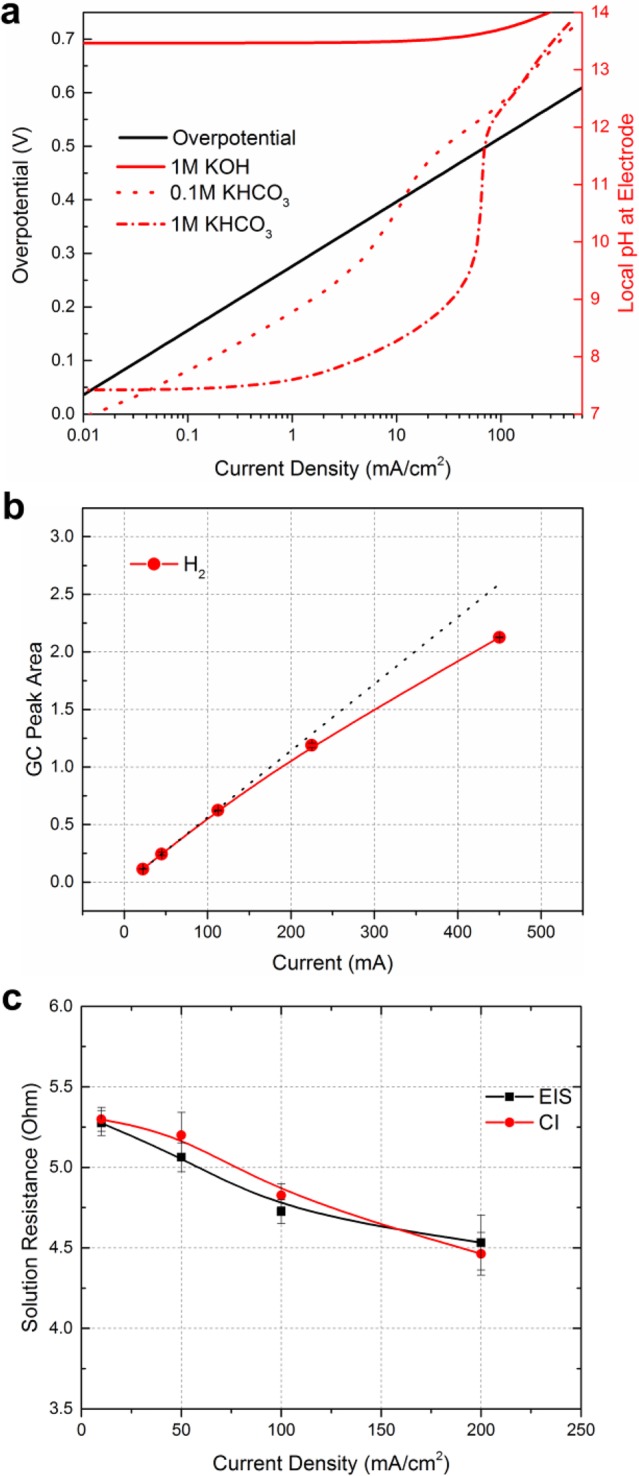
Examples of important considerations when benchmarking catalytic CO2 reduction performance under high current density operation. (a) Comparative effect of current density on the ideal required overpotential and the local reaction environment, showing how the system behaves differently as a function of current density. (b) Effect of high current operation on the measured peak area of H2 as recorded by a gas chromatograph (GC), showing deviation from the limit of linearity (dashed) at high concentrations (2.25 cm2 geometric area). (c) Measured solution resistance between the working and reference electrodes as a function of applied current density using both current interrupt (CI) and electrochemical impedance spectroscopy (EIS) (1 M KHCO3 is used as an electrolyte). This illustrates changes in the electrolyte conductivity as a function of operation and current density.
Assembly of the entire cell for electrochemical testing can be seen step-by-step in the Supporting Information, while a video showing cell operation is also available (Supporting Video 3). Here and in Figure 1, we can see the overall simplicity of the cell design but also the typical ancillary equipment needed to run the device such as a pump to flow liquid through the electrolyte channels and a GC to measure the formed gaseous products. In high current density experiments, it is also important to note that the concentration of product gases entering the GC increases significantly as compared to H-cell testing, which can impact product measurement. Multipoint calibration of a GC across the concentration range of all of the expected gas products (H2, CO, CH4, C2H4) is then necessary as the concentrations produced may no longer be within a linear range, known as the Limit of Linearity (illustrated using H2 in Figure 2b). This can lead to overall Faradaic efficiencies (FEs) greater than 100% if the lower-concentration calibrations typically used in H-cell testing are extrapolated. This can be particularly misleading if liquid products are not measured or reported in the total FE measurement as the observed total FE of gas products may still be below 100%. While it is possible to increase the gas flow rate to reduce the GC’s measured concentrations back to a linear calibration range, this may cause pressure imbalances between the gas/liquid phases within the GDL while reducing the single-pass conversion efficiency of CO2. Similarly, because the GC peak areas are related to the total applied current, the overall geometric catalyst area can also be reduced. Either way, it is essential that the GC is properly calibrated within the range of concentrations that are produced, instead of extrapolating from lower-concentration calibration data.
During operation of a GDL or membrane–electrode assembly, other operational factors may cause instability or complicate analysis of electrochemical behavior if not taken into consideration. At higher current densities, much higher ohmic drops between the working and reference electrodes can be expected due to the larger charge passed through the electrolyte. Not only can this be a major contributing factor to the overall cell potential in a two-electrode setup but it must be taken into account when correcting for the potential of the working CO2 reduction cathode in a three-electrode system. While electrochemical impendence spectroscopy (EIS) is typically performed even in H-cells to determine a system’s ohmic drop between the working and reference electrodes, the measured electrolyte resistance value between the reference and working electrodes can also be shown to vary as a function of the current density (Figure 2c). Here, using two different methods and several replicates, the solution resistance is found to decrease with the applied current density. This means that iR determination and correction may require resistance measurements at various conditions to capture the real working potential of a cathode. In the case of the 1 M KHCO3 electrolyte used to acquire this data, the change in electrolyte resistance is likely due to an overall increase in electrolyte conductivity due to the generation of hydroxide at the cathode surface. This will change the electrical properties of the electrolyte within the diffusion region but can also change the overall pH and conductivity of the bulk catholyte over time, particularly if the buffer breaks down. Alternatively, if 1 M KOH is used as a catholyte, long exposure to gaseous CO2 via the GDL may cause the pH to steadily decline due to the spontaneous formation of bicarbonate, along with a corresponding reduction in the conductivity of the electrolyte. Unless accounted for, these factors can cause the determined working potential of the cathode to differ from an iR correction performed at 0 mA/cm2.
Additionally, the large ohmic drop throughout the system driven by high current density operation can result in large temperature changes to the electrolyte, further affecting solution conductivity, electrode activity (via Arrhenius’ Law), and the solubility of CO2 diffusing across the gas–liquid interface. Without a sufficient electrolyte volume and passive cooling of the electrolyte chambers to mitigate the heating in the system, these temperature changes will affect the observed electrochemical results. Even worse, over a multihour stability test, the temperature can increase gradually, providing transient operating conditions. Additional strategies to avoid this include minimizing the electrolyte distance between the anode and cathode and using a high-conductivity electrolyte to facilitate charge transport.
A final complexity of operating GDLs pertains to the delicate gas–liquid interface that provides gaseous CO2 in close proximity to the catalyst. Even slight overpressures on either the gas or liquid side of the GDL can cause gas to bubble into the liquid phase or result in flooding of the GDL. If possible, the pressure of both phases across the GDL should then be regulated to ensure that catalytic activity can be determined without additional uncertainties introduced by stability of the gas–liquid interface. One notable source of pressure imbalance can even come from in-line GCs connected to the cell. Here, a constant backpressure from the GC, as well as pressure increases during injections, can cause gas to enter the liquid phase if the pressure spike is too high. For GCs that use syringe injections, however, the CO2 gas pressure is easier to maintain near atmospheric pressure.
In summary, this Viewpoint discusses several operational intricacies of using GDLs for electrochemical CO2 reduction, which is becoming increasingly important as the number of reports at higher current densities grows. The differences in testing/optimizing catalyst performance between traditional aqueous H-cells and gas diffusion electrodes is not trivial, and many new protocols must be used to ensure proper sample preparation, recording of data, and product identification. However, we believe that, with proper care and attention, the large field of catalyst researchers working on CO2 electroreduction can leverage existing infrastructures to expedite the scientific development of this technology. We hope this article acts as a source of information for catalyst-focused researchers looking to move to high current density catalyst testing and reinforces the need for fundamental research performed under practical conditions.
Acknowledgments
This project received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme (Grant Agreement No. 759743 – WUTANG). K.L. would also like to acknowledge funding from the China Scholarship Council (CSC). The authors thank Minouk van Oorschot for the descriptive cell drawings in Figure 1. The authors greatly acknowledge the experimental, design, and conceptual work from current and former colleagues that cumulatively led to the presented work.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsenergylett.9b00137.
Step-by-step pictures of the assembly of an electrochemical cell for CO2 reduction using a gas-diffusion layer, a description of the EIS and CI experiments to determine the solution resistance, and a description of the parameters used for the model data in Figure 2a (PDF)
Video (with audio) of the wettability of GDEs and catalysts (MP4)
Video (with audio) of the application of conducting tape to the back of a GDE (MP4)
Video (with audio) of a short overview of an electrochemical cell in operation (MP4)
Views expressed in this Viewpoint are those of the authors and not necessarily the views of the ACS.
The authors declare no competing financial interest.
Supplementary Material
References
- Salvatore D. A.; Weekes D. M.; He J.; Dettelbach K. E.; Li Y. C.; Mallouk T. E.; Berlinguette C. P. Electrolysis of Gaseous CO2 to CO in a Flow Cell with a Bipolar Membrane. ACS Energy Lett. 2018, 3 (1), 149–154. 10.1021/acsenergylett.7b01017. [DOI] [Google Scholar]
- Li Y. C.; Zhou D.; Yan Z.; Gonçalves R. H.; Salvatore D. A.; Berlinguette C. P.; Mallouk T. E. Electrolysis of CO2 to Syngas in Bipolar Membrane-Based Electrochemical Cells. ACS Energy Lett. 2016, 1 (6), 1149–1153. 10.1021/acsenergylett.6b00475. [DOI] [Google Scholar]
- Ripatti D. S.; Veltman T. R.; Kanan M. W. Carbon Monoxide Gas Diffusion Electrolysis That Produces Concentrated C2 Products with High Single-Pass Conversion. Joule 2019, 3 (1), 240–256. 10.1016/j.joule.2018.10.007. [DOI] [Google Scholar]
- Kaczur J. J.; Yang H.; Liu Z.; Sajjad S. D.; Masel R. I. Carbon Dioxide and Water Electrolysis Using New Alkaline Stable Anion Membranes. Front. Chem. 2018, 10.3389/fchem.2018.00263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vennekoetter J.-B.; Sengpiel R.; Wessling M. Beyond the Catalyst: How Electrode and Reactor Design Determine the Product Spectrum during Electrochemical CO2 Reduction. Chem. Eng. J. 2019, 364, 89. 10.1016/j.cej.2019.01.045. [DOI] [Google Scholar]
- Dinh C.-T.; García de Arquer F. P.; Sinton D.; Sargent E. H. High Rate, Selective, and Stable Electroreduction of CO2 to CO in Basic and Neutral Media. ACS Energy Lett. 2018, 3 (11), 2835–2840. 10.1021/acsenergylett.8b01734. [DOI] [Google Scholar]
- Jhong H.-R.; Tornow C. E.; Kim C.; Verma S.; Oberst J. L.; Anderson P. S.; Gewirth A. A.; Fujigaya T.; Nakashima N.; Kenis P. J. A. Gold Nanoparticles on Polymer-Wrapped Carbon Nanotubes: An Efficient and Selective Catalyst for the Electroreduction of CO2. ChemPhysChem 2017, 18 (22), 3274–3279. 10.1002/cphc.201700815. [DOI] [PubMed] [Google Scholar]
- Zhuang T.-T.; Liang Z.-Q.; Seifitokaldani A.; Li Y.; De Luna P.; Burdyny T.; Che F.; Meng F.; Min Y.; Quintero-Bermudez R.; et al. Steering Post-C–C Coupling Selectivity Enables High Efficiency Electroreduction of Carbon Dioxide to Multi-Carbon Alcohols. Nat. Catal. 2018, 1 (6), 421–428. 10.1038/s41929-018-0084-7. [DOI] [Google Scholar]
- Kibria M. G.; Dinh C.-T.; Seifitokaldani A.; De Luna P.; Burdyny T.; Quintero-Bermudez R.; Ross M. B.; Bushuyev O. S.; Garcia de Arquer F. P.; Yang P.; et al. A Surface Reconstruction Route to High Productivity and Selectivity in CO2 Electroreduction toward C2+ Hydrocarbons. Adv. Mater. 2018, 30 (49), 1804867. 10.1002/adma.201804867. [DOI] [PubMed] [Google Scholar]
- Cook R. L.; MacDuff R. C.; Sammells A. F. High Rate Gas Phase CO 2 Reduction to Ethylene and Methane Using Gas Diffusion Electrodes. J. Electrochem. Soc. 1990, 137 (2), 607–608. 10.1149/1.2086515. [DOI] [Google Scholar]
- Park S.; Lee J.-W.; Popov B. N. A Review of Gas Diffusion Layer in PEM Fuel Cells: Materials and Designs. Int. J. Hydrogen Energy 2012, 37 (7), 5850–5865. 10.1016/j.ijhydene.2011.12.148. [DOI] [Google Scholar]
- Kim B.; Hillman F.; Ariyoshi M.; Fujikawa S.; Kenis P. J. A. Effects of Composition of the Micro Porous Layer and the Substrate on Performance in the Electrochemical Reduction of CO2 to CO. J. Power Sources 2016, 312, 192–198. 10.1016/j.jpowsour.2016.02.043. [DOI] [Google Scholar]
- Dinh C.-T.; Burdyny T.; Kibria M. G.; Seifitokaldani A.; Gabardo C. M.; Garcia de Arquer F. P.; Kiani A.; Edwards J. P.; De Luna P.; Bushuyev O. S.; et al. CO2 Electroreduction to Ethylene via Hydroxide-Mediated Copper Catalysis at an Abrupt Interface. Science 2018, 360 (6390), 783–787. 10.1126/science.aas9100. [DOI] [PubMed] [Google Scholar]
- Jeanty P.; Scherer C.; Magori E.; Wiesner-Fleischer K.; Hinrichsen O.; Fleischer M. Upscaling and Continuous Operation of Electrochemical CO2 to CO Conversion in Aqueous Solutions on Silver Gas Diffusion Electrodes. J. CO2 Util. 2018, 24, 454–462. 10.1016/j.jcou.2018.01.011. [DOI] [Google Scholar]
- Burdyny T.; Smith W. A. CO2 Reduction on Gas-Diffusion Electrodes and Why Catalytic Performance Must Be Assessed at Commercially-Relevant Conditions. Energy Environ. Sci. 2019, 10.1039/C8EE03134G. [DOI] [Google Scholar]
- Clark E. L.; Resasco J.; Landers A.; Lin J.; Chung L.-T.; Walton A.; Hahn C.; Jaramillo T. F.; Bell A. T. Standards and Protocols for Data Acquisition and Reporting for Studies of the Electrochemical Reduction of Carbon Dioxide. ACS Catal. 2018, 8 (7), 6560–6570. 10.1021/acscatal.8b01340. [DOI] [Google Scholar]
- Raciti D.; Mao M.; Park J. H.; Wang C. Mass Transfer Effects in CO2 Reduction on Cu Nanowire Electrocatalysts. Catal. Sci. Technol. 2018, 8 (9), 2364–2369. 10.1039/C8CY00372F. [DOI] [Google Scholar]
- Lv J.-J.; Jouny M.; Luc W.; Zhu W.; Zhu J.-J.; Jiao F. A Highly Porous Copper Electrocatalyst for Carbon Dioxide Reduction. Adv. Mater. 2018, 30, 1803111. 10.1002/adma.201803111. [DOI] [PubMed] [Google Scholar]
- Ma S.; Sadakiyo M.; Luo R.; Heima M.; Yamauchi M.; Kenis P. J. A. One-Step Electrosynthesis of Ethylene and Ethanol from CO2 in an Alkaline Electrolyzer. J. Power Sources 2016, 301, 219–228. 10.1016/j.jpowsour.2015.09.124. [DOI] [Google Scholar]
- Jouny M.; Luc W.; Jiao F. General Techno-Economic Analysis of CO2 Electrolysis Systems. Ind. Eng. Chem. Res. 2018, 57 (6), 2165–2177. 10.1021/acs.iecr.7b03514. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.