

Abstract
PEGylated polylysine peptides represent a new class of scavenger receptor inhibitors that may find utility at inhibiting DNA nanoparticle uptake by Kupffer cells in the liver. PEG-peptides inhibit scavenger receptors in the liver by a novel mechanism involving in situ formation of albumin nanoparticles. The present study developed a new in vivo assay used to explore the structure-activity-relationships of PEG-peptides to find potent scavenger receptor inhibitors. Radio-iodinated PEG-peptides were dosed i.v. in mice and shown to saturate liver uptake in a dose-dependent fashion. The inhibition potency (IC50) was dependent on both the length of a polylysine repeat and PEG molecular weight. PEG30kda-Cys-Tyr-Lys25 was confirmed to be a high molecular weight (33.5 kDa) scavenger receptor inhibitor with an IC50 of 18 μM. Incorporation of multiple Leu residues compensated, to allow a decrease in PEG MW and Lys repeat, resulting in PEG5kda-Cys-Tyr-Lys-(Leu-Lys4)3-Leu-Lys that inhibited scavenger receptors with an IC50 = 20 μM. A further decrease in PEG MW to 2 kDa increased potency, resulting in a low molecular weight (4403 g/mol) PEG-peptide with an IC50 of 3 μM. Optimized low molecular weight PEG-peptides also demonstrated potency when inhibiting the uptake of radio-iodinated DNA nanoparticles by the liver. This study demonstrates an approach to discover low molecular weight PEG-peptides that serve as potent scavenger receptor inhibitors to block nanoparticle uptake by the liver.
Keywords: Nanoparticle, Scavenger Receptor, Biodistribution, Liver, Inhibitor
Introduction
The reticuloendothelial system (RES) is a network of cells located in the liver, spleen and bone marrow composed of Kupffer cells and fenestrated endothelial cells that recognizes and removes foreign molecules and nanoparticles from the bloodstream.2 The capture of drug delivery nanoparticles by RES decreases the percent of dose reaching remote tissues and results in activation of the innate immune system.3–7 Kupffer cells and fenestrated endothelial cells express scavenger receptors that bind anionic plasmid DNA, liposomes, viruses, modified albumin, oxidized low-density lipoproteins and many other drug delivery nanoparticles.5, 8–13
DNA nanoparticles generated by combining plasmid DNA with cationic polymers such as polylysine, polyethylene imine, dendrimers or peptides,14–18 have been optimized to maximize in vitro transfection.19 However, when dosed i.v., they bind and aggregate serum proteins, resulting in lung biodistribution, embolisms and death.20 Covalent attachment of polyethylene glycol (PEG) results in DNA nanoparticles that avoid biodistribution to the lung, but are instead taken-up by the RES of the liver.21–22
Despite possessing a stealth layer to mask the underlying positive charge, DNA nanoparticles still bind albumin and undergo charge reversal,23 resulting in the rapid capture of these anionic nanoparticles by scavenger receptors in the liver.24 The percent of dose captured by liver is highly dependent on the density and length of PEG. Nanoparticles shielded with 30 kDa PEG, possessing a zeta potential (δ) of +3 mV, avoid nearly all liver uptake.25 Conversely, nanoparticles possessing either 5 or 2 kDa PEG, and a zeta potential +15 or +30 mV respectively, are captured with 50 and 70% of dose recovered in the liver after a 5 min biodistribution.25 Liver uptake of PEGylated DNA nanoparticles can be significantly inhibited by co-administering excess PEG-peptide.24 The proposed mechanism involves the binding of PEG-peptides to serum proteins to form small albumin nanoparticles that saturate scavenger receptors (Scheme 1).
Scheme 1. Mechanism of Scavenger Receptor Inhibition.

The i.v. administration of a PEG-peptide leads to in situ formation of albumin nanoparticles that saturate scavenger receptors. The peptide sequence and PEG length influence the potency of albumin nanoparticles at inhibiting scavenger receptors.
Previously published structure-activity-relationship studies established that PEG30kda-Cys-Trp-Lys25 was a potent scavenger receptor inhibitor with an in vivo IC50 of 2 μM when used to inhibit 125I-DNA nanoparticles.1 The inhibition potency was dependent on the length of the polylysine repeat and PEG molecular weight with maximal potency achieved with a Lys repeat of 25 or 30 modified with a single 30kDa PEG.1 Shorter polylysine peptides of 10–20 residues were less potent and attempted reduction in molecular weight by substitution with a 5 kDa PEG resulted in an inactive analogue.
In the present study, PEG-peptides were directly radio-iodinated and dosed i.v. tail vein in mice. Dose escalation resulted in the in situ formation of protein nanoparticles that saturated scavenger receptors, resulting in a decrease in the percent of dose in the liver. Scavenger receptor inhibition potency was dependent on the size and charge of the PEG-peptide protein nanoparticles. Based on this new approach, we report that substitution of polylysine peptides with Leu allowed reduction in both the number of Lys and PEG molecular weight resulting in an increase in potency and a decrease in MW of nearly 10-fold. The following study defines structural features of PEG-peptides resulting in potent low molecular weight PEG-peptide scavenger receptor inhibitors that function in vivo.
Results
In a previously published study, polylysine PEG-peptides 16–19 (Table 1) were combined with 125I-DNA and delivered i.v. tail vein in triplicate mice. PEG-peptide dose escalation resulted in saturation of scavenger receptors, leading to inhibition of 125I-DNA nanoparticle uptake in liver.1 The PEG-peptide inhibition concentration to block liver uptake by 50% (IC50) was dependent on the number of repeating Lys residues, with maximum potency (IC50 of 2.1 μM) achieved with 19 (PEG30kDa-Cys-Trp-Lys25) (Fig. 1, Table 1). While these studies demonstrated PEG-peptides could saturate scavenger receptors, the apparent IC50 was also dependent on PEG-peptide binding affinity for DNA. PEG-peptides with longer Lys repeats possess greater affinity for DNA which could contribute to the observed inhibition potency.26
Table 1.
Structure and Characterization of Peptides and PEG-Peptides
| Peptide and PEG-Peptides | Yielda | Mass (calc/obs)b |
|---|---|---|
| 1. Cys-Tyr-Lys10 | 51 | 1566.1/1565.9 |
| 2. Cys-Tyr-Lys15 | 44 | 2206.9/2206.8 |
| 3. Cys-Tyr-Lys20 | 40 | 2847.8/2847.2 |
| 4. Cys-Tyr-Lys25 | 68 | 3488.6/3488.7 |
| 5. Tyr-(Acr-Lys4)3-Acr-Lys-Cys | 77 | 3172.1/3172.0 |
| 6. Lys-(Tyr-Lys4)3-Tyr-Lys-Cys | 73 | 2568.6/2568.0 |
| 7. Tyr-(Phe-Lys4)3-Phe-Lys-Cys | 16 | 2539.3/2540.1 |
| 8. Cys-Tyr-(Trp-Lys3)4 | 28 | 2567.3/2567.4 |
| 9. Cys-Tyr-(Leu-Lys4)3-Leu-Lys | 43 | 2403.2/2403.0 |
| 10. Cys-Tyr-(Leu-Lys)8 | 12 | 2343.1/2343.8 |
| 11. Cys-Tyr-(Leu-Lys)6 | 20 | 1732.3/1731.6 |
| 12. Cys-Tyr-(Leu-Lys)4 | 36 | 1249.7/1249.0 |
| 13. Cys-Tyr-(Leu-Arg)8 | 10 | 2437.6/2437 |
| 14. Cys-Tyr-Lys4-(Leu-Lys)4-Leu4 | 11 | 2215.0/2212.6 |
| 15. Cys-Tyr-Leu4-(Leu-Lys)4-Lys4 | 17 | 2215.0/2215.0 |
| 16. PEG30kDa-Cys-Trp-Lys10 | 80 | 31589 |
| 17. PEG30kDa-Cys-Trp-Lys15 | 89 | 32230 |
| 18. PEG30kDa-Cys-Trp-Lys20 | 80 | 32871 |
| 19. PEG30kDa-Cys-Trp-Lys25 | 75 | 33512 |
| 20. PEG30kDa-Cys-Tyr-Lys10 | 36 | 31566 |
| 21. PEG30kDa-Cys-Tyr-Lys15 | 34 | 32206 |
| 22. PEG30kDa-Cys-Tyr-Lys20 | 57 | 32847 |
| 23. PEG30kDa-Cys-Tyr-Lys25 | 50 | 33488 |
| 24. PEG20kDa-Cys-Tyr-Lys25 | 50 | 23488 |
| 25. PEG10kDa-Cys-Tyr-Lys25 | 97 | 13488 |
| 26. PEG5kDa-Cys-Tyr-Lys25 | 14 | 8488 |
| 27. (Acr-Lys4)3-Acr-Lys-Cys- | 33 | 8009 |
| 28.Tyr-(Acr-Lys4)3-Acr-Lys-Cys- | 44 | 8172 |
| 29.Lys-(Tyr-Lys4)3-Tyr-Lys-Cys- | 55 | 7568 |
| 30.Tyr-(Phe-Lys4)3-Phe-Lys-Cys- | 72 | 7539 |
| 31. PEG5kDa-Cys-Tyr-(Trp-Lys3)4 | 60 | 7567 |
| 32. PEG5kDa-Cys-Tyr-(Leu-Lys4)3− | 25 | 7403 |
| 33. PEG2kDa-Cys-Tyr-(Leu-Lys4)3− | 75 | 4403 |
| 34. Alk-Cys-Tyr-(Leu-Lys4)3-Leu- | 25 | 2459 |
| 35. PEG5kDa-Cys-Tyr-(Leu-Lys)8 | 45 | 7343 |
| 36. PEG5kDa-Cys-Tyr-(Leu-Lys)6 | 50 | 6732 |
| 37. PEG5kDa-Cys-Tyr-(Leu-Lys)4 | 66 | 6249 |
| 38. PEG5kDa-Cys-Tyr-(Leu-Arg)8 | 38 | 7437 |
| 39. PEG5kDa-Cys-Tyr-(Lys)4-Leu- | 65 | 7215 |
| 40. PEG5kDa-Cys-Tyr-Leu4-(Leu- | 75 | 7215 |
The isolated yield of purified peptides was determined by absorbance. The reported isolated yield for PEG-peptides is for conversion from the peptide.
The calculated mass closely matches the observed mass determined by ESI-MS for peptides. The calcualted mass for PEG-peptides was obtained by addition of the average mass for PEG reported by the vendor with the observed mass determined for peptides.
Figure 1. Inhibition of DNA Nanoparticle Uptake by the Liver.

The percent of a 1 μg (0.6 μCi) 125I-DNA nanoparticle dose recovered from the liver at a biodistribution time of 5 min following co-administration of 1−80 nmols of PEG-peptide to triplicate mice is illustrated. The IC50 is determined from the PEG-peptide concentration resulting in 50% inhibition, assuming a 2 ml blood volume. This data is reproduced with permission from Baumhover et.al. 2015.1
To directly evaluate the potency of PEG-peptides to saturate scavenger receptors, independent of DNA binding affinity, Trp was replaced with Tyr resulting in PEG-peptides 20–23 (PEG30 kDa-Cys-Tyr-Lys10, 15, 20, and 25) (Table 1). Iodination of each PEG-peptide followed by tail vein dosing established the liver was the primary organ of biodistribution at 5 min. The percent of dose captured by liver was dependent on both the length of the polylysine repeat and dose (Fig. 2). A plot of the PEG-peptide dose versus the percent of dose captured by the liver resulted a saturation curve used to calculate the IC50 in μM (assuming a 2-mL blood volume in mice) (Fig. 2). Following administration of a 1 nmol dose of 20 (PEG30kDa-Cys-Tyr-Lys10), only 18% of the dose was captured by the liver at 5 min. Escalating the dose to 80 nmol partially saturated scavenger receptors, resulting in 11% of dose captured by the liver and an apparent IC50 of 1.5 mM (Fig. 2).
Figure 2. Saturation of Liver Uptake by PEG-Peptides as a Function of Lys Repeat.
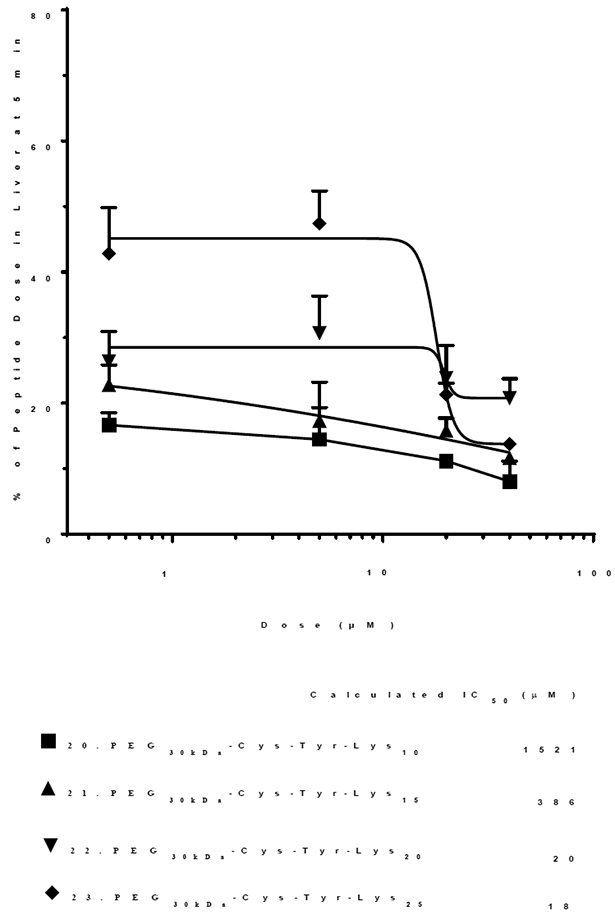
The percent of 125I-PEG-peptide recovered from the liver at a biodistribution time of 5 min following i.v. administration of triplicate mice under dose escalation of 1–80 nmol is illustrated. The IC50 is determined from the PEG-peptide concentration resulting in 50% saturation of liver uptake, assuming a 2 ml blood volume. The IC50 values ranging from 18–1521 μM were derived from fitting the results with R2 =0.63, 0.63, 0.39 and 0.93 for 20–23. A value of p < 0.05 was determined for each PEG-peptides 20, 21 and 23 when comparing 1 and 80 nmol dose.
Increasing the lysine repeat in 21 (PEG30kDa -Cys-Tyr-Lys15), increased liver capture to 23% for a 1 nmol dose which decreased to 12% upon saturation with 80 nmol resulting an apparent IC50 of 390 μM (Fig. 2). A 1 nmol dose of 22 (PEG30kDa -Cys-Tyr-Lys20) resulted in 40% of dose captured by the liver at 5 min, whereas a saturating dose of 80 nmol decreased liver uptake to 20% resulting in an IC50 of 17 μM (Fig. 2). Administration of 1 nmol of 23 (PEG30kDa-Cys-Tyr-Lys25) resulted in 42% of the dose captured by the liver. Saturation of liver uptake with an 80 nmol dose decrease the percent of dose in liver to 35% resulting in an IC50 of 18 μM (Fig. 2).
The results of figure 1 and 2 both establish that increasing the lysine repeat increases scavenger receptor inhibitory potency resulting in a lower IC50.1 However, when monitoring 125I-DNA nanoparticle biodistribution, a 1 nmol dose results in a consistent 65% captured by liver in 5 min regardless of which PEG-peptide was dosed (Fig. 1). In contrast, when dosing 125I-PEG-peptides, the percent of dose captured by the liver in 5 min ranges from 18–42% and, was proportional to the polylysine repeat (Fig. 2). Although, the saturation range (65% reduced to 10%) is greater when monitoring 125I-DNA nanoparticles (Fig. 1), compared to a variable saturation range determined for 125I-PEG-peptides (Fig. 2), the IC50 values determined when monitoring 125I-PEG-peptide liver uptake in the absence of DNA are more accurate. This is evident for 20 and 21 (PEG30kDa-Cys-Tyr-Lys10 or 15) where binding to 125I-DNA leads to an over estimation of the IC50 (Fig. 1), relative to that determined in the absence of DNA (Fig. 2). As the inhibitory potency increases, the IC50 derived from the two approaches converge. Consequently, dosing 125I-PEG-peptides provides a more direct comparison of the relative scavenger receptor inhibitor potency of structurally diverse PEG-peptides, independent of DNA binding affinity.
In addition to evaluating biodistribution, the pharmacokinetics of 125I-PEG-peptides 20–23 were evaluated (Fig. 3, Table 1). The results established that 22 and 23, possessing 20 or 25 Lys, exhibit a longer terminal half-life of 22–36 hrs compared to a shorter half-life of 5–6 hrs for 20 and 21, possessing a shorter polylysine repeat of 15 or 10 (Fig. 3). These results correlate with the percent of dose captured by the liver and the scavenger receptor IC50 potency (Fig. 2). The difference in pharmacokinetic half-life for PEG-peptides most likely reflects differences in albumin binding affinity, with longer Lys repeats binding with higher affinity resulting in more stable and long-circulating albumin nanoparticles, and not a difference in renal filtration since the molecular weight (31–33 kDa) of PEG-peptides 20–23 is not appreciable different (Table 1).
Figure 3. Pharmacokinetic Analysis of PEG-Peptides as a Function of Lys Repeat.
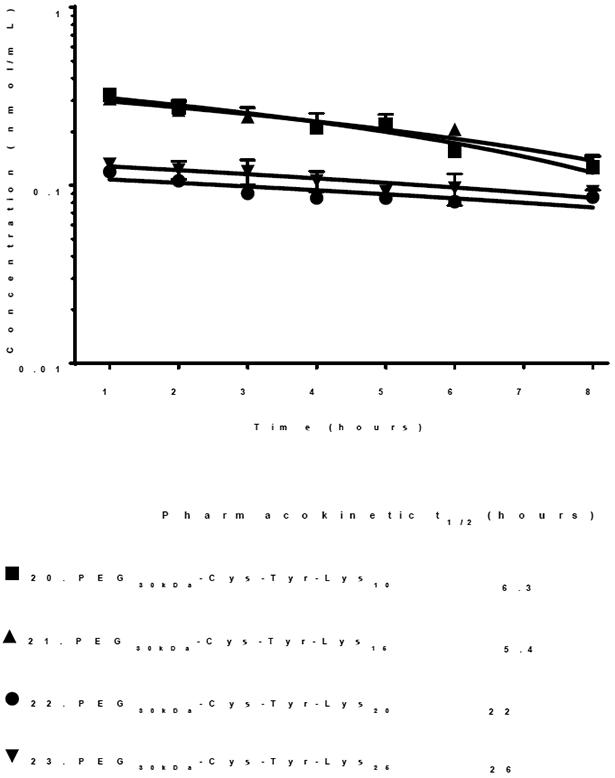
A 1 nmol (0.6 μCi) dose of 125I-PEG-peptide was administered via the tail vein of triplicate mice. At time points of 1–8 hrs, 10 μl of blood was serially sampled from the tail vein and directly gamma counted to determine the concentration 125I-PEG-peptide in blood. The data were fit via Phoenix WinNonlin to determine the β-half-life.
In addition to the length of polylysine repeat, potency is also dependent on PEG MW.1 While 19 (PEG30kDa-Cys-Trp-Lys25) was the most potent at blocking 125 I-DNA nanoparticle uptake by the liver, an analogue substituted with a 5 kDa PEG was completely inactive.1 To examine the effect of PEG MW on saturating liver uptake and pharmacokinetic half-life in the absence of DNA, 125I-PEG-peptides 24–26 were prepared that possess an invariable polylysine repeat of 25 residues but varied in PEG MW from 20–5 kDa (Table 1).
Decreasing the PEG MW from 30 to 5 kDa in 23-26 (PEGkDa-Cys-Tyr-Lys25) led to a systematic decrease in liver uptake of a 1 nmol dose of PEG-peptides (Fig. 4). The percent of dose captured by the liver in 5 min was 40% when dosing 23 (PEG30kDa-Cys-Tyr-Lys25) compared to 28% for 24 by substitution with PEG20kDa and 5% for 25 and 26, when either a PEG10 or 5 kDa was attached to Cys-Tyr-Lys25 (Fig. 4). In contrast to the results presented in figure 3, the long pharmacokinetic half-life of 22–33 hours determined for 23–26 suggests these PEG-peptides bind albumin with comparable affinity (Fig. 5). The inability of 25 and 26 (PEG10 or 5 kDa-Cys-Tyr-Lys25) to appreciably accumulate in liver (Fig. 4) is thereby likely due to the physical properties of the resultant albumin nanoparticles. Decreasing the PEG MW in 25 and 26 would influence albumin nanoparticle charge, which could decrease binding affinity to scavenger receptors. In addition, the percent of PEG-peptide 25 and 26 dose recovered in the kidney increased to 10%, suggesting increased renal filtration (Fig. 4). These results establish that in addition to the length of the Lys repeat, PEG MW influences scavenger receptor inhibition potency.
Figure 4. Saturation of Liver Uptake by PEG-Peptides as a Function of PEG MW.

The percent of 125I-PEG-peptide recovered from the liver at a biodistribution time of 5 minutes following i.v. administration of triplicate mice under dose escalation of 1–80 nmol is illustrated. The IC50 is determined from the PEG-peptide concentration resulting in 50% saturation of liver uptake, assuming a 2 ml blood volume.
Figure 5. Pharmacokinetic Analysis of PEG-Peptides as a Function of PEG MW.

A 1 nmol (0.6 μCi) dose of 125I-PEG-peptide was administered via the tail vein of triplicate mice. At time points of 1–8 hours, 10 μl of blood was serially sampled from the tail vein and directly gamma counted to determine the concentration PEG-peptide in blood. The data were fit via Phoenix WinNonlin to determine the β-half-life.
Development of Low Molecular Weight PEG-Peptide Scavenger Receptor Inhibitors.
We have previously reported the development of a novel DNA binding PEG-peptide 27 ((Acr-Lys4)3Acr-Lys-Cys-PEG5kDa), where Acr represents acridine attached to the ε-amine of Lys.27–28 Four optimally spaced Acr residues dramatically increased DNA binding affinity due to poly-intercalation resulting in stabilized DNA nanoparticles possessing a long circulatory half-life following i.v. dosing25. Dose escalation of 27 with 125I-DNA nanoparticle results in the formation of albumin nanoparticles that inhibit scavenger receptors and delayed DNA nanoparticle metabolism in the liver.28 Since 27 possesses a 5 kDa PEG, we hypothesized that other peptide analogues containing hydrophobic residues may also form albumin nanoparticles and demonstrate scavenger receptor inhibitory potency. Consequently, PEG-peptides 29–40 (Table 1) were prepared and used to evaluate the influence of amino acid substitution on scavenger receptor inhibitor potency. PEG-peptides are proposed to bind serum proteins in the blood.1 Since albumin is the major serum protein with a concentration of 50 mg/ml, we hypothesize that it is primarily albumin nanoparticles that are responsible for saturating liver scavenger receptors.29 The particle sizes of PEG-peptide albumin nanoparticles was investigated using dynamic light scattering (Fig. 6A). Compared to albumin, which possesses a diameter of 5 nm, PEG-peptides 29–40 formed albumin nanoparticles that ranged from 10–30 nm, with the exception of 35 (PEG5kDa-Cys-Tyr-(Leu-Lys)8) and 38 (PEG5kDa-Cys-Tyr-(Leu-Arg)8) that produced significantly larger particles of 60 and 120 nm (Fig. 6A). Likewise, alkylated peptide 34 formed albumin nanoparticles that were 514 nm, establishing that PEG is necessary to form small albumin nanoparticles.
Figure 6. Particle Size Analysis of PEG-Peptide Albumin Nanoparticles and Micelles.

The particles size of albumin nanoparticles was determined by dynamic light scattering following the addition of 40 nmols of PEG-peptide with 5 mg of BSA in a total volume of 1 ml of 5 mM Hepes pH 7.4 (Panel A). The mean and standard error was determined from ten analysis. The albumin nanoparticle size ranged from 10–120 nm in diameter depending of PEG-peptide structure. Zeta potential of albumin nanoparticles prepared with 32 (PEG5kDa-Cys-Tyr-(Leu-Lys4)3-Leu-Lys) and 33 (PEG2kDa-Cys-Tyr-(Leu-Lys4)3-Leu-Lys) are compared to albumin (Panel A inset). PEG-peptides were independently analyzed for micelle formation by dynamic light scattering at increasing concentrations of 1–40 nmol in 1 ml of 150 mM sodium chloride, 50 mM sodium phosphate pH 7.4 (Panel B and C). PEG-peptide with a closed bar in panel A failed to form micelles whereas PEG-peptides with open bar are represented in panel B and C.
To study the influence of scavenger receptor inhibitory potency, a Tyr was incorporated as the N-terminal residue in 28 (Tyr-(Acr-Lys4)3Acr-Lys-Cys-PEG5kDa) to allow direct radio iodination and biodistribution analysis, independent of DNA dosing. At a low dose of 1 nmol of PEG-peptide, 25% was captured by the liver in 5 min. Increasing the dose to 80 nmol, decreased liver uptake to 15%, resulting in an IC50 of 5 μM, in close agreement with the previously reported IC50 of 8 mM for 27 ((Acr-Lys4)3Acr-Lys-Cys-PEG5kDa) inhibition of 125I-DNA nanoparticle uptake by the liver25(Fig 7A). Substitution of Acr with Tyr resulted in 29 (Lys-(Tyr-Lys4)3Tyr-Lys-Cys-PEG5kDa). Biodistribution analysis of a 1 nmol dose of 29 established 30% was captured by the liver in 5 min, which decreased to 20% when escalating the dosing to 80 nmols, resulting in loss of activity with an IC50 of 40 μM (Fig. 7B). Substitution of Acr with Leu and reversal of the PEG from the C-terminus to the N-terminus resulted in 32 (PEG5kDa-Cys-Tyr-Lys-(Leu-Lys4)3-Leu-Lys). Biodistribution analysis of a 1 nmol dose established that 20% was captured by the liver at 5 min. Increasing the dose to 80 nmol decreased liver uptake to 15% resulting in an IC50 of 20 μM (Fig. 7C). Further decreasing the PEG-length from 5 to 2 kDa in 33 (PEG2kDa-Cys-Tyr-Lys-(Leu-Lys4)3-Leu-Lys) resulted in a 6-fold increase in the potency with an IC50 of 3 μM (Fig. 7D). This increase in potency directly correlated with an increase in the zeta potential from −7 mV to −13 mV when comparing 32 and 33 in which the PEG MW decreased to 2kDa (Fig. 6A inset). However, complete removal of PEG not only resulted in very large particles. In addition, biodistribution analysis of alkylated peptide 34 resulted in only 4% of the dose recovered from the liver, which was unchanged by dose escalation.
Figure 7. Optimized Low Molecular Weight Scavenger Receptor Inhibitor PEG-Peptides.

The percent of 125I-PEG-peptide recovered from the liver at a biodistribution time of 5 min following i.v. administration of triplicate mice under dose escalation of 1–80 nmol is illustrated. The IC50 is determined from the PEG-peptide concentration resulting in 50% saturation of liver uptake, assuming a 2 ml blood volume. The IC50 values of 3–40 μM were derived from fitting the results with R2 =0.93, 0.7, 0.82 and 0.82 for panels A-D. A value of p < 0.05 was determined for each PEG-peptides 28, 29, 32, and 33 when comparing 1 and 80 nmol dose.
PEG-peptide analogues 30, 31, 39 and 40 possessing substitutions with Phe or Trp, and C or N-terminal Leu clusters not only failed to saturate the liver upon dose escalation, but instead, resulted in an increase in the percent of dose captured by the liver (Fig. 8). Substitution with Phe resulted in 30 (Tyr-(Phe-Lys4)3Phe-Lys-Cys-PEG5kDa), demonstrating increased liver uptake at higher doses (Fig. 8A). Likewise, substitution with Trp resulted in 31 (PEG5kDa-Cys-Tyr-(Trp-Lys3)4) which increased from 12% to 20% in liver when dose escalated (Fig. 8B). Attempts to cluster Leu residues on either the N or C-terminus resulted in 39 (PEG5kDa-Cys-Tyr-(Lys)4-Leu-Lys4-Leu4) and 40 (PEG5kDa-Cys-Tyr-Leu4-(Leu-Lys)4-Lys4) both demonstrated increased liver biodistribution on dose escalation (Fig. 8C and D). Based on their hydrophobicity and surface-active properties, we hypothesized that certain PEG-peptides formed micelles at higher concentrations that accounted for the increase in liver uptake. In support of this hypothesis, the particle size was determined as a function of concentration (Fig. 6B). At concentrations above the critical-micelle-concentration of approximately 2–5 μM, 35, 36, 38 and 39 formed micelles with an average size of approximately 100 nm (Fig. 6B), whereas 37 and 40 formed even larger 300–400 nm micelles (Fig. 6C).
Figure 8. Biodistribution of Micelle Forming Low Molecular Weight PEG-Peptides.

The percent of 125I-PEG-peptide recovered from the liver at a biodistribution time of 5 min following i.v. administration of triplicate mice under dose escalation of 1–80 nmol is illustrated. Each PEG-peptide showed an increase in the percent of dose in liver upon dose escalation suggesting the formation of PEG-peptide micelles that failed to saturate liver uptake. However, only 39 and 40 produced measurable micelles (Fig. 6B,C). The curve fitting resulted in an R2 =0.5, 0.74, 0.76 and 0.78 for panels A-D. A value of p < 0.05 was determined for PEG-peptides 30, 31, 39, and 40 when comparing 1 and 80 nmol dose.
Since the Leu substituted peptide 32 (PEG5kDa-Cys-Tyr-Lys-(Leu-Lys4)3-Leu-Lys) was a potent scavenger receptor inhibitor that avoided micelle formation, the number of Leu residues was increased in analogue 35 (PEG5kDa-Cys-Tyr-(Lys-Leu)8) in an attempt to increase potency. Biodistribution analysis established 38% of a 1 nmol dose accumulated in the liver at 5 min whereas saturation resulted in 16% of the dose recovered in liver, with a loss in potency and a IC50 of 39 μM (Fig. 9A). Attempts to decrease the number of Leu-Lys repeats resulted in 36 (PEG5kDa-Cys-Tyr-(Lys-Leu)6) and 37 (PEG5kDa-Cys-Tyr-(Lys-Leu)4) that formed micelles (Fig. 6C) and proved to be inactive as scavenger receptor inhibitors (Fig. 9B and C). The scavenger receptor inhibition potency was recovered by substituting Arg for Lys, resulting in 38 (PEG5kDa-Cys-Tyr-(Leu-Arg)8) of which 45% of a 1 nmol dose was captured by the liver at 5 min (Fig. 9D). Even though 38 formed large 120 nm albumin nanoparticles (Fig. 6A), dose escalation led to liver saturation with an IC50 of 20 μM (Fig. 9D). However, this was also accompanied by an increase in lung accumulation, exposing a potentially toxicity for larger albumin nanoparticles.
Figure 9. Influence of PEG-Peptide Size on Scavenger Receptor Inhibition.

The percent of 125I-PEG-peptide recovered from the liver at a biodistribution time of 5 min following i.v. administration of triplicate mice under dose escalation of 1–80 nmol is illustrated. The IC50 values of 20–30 μM were derived from fitting the results with R2 =0.58 and 0.6 for panels A and D. A value of p < 0.05 was determined for PEG-peptides 35 and 38 when comparing 1 and 80 nmol dose.
In summary, PEG-peptides 32 and 33 avoided micelle formation, successfully formed 13–16 nm albumin nanoparticles, and were able to saturate liver uptake (Fig. 7C, D). Conversely, PEG-peptides 36, 37, 39 and 40 formed micelles and failed to saturate liver uptake. Only 35 (PEG5kDa-Cys-Tyr-(Leu-Lys)8) and 38 (PEG5kDa-Cys-Tyr-(Leu-Arg)8) formed micelles and saturated liver uptake (Fig 9A, D). PEG-peptides 30 and 31 avoided the formation of detectable micelles, but were unable to affect liver saturation upon dose escalation (Fig. 8A, B).
Inhibition of PEG-peptide DNA Nanoparticle Uptake in Liver.
Experiments were conducted to validate that scavenger receptor inhibitor PEG-peptides could also inhibit DNA nanoparticle uptake. PEGylated polyacridine peptide 27 ((Acr-Lys4)3Acr-Lys-Cys-PEG5kDa) was combined with 125I-DNA to form stable nanoparticles28. 125IDNA nanoparticles were co-administered with an escalating dose of 1–80 nmols of 32 (PEG5kda-Cys-Tyr-Lys-(Leu-Lys4)3-Leu-Lys), 33 (PEG2kda-Cys-Tyr-Lys-(Leu-Lys4)3-Leu-Lys), and 38 (PEG5kDa-Cys-Tyr-(Leu-Arg)8), (Fig 10). Approximately 55% of the dose was recovered in liver at a biodistribution time of 5 min which decreased as a function of increasing PEG-peptide dose (Fig. 10). Nanoparticles co-administered with 32 (PEG5kda-Cys-Tyr-Lys-(Leu-Lys4)3-Leu-Lys) or 33 (PEG2kda-Cys-Tyr-Lys-(Leu-Lys4)3-Leu-Lys) resulted in an IC50 of 11 or 3 μM respectively (Fig. 10A, B), whereas co-administration with 38 (PEG5kDa-Cys-Tyr-(Leu-Arg)8) resulted in an IC50 of 15 μM (Fig. 10C).
Figure 10. Inhibition of DNA Nanoparticle Uptake by the Liver with Optimized PEG-Peptide Scavenger Receptor Inhibitors.

Stable nanoparticles were prepared with 0.8 nmol of (Acr-Lys4)3-Acr-Lys-Cys-PEG5kDa added to 1 μg (0.6 μCi) of 125I-DNA followed by co-administration with a escalating (1–80 nmol) dose of scavenger receptor inhibiting PEG-peptides. The percent of 125I-DNA nanoparticle recovered from the liver at a biodistribution time of 5 min following i.v. administration of triplicate mice is illustrated. The IC50 values of 5–15 μM were derived from fitting the results with R2 =0.90, 0.79 and 0.89 for panels A-C. A value of p < 0.05 was determined for PEG-peptides 32, 33 and 38 when comparing 1 and 80 nmol dose.
Discussion
Following i.v. dosing, many nanoparticles biodistribute to the liver.5, 7, 25, 30–34 A large percent of a DNA nanoparticle dose (60–80%) is immediately captured by SR-A1 on liver non-parenchymal cells.5–6, 9, 35–37,38 SR-A1 is found on both Kupffer cells and fenestrated endothelial cells and is the most abundant member of a large family of scavenger receptors that bind diverse anionic ligands such as oxidized LDL and HDL, acetylated LDL, maleylated and malondialdehyde BSA, fucoidan, dextran sulfate, polyinosinic acid (Poly-I), polyguanylic acid (Poly-G) and gram-positive and negative bacteria.39
Phagocytosis of foreign polymers and particles by SR-A1 results in activation of the innate immune response with release of TNF-α into the blood.40 The i.v. administration of plasmid DNA and lipoplexes41 also activates the innate immune response, as does intrabiliary administration of plasmid DNA, chitosan-DNA and PEI-DNA which significantly increase the serum concentration of TNF-α 10−fold.42 The realization that adenovirus is primarily taken up by Kupffer cells via scavenger receptors, and that higher doses of adenovirus (1012) leads to Kupffer cell death and the release of serum lactate dehydrogenase,6 prompted the use of pre-administered Poly-I to inhibit adenovirus accumulation in Kupffer cells and to improve hepatocyte transfection.5 While Poly-I is reportedly not toxic to mice, co-administration of Poly-I with adenovirus decreases the lethal threshold for adenovirus in mice,5 making Poly-I inhibition of scavenger receptors a clinically unacceptable approach to improve viral mediated gene delivery.5 The high molecular weight and polydispersity of Poly-I makes it too complex to optimize chemically to remove its toxicity. There is a need for new low molecular weight, potent and safe scavenger receptor inhibitors that are fully compatible with i.v. dosed gene delivery vectors and nanoparticles to increase their potential for clinical translation.
There are relatively few reports of polypeptides used as scavenger receptor inhibitors.40, 43–44 One study found that pre-administration of 150 μg of high molecular weight polylysine increased the expression of adeno-associated virus (AAV2) in liver of mice 12-fold.43 The increased expression was dependent on using high MW polylysine, which was also lethal in 20% of the mice. A second set of studies dosed Fabs directed against SRA1 and SREC-I and demonstrated increased expression of helper-dependent adenovirus.40 Using phage display, this group also developed peptides that inhibit SRA1 and SREC-I.44 Pre-administration of 1–10 nmols of inhibitory peptide resulted in a significant 3–4 fold increase in gene expression from advenovirus.44 A report from our group established that co-administration of excess PEG-peptide, extends the circulatory stability and transfection competency of a DNA nanoparticle from 4 to 12 hours.24
In an effort to better understand how PEG-peptides function as scavenger receptor inhibitors to improve potency, we have previously reported that an escalating dose of 16–19 (Table 1) with a constant dose of 125I-DNA provided a saturation curve for nanoparticle uptake by the liver(Fig. 1).1, 24 However, PEG-peptides bind both DNA and albumin with different affinities to form both DNA and albumin nanoparticles of different sizes. A major advancement reported in the present study is the use of radiolabeled PEG-peptides to directly measure the scavenger receptor inhibition potency in vivo in the absence of a DNA nanoparticle. Removal of the requirement of DNA binding allowed direct evaluation of PEG-peptides for their ability to aggregate albumin and undergo capture by the liver. This approach is validated by comparing the magnitude of the IC50 determined when saturating liver uptake using 28 (125I-Tyr-(Acr-Lys4)3-Acr-Lys-Cys-PEG5kDa) producing an IC50 of 5 μM approximating the IC50 of 8 μM determined for 27 ((Acr-Lys4)3-Acr-Lys-Cys-PEG5kDa) to inhibit 125I-DNA nanoparticle uptake.
Using this approach, radiolabeled PEG-peptides of varying polylysine and PEG MW were compared for scavenger receptor inhibitory potency. As predicted by previously published results, PEG-peptide 23 had appreciable scavenger receptor inhibition potency resulting in an IC50 of 18 μM.1 However, compared to 28, its molecular weight was four times greater. Thereby, an equivalent dose based on mols of PEG-peptide results in four time’s greater dose in mg/kg. Attempts to decrease the molecular weight of 28 by decreasing the PEG MW resulted in partial or complete loss of activity.
The mechanism by which PEG-peptides inhibit scavenger receptors involves the in situ formation of albumin nanoparticles, and thereby PEG-peptide binding affinity for albumin plays a key role. While it is likely that other serum proteins are also incorporated into PEG-peptide nanoparticles, given its concentration in blood (50 mg/ml), albumin is undoubtedly the primary component of PEG-peptide nanoparticles. Albumin binds and transports many hydrophobic drugs in the circulation.45 In doing so, it influences the biodistribution, rate of clearance, volume of distribution and half-life of iv dosed nanoparticles and drugs.46 Consequently, the potency of 28 (Tyr-(Acr-Lys4)3-Acr-Lys-Cys-PEG5kDa), which possesses only 13 Lys residues and a short 5 kDa PEG, is dependent on four hydrophobic acridine-lysine (Acr) residues contributing to its albumin binding. However, the Lys-acridine (Acr) residues in 28 also substantially increase the binding affinity for DNA.27–28 We thereby attempted to substitute Acr residues with natural hydrophobic amino acid residues to maintain albumin binding and scavenger receptor inhibition potency while decreasing DNA binding.
Substitution of Acr with either Phe or Trp resulted in amphiphilic PEG-peptides 30 and 31 that failed to saturate liver uptake, and instead, exhibited an increase in liver uptake on dose escalation, which is indicative of micelle formation (Fig. 8). Alternatively, substitution of Acr with either Tyr or Leu resulted in PEG-peptides 29 or 32 that saturated liver uptake with an apparent IC50 of 40 or 20 μM, respectively (Fig. 7B, C). A further decrease in the PEG MW to 2 kDa resulted in 33, which possessed an IC50 of 3 μM (Fig. 7D). Thereby, decreasing the PEG length, increased the negative charge on albumin nanoparticles, which increased the in vivo potency 6-fold. Substitution with Leu also afforded a PEG-peptide with much lower binding affinity for DNA due to removal of polyintercalating Acr residues. The spacing of Leu is important since clustering Leu toward the C or N terminus resulted in PEG-peptides 39 and 40 exhibiting increased accumulation in liver on dose escalation, consistent with micelle formation (Fig. 6 and 8). An attempt to increase the hydrophobicity by increasing the number Leu and decrease the number of Lys while avoiding micelle formation resulted in 35 (PEG5kDa-Cys-Tyr-(Leu-Lys)8) that possessed decreased potency with an IC50 of 38 μM. Substitution of Lys with Arg to generate 38 (PEG5kDa-Cys-Tyr-(Leu-Arg)8), resulted in a more potent scavenger receptor inhibitor possessing an IC50 of 20 μM (Fig. 8D).
Scavenger receptor PEG-peptides derived from this optimization were able to inhibit DNA nanoparticle uptake with good potency (Fig. 10). This result validates the approach of direct radiolabeling of PEG-peptides followed by dose escalation in vivo. It suggests that PEG-peptides of widely varying sequences of cationic and hydrophobic residues can be tailored to inhibit the liver uptake of diverse nanoparticles.
Materials and Methods.
Unsubstituted Wang, Fmoc-protected amino acids, N-hydroxybenzotriazole (HOBt), O-(benzotr1-yl)-N,N,N′,N′-tetramethyluronium hexa-fluorophosphate (HBTU), and N,N dimethylformamide (DMF) were from AAPPTec (Louisville, KY, U.S.A.). Trifluoroacetic acid (TFA) and acetonitrile were obtained from Fisher Scientific (Pittsburgh, PA, U.S.A.). Diisoproplyethylamine (DIPEA), piperidine, acetic anhydride, Sephadex G-25 and G-10 were purchased from Sigma Chemical Co. (St Louis, MO, U.S.A.). Maleimide- mPEG 2, 5, 10, 20 and 30 kDa were obtained from Laysan Bio (Arab, AL, U.S.A.). Sodium 125Iodine was obtained from Perkin Elmer (Waltham, MA, U.S.A.). Pierce iodogen tubes were obtained from ThermoFisher (Waltham, MA, U.S.A.). pGL3 control vector, a 5.3-kbp luciferase plasmid containing a SV40 promoter and enhancer, was obtained from Promega (Madison, WI, U.S.A.). pGL3 was amplified in a DH5α strain of Escherichia coli and purified using a Qiagen giga prep (Germantown, MD) according to the manufacturer’s instructions.
Synthesis and Characterization of PEGylated Peptides.
Peptides 1–15 illustrated in Table 1 were prepared by solid-phase peptide synthesis on a 30 μmol scale using an APEX 396 synthesizer (AAPPTec, Louisville, KY, U.S.A.) with standard Fmoc procedures as described previously.1 Peptides were purified by injecting 2 μmols onto a XB-C18 semi-preparative RP-HPLC column (2.5 × 2.1 cm) (Phenomenex, Torrance, CA) eluted at 10 ml per min with 0.1% TFA and a 10–30% acetonitrile gradient over 30 min while monitoring Abs 280 nm for Tyr or Trp or acridine (Acr) at 409 nm. Fractions from multiple runs were pooled, concentrated by rotary evaporation and freeze dried. The mass of peptides was determined on an Agilent 1100 LC-MS. Peptides were resolved on an analytical (0.47 × 25 cm) XB-C18 analytical RP-HPLC column eluted at 0.7 ml/min with 0.1% TFA and a 30 min 10–30% acetonitrile gradient and detected by positive mode ESI-MS ion trap.
Purified and characterized peptides 1-15 were covalently modified by attaching a single maleimide polyethylene glycol (PEG) to Cys, resulting in PEG-peptides 16-33 and 35–40, and alkylated peptide 34 (Table 1). The Cys residue was PEGylated by reacting 1 μmol of peptide with 1.1 μmol of maleimide mPEG (2, 5, 10, 20 or 30 kDa) in 1 ml of 100 mM ammonium acetate buffer pH 7 for 2 hrs at RT. PEGylation resulted in a greater retention time on RP-HPLC. Alkylated peptide 34 was prepared by reaction of 9 with 50 mol-equivalents of iodo-acetamide. PEGylated peptides, and alkylated peptide 34, were purified by semi-preparative RP-HPLC eluted at 10 ml/min with 0.1% TFA with a 10−65% acetonitrile gradient over 30 min. The major peak was collected and pooled from multiple runs and concentrated by rotary evaporation, lyophilized and stored frozen. The TFA counter ion was exchanged by two freeze-drying cycles with 0.1 (v/v) % acetic acid. PEG-peptides and alkylated peptide 34 were reconstituted in water and quantified by absorbance, Tyr ε280nm = 1350 M−1cm−1, Acr (acridine) ε409nm = 9266 M−1cm−1 and Trp ε280nm = 5600 M−1cm−1.
Particle size and Zeta Potential Analysis.
The size and charge of PEG-peptide albumin nanoparticles were determined on a Brookhaven ZetaPlus (Holtsville, NY) by combining 80 nmols of peptide with 1 ml of 5 mg/ml BSA in 5 mM Hepes pH 7.4. Zeta potential measurements of albumin nanoparticles are reported as the mean and standard error following ten measurements. The critical micelle concentration (CMC) for PEG-peptides in the absence of albumin was determined by measuring the particle size of 1–40 nmols of PEG-peptide in 1 ml of 150 mM sodium chloride, 50 mM sodium phosphate pH 7.4. The reported sizes are the means and standard error of ten measurement following deconvolution by intensity averaged multimodal distribution.
PEG-Peptide Iodination and Purification.
PEG-peptides were iodinated using Pierce iodogen tubes. A PEG-peptide (5 nmol) prepared in 100 μl of 0.5 M sodium phosphate pH 7 was added to the iodogen tube followed by 100 μCi (10 μl) of Na125I. The reaction proceeded at RT for 15 min followed by purification by gel filtration on Sephadex G10 (0.25 × 10 cm) eluted with 0.15 M sodium chloride. 125I-PEG-peptides eluting as a peak at 5–6 ml were collected and gamma (γ) counted resulting in the recovery of 20 μCi with a specific activity of 4 μCi/nmol of PEG-peptide, assuming quantitative recovery. 125I-PEG-peptides were stored at 4°C in 0.15 M sodium chloride.
PEG-Peptide Biodistribution.
PEG-peptide doses were prepared by combining a tracer dose of 0.6 μCi 125I-PEG-peptide with 1, 10, 40, 80 nmols of unlabeled PEG-peptide in a total volume of 100 μl of 0.15 M sodium chloride. PEG-peptide doses were gamma counted prior to tail vein dosing in triplicate 30 g ICR male mice. At 5 min post-administration, mice were anesthetized by intraperitoneal injection of 100 μl of ketamine (100 mg/kg) and xylazine (10 mg/kg), then euthanized by cervical dislocation. The major organs (liver, lung, spleen, stomach, kidney, heart, small intestine, thyroid and large intestine) were harvested, rinsed with saline, and gamma counted to determine the percentage of dose in the organ.
PEG-Peptide Pharmacokinetics.
125I-PEG-peptide doses of 1 nmol (0.6 μCi in 100 μl of 0.15 M sodium chloride) were dosed tail vein in triplicate 30 g ICR male mice. Blood (10 μl) time points were serially sampled at 1–8 hrs from the tail vein and gamma counted to determine the blood concentration (nmol/ml) of PEG-peptide based on the specific activity. The mean concentration values from triplicate time points were fit to determine the half-life using Phoenix WinNonlin (Version 7.0, Certara USA, Inc. Princeton, NJ).
Inhibition of PEG-Peptide DNA Nanoparticle Uptake by the Liver.
125I-DNA was prepared as previously reported47. PEGylated DNA nanoparticles were prepared by combining 0.8 nmol of polyacridine PEG-peptide 27 ((Acr-Lys4)3Acr-Lys-Cys-PEG5kDa) with 1 μg (0.6 μCi) of 125I-DNA to form DNA nanoparticles. 125I-DNA nanoparticles were co-administered via the tail vein with 1, 10, 40, 80 nmol of scavenger receptor inhibitor PEG-peptide. At a biodistribution time of 5 min, mice were euthanized and the major tissues were harvested and gamma counted as described above.
Statistical Analysis
Inhibition curves were plotted and analyzed using Graphpad Prism 7.03 (San Diego, CA). The quality of the IC50 was judged by the goodness of fit (R2). A two-tailed unpaired T-test was used to determine statistical significance between the percent of dose in liver at the lowest and highest dose.
Acknowledgement.
Support for this work by NIH grants GM117785, T32 GM067795 and R25 GM058939 is gratefully acknowledged. The authors thank G. An and R. D’Cunha for assistance in analyzing pharmacokinetic data.
References
- 1.Baumhover NJ; Duskey JT; Khargharia S; White CW; Crowley ST; Allen RJ; Rice KG; , Structure–Activity Relationship of PEGylated Polylysine Peptides as Scavenger Receptor Inhibitors for Non-Viral Gene Delivery. Molecular Pharmaceutics 2015, 12, 4321–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tsoi KM; MacParland SA; Ma XZ; Spetzler VN; Echeverri J; Ouyang B; Fadel SM; Sykes EA; Goldaracena N; Kaths JM; Conneely JB; Alman BA; Selzner M; Ostrowski MA; Adeyi OA; Zilman A; McGilvray ID; Chan WC, Mechanism of hard-nanomaterial clearance by the liver. Nat Mater 2016, 15, 1212–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu YP; Tong C; Dispenzieri A; Federspiel MJ; Russell SJ; Peng KW, Polyinosinic acid decreases sequestration and improves systemic therapy of measles virus. Cancer Gene Ther 2012, 19, 202–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Haisma HJ; Kamps JAAM; Kamps GK; Plantinga JA; Rots MG; Bellu AR, Polyinosinic acid enhances delivery of adenovirus vectors in vivo by preventing sequestration in liver macrophages. Journal of General Virology 2008, 89, 1097–1105. [DOI] [PubMed] [Google Scholar]
- 5.Xu Z; Tian J; Smith JS; Byrnes AP, Clearance of adenovirus by Kupffer cells is mediated by scavenger receptors, natural antibodies, and complement. J Virol 2008, 82, 11705–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Manickan E; Smith JS; Tian J; Eggerman TL; Lozier JN; Muller J; Byrnes AP, Rapid Kupffer cell death after intravenous injection of adenovirus vectors. Mol Ther 2006, 13, 108–17. [DOI] [PubMed] [Google Scholar]
- 7.van Dijk R; Montenegro-Miranda PS; Riviere C; Schilderink R; Ten Bloemendaal L; van Gorp J; Duijst S; de Waart DR; Beuers U; Haisma HJ; Bosma PJ, Polyinosinic Acid blocks adeno-associated virus macrophage endocytosis in vitro and enhances adeno-associated virus liver-directed gene therapy in vivo. Hum Gene Ther 2013, 24, 807–13. [DOI] [PubMed] [Google Scholar]
- 8.van Etten EW; ten Kate MT; Snijders SV; Bakker-Woudenberg IA, Administration of liposomal agents and blood clearance capacity of the mononuclear phagocyte system. Antimicrob Agents Chemother 1998, 42, 1677–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Takakura Y; Takagi T; Hashiguchi M; Nishikawa M; Yamashita F; Doi T; Imanishi T; Suzuki H; Kodama T; Hashida M, Characterization of plasmid DNA binding and uptake by peritoneal macrophages from class A scavenger receptor knockout mice. Pharm Res 1999, 16, 503–8. [DOI] [PubMed] [Google Scholar]
- 10.Hisazumi J; Kobayashi N; Nishikawa M; Takakura Y, Significant role of liver sinusoidal endothelial cells in hepatic uptake and degradation of naked plasmid DNA after intravenous injection. Pharm Res 2004, 21, 1223–8. [DOI] [PubMed] [Google Scholar]
- 11.Platt N; Gordon S , Multiligand receptors Is the class A macrophage scavenger receptor ( SR-A ) multifunctional? The mouse’s tale. The Journal of Clinical Investigation 2001, 108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Platt N; Gordon S, Is the class A macrophage scavenger receptor (SR-A) multifunctional? — The mouse’s tale. Journal of Clinical Investigation 2001, 108, 649–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.GOLDSTEIN JL; Ho YK; BASU SK; S.BR M, Binding site on macrophages that mediates uptake and degradation of acetylated lowdensity lipoprotein, producing massive cholesterol deposition. PNAS 1979, 76, 333–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McKee TD; DeRome ME; Wu GY; Findeis MA, Preparation of Asialoorosomucoid-Polylysine Conjugates. Bioconjug. Chem 1994, 5, 306–311. [DOI] [PubMed] [Google Scholar]
- 15.Boussif O; Lezoualc’h F; Zanta MA; Mergny MD; Scherman D; Demeneix B; Behr JP, A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: polyethylenimine. Proceedings of the National Academy of Sciences of the United States of America 1995, 92, 7297–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee SH; Castagner B; Leroux J-C, Is there a future for cell-penetrating peptides in oligonucleotide delivery? European Journal of Pharmaceutics and Biopharmaceutics 2013, 85, 5–11. [DOI] [PubMed] [Google Scholar]
- 17.Kwok A; Eggimann GA; Reymond JL; Darbre T; Hollfelder F, Peptide dendrimer/lipid hybrid systems are efficient DNA transfection reagents: structure--activity relationships highlight the role of charge distribution across dendrimer generations. ACS Nano 2013, 7, 4668–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Raad M. d.; Teunissen EA; Lelieveld D; Egan DA; Mastrobattista E, High-content screening of peptide-based non-viral gene delivery systems. Journal of Controlled Release 2012, 158, 433–442. [DOI] [PubMed] [Google Scholar]
- 19.Eliyahu H; Barenholz Y; Domb AJ, Polymers for DNA delivery Molecules (Basel, Switzerland: ) 2005, 10, 34–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Merdan T; Kunath K; Petersen H; Bakowsky U; Voigt KH; Kopecek J; Kissel T, PEGylation of poly(ethylene imine) affects stability of complexes with plasmid DNA under in vivo conditions in a dose-dependent manner after intravenous injection into mice. Bioconjug Chem 2005, 16, 785–92. [DOI] [PubMed] [Google Scholar]
- 21.Ogris M; Brunner S; Schuller S; Kircheis R; Wagner E, PEGylated DNA/transferrin-PEI complexes: reduced interaction with blood components, extended circulation in blood and potential for systemic gene delivery. Gene Ther 1999, 6, 595–605. [DOI] [PubMed] [Google Scholar]
- 22.Mosqueira VC; Legrand P; Morgat JL; Vert M; Mysiakine E; Gref R; Devissaguet JP; Barratt G, Biodistribution of long-circulating PEG-grafted nanocapsules in mice: effects of PEG chain length and density. Pharm Res 2001, 18, 1411–9. [DOI] [PubMed] [Google Scholar]
- 23.Oupický D; Koňák Č; Dash PR; Seymour LW; Ulbrich K, Effect of Albumin and Polyanion on the Structure of DNA Complexes with Polycation Containing Hydrophilic Nonionic Block. Bioconjugate Chemistry 1999, 10, 764–772. [DOI] [PubMed] [Google Scholar]
- 24.Khargharia S; Baumhover NJ; Crowley ST; Duskey J; Rice KG, The Uptake Mechanism of PEGylated DNA Polyplexes by the Liver Influences Gene Expression. Gene Therapy 2014, 21, 1021–1028. [DOI] [PubMed] [Google Scholar]
- 25.Khargharia S; Kizzire K; Ericson MD; Baumhover NJ; Rice KG, PEG length and chemical linkage controls polyacridine peptide DNA polyplex pharmacokinetics, biodistribution, metabolic stability and in vivo gene expression. J Control Release 2013, 170, 325–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wadhwa MS; Collard WT; Adami RC; McKenzie DL; Rice KG, Peptide-mediated gene delivery: influence of peptide structure on gene expression. Bioconjugate Chemistry 1997, 8, 81–8. [DOI] [PubMed] [Google Scholar]
- 27.Fernandez CA; Baumhover NJ; Duskey JT; Khargharia S; Kizzire K; Ericson MD; Rice KG, Metabolically stabilized long-circulating PEGylated polyacridine peptide polyplexes mediate hydrodynamically stimulated gene expression in liver. Gene Ther 2011, 18, 23–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kizzire K; Khargharia S; Rice KG, High-affinity PEGylated polyacridine peptide polyplexes mediate potent in vivo gene expression. Gene Ther 2013, 20, 407–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu Z; Jiao Y; Wang T; Zhang Y; Xue W, Interactions between solubilized polymer molecules and blood components. J Control Release 2012, 160, 14–24. [DOI] [PubMed] [Google Scholar]
- 30.Kamps JA; Scherphof GL, Biodistribution and uptake of liposomes in vivo. Methods Enzymol 2004, 387, 257–66. [DOI] [PubMed] [Google Scholar]
- 31.Kamps JA; Scherphof GL, Receptor versus non-receptor mediated clearance of liposomes. Adv Drug Deliv Rev 1998, 32, 81–97. [DOI] [PubMed] [Google Scholar]
- 32.Takakura Y; Mahato RI; Hashida M, Extravasation of macromolecules. Advanced drug delivery reviews 1998, 34, 93–108. [DOI] [PubMed] [Google Scholar]
- 33.Smith JS; Xu Z; Tian J; Stevenson SC; Byrnes AP, Interaction of systemically delivered adenovirus vectors with Kupffer cells in mouse liver. Hum Gene Ther 2008, 19, 547–54. [DOI] [PubMed] [Google Scholar]
- 34.Haisma HJ; Kamps JA; Kamps GK; Plantinga JA; Rots MG; Bellu AR, Polyinosinic acid enhances delivery of adenovirus vectors in vivo by preventing sequestration in liver macrophages. J Gen Virol 2008, 89, 1097–105. [DOI] [PubMed] [Google Scholar]
- 35.Kawabata K; Takakura Y; Hashida M, The Fate of Plasmid DNA After Intravenous Injection in Mice: Involvement of Scavenger Receptors in Its Hepatic Uptake. Pharm. Res 1995, 12, 825–830. [DOI] [PubMed] [Google Scholar]
- 36.Collard WT; Yang Y; Kwok KY; Park Y; Rice KG, Biodistribution, metabolism, and in vivo gene expression of low molecular weight glycopeptide polyethylene glycol peptide DNA co-condensates. Journal of Pharmaceutical Sciences 2000, 89, 499–512. [DOI] [PubMed] [Google Scholar]
- 37.Liu F; Shollenberger LM; Conwell CC; Yuan X; Huang L, Mechanism of naked DNA clearance after intravenous injection. J Gene Med 2007, 9, 613–9. [DOI] [PubMed] [Google Scholar]
- 38.Platt N; Gordon S, Is the class A macrophage scavenger receptor (SR-A) multifunctional? - The mouse’s tale. J Clin Invest 2001, 108, 649–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Whelan FJ; Meehan CJ; Golding GB; McConkey BJ; Bowdish DM, The evolution of the class A scavenger receptors. BMC Evol Biol 2012, 12, 227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Piccolo P; Vetrini F; Mithbaokar P; Grove NC; Bertin T; Palmer D; Ng P; Brunetti-Pierri N, SR-A and SREC-I are Kupffer and endothelial cell receptors for helper-dependent adenoviral vectors. Mol Ther 2013, 21, 767–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kako K; Nishikawa M; Yoshida H; Takakura Y, Effects of inflammatory response on in vivo transgene expression by plasmid DNA in mice. J Pharm Sci 2008, 97, 3074–83. [DOI] [PubMed] [Google Scholar]
- 42.Dai H; Jiang X; Leong KW; Mao HQ, Transient depletion of kupffer cells leads to enhanced transgene expression in rat liver following retrograde intrabiliary infusion of plasmid DNA and DNA nanoparticles. Hum Gene Ther 2011, 22, 873–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Moulay G; Boutin S; Masurier C; Scherman D; Kichler A, Polymers for improving the in vivo transduction efficiency of AAV2 vectors. PLoS One 2010, 5, e15576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Piccolo P; Annunziata P; Mithbaokar P; Brunetti-Pierri N, SR-A and SREC-I binding peptides increase HDAd-mediated liver transduction. Gene Ther 2014, 21, 950–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Larsen MT; Kuhlmann M; Hvam ML; Howard KA, Albumin-based drug delivery: harnessing nature to cure disease. Mol Cell Ther 2016, 4, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Krenzel ES; Chen Z; Hamilton JA, Correspondence of fatty acid and drug binding sites on human serum albumin: a two-dimensional nuclear magnetic resonance study. Biochemistry 2013, 52, 1559–67. [DOI] [PubMed] [Google Scholar]
- 47.Joseph Terebesi KYK, and Rice Kevin G., Iodinated Plasmid DNA as a Tool for Studying Gene Delivery. Analytical Biochemistry 1998, 120–123. [DOI] [PubMed] [Google Scholar]