

Abstract
The developing brain is sensitive to a variety of insults. Epidemiological studies have identified prenatal exposure to infection as a risk factor for a range of neurological disorders, including autism spectrum disorder and schizophrenia. Animal models corroborate this association and have been used to probe the contribution of gene-environment interactions to the etiology of neurodevelopmental disorders. Here we review the behavior and brain phenotypes that have been characterized in MIA offspring, including the studies that have looked at the interaction between maternal immune activation and genetic risk factors for autism spectrum disorder or schizophrenia. These phenotypes include behaviors relevant to autism, schizophrenia, and other neurological disorders, alterations in brain anatomy, and structural and functional neuronal impairments. The link between maternal infection and these phenotypic changes is not fully understood, but there is increasing evidence that maternal immune activation induces prolonged immune alterations in the offspring’s brain which could underlie epigenetic alterations which in turn may mediate the behavior and brain changes. These concepts will be discussed followed by a summary of the pharmacological interventions that have been tested in the maternal immune activation model.
1. Introduction
Brain development is a complex organization of processes under genetic, environmental, and immune regulation, and consequently is vulnerable to a variety of insults (Garay and McAllister, 2010; Stiles and Jernigan, 2010). Insults during specific windows of development can alter the normal trajectory of brain development, leading to disorders that have developmental origins, including autism spectrum disorder (ASD) and schizophrenia (Bale, 2015; Ploeger et al., 2010; Rapoport et al., 2012). One such insult is maternal infection, which is particularly detrimental to neurodevelopment when it occurs early in gestation (Estes and McAllister, 2016; Knuesel et al., 2014; Meyer, 2014; Meyer et al., 2007; Reisinger et al., 2015). Numerous epidemiological reports suggest an association between ASD or schizophrenia and prenatal exposure to viral or bacterial pathogens (Atladottir et al., 2010; Brown and Derkits, 2010; Brown, 2012; Hagberg et al., 2012; Jiang et al., 2016; Patterson, 2009; Zerbo et al., 2013; Zerbo et al., 2015). Maternal infection has also been associated with increased risk for bipolar disorder, major depression, epilepsy, and cerebral palsy in the offspring (Brown and Derkits, 2010; Knuesel et al., 2014; Reisinger et al., 2015). Maternal infection disrupts the delicate immune balance between the maternal and fetal environments, resulting in an altered immune profile in the developing brain. Under normal conditions, immune molecules have regulatory roles throughout neurodevelopment, beginning with neural induction (Deverman and Patterson, 2009). They are also involved in the proper formation of neural circuits through their regulation of synaptic refinement, transmission, and plasticity (Garay and McAllister, 2010).
Animal models have helped to further establish the relationship between maternal infection and neurodevelopmental disorders. It is well established that it is the maternal immune response, not a specific pathogen, which is a risk factor for neurodevelopmental disorders (Hagberg et al., 2012). Likewise, different agents have been used to induce maternal immune response during gestation in animal models of maternal immune activation (MIA) (Meyer, 2014). The most commonly used agents are polyinosinic-polycytidylic acid (Poly (I:C)) and lipopolysaccharide (LPS), which mimic viral and bacterial maternal infections by activating the Toll-like receptor 3 and Toll-like receptor 4 pathways, respectively (Meyer, 2014; Smith et al., 2010). A side-by-side comparison demonstrated that certain MIA-induced phenotypes are observed with either agent, while other phenotypes appear to be specific to the immunogen used (Arsenault et al., 2014). Strains of influenza have also been used, with the main advantage being that they elicit a full spectrum of immune responses (Meyer, 2014; Smith et al., 2010). Poly (I:C) and LPS, on the other hand, induce a limited, well-defined immune response, and allow the time and intensity of MIA to be more precisely controlled (Meyer and Feldon, 2012). The ability to restrict the immune response to a specific time point in gestation is important because different phenotypes emerge in the offspring depending on which developmental processes were disrupted by MIA (Fortier et al., 2007; Meyer et al., 2006b; Meyer et al., 2008c).
It has been suggested that the etiology of neurodevelopmental disorders is a combination of environmental factors and genetic predisposition (Clarke et al., 2009). MIA may be one of two hits that together result in ASD or schizophrenia. The other hits may be genetic, for example, mutations in ASD or schizophrenia risk genes (Abazyan et al., 2010; Ehninger et al., 2012; Lipina et al., 2013), or environmental, such as gestational diabetes mellitus (Money et al., 2017), maternal iron deficiency (Boksa et al., 2016; Li et al., 2018), or stress around the time of puberty (Giovanoli et al., 2016a; Giovanoli et al., 2013). The synergism of gene-environment or environment-environment risk factors has been evaluated in multiple studies using either a full dose of Poly (I:C) or a physiologically relevant, subthreshold dose of Poly (I:C), such that MIA alone does not produce disease phenotypes in the offspring, but offspring exposed to a second hit (genetic or environmental) display disease-relevant behavioral abnormalities (Abazyan et al., 2010; Giovanoli et al., 2013; Lipina et al., 2013). These studies offer valuable insight into the complex etiology of neurodevelopmental disorders.
With MIA being used as a model for multiple disorders, MIA paradigms are quite heterogeneous. As such, there is variation in experimental parameters including the time of immune insult, the age of the offspring, and the brain regions studied, each having an effect on the phenotypes observed (e.g. Garay et al., 2013; Meyer et al., 2006b; Patrich et al., 2016b; Shin Yim et al., 2017). Additional factors such as the caging system (Mueller et al., 2018) or the mouse strain (Babri et al., 2014; Morais et al., 2018; Schwartzer et al., 2013) also influence experimental outcomes.
In this review, we present the major alterations in MIA offspring, including, behavioral impairments, alterations in brain structure, impairments in synaptic function and transmission, and immune alterations in the developing brain. We propose that such diverse effects could be mediated by epigenetic alterations that occur following exposure to MIA (Figure 1). Increasing evidence suggests an association between prenatal exposure to environmental insults, epigenetic modifications, and neurodevelopmental disorders (reviewed by Kundakovic and Jaric, 2017). During normal neurodevelopment, gene expression is regulated by a pattern of epigenetic changes including histone modifications, DNA methylation, and microRNA expression. Acetylation of histones induces a looser chromatin structure and facilitates transcription (Eberharter and Becker, 2002). In contrast, DNA methylation within the gene promoter represses transcription while DNA methylation in the gene body is associated with transcription (Jones, 2012). Gene expression can also be regulated by microRNAs, short noncoding nucleic acids that bind to mRNA and either block translation or facilitate degradation of the target mRNA. All of these epigenetic mechanisms have been implicated in regulating proper neuronal migration, neurite outgrowth, dendritic development (Mehler, 2008; Sun and Shi, 2015; Trakhtenberg and Goldberg, 2012; Zibetti et al., 2010), and synapse formation and function (Nelson et al., 2008; Sando et al., 2012). Epigenetic modifications are also associated with many of the behavior changes that have been observed in MIA offspring including social deficits, impairments in learning and memory, and anxiety- and depression-like behaviors (Allan et al., 2008; Guan et al., 2015; Sun et al., 2013). The timing of epigenetic changes may be related to certain developmental windows of susceptibility, time periods during which offspring are particularly vulnerable to environmental insults (Bale, 2015). Epigenetic studies are emerging in attempts to provide a link between prenatal exposure to maternal immune activation and the phenotypic changes in MIA offspring. These studies address how epigenetic alterations in MIA offspring affect gene expression and behavior and the plausible relationship between the two.
Figure 1: The maternal immune activation model.
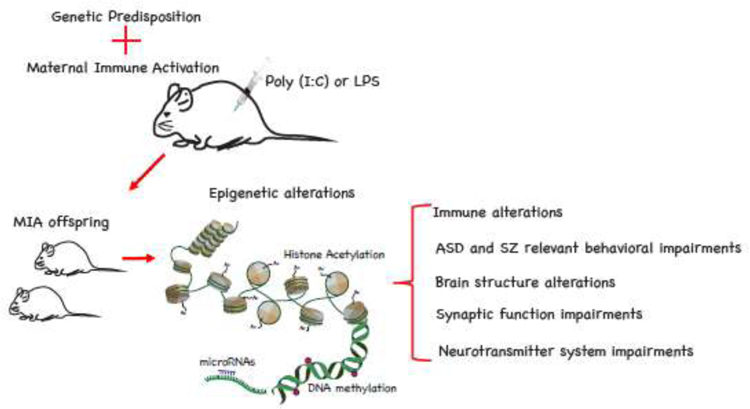
Strong immunological stimulation of the gestating dam with immune-stimulating reagents such as Poly (I:C) or LPS (or a mild stimulus that interacts with maternal genetic susceptibility) result in epigenetic alterations including histone acetylation, DNA methylation, and microRNA expression that contribute to a spectrum of molecular alterations, neuronal dysfunctions, and behavioral phenotypes in the MIA offspring.
2. Behavioral changes
Behavior tests are key in determining the face validity of animal models of CNS diseases, especially disorders for which behavioral symptoms are an important component of the diagnosis. MIA offspring exhibit several behavioral alterations that are relevant for ASD and schizophrenia. The core symptoms of ASD fit into three main categories: abnormal social interactions, communication deficits, and repetitive behavior while schizophrenia symptoms are often classified as positive (behaviors not seen in healthy people, such as hallucinations or racing thoughts), negative (disrupted normal emotions or behaviors), or cognitive (e.g. poor executive functioning or trouble focusing; https://www.nimh.nih.gov/health/topics/schizophrenia/index.shtml). In addition to the core symptoms of ASD, there are several associated symptoms that are present in a subset of ASD patients including seizures, anxiety, intellectual disability, and hyperreactivity or hyporeactivity to sensory stimuli (Silverman et al., 2010). Some of these symptoms overlap with symptoms of schizophrenia.
Maternal separation-induced production of ultrasonic vocalizations (USVs), which is often interpreted in the context of communicative ability (Scattoni et al., 2009), but can also be an indication of stress response, altered bonding with the mother (Malkova et al., 2012), or anxiety (Kessler et al., 2011), is the earliest ASD-like behavior that can be tested in rodents. Abnormalities in USV production by MIA offspring emerged within the first two postnatal weeks, a period that is typically characterized by a peak in USV production between P8 and P10 and a decline towards no USVs produced on P14 (Scattoni et al., 2009). Both rats and mice exposed to MIA, induced either by Poly (I:C) or LPS, exhibited abnormalities in USV production, but the exact differences observed in independent studies varied. Several studies reported a decrease in the number or duration of USVs (Baharnoori et al., 2012; Fernandez de Cossio et al., 2017; Malkova et al., 2012; Pendyala et al., 2017) while some found an increase in the number of USVs produced (Choi et al., 2016; Pendyala et al., 2017; Shin Yim et al., 2017). A number of factors may contribute to these differences including species (rat or mouse), immunogen used, timing of immune insult, and pup age during testing. Altered USV production was not observed when a single Poly (I:C) injection was administered later than E12.5 (Shin Yim et al., 2017), but LPS administration later in gestation resulted in defects in mice and rats (Baharnoori et al., 2012; Fernandez de Cossio et al., 2017). Repeated testing of pups revealed age-dependent effects of MIA on USV production (Baharnoori et al., 2012; Malkova et al., 2012; Pendyala et al., 2017). The type of USV syllables was also altered in MIA offspring, although different syllable types were altered when Poly (I:C) was administered once compared to a milder dose three times on alternate days (Malkova et al., 2012; Shin Yim et al., 2017). MIA offspring that were tested for both USVs and reflex development had normal reflexes at this time, suggesting that the USV deficit is not due to general developmental delays (Fernandez de Cossio et al., 2017; Malkova et al., 2012). However, others reported modest reflex impairments in LPS-induced MIA offspring and larger impairments in Poly (I:C)-induced MIA offspring when the immunogen was administered daily for three days near the end of gestation (Arsenault et al., 2014). USVs produced by adult animals in social contexts have also been used as indication of communication capabilities. Adult male MIA offspring produced a smaller number and altered type of USVs in the presence of either an unfamiliar male or female (Malkova et al., 2012). In addition, male MIA offspring exhibited a deficit in chemical communication, as seen in their tendency to leave fewer urinary scent marks in response to female urine in the absence of disrupted olfactory sensitivity (Ehninger et al., 2012; Malkova et al., 2012).
A behavior tested in many MIA studies that is relevant to both ASD and schizophrenia is social interaction. Social interaction deficits are a key phenotype of ASD and are one of the negative symptoms of schizophrenia. The following tests take advantage of the natural sociability of mice to assess abnormal social interactions in the MIA model. One indication of a deficit in social interaction, specifically social affiliation, is an obvious preference for a novel object or empty cage over a novel conspecific in a three-chamber apparatus. MIA offspring often spent more or equal time in the chamber with the object compared to the chamber with the conspecific (Bitanihirwe et al., 2010a; Choi et al., 2016; Fernandez de Cossio et al., 2017; Labouesse et al., 2015; Malkova et al., 2012; Mattei et al., 2017; Shin Yim et al., 2017; Smith et al., 2007). This social deficit was not observed in adolescent mice (O’Leary et al., 2014). MIA did not exacerbate the social interaction deficits observed in mice heterozygous for Nrg1, a candidate gene for schizophrenia (O’Leary et al., 2014). Subthreshold MIA combined with expression of mutant Tsc2 or Disc1, genes implicated in ASD and schizophrenia respectively, led to reduced preference for the conspecific (Abazyan et al., 2010; Ehninger et al., 2012; Lipina et al., 2013).
There is also a social recognition deficit in MIA offspring. Mice tend to spend more time near a novel mouse than a familiar mouse, but given the choice between a novel mouse and a familiar mouse, MIA offspring will spend more time with the familiar mouse or equal amounts of time with both mice (O’Leary et al., 2014). Abnormal social interactions are also seen when MIA offspring are allowed to interact directly with a novel mouse (Hava et al., 2006; Pendyala et al., 2017; Zhu et al., 2014), but not when a cage prevented direct interaction with the novel mouse (Li et al., 2018). MIA in rhesus monkeys did not affect social interaction as measured by time spent with the novel conspecific, but they did show abnormal social behaviors such as increased cooing and interactions with the novel monkey (Bauman et al., 2014). In addition, they showed abnormal patterns of eye fixation, preferring to fixate on the mouth, rather than the eyes, in a picture of a grimacing monkey (Machado et al., 2015).
ASD is also characterized by repetitive or stereotypic behaviors or an insistence on sameness. Marble burying is commonly used to test repetitive behaviors. Increased digging results in an increased number of buried marbles. MIA offspring consistently buried more marbles (Choi et al., 2016; Coiro et al., 2015; Fernandez de Cossio et al., 2017; Malkova et al., 2012; Pendyala et al., 2017; Shin Yim et al., 2017; Wu et al., 2015). Other repetitive/stereotypic behaviors in MIA offspring include excessive grooming or head bobbing (Fernandez de Cossio et al., 2017; Kirsten and Bernardi, 2017; Malkova et al., 2012). Non-human primates also exhibited stereotypic behaviors such as repetitive pacing, spinning, and bouncing (Bauman et al., 2014).
When given the choice between a familiar object and a novel object or displaced and nondisplaced objects, MIA offspring showed a preference for the familiar or nondisplaced object (Coyle et al., 2009; Li et al., 2014; Lipina et al., 2013; Ozawa et al., 2006; Wischhof et al., 2015b), in contrast to the typical response in which a rodent will spend more time exploring the novel object. Novel object recognition was impaired by an interaction of subthreshold MIA and a point mutation in Disc1 (Lipina et al., 2013). This test is often interpreted in the context of working memory or cognitive impairment (Leger et al., 2013), but preference for the familiar object could also indicate restricted interests, which are observed in patients with ASD (Silverman et al., 2010). Both interpretations are corroborated by other tests for repetitive behavior or cognitive ability in MIA offspring as described above and in the following paragraphs.
In maze tests (T maze, Y maze, Morris water maze, and dry maze) for working and/or spatial memory MIA offspring were found to have impaired memory (Giovanoli et al., 2015; Labouesse et al., 2015; MacDowell et al., 2017; O’Leary et al., 2014; Vuillermot et al., 2012; Zhang and van Praag, 2015). However, in the Morris water maze, memory deficits only emerged following a longer interval between trials (Meyer et al., 2005; Meyer et al., 2008c) or in adult males (Batinic et al., 2016) while others found no cognitive deficits in MIA offspring (Abazyan et al., 2010). The timing of the prenatal immune insult and the caging system were both found to affect performance in the Y-maze (Mueller et al., 2018). MIA offspring were further shown to have impaired working memory capacity in the odor span test (Murray et al., 2017).
Latent inhibition, another measure of cognitive ability, was abolished in adult MIA offspring (Garay et al., 2013; Giovanoli et al., 2013; Lipina et al., 2013; Meyer et al., 2005; Meyer et al., 2008a; Meyer et al., 2006c; Smith et al., 2007), except when MIA occurred late in prenatal development (Meyer et al., 2006a). This emerged in adult, but not young mice, regardless of the conditioning paradigm used (Meyer et al., 2006c; Zuckerman et al., 2003) which is appropriate because latent inhibition pertains to the cognitive symptoms of schizophrenia which tend to manifest in adulthood in humans. Rearing conditions also appear to impact the presence or absence of latent inhibition. Mice that were not prenatally exposed to immune activation, but were cross-fostered to surrogate mothers who had an immune activation during pregnancy also lacked latent inhibition which would suggest that both prenatal and postnatal events can affect the emergence of latent inhibition (Meyer et al., 2006c). Additional indicators of cognitive ability including temporal perception and set-shifting were also impaired in MIA offspring (Canetta et al., 2016; Deane et al., 2017; Zhang et al., 2012). Optogenetic inhibition of parvalbumin (PV) interneurons in non-immune challenged mice recapitulated the set-shifting impairments (Canetta et al., 2016), while transplantation of stem cell-derived PV interneurons into the mPFC improved cognitive flexibility (Donegan et al., 2018), suggesting that loss or dysfunction of PV interneurons contributes to cognitive impairments in MIA offspring.
Another schizophrenia relevant behavior observed in MIA offspring is increased locomotor activity induced by amphetamine, an indirect dopamine receptor agonist, or dizocilpine (MK-801), an NMDA receptor antagonist (Luan et al., 2018; Meyer et al., 2005; Meyer et al., 2008c; Ozawa et al., 2006; Zuckerman et al., 2003). The sensitivity to MK-801 is age dependent (Meyer et al., 2008b). Subthreshold MIA combined with peripubertal stress also elicited this response (Giovanoli et al., 2016a; Giovanoli et al., 2013).
A widely used test in the MIA model is a test for prepulse inhibition (PPI). A decrease in the percent PPI is indicative of deficits in sensorimotor gating, a characteristic of patients with schizophrenia. The majority of studies demonstrated PPI deficits in MIA offspring (Garay et al., 2013; Giovanoli et al., 2016b; Hadar et al., 2017; Lipina et al., 2013; Mattei et al., 2017; Meehan et al., 2017; Meyer et al., 2005; Meyer et al., 2008c; Ozawa et al., 2006; Smith et al., 2007; Wischhof et al., 2015a; Wischhof et al., 2015b; Wu et al., 2015; Zhang and van Praag, 2015; Zhu et al., 2014). However, the emergence of PPI deficits depends on the caging system used and the timing of the prenatal insult (Mueller et al., 2018). In addition, young MIA offspring (Lipina et al., 2013; Ozawa et al., 2006; Wischhof et al., 2015b), MIA offspring that overexpress IL-10 in macrophages (Meyer et al., 2008a) and MIA offspring exposed to inflammation near the end of gestation (Meyer et al., 2006b) did not exhibit PPI deficits. Variability also arose between males and females (Meehan et al., 2017; Wischhof et al., 2015b), with different prepulse intensities (Missault et al., 2014), and with different interstimulus intervals (Wischhof et al., 2015a). A low dose of Poly(I:C) did not cause deficits in PPI (Abazyan et al., 2010), but a deficit emerged when a mild immune activation was combined with peripubertal stress (Giovanoli et al., 2016a; Giovanoli et al., 2013) or mutations in candidate schizophrenia genes including Disc1 and Nurr1 (Lipina et al., 2013; Vuillermot et al., 2011; Vuillermot et al., 2012).
MIA offspring exhibit some depressive-like behaviors in a strain-specific manner. MIA offspring from the NMRI strain, an outbred strain of mice, were immobile for a greater period of time in the forced swim and tail suspension tests but inbred C57BL6 mice exposed to MIA did not exhibit behavioral alterations in these tests (Babri et al., 2014). However, MIA offspring expressing mutant human DISC1 on a C57BL6 background spent more time immobile in the forced swim and tail suspension tests (Abazyan et al., 2010). Reduced sucrose preference, an indicator of anhedonia and depressive-like behavior, was consistently observed in MIA offspring (Bitanihirwe et al., 2010a; Missault et al., 2014; Reisinger et al., 2016). In addition to the behaviors mentioned above, other behaviors that are not specifically related to ASD or schizophrenia have been observed in MIA offspring (Table 1). These other behaviors suggest that prenatal infection causes diverse neurological dysfunctions and may be risk factor for other neurological disorders (reviewed by Knuesel et al., 2014).
Table 1:
Altered behaviors in MIA offspring
| P0–7 | P8–21 | P22–35 | P36–60 | >P60 | |
|---|---|---|---|---|---|
| Communication deficits | 6 | 14, 21, 47, 63, 70 | ND | ND | 8, 47 |
| Altered social behavior | ND | ND |
Affiliation 80 Recognition 61, 80 Interaction 34, 58 |
Affiliation 80 Recognition 80 Interaction 82 |
Affiliation 2, 3, 8, 10, 14, 20, 21, 34, 37, 42, 47, 48, 58, 59, 61, 65, 70, 71 Recognition 21, 42, 58, 61, 65 Interaction 8, 33, 63 |
| Stereotypic/repetitive behavior | ND | ND | ND | 58, 63 | 8, 14, 15, 21, 36, 47, 70, 79 |
| Impaired object recognition | ND | ND | 24, 32 | 42 | 17, 24, 32, 40, 42, 48, 59, 62, 78 |
| Impaired learning and memory | ND | ND | 27, 38, 39 | 7 | 16, 27, 37, 45, 50, 56, 60, 61, 65, 68,74, 83 |
| Altered latent inhibition | ND | ND | ND | 42, 53 | 25, 26, 42, 50, 51, 53, 54, 71, 74, 83 |
| Altered drug sensitivity | ND | ND |
Amphetamine 55 |
ND |
Amphetamine 12, 22, 26, 28, 50, 54–56, 83 Methamphetamine 62 MK-801 26, 28, 54–56 |
| PPI deficits | ND | ND | ND | 42, 69, 78 | 11, 23, 25, 26, 28, 29, 31, 32, 42, 44, 48–50, 54, 56, 59 62, 66, 67, 71, 73, 74, 77–79, 81 |
| Depressive-like behavior | ND | ND | 80 | 80 | 2, 5, 9, 10, 18, 57, 58, 64 |
| Altered behavior in open field | ND | ND | 78 | 42, 72, 78 | 35, 40–43, 72, 74, 75 |
| Anxiety-like behavior | ND | ND | 34, 78, 80 | 34, 78 | 2, 13, 18, 33, 50, 52, 54, 62, 68, 70, 71, 75 |
| Impaired motor coordination | ND | ND | 30 | 1, 30 | 1, 81 |
| Altered food hoarding | ND | ND | ND | ND | 27, 75 |
|
Other P0–7: Reflex impairments 4 Delayed acquisition of the forelimb grasp reflex 6 P8–21: ↓ grip strength; ↓approaches toward maternal nest; impaired associative learning 6 P36–60: ↑ risk assessment in elevated plus maze 42 >P60: Faster acquisition of the delayed alternation task; impaired set shifting 13 Impaired temporal perception 19 ↑ ethanol intake and preference 43 Perseverative behavior 52 ↓ basal home cage activity 68 Altered burrowing and food consumption; impaired balance 75 ↑ latency to initiate sexual behavior 76 | |||||
ND: not determined
References:
Baharnoori et al., 2010 (L)
Borrell et al., 2002 (L)
Fortier et al., 2004 (L)
Girard et al., 2009 (L)
Lanté et al., 2007 (L)
Ling et al., 2009 (L)
Liu et al., 2004 (L)
Romero et al., 2007 (L)
Romero et al., 2010 (L)
Shi et al., 2003
Wang et al., 2010 (L)
Wijkstra et al., 1991 (L)
Wu et al., 2018 (L)
References noted with (L) used LPS; all others used Poly (I:C) except Arsenault et al., 2014 which compared both immunogens. (G) indicates gene-environment interaction studies. (S) indicates MIA offspring were exposed to peripubertal stress.
3. Structural and functional brain changes
3.1. Anatomical abnormalities
Given the abundance and diversity of atypical behaviors observed in MIA offspring, an important question is: what are the underlying brain abnormalities and how do they relate to brain function?
MRI studies have identified enlarged ventricles and reductions in the volume of several brain regions in adult rodent and nonhuman primate MIA offspring (da Silveira et al., 2017; Fatemi et al., 2008; Patrich et al., 2016b; Piontkewitz et al., 2011; Short et al., 2010) although a longitudinal study found that a decrease in ventricle volume emerged in older mice (Crum et al., 2017). Another study found no change in total brain volume, but the relative volume of several regions was increased or decreased unilaterally (Richetto et al., 2017a). Subthreshold MIA combined with low or high expression of mutant human DISC1 also reduced brain volume and increased ventricular volume (Abazyan et al., 2010). Full MIA also decreased the volume of the central nucleus of the amygdala in P14 MIA offspring (O’Loughlin et al., 2017). The extent of these changes in volume is correlated with the maternal immune response to influenza (Short et al., 2010).
Histological experiments suggest that underlying the overall decrease in brain volume is a change in neuronal density. In a porcine model of MIA, the total neuron density was decreased in the dentate gyrus and subiculum of fetal MIA offspring (Antonson et al., 2018). In contrast, MIA induced late in gestation caused a slight increase in the density of neurons in the corpus callosum of MIA offspring, accompanied by an increase in the density of somatostatin interneurons (Duchatel et al., 2016). A common finding is a decrease in the density of GABAergic neurons, specifically Purkinje cells in the cerebellum (Naviaux et al., 2013; Shi et al., 2009) and PV-positive and reelin-positive neurons in the hippocampus and cortex (Donegan et al., 2018; Fatemi et al., 1999; Matsuura et al., 2018; Meyer et al., 2008c; Wischhof et al., 2015b; Zhang and van Praag, 2015). The Purkinje cell deficit was restricted to lobule VII and some Purkinje cells in this lobule were also heterotopic (Naviaux et al., 2013; Shi et al., 2009). However, the timing of MIA is critical. When mice were exposed to MIA after the height of mitosis in Purkinje precursor cells (Miale and Sidman, 1961), an increase in cerebellum size and number of Purkinje cells was observed (Aavani et al., 2015). Similar to Purkinje cells, the loss of PV-positive cells appears to depend on the time of MIA. Interneurons begin to migrate from the ganglionic eminence at E12.5 in rodents (Corbin and Butt, 2011) and only MIA after this time consistently resulted in loss of PV-positive neurons in adult offspring (Canetta et al., 2016; Matsuura et al., 2018; Meyer et al., 2008c; Wischhof et al., 2015b). However, in young MIA offspring an increase in the number of PV interneurons was observed in the dorsolateral PFC, mPFC, and ventral subiculum at P14, but the number of PV neurons normalized by P28 (Boksa et al., 2016). Interestingly, this increase in PV neuron density was abolished when the mother had iron deficiency. One study reported a significant main effect of prenatal treatment on the density of PV neurons across multiple time points in the frontal association cortex and amygdala (Paylor et al., 2016). In the same study, the proportion of PV neurons ensheathed by perineuronal nets was evaluated. While there was no significant effect of MIA on the percentage of PV neurons surrounded by perineuronal nets in the regions with fewer PV neurons, a smaller proportion of PV neurons were surrounded by perineuronal nets in the medial prelimbic cortex (Paylor et al., 2016). In support of impaired neuronal migration in MIA offspring, gene expression of several proteins involved in tangential migration of interneurons was reduced in the fetal brain four hours after LPS administration (Oskvig et al., 2012). GABAergic neurons may be particularly susceptible to MIA given that DNA methylation patterns are altered in MIA offspring (Basil et al., 2014; Basil et al., 2018; Labouesse et al., 2015; Richetto et al., 2017b) and the DNA of GABAergic neurons is more highly methylated compared to that of glutamatergic neurons or glia (Jang et al., 2017).
Migration of pyramidal neurons may also be impaired by MIA. Reduced production of reelin, a glycoprotein involved in the migration of both embryonic-born and adult-born neurons (Fatemi et al., 2008; Teixeira et al., 2012), was observed in the neonatal (Fatemi et al., 1999), developing (Harvey and Boksa, 2012; Nouel et al., 2012), and adult (Meyer et al., 2008c) brain of MIA offspring. Abnormalities in cortical neurons have been reported as early as E14.5, two days after MIA induction with Poly (I:C), or E18.5, four days after MIA induction with LPS. These abnormalities were characterized by loss of neurons expressing special AT-rich sequence binding protein 2 (SATB2) and later by disorganized expression of the layer-specific neuronal markers SATB2 and T-brain-1 (TBR1) (Choi et al., 2016; Wu et al., 2018). In Poly (I:C) offspring, patches of cortical disorganization were observed throughout the cortex of adult MIA offspring, but the majority of patches were located in the primary somatosensory cortex, secondary motor cortex, and temporal association cortex (Shin Yim et al., 2017). These patches were sensitive to the time of MIA and the size of patches in the primary somatosensory cortex was correlated with behaviors characteristic of MIA offspring. These studies suggest that MIA at discrete developmental stages impairs neuronal migration.
While the number of inhibitory neurons is affected by MIA, the dendritic structure and synaptic formation of excitatory neurons is altered (Figure 3). The length and complexity of dendrites were reduced in an age-specific manner in the cortex and hippocampus of rodents (Baharnoori et al., 2009; Fernandez de Cossio et al., 2017; Li et al., 2014; Zhang and van Praag, 2015), but dendrite complexity in the basolateral amygdala was not altered by MIA (Li et al., 2018). However, in rhesus monkeys, no change in dendrite complexity was observed, but the apical dendrites were thinner in MIA offspring (Weir et al., 2015). In addition, the density of dendritic spines on pyramidal neurons in the cortex (Baharnoori et al., 2009; Coiro et al., 2015; Li et al., 2014) and granule cells in the DG (Abazyan et al., 2010; Li et al., 2014) was reduced in MIA offspring. Subthreshold MIA combined with expression of mutant human DISC1 also caused spine deficits on granule cells (Abazyan et al., 2010). On apical dendrites in the medial prefrontal cortex (mPFC), the decreased spine density was limited to dendrite segments proximal to the soma but the spine deficit was observed on first, second, and third order basilar dendrites (Li et al., 2014). The spine density on cortical neurons at 3 months was inversely correlated with marble burying behavior (Coiro et al., 2015). In vivo time-lapse imaging of dendritic spines in P17 mice demonstrated reduced rate of spine turnover in MIA offspring, due to a lower rate of both spine gain and spine loss, suggesting an impairment in the normal dynamic properties of dynamic spines (Coiro et al., 2015). Reduced synapse density on cultured cortical neurons was shown to be caused by increased expression of MEF2 transcription factors and surface MHC1 (Elmer et al., 2013). However, one group reported an increase in the number of spines on granule cells at the peak of synaptic pruning, accompanied by decreased expression of CX3CR1, the microglial fractalkine receptor, which is involved in synaptic pruning (Fernandez de Cossio et al., 2017). A reduction in spine density was not observed in primates (Weir et al., 2015). Dendritic spines are often an appropriate proxy for synapses, but spine density alone is insufficient to fully describe synaptic alterations in MIA offspring. In the cortex, although there are fewer spines, there is a greater percentage of spines that are contacted by excitatory or inhibitory presynaptic input (Coiro et al., 2015). On the other hand, fewer spines on Purkinje cells form structural synapses (Pendyala et al., 2017).
Figure 3: Structural and functional neuronal impairments in MIA offspring.
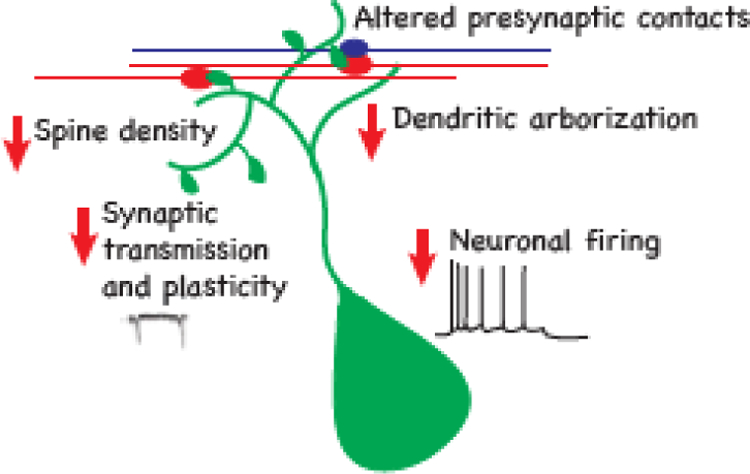
Reduced dendritic branching (Baharnoori et al., 2009; Fernandez de Cossio et al., 2017; Li et al., 2014; Zhang and van Praag, 2015) and dendritic spine density have been observed in MIA (Coiro et al., 2015). Altered innervation of dendritic spines was observed in the cortex and the cerebellum (Coiro et al., 2015; Pendyala et al., 2017). Impaired synaptic transmission, mainly in the form of reduced synaptic transmission (Canetta et al., 2016; Coiro et al., 2015; Ito et al., 2010; Oh-Nishi et al., 2010; Patrich et al., 2016a; Shin Yim et al., 2017; Zhang and van Praag, 2015) has been observed in the cortex and hippocampus. In addition to the synaptic impairments, neurons in the hippocampus have altered membrane properties that result in reduced excitability (Patrich et al., 2016b).
Consistent with the reduction in the structural unit of synapses, there is a reduction in the expression of presynaptic proteins (cerebellin-1, bassoon, and synaptophysin) and post synaptic proteins (GluRδ2, PSD-95, and SynGAP) in the cerebellum and hippocampus of MIA offspring (Giovanoli et al., 2015; Giovanoli et al., 2016b; Pendyala et al., 2017). A decrease in brain-derived neurotrophic factor (BDNF) expression and signaling have also been observed in MIA offspring (Giovanoli et al., 2015; Han et al., 2017b; Schaafsma et al., 2017) and this may contribute to the reduced expression of some synaptic proteins, such as synaptophysin, that are downstream targets of BDNF signaling (Poo, 2001). In the cerebellum, the reduction in expression of synaptic proteins is not necessarily because there are fewer spines. In the case of glutamate receptor delta 2 (GluRδ2) expression on Purkinje cells, the proportion of spines in which GluRδ2 is expressed is lower in MIA offspring compared to controls (Pendyala et al., 2017). Taken together, these data suggest that synaptic function is impaired in MIA offspring. This concept will be further discussed in the following paragraphs.
3.2. Altered neuronal function
The effect of MIA on the excitatory and inhibitory drive of pyramidal cells in cortical and hippocampal slices was investigated by recording miniature excitatory or inhibitory postsynaptic currents (mEPSC and mIPSC). A common finding was reduced mEPSC frequency while the amplitude was either unchanged or increased (Coiro et al., 2015; Ito et al., 2010). Recordings from granule cells in the dentate gyrus of the hippocampus detected no differences in mEPSC frequency or amplitude in either adult-born or embryonic-born granule cells (Zhang and van Praag, 2015). The reduction in mEPSC frequency is unlikely to be due to altered presynaptic release properties as, with the exception of one study (Oh-Nishi et al., 2010), no difference in presynaptic release properties was observed in either brain regions (Ito et al., 2010; Patrich et al., 2016a; Zhang and van Praag, 2015) nor in the mPFC to basolateral amygdala (BLA) projections (Li et al., 2018). The reported effects of MIA on inhibitory drive were also variable. mIPSC frequency was unchanged in the somatosensory cortical neurons (Coiro et al., 2015), hippocampal pyramidal neurons (Inestrosa and Varela-Nallar, 2015; Ito et al., 2010; Patrich et al., 2016a) and in adult born granule cells in the hippocampus while being reduced in embryonically born granule cells (Zhang and van Praag, 2015), mPFC (Canetta et al., 2016) and in cortical patches of the somatosensory cortex (Shin Yim et al., 2017). mIPSC amplitude was found to be unaffected in most studies in the cortex (Canetta et al., 2016; Shin Yim et al., 2017) and the hippocampus (Ito et al., 2010; Zhang and van Praag, 2015) although increases were also reported (Coiro et al., 2015; Patrich et al., 2016a).
Consistent with the findings of unitary synaptic responses, evoked synaptic responses were also found to be altered in MIA offspring. Stimulation of Schaffer collaterals while recording field-EPSPs in CA1 pyramidal neurons revealed reduced excitatory transmission in MIA offspring with female mice exhibiting a delay in the expression of this phenotype (Patrich et al., 2016b). In the mPFC, connectivity between various GABAergic interneurons and pyramidal cells was probed with cell specific expression of the light-activated cation channel, channelrhodopsin2 (Canetta et al., 2016). It was found that in MIA offspring there was reduced light-evoked-IPSCs, suggesting reduced connectivity between pyramidal neurons and PV neurons. This deficit was specific to PV neurons as GABAergic transmission from calretinin or somatostatin interneurons onto pyramidal neurons was unchanged. In this study, the decreased connectivity has been shown not to be mediated by altered number of PV neurons or synaptic contacts but rather by reduced presynaptic release probability (Canetta et al., 2016). This is in contrast to excitatory synaptic inputs where the majority of studies found no change in release properties with MIA (Ito et al., 2010; Patrich et al., 2016a; Zhang and van Praag, 2015). However, analysis of mPFC projections to the amygdala demonstrated an increase in glutamatergic synaptic transmission with MIA on both interneurons and principle neurons (Li et al., 2018).
In addition to the finding of alterations in basic synaptic properties a few studies have examined how synaptic plasticity is impacted by MIA. Measurements of long-term potentiation (LTP), a cellular mechanism of learning and memory has demonstrated that LTP is reduced in the CA1 hippocampal regions when measured in slices (Ito et al., 2010; Oh-Nishi et al., 2010). However, in vivo recordings in MIA offspring demonstrated increased maintenance of LTP with no discernable difference in magnitude (Savanthrapadian et al., 2013). The effect of MIA on long-term depression has been examined in LPS injected rats demonstrating the acceleration of the normal developmentally regulated reduction of NMDAR mediated long-term depression (LTD) in the hippocampus (Escobar et al., 2011).
Neuronal function is determined not only by synaptic properties but also by the membrane properties that regulate the intrinsic excitability of neurons and thus affect their firing properties. The intrinsic excitability of neurons in the MIA model has not been extensively studied but in hippocampal cultures and in slices, MIA offspring displayed reduced intrinsic excitability manifested as increased current necessary to evoke a single action potential as well as reduced spiking frequency (Patrich et al., 2016a). Together these studies indicate that both synaptic and intrinsic properties of neurons are impaired in MIA offspring.
So far a handful of studies analyzed how MIA affects neuronal activity in vivo in awake behaving mice. Recordings of hippocampal place cells (pyramidal neurons that fire in specific locations in the environment) has not detected a difference in basal firing properties such as firing rates but did detect smaller place fields in the MIA offspring which could underlie the spatial memory deficits reported in these mice (Wolff and Bilkey, 2015). In addition the same group has investigated how synchrony in neuronal activity between brain regions was impacted by MIA. Using EEG recordings they found reduced mPFC-hippocampus coherence suggesting reduced long-range functional connectivity in MIA offspring (Dickerson et al., 2010). In future studies it will be critical to examine neuronal activity in the intact brain and in relevant brain regions in order to further understand how altered neuronal activity affects behaviors impaired in MIA offspring.
3.3. Neurotransmitter systems
In line with the altered neuronal function described above, components of neurotransmitter systems (neurotransmitters, receptors, and transporters) are dysregulated in MIA offspring. Affected systems include the glutamatergic, GABAergic, dopaminergic, serotonergic, and cholinergic systems (see Table 2). Expression of the genes encoding many of the proteins involved in these signaling systems is regulated by microRNA or histone modifications (Shrestha and Offer, 2016; Stadler et al., 2005; Sun and Shi, 2015).
Table 2:
Alterations in neurotransmitter systems
| Age | Alteration | Immunogen | Reference |
|---|---|---|---|
| <P0 | ↑ choline acetyltransferase (basal forebrain) | Poly (I:C) | Pratt et al., 2013 |
| ↑ mGluR5 (whole brain) | Poly (I:C) or LPS | Arsenault et al., 2014 | |
| ↑ GABA; ↓ Glutamate (whole brain) | LPS | Batinić et al., 2017 | |
| P0–7 | ↓ 5HT1A and 5HT1B mRNA (frontal cortex) | LPS | Baharnoori et al., 2010 |
| P8–21 | ↑ mGluR5 (right hemisphere) | Poly (I:C) | Arsenault et al., 2014 |
| P22–35 | ↑ GABAA receptor α2 and α4 subunits mRNA (mPFC) | Poly (I:C) | Richetto et al., 2014 |
| ↓ Gria1, Gria2, and
Slc17a7 mRNA (cortex); ↑ Gria1, Gria2, and Slc17a7 mRNA (hippocampus) |
Poly (I:C) | Tang et al., 2013 | |
| ↓ Grin1, Grin2a,
Grin2b mRNA (hippocampus); ↓ GABA (frontal cortex) |
Poly (I:C) | Fujita et al., 2016 | |
| ↓ DAT immunoreactivity (caudate putamen, nucleus accumbens) | Poly (I:C) | Vuillermot et al., 2010 | |
| ↓ D2R (PFC); ↓ DAT (nucleus accumbens core and shell) |
LPS | Baharnoori et al., 2010 | |
| >P60 | ↓ GABAA receptor α2,
α4, and α5 subunits mRNA, ↑ GABAA receptor α4 subunit mRNA (mPFC) |
Poly (I:C) | Richetto et al., 2014 |
| ↓ GAD67, parvalbumin (dorsal hippocampus) | Poly (I:C) | Luoni et al., 2017 | |
| ↓ Gria1 mRNA (cortex and hippocampus); | Poly (I:C) | Tang et al., 2013 | |
| ↓ Gria2 mRNA (striatum) | |||
| ↓ EAAT3 expression | LPS | Kentner et al., 2016 | |
| ↑ NMDAR channels (cingulate cortex,
striatum); ↑ NR2A (hippocampus, cortex, striatum); ↑ NR2A:NR2B ratio (hippocampus); ↑ Grin1 mRNA (nucleus accumbens shell);↑ Grin2a mRNA (cortex, DG, CA1) |
Poly (I:C) | Rahman et al., 2017 | |
| ↑ GABAA receptor α1 and α2 subunits mRNA (ventral hippocampus); ↑ α3 subunit mRNA (cortex) | Poly (I:C) | Richetto et al., 2015 | |
| ↑ dopamine (nucleus accumbens) | Poly (I:C) | Giovanoli et al., 2013 | |
| ↑ dopamine receptor 1 mRNA (hippocampus) | Poly (I:C) | Buschert et al., 2016 | |
| ↓ D1R and D2R immunoreactivity
(mPFC); ↓ GluR1 immunoreactivity (nucleus accumbens shell) |
Poly (I:C) | Meyer et al., 2008b | |
| ↑ alpha 7 nicotinic acetylcholine receptor mRNA (hippocampus) | Poly (I:C) | Wu et al., 2015 | |
| ↑ Acetylcholine (striatum); ↑ glutamate (ventral hippocampus) | LPS | Batinić et al., 2017 | |
| ↑ D1R immunoreactivity (caudate putamen, nucleus accumbens shell); ↑ D2R immunoreactivity (nucleus accumbens shell) | Poly (I:C) | Vuillermot et al., 2010 | |
| ↑ dopamine (PFC, lateral globus
pallidus); ↓ serotonin (nucleus accumbens, lateral globus pallidus, hippocampus) |
Poly (I:C) | Winter et al., 2009 | |
| ↑ 5-HT2A (males, PFC) | LPS | Wischhof et al., 2015a | |
| ↑ Serotonin transporter (hippocampus) | Poly (I:C) | Reisinger et al., 2016 | |
| ↓ D1R immunoreactivity
(mPFC); ↓ NR1 immunoreactivity (dorsal hippocampus); ↑ GABAA receptor α2 subunit immunoreactivity (ventral hippocampus) |
Poly (I:C) | Meyer et al., 2008c | |
| ↑ D1R mRNA (nucleus accumbens) | Poly (I:C) | Meehan et al., 2017 | |
| ↓ D2R expression (PFC) | LPS | Baharnoori et al., 2013 | |
| ↓ dopamine (striatum) | LPS | Kirsten et al., 2010 |
Abbreviations: 5HT: 5-hydroxytryptamine; D1R/D2R: dopamine receptor 1/2; DAT: dopamine transporter; EAAT: excitatory amino acid transporter
Altered expression of components of the glutamatergic and GABAergic signaling pathways provide a molecular basis for the effect of MIA on excitatory and inhibitory drive as well as behavior impairments described above. Of note is the increased NR2A:NR2B ratio in the hippocampus of MIA offspring (Rahman et al., 2017). The proper balance of these NMDA receptor subunits is critical for activity-dependent plasticity. Additional evidence for impaired NMDA receptor signaling was observed in juvenile and adult offspring (Fujita et al., 2016). Levels of the NMDA receptor agonist glutamate and/or glycine, an NMDA receptor co-agonist, were decreased in multiple brain regions of juvenile MIA offspring while the NMDA receptor co-agonist D-serine and its precursor L-serine were decreased in adult MIA offspring (Fujita et al., 2016).
Another mechanism that affects the excitatory/inhibitory balance is the developmental excitatory-inhibitory switch of GABA. Adult MIA offspring appear to have an immature GABAergic system. Increased intracellular chloride levels due to reduced expression of the K+-Cl− cotransporter 2 (KCC2) and elevated expression of the Na+-K+-Cl− cotransporter 1 results in a delay in the switch of the inhibitory action of GABA and is likely to underlie the increased susceptibility to seizures observed in MIA offspring (Corradini et al., 2018; Richetto et al., 2014). Reduced expression of KCC2 is attributed to increased binding of two transcription factors, RE1-silencing transcription factor and methyl-CpG-binding protein 2, to the Kcc2 promoter, thus implicating epigenetic modifications in the delayed neuronal inhibition by GABA.
Aberrations in the serotonergic system were observed as early as 48 hours after Poly (I:C) administration. Levels of serotonin were increased in the forebrain of MIA offspring, likely due to elevated serotonin output from the placenta corresponding to an increase in tryptophan hydroxylase activity in the placenta of Poly (I:C)-injected females (Goeden et al., 2016). In addition, the density of serotonergic axons was significantly decreased in the fetal brain (Goeden et al., 2016).
The dopaminergic system has been widely studied in the context of MIA as imbalances in this signaling pathway are pertinent to schizophrenia. Quantification of dopaminergic neurons on the mesencephalon revealed a reduction in the number of dopaminergic precursor cells in the MIA fetus two days following MIA as well as altered positioning of post-mitotic and mature dopaminergic neurons (Luan et al., 2018). In contrast, levels of tyrosine hydroxylase, the rate limiting enzyme in dopamine synthesis, were increased in the striatum in an age and region specific manner (Meyer et al., 2008b; Vuillermot et al., 2010). The number of tyrosine hydroxylase-positive neurons, as well as the number of neurons expressing Nurr1, a transcription factor critical for the development of the dopaminergic system, were increased in the ventral tegmental area (VTA) (Li et al., 2014; Vuillermot et al., 2012; Vuillermot et al., 2010). However, MIA late in gestation caused a decrease in the number of tyrosine hydroxylase-positive neurons in the VTA (Vuillermot et al., 2012). Levels of dopamine and its metabolites have been found to be elevated in some brain regions, but decreased or not changed in other regions in MIA offspring (Abazyan et al., 2010; Giovanoli et al., 2013; Kirsten et al., 2010; Ozawa et al., 2006; Winter et al., 2009). Peripubertal stress increased the level of dopamine in the hippocampus of MIA offspring (Giovanoli et al., 2013). In addition, stimulated dopamine release was greater in the striatum of MIA offspring (Zuckerman et al., 2003). There was also altered expression of dopamine receptors D1R and D2R throughout development (Baharnoori et al., 2013; Buschert et al., 2016; Meehan et al., 2017; Meyer et al., 2008b; Meyer et al., 2008c; Ozawa et al., 2006; Vuillermot et al., 2010). MIA also caused a decrease in D2R expression in mice heterozygous for Nurr1 (Vuillermot et al., 2012). Expression and activation of glycogen synthase kinase 3 beta (GSK3β) and protein kinase B (AKT), two kinases that are involved in dopaminergic signaling, were altered in MIA offspring as well (Bitanihirwe et al., 2010b; Willi et al., 2013). Inhibiting GSK3β normalized some behaviors in MIA offspring (Willi et al., 2013) and cognitive performance was correlated with the number of cells that express AKT1 in the mPFC (Bitanihirwe et al., 2010b). There was also a decrease in the expression of dopamine transporter (DAT) in MIA offspring (Baharnoori et al., 2013; Vuillermot et al., 2010). Accordingly, the absence of latent inhibition and hypersensitivity to amphetamine in MIA offspring are thought to be a result of elevated dopamine transmission in the mesolimbic pathway (Zuckerman et al., 2003). Taken together, these results suggest that dopaminergic signaling is sensitive to a prenatal immune insult, but the exact alterations are age and brain region specific.
4. Immune alterations
Anomalies in MIA offspring, specifically in terms of behavior, anatomy, physiology, and neurotransmitter systems are becoming increasingly well characterized, but much is still unclear, especially the link between maternal inflammation and the phenotypes in the offspring described above. The next two sections will describe the major immune changes in the mother and the offspring (Figure 4) and the epigenetic mechanisms that may mediate the effects of MIA.
Figure 4: Maternal and MIA offspring immune changes.
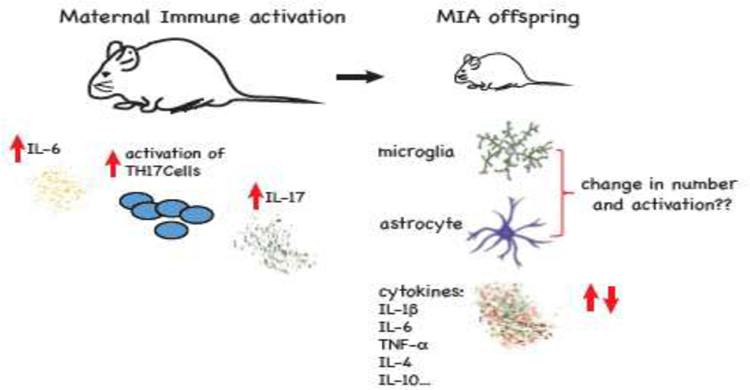
Immunological response includes increase in pro-inflammatory cytokines IL-6 and IL-17 and activation of TH17 cells. In addition to peripheral immune alterations, long lasting changes in the expression of multiple cytokines were observed in the brain of the MIA offspring.
4.1. Cytokines
Cytokines are small signaling molecules of the innate immune system that also have defined roles in neurodevelopment (Garay and McAllister, 2010). Altered cytokine levels may both contribute to and be a result of epigenetic alterations in MIA offspring. Examples of epigenetic regulation by cytokines include regulation of DNA methyltransferase 1 expression and subcellular localization by interleukin-6 (IL-6) (Hodge et al., 2007; Hodge et al., 2001) and inhibition of histone deacetylase activity by IL-17a (Zijlstra et al., 2012). Conversely, expression of cytokines such as IL-1β and tumor necrosis factor α (TNF-α) are regulated by epigenetic mechanisms including DNA methylation and microRNA expression (Cheray and Joseph, 2018).
In all MIA models, there is an initial increase in circulating cytokine levels in the mother, as would be expected following an immune insult. Two specific maternal cytokines appear to be critical for mediating the behavioral abnormalities in MIA offspring: (IL-6) and IL-17a. IL-6, a pro-inflammatory cytokine, reaches peak levels 3 hours after exposure to Poly (I:C) (Meyer et al., 2006b). It activates the JAK/STAT3 pathway in fetal cells of the placenta, leading to altered production of placental factors that are important in fetal development (Hsiao and Patterson, 2011). Maternal administration of recombinant IL-6, in the absence of any other infection, was sufficient to induce a deficit in PPI and lack of latent inhibition in the offspring, behaviors which are relevant for ASD and schizophrenia (Smith et al., 2007). Further supporting the role of IL-6, Smith and colleagues also found that offspring of dams who received anti-IL-6 antibodies in addition to Poly (I:C) did not have aberrant gene expression or behaviors in tests for latent inhibition, PPI, anxiety, and social interaction. Offspring of immune-challenged IL-6 knockout mice also did not develop behavioral abnormalities. Elevated IL-6 appears to be detrimental because of the activation of the IL-6 pathway in the placenta. Deletion of IL-6 receptor IL-6Rα in placental trophoblasts prevents MIA-induced neuron loss and behavioral impairments in the offspring (Wu et al., 2017).
Downstream of IL-6, maternal IL-17a is also essential for MIA-induced phenotypes in the offspring. IL-17a was elevated in the placenta and maternal serum following immune activation, leading to an increase in IL-17a receptor subunit A (IL-17Ra) in the fetal brain (Choi et al., 2016). Blocking IL-17a with IL-17a antibodies prevented cortical malformations and the upregulation of IL-17Ra in the fetal brain as well as the emergence of abnormal behaviors in adult MIA offspring (Choi et al., 2016). The role of IL-17a in MIA led to the discovery that the maternal gut microbiome is a critical factor in the development of brain and behavioral abnormalities in MIA offspring. IL-17a is elevated in the maternal serum if segmented filamentous bacteria are present in the maternal microbiota. These bacteria are major contributors to the differentiation of TH17 cells, which produce IL-17a. Behavioral abnormalities and cortical patches were not observed in offspring of immune challenged mothers that lacked segmented filamentous bacteria (Kim et al., 2017).
Other maternal cytokines that are increased in the periphery following infection during pregnancy include tumor necrosis factor-α (TNF-α), IL-10, IL-1β, IL-4, and IFN-β (Ashdown et al., 2006; Ballendine et al., 2015; Choi et al., 2016; Giovanoli et al., 2013; Giovanoli et al., 2015; Money et al., 2017; Wu et al., 2015) (Table 3). While altered levels of these maternal cytokines may impact neurodevelopment and contribute to aberrant phenotypes in MIA offspring, they are not sufficient to induce changes. Maternal administration of TNF-α alone was not sufficient to induce PPI deficits like those seen in MIA offspring (Smith et al., 2007). In addition, induction of MIA in TNF-knockout mothers did not prevent the emergence of MIA-induced behavioral impairments in the offspring (Konefal and Stellwagen, 2017). The increase in maternal IL-10, which is considered to be anti-inflammatory, may actually attenuate the effects of IL-6 and IL-17a considering that overexpression of IL-10 in macrophages prevented the emergence of MIA-induced behavior, while IL-10 overexpression in the absence of an inflammatory stimulus was associated with deficits in spatial exploration and associative learning (Meyer et al., 2008a). In addition, IL-10 administration prevented MIA-induced fetal loss and white matter damage (Pang et al., 2005; Robertson et al., 2006).
Table 3:
Alterations in maternal cytokines
| Time after MIA | Alteration | Immunogen | Reference |
|---|---|---|---|
| 1 hour | ↑ IL-1β, IL-2, IL-4, IL-6, IL-10, IL- 15, IL-17A, IL-27p28, TNF-α, CXCL1, CXCL2, MCP-1(1) | 1 or 5 mg/kg Poly (I:C), i.v. | Mueller et al., 2018 |
| 1.5 hours | ↑ IL-10, IL-6, TNF-α (wild type); ↑ IL-10; ↓ IL-6, TNF-α (transgenic mice overexpressing IL-10 in macrophages, compared to wild type) (2) | 2 mg/kg Poly (I:C), i.v. | Meyer et al., 2008a |
| 2 hours | ↑ IL-6, TNF-α (3) | 0.05 mg/kg LPS, i.p. | Ashdown et al., 2006 |
| ↑ IL-17a (3) | 75 μg/kg LPS, i.p. | Wu et al., 2018 | |
| ↑ IL-6, IL-10, TNF-α (wild type); ↓ IL-6, IL-10, TNF-α (in Nurr1 heterozygous mice compared to wild type) (2) | 2 mg/kg Poly (I:C), i.v. | Vuillermot et al., 2012 | |
| 3 hours | ↑ IL-1β, IL-6, IL-10, TNF-α (2) | 5 mg/kg Poly (I:C), i.v. | Meyer et al., 2006b |
| ↑ IL-6, TNF-α (3) | 5 mg/kg Poly (I:C), i.v. | Connor et al., 2012 | |
| ↑ IL-1β, TNF-α (4) | 4 mg/kg Poly (I:C), i.v. | Ballendine et al., 2015 | |
| ↑ IL-1β, IL-4, IL-6, TNF-α (1) | 5 mg/kg Poly (I:C), i.v. | Giovanoli et al., 2015 | |
| ↑ IFN-β, IL-1β, IL-6, TNF-α (3) | 20 mg/kg Poly (I:C), i.p. | Choi et al., 2016 | |
| ↑ Eotaxin, IL-6, IL-10, IL-12p40, IL-17, MCP-1, RANTES, TNF-α (4) | 20 mg/kg Poly (I:C), i.p. | Money et al., 2017 | |
| ↑ TNF-α (3) | 0.25, 0.1, and 0.05 mg/kg LPS, i.p. (consecutive days) | Schaafsma et al., 2017 | |
| ↑ IL-1β, IL-6 (3) | 0.05 mg/kg LPS, i.p. | Ashdown et al., 2006 | |
| 4 hours | ↑ IL-6, IL-10, TNF-α (2) | 1 mg/kg Poly (I:C), i.v. | Giovanoli et al., 2013 |
| ↑ IL-10, IL-6, TNF-α (wild type); ↑ IL-10; ↓ IL-6, TNF-α (transgenic mice overexpressing IL-10 in macrophages, compared to wild type) (2) | 2 mg/kg Poly (I:C), i.v. | Meyer et al., 2008a | |
| 5 hours | ↑ IL-10/TNF-α ratio (2) | 5 mg/kg Poly (I:C), i.v. | Meyer et al., 2006a |
| 6 hours | ↑ IL-6, IL-10 (2) | 5 mg/kg Poly (I:C), i.v. | Meyer et al., 2006b |
| ↑ IL-1β (5), TNF-α (3, 5) | 4 mg/kg Poly (I:C), s.c. | Missault et al., 2014 | |
| ↑ IL-1β, IL-2, IL-6, IL-10, IL-15, IL- 17A, IL-27p28, TNF-α, CXCL1, CXCL2, MCP-1; ↑/↓ IL-4 (depending on caging conditions) (1) | 1 or 5 mg/kg Poly (I:C), i.v. | Mueller et al., 2018 | |
| ↑ IFN-β, IL-1β, IL-17a, TNF-α (3) | 20 mg/kg Poly (I:C), i.p. | Choi et al., 2016 | |
| >24 hours | ↑ IL-6 (24 hours; no difference at 48 hours) (6) | 2 mg/kg Poly (I:C), i.p. | Goeden et al., 2016 |
Superscripts indicate methods used to measure cytokines:
V-Plex electrochemiluminescence assay
Multiplexed particle-based flow cytometry assay
Enzyme-linked immunosorbent assay
Bead- based multiplex assay
qPCR
Cytometric bead array
For the studies in this table, the immunogen was administered via either intraperitoneal (i.p.), intravenous (i.v.) or subcutaneous (s.c.) injection.
Many cytokines are also dysregulated in the serum of MIA offspring throughout development (Table 4). In a comparison of LPS versus Poly (I:C)-induced MIA, only Poly (I:C)-induced MIA offspring exhibited significant increases in IL-2, IL-5, and IL-6 (Arsenault et al., 2014), however a direct measure of the magnitude of the maternal immune response, such as maternal serum levels of IL-6, was not reported, so it is uncertain whether LPS and Poly (I:C) induced comparable maternal immune responses in this study. A long term study in rhesus monkeys showed a shift in circulating cytokines from alterations in cytokine levels primarily of the innate immune system at 1 year of age to a more TH2 cytokine phenotype in 4 year old MIA offspring (Rose et al., 2017). Adult mice exposed to prenatal Poly (I:C) were also shown to have an altered peripheral cytokine profile (Hsiao et al., 2012).
Table 4:
Alterations in peripheral cytokines in MIA offspring
| Age | Alteration | Immunogen | Reference |
|---|---|---|---|
| P0–7 | ↑ G-CSF, IFNγ, IL-1β, IL-3, IL-6, IL-12(p40), TNF-α; ↓ GM-CSF, IL-1α, IL- 2, IL-3, IL-12(p70) (1) | 20 mg/kg Poly (I:C), i.p. | Garay et al., 2013 |
| P8–21 | ↑ TNF-α (1) | 20 mg/kg Poly (I:C), i.p. | Garay et al., 2013 |
| ↑ IL-2, IL-5, IL-6 (2) | 5 mg/kg Poly (I:C), i.v. (3 consecutive days) | Arsenault et al., 2014 | |
| P22–35 | ↑ IL-1β, IL-6, IL-9; Decreased IL-3 (1) | 20 mg/kg Poly (I:C), i.p. | Garay et al., 2013 |
| ↓ IL-6, TNF-α (3) | 5 mg/kg Poly (I:C), i.v. | Pacheco-López et al., 2011 | |
| ↑ IL-1β, IL-6 (4) | 25, 25, and 50 μg/kg LPS on 3 consecutive days | Hsueh et al., 2018 | |
| P36–60 | ↑ IL-1β, IL-6, IL-10 (4) | 25, 25, and 50 μg/kg LPS on 3 consecutive days | Hsueh et al., 2018 |
| >P60 | ↓ IFN-γ, IL-2 (3) | 5 mg/kg Poly (I:C), i.v. | Pacheco-López et al., 2011 |
| ↑ CCL2, G-CSF, IFNγ, IL-2, IL-6, IL- 13, TNF-α (1) | 3 doses of 0.25 mg/kg Poly I:C-LC, i.v. | Rose et al., 2017 | |
| ↑ IL-6, IL-17 released by splenic CD4+ T cells upon stimulation (2) | 20 mg/kg Poly (I:C), i.p. | Hsiao et al., 2012 |
Abbreviations: CCL: chemokine (C-C motif) ligand; G-CSF: granulocyte colony-stimulating factor; GM-CSF: granulocyte-macrophage colony-stimulating factor; IFN: interferon
Superscripts indicate methods used to measure cytokines:
Bead-based multiplex assay
Enzyme- linked immunosorbent assay
Multiplexed particle-based flow cytometry assay
Cytometric bead array
For the studies in this table, the immunogen was administered via either intraperitoneal (i.p.) or intravenous (i.v.) injection.
Cytokine expression in the developing brain varies across regions (Table 5). The initial dysregulation occurs as early as three hours after Poly (I:C) injection and is characterized by elevated levels of IL-6 in the fetal brain and an increase in STAT3 activation (downstream of IL-6 receptor activation), predominantly in the prepontine hindbrain and pontine hindbrain (Wu et al., 2017). Evidence for immune activation in the fetal brain was also observed in the LPS model of MIA. Twenty-four hours after administration of the third dose of LPS, several interferon-related genes were overexpressed (Hsueh et al., 2018). Immune dysregulation in the brain of MIA offspring is persistent, as evidenced by the upregulation of several inflammatory markers prenatally and in adulthood in the amygdala of MIA offspring (O’Loughlin et al., 2017). A pattern of cytokine dysregulation in MIA offspring emerged in the frontal and cingulate cortices where there was an increase in pro-inflammatory cytokines at birth, reduced levels when critical neurodevelopmental processes such as synaptogenesis and neuronal plasticity are at their peaks, and a return to increased levels of pro-inflammatory cytokines in adult MIA offspring (Garay et al., 2013). There is not a clear pattern of cytokine alterations in the hippocampus, but the majority of alterations appeared to occur more in the developing MIA offspring than in adults when there was a single immune insult (Garay et al., 2013; Giovanoli et al., 2016a; Giovanoli et al., 2015). Increased IL-6 persisted in the hippocampus for 6 months when IL-6 was administered three times during embryonic development (Samuelsson et al., 2006). Synergism between MIA and peripubertal stress was manifested in the hippocampus as an increase in levels of IL-1β and TNF-α compared to control offspring or offspring exposed only to MIA (Giovanoli et al., 2013). Among the cytokine alterations in the cerebellum of MIA offspring, increased expression of TNF-α and its receptor TNFRI at P14 stand out because of their involvement in regulating synapses (Pendyala et al., 2017). These changes in the brain are not caused by infiltration of circulating cytokines, as it was demonstrated that the blood brain barrier is not compromised and there is no immune cell infiltration in the brain of MIA offspring (Garay et al., 2013). However, we cannot dismiss the possibility that peripheral immune changes induce some of the behavioral phenotypes commonly observed in MIA offspring by activation of afferent nerve fibers, a mechanism known to transmit immune signals from the periphery to the brain, resulting in impairments in cognition as well as social and exploratory behavior (Dantzer, 2001; Larson and Dunn, 2001).
Table 5:
Alterations in brain cytokines during development of MIA offspring
| Age | Alteration | Immunogen | References |
|---|---|---|---|
| <P0 | ↑ IL-1β and TNF-α mRNA (whole brain) (1) | 500 μg/kg LPS, i.p. on 2 consecutive days | Cai et al., 2000 |
| ↓ Fas ligand, fractalkine, IL-3, IL-4, IL-5, lympotactin, M-CSF, MIG, MIP-1α, TARC, TECK, TNF-α,VCAM-1 (whole brain) (2) | 20 mg/kg Poly (I:C), i.p. | Ehninger, 2014 | |
| ↑ TNF-α (whole brain) (3) | 5 mg/kg Poly (I:C), i.v. or 120 μg/kg LPS, i.p. (3 consecutive days) | Arsenault et al., 2014 | |
| ↑ IlL-6 mRNA (whole brain) (4) | 20 mg/kg Poly (I:C), i.p. | Wu et al., 2015 | |
| ↑ Il6 mRNA (whole brain) (4); ↑ IL-4, IL-6, IP-10 (whole brain, IL-6+/− offspring, WT mother) (5) | 20 mg/kg Poly (I:C), i.p. | Wu et al., 2017 | |
| ↑ IL-1β, IL-6, TNF-α, IL-10 mRNA (whole brain) (4) | 50 μg/kg LPS, i.p. | O’Loughlin et al., 2017 | |
| P0–7 | ↑ IL-1β (P1); ↓ TNF-α (P7) mRNA (whole brain) (4) | 300 μg/kg LPS, i.p. (2 consecutive days) | Rousset et al., 2006 |
| ↑ GM-CSF, G-CSF, IL-1β, IL-10,
IL- 12(p70); ↓ IL-2, IL-4, IL-5, IL-10, IL-12(p40) (frontal cortex); ↑ IFNγ, IL-12(p70), IL-17; ↓ IL-2, IL-5, IL-6, IL-10 (cingulate cortex); ↑ IL-6, IL-9; ↓ IL-1β, IL-2, IL-3, IL-4, IL-5, IL-10 (hippocampus) (5) |
20 mg/kg Poly (I:C), i.p. | Garay et al., 2013 | |
| ↑ FasL, IL-2, IL-3, IL-6; ↓ Eotaxin-2 (cerebellum) (2) | 20 mg/kg Poly (I:C), i.p. | Pendyala et al., 2017 | |
| ↑ IL-1β mRNA (amygdala) (4) | 50 μg/kg LPS, i.p. | O’Loughlin et al., 2017 | |
| ↓ GM-CSF, IFNγ, IL-1α, IL-1β, IL-2, IL-5, IL-9, IL-10, IL-13 (frontal cortex); ↓ GM- CSF, IFNγ, IL-1β, IL-10, IL-17 (cingulate cortex); ↑ IL-1α, IL-6; ↓ IL-2, IL-5, IL-9 (hippocampus) (5) | 20 mg/kg Poly (I:C), i.p. | Garay et al., 2013 | |
| P8–21 | ↑ MIP-1γ; ↓ ICAM-1, IL-10 (2); ↑ TNF-α (2,3) (cerebellum) | 20 mg/kg Poly (I:C), i.p. | Pendyala et al., 2017 |
| ↓ G-CSF, GM-CSF, IL-1β, IL-3,
IL-5, IL- 6, IL-10, IL-12(p40), IL-12(P70) (frontal cortex); ↓ G-CSF, IL-1β, IL-3, IL-4, IL-5, IL-6, IL-10, IL-12(p40), IL-12(p70), IL-17 (cingulate cortex); ↓ IL-6 (hippocampus) (5) |
20 mg/kg Poly (I:C), i.p. | Garay et al., 2013 | |
| P22–35 | ↑ MIP-1γ; ↓ IFNγ, IL-17 (cerebellum) (2) | 20 mg/kg Poly (I:C), i.p. | Pendyala et al., 2017 |
| ↑ IL-1α, IL-6, IL-9, IL-10 (frontal cortex); ↑ IFNγ, IL-10 (cingulate cortex) (5) | 20 mg/kg Poly (I:C), i.p. | Garay et al., 2013 | |
| >P60 | Increased IL-1β (hippocampus) (6) | 5 mg/kg Poly (I:C), i.v. | Giovanoli et al., 2016b |
Abbreviations: FasL: fas ligand; G-CSF: granulocyte colony-stimulating factor; GM-CSF: granulocyte- macrophage colony-stimulating factor; IFN: interferon; M-CSF: macrophage colony-stimulating factor; MIG: monokine induced by interferon-gamma; MIP: macrophage inflammatory protein; TARC: thymus and activation-regulated chemokine; TECK: thymus expressed chemokine; VCAM: vascular cell adhesion molecule
Superscripts indicate methods used to measure cytokines:
RT-PCR
Cytokine antibody array
Western blot
qRT-PCR
Bead-based multiplex assay
Meso-Scale Discovery V-Plex electrochemiluminescence assay
For the studies in this table, the immunogen was administered via either intraperitoneal (i.p.) or intravenous (i.v.) injection.
The recent observations that IL-1 receptor type I knock out embryos implanted in wild type mothers exhibited attenuated MIA-induced cytokine changes and normal KCC2 expression (Corradini et al., 2018), suggest that the effects of MIA depend not only on maternal cytokine elevations, but also on cytokine dysregulation in the fetal brain.
4.2. Microglia
Microglia, the resident immune cells in the brain, were once considered to only play a role during brain injury and disease, but are now known to have critical roles in neurodevelopment and homeostasis in the brain. Microglia begin to colonize the brain around E9 in rodents and at the beginning of the second trimester in humans (Chan et al., 2007) where they are involved in both the support and elimination of neurons and the activity-dependent pruning of synapses (Bilimoria and Stevens, 2015). Given this evidence, it is clear that it is crucial for microglia to function properly during neurodevelopment and throughout life. Despite evidence for alterations in cytokine production in the brain, many studies reported no change in the density or activation state of microglia in MIA offspring (Buschert et al., 2016; Corradini et al., 2018; Garay et al., 2013; Giovanoli et al., 2013; Giovanoli et al., 2015; Giovanoli et al., 2016b; Li et al., 2018; Missault et al., 2014; Paylor et al., 2016; Pratt et al., 2013; Smolders et al., 2015). However, an almost equal number of studies found that MIA increased microglia density in various brain regions (Hadar et al., 2017; Juckel et al., 2011; Li et al., 2014; Mattei et al., 2014; Mattei et al., 2017; Van den Eynde et al., 2014; Wu et al., 2018; Zhu et al., 2014). On postnatal day 2, MIA offspring exhibit increased density of microglia in the supraventricular corpus callosum, a region where microglia accumulate before migrating to other brain regions (Zhang et al., 2018). In contrast, regions to which microglia migrate from the supraventricular corpus callosum including the corpus callosum, striatum, somatosensory cortex, and hippocampus of MIA offspring have lower density of microglia compared to controls. This suggests that MIA induces a delay in tangential microglial migration. In addition, a greater proportion of microglia in the hippocampus had the more immature amoeboid morphology, suggesting a delay in microglia maturation as well as migration (Zhang et al., 2018).
When there is an increase in the number of microglia, there is also often an increase in the number of amoeboid microglia, indicative of activated microglia (Juckel et al., 2011; Missault et al., 2014; O’Loughlin et al., 2017; Van den Eynde et al., 2014; Zhu et al., 2014). Amoeboid microglia have been observed as early as P7 (O’Loughlin et al., 2017) and as late as P180 (Van den Eynde et al., 2014) in various brain regions. Activated microglia can also be identified by cell surface proteins such as MHCII and CD68. Microglia were found to have increased MHCII expression in the medial prefrontal cortex of MIA offspring (Hadar et al., 2017) and a greater number of microglia expressed CD68 in the embryonic brain (Wu et al., 2018). Others found that MIA alone did not increase microglia activation (Antonson et al., 2017; Giovanoli et al., 2015; Giovanoli et al., 2016b; Smolders et al., 2015), but MIA followed by exposure to peripubertal stress transiently increased expression of CD68 and CD11b (markers for activated microglia) in the hippocampus and medial prefrontal cortex (Giovanoli et al., 2016a; Giovanoli et al., 2013). Binding of radioactive ligands to translocator protein, which is often used to measure glia activation in schizophrenia studies, also showed increased binding in the hippocampus of MIA offspring with a confirmed deficit in PPI (Mattei et al., 2017). This would suggest an increase in the number of activated glia, but a phagocytic activity assay, which measured the number of microglia that phagocytosed fluorescent beads, showed that cultured hippocampal microglia from MIA offspring have reduced phagocytic activity. However, it is important to note that translocator protein is not solely expressed by microglia (Cosenza-Nashat et al., 2009) and that microglia behave differently in vitro than in vivo (Jeong et al., 2013).
The variations reported above might be less surprising considering that there are also variable findings in schizophrenia studies; some have reported increases in microglia density and activation while other studies have reported no change (Laskaris et al., 2016). However, there is evidence that aspects of experimental design could explain these differences. Poly (I:C) administered to rats on E10 had no effect on IBA1 immunoreactivity while Poly (I:C) on E19 increased IBA1 immunoreactivity (Duchatel, 2018). The age of the animals at the time of testing also matters, as there was an increase in the number of microglia in the hippocampus of P2, but not P80 MIA offspring (Li et al., 2014). The brain region studied may also determine whether or not microglia density is affected by MIA, as within the same study, microglia density was increased in the nucleus accumbens, but the other regions investigated showed no change in microglia density (Mattei et al., 2014). In addition, a Poly (I:C) injection on E19 increased IBA1 immunoreactivity in the corpus callosum, but not the cingulate cortex (Duchatel, 2018). However, the main effect of MIA on microglia may be that of priming (Knuesel et al., 2014), resulting in abnormal responses of microglia to subsequent insults. For example, MIA alone did not visibly affect microglia, but microglia were activated by the combination of MIA and peripubertal stress (Giovanoli et al., 2013). Another study, in which MIA offspring received an injection of LPS in adulthood, demonstrated that microglia from the hippocampus of MIA offspring express higher levels of IL-1β following the second LPS insult compared to control offspring that received an LPS injection. However, microglia isolated from the whole brain of MIA offspring exhibited reduced cytokine production following LPS injection (Schaafsma et al., 2017).
The effect of these microglial alterations on neuron-microglia interactions is not clear, but there is support for a reduction in neuron-mediated suppression of microglia activity as well as altered glutamatergic transmission. MIA alone or when followed by peripubertal stress caused alterations in CD200R and CD172a expression. These molecules interact with neuronal CD200 and CD47, respectively, which also have altered expression in MIA, suggesting that there is aberrant neuron-microglia inhibitory signaling (Giovanoli et al., 2013). It is also suggested that MIA-induced microglia activation and neuroinflammation during prenatal development causes increased AMPAR-mediated evoked EPSCs in adult MIA offspring (Roumier et al., 2008). This is based on the observation that mice with a loss of function of the DAP12 gene, a gene that is transiently expressed by microglia during prenatal development, exhibited microglia activation and mild neuroinflammation at P0, followed by increased AMPAR-mediated neurotransmission in adults (Roumier et al., 2008).
4.3. Astrocytes
Astrocytes, under normal conditions, perform many signaling and support functions in the brain. Under inflammatory conditions, they enter a state of astrogliosis which is characterized by functional, structural, and genetic changes. Astrogliosis has been observed to varying degrees in models of MIA with several groups observing an increase in the expression of common astrocyte markers such as glial fibrillary acidic protein (GFAP) and S100β in the brain and cerebral spinal fluid of MIA offspring when MIA is induced with LPS (Cai et al., 2000; de Souza et al., 2015; Hao et al., 2010; O’Loughlin et al., 2017; Rousset et al., 2006). A prenatal IL-6 injection led to increased astrocyte density and branching in MIA offspring (Samuelsson et al., 2006). But administration of Poly(I:C) during gestation did not appear to induce astrogliosis in MIA offspring (Giovanoli et al., 2015; Giovanoli et al., 2016b; Paylor et al., 2016) or in MIA offspring subjected to peripubertal stress (Giovanoli et al., 2013). The presence or absence of astrogliosis could be justified by considering the intensity of MIA. Astrogliosis was observed when pregnant dams were repeatedly exposed to LPS, but a single Poly (I:C) injection did not induce astrogliosis. Histological analysis of astrocytes in MIA is a good starting point, but functional studies to address the roles of astrocytes in the MIA model are still needed.
5. Epigenetic reprogramming of brain development induced by MIA
To date, the epigenetic alterations investigated in MIA offspring include histone modifications, DNA methylation, and microRNA expression. Here we will discuss these MIA-induced epigenetic modifications and their relationship to MIA-induced brain and behavior phenotypes described above.
Consistent with the concept of windows of susceptibility, alterations in histone acetylation depend on the time of MIA or the age of the animal. For example, global acetylation of histones H3 and H4 was reduced in the cortex of juvenile, but not adult, MIA offspring (Tang et al., 2013). In the hippocampus, MIA in mid-gestation had no effect on histone acetylation (Tang et al., 2013), but MIA early in the second half of gestation induced an increase in global acetylation of histone H3 and a decrease in global acetylation of histone H4 (Reisinger et al., 2016). The increased acetylation of histone H3 is likely due to the observed decrease in levels of histone deacetylase 1 (Reisinger et al., 2016). In contrast to global changes in acetylation, modifications at specific gene promoters are directly related to altered gene expression. Histones H3 and H4 were both hyperacetylated at the serotonin transporter (SERT) promoter in MIA offspring, resulting in increased SERT expression (Reisinger et al., 2016). In the cortex of juvenile and adult MIA offspring, hypoacetylation of histone H3 at lysine residues 9 and 14 was observed in the promoter regions of several genes that are implicated in the pathology of schizophrenia, many of which are involved in neurodevelopment or glutamatergic neurotransmission (Tang et al., 2013). Hypoacetylation in these promoter regions was associated with decreased gene expression. In the hippocampus, acetylation of histone H3 was increased in the promoter regions for a number of schizophrenia-related genes that are overexpressed in the hippocampus of juvenile MIA offspring (Tang et al., 2013).
DNA hypomethylation and hypermethylation have both been observed in MIA offspring (Basil et al., 2014; Basil et al., 2018; Labouesse et al., 2015; Richetto et al., 2017b). In the first study to identify altered DNA methylation in MIA offspring, methylation of long interspersed element 1 (LINE1) was observed to be decreased in in the hypothalamus of adolescent female but not male MIA offspring (Basil et al., 2014). LINE1 activity may be further dysregulated in the hypothalamus of female MIA offspring given that the authors also found hypomethylation in the promoter region of methyl-CpG-binding protein 2 (Mecp2), which regulates LINE1 activity (Muotri et al., 2010). Hypomethylation of LINE1 has not been investigated in other brain regions of MIA offspring, but given that methylation of LINE1 represses its transcription (Muotri and Gage, 2006), hypomethylation could be responsible for the overexpression of LINE1 in the prefrontal cortex of MIA offspring (Bundo et al., 2014). Altered expression of LINE1 has implications for neurodevelopment, given that LINE1 is a retrotransposon and can modulate gene expression of neuron-associated genes (Muotri and Gage, 2006).
Analysis of methylation changes across the entire genome at single nucleotide resolution revealed that MIA induces hypomethylation as well as hypermethyaltion in the adult prefrontal cortex (Richetto et al., 2017b). Hypomethylation was more prominent, regardless of whether MIA was induced during middle or late gestation. However, only a fraction of the differentially methylated sites in the two groups overlapped. Gene ontology term enrichment analysis of the genes located in regions of differential methylation identified neuronal differentiation as having a high enrichment score in both MIA groups. However, enrichment of subterms within this category differed between the two groups; a greater number of genes involved in GABAergic differentiation were differentially methylated in offspring exposed to MIA late in gestation. Despite the abundance of changes in methylation in adult MIA offspring, when select genes were tested for methylation and expression the day after birth, no changes were found (Richetto et al., 2017b), suggesting a potentially delayed effect of MIA on the mechanisms regulating DNA methylation.
Consistent with the known function of DNA methylation, MIA-induced changes in methylation of specific genes were accompanied by altered levels of mRNA (Richetto et al., 2017b). Of significance, increased methylation and/or hydroxymethylation of cytosines in the promoter regions of GAD1 and GAD2 enhanced MeCP2 binding and consequently decreased GAD67 and GAD65 mRNA expression in the mPFC of MIA offspring (Labouesse et al., 2015). Hypermethylation in two segments of the promoter region of the myelin-associated oligodendrocytic basic protein was associated with reduced gene and protein expression in adult MIA offspring (Richetto et al., 2017a).
A study of microRNA expression in the entorhinal cortex found that MIA offspring have 21 differentially expressed microRNAs (Hollins et al., 2014). Some of the same microRNA were affected when MIA offspring were exposed to HU210, a synthetic cannabinoid, in adolescence, but this second hit also altered the expression of different microRNAs. An interesting finding in this study is that this combination of pre- and post-natal insults affected the left and right hemispheres differently, in terms of microRNA expression. Many of the differentially expressed microRNAs are located in the long arm of chromosome 6, a region under the control of Mef2 transcription factors. This study found a decrease in Mef2c and Mef2d mRNA in the entorhinal cortex of rats following MIA and synthetic cannabinoid exposure, but an increase in Mef2a and Mef2d expression in cultured cortical neurons has also been reported (Elmer et al., 2013). In addition, expression of several microRNAs with relevance to neuro-immune interactions or models of depression were found to be upregulated in the hippocampus of MIA offspring (Berger et al., 2018).
An interesting question addressed by many of these studies is whether epigenetic alterations are associated with behavioral abnormalities. Some epigenetic changes may precede the emergence of abnormal behaviors given that many epigenetic changes occurred in juvenile MIA offspring while hyperactivity and decreased exploratory behavior were observed only in adult MIA offspring (Tang et al., 2013). Altered histone acetylation can also be concurrent with behavioral abnormalities such as reduced sucrose preference (Reisinger et al., 2016). On the other hand, methylation of histone H3 was not significantly altered in the cortex of adult MIA offspring, but these animals had cognitive deficits (Connor et al., 2012). There is a correlation between increased methylation of the GAD1 promoter and working memory deficits and impairments in social interaction, but methylation of the GAD2 promoter was not found to be significantly correlated with working memory performance or levels of social interaction. Spatial recognition memory was also negatively correlated with methylation of CpG343 in the promoter region of myelin-associated oligodendrocytic basic protein in the mPFC of adult MIA offspring (Richetto et al., 2017a). Taken together, these findings suggest that epigenetic alterations at specific sites may impact behavior (Labouesse et al., 2015).
Some of these epigenetic effects are proposed to be heritable, given that two studies of inbred mice found persistent behavioral abnormalities and altered gene expression in MIA offspring (F1) as well as in the second (F2) and third (F3) generations of offspring of MIA offspring (Ronovsky et al., 2017; Weber-Stadlbauer et al., 2017). The F2 generation in an outbred mouse line also displayed behavioral abnormalities, though not in the same direction as the F1 offspring (Berger et al., 2018). Distinct behavioral and genetic characteristics were passed through the maternal and paternal lines, but the effects tended to be greater when both parents were MIA offspring.
Whether these epigenetic alterations induced by MIA directly induce the brain and behavioral changes described below remains to be determined. Thus far, only correlation, not causation, has been shown in the context of MIA-induced epigenetic and behavioral alterations. Gene expression profiling studies have identified hundreds of differentially expressed genes in the brain of developing MIA offspring (Fatemi et al., 2005; Garbett et al., 2012; Lombardo et al., 2018; Richetto et al., 2017a; Smith et al., 2007), a subset of which have been associated with specific epigenetic changes. Expression of additional dysregulated genes can likely be linked to MIA-induced epigenetic alterations, thus providing a link between epigenetic dysregulation and brain and behavior changes. Future work should address additional epigenetic mechanisms that are known to contribute to the phenotypes that have been observed in MIA offspring.
6. Pharmacological interventions
A number of therapeutic strategies have been tested on MIA offspring. Antipsychotics that are currently on the market have been shown to be effective for treating MIA-induced phenotypes. Paliperidone was shown to regulate Toll-like receptor 3 signaling and improve performance in the T maze (MacDowell et al., 2017). Clozapine improved long-range synchrony, latent inhibition, working memory, and novel object recognition in MIA offspring, but impaired working memory in control offspring (Dickerson et al., 2012; Meyer et al., 2010a; Ozawa et al., 2006; Zuckerman et al., 2003). Chronic administration of lurasidone in adulthood restored levels of GAD67 and parvalbumin protein in the dorsal hippocampus (Luoni et al., 2017). The effects of risperidone as preventive treatment were tested by administering risperidone during adolescence, before most symptoms emerge. Risperidone treatment prevented latent inhibition deficiency, hypersensitivity to amphetamine, and the loss of PV-positive cells (Piontkewitz et al., 2012; Richtand et al., 2011). Haloperidol, clozapine, or fluoxetine can also effectively prevent MIA-induced behaviors (Meyer et al., 2010b; Richtand et al., 2012).
Anti-inflammatory drugs have also been effective in treating phenotypes of MIA offspring. Different minocycline treatment regimens have produced similar results. Minocycline reduced microglia activation and density and rescued behaviors such as PPI and social interaction/preference (Mattei et al., 2014; Mattei et al., 2017; Zhu et al., 2014). It also restored neurogenesis and normalizes the expression of many microglial genes differentially expressed following MIA (Mattei et al., 2014; Mattei et al., 2017). In a two-hit model using subthreshold MIA and peripubertal stress in which minocycline was administered in the drinking water one day before and throughout the stress paradigm, microglia activation and behaviors that emerged in stressed MIA offspring were blocked with minocycline treatment (Giovanoli et al., 2016a). However, anxiety-like behavior displayed by MIA offspring not exposed to stress was not ameliorated with minocycline treatment. Ibudilast, a type IV-phosphodiesterase that suppresses the production of pro-inflammatory cytokines, prevented spine loss and reduced marble burying in MIA offspring when administered to MIA offspring through the milk of lactating dams (Coiro et al., 2015). In addition, suppression of the kynurenine pathway during puberty with celecoxib, a cyclo-oxygenase-2 inhibitor, prevented hypersensitivity to MK-801 in adult MIA offspring (Zavitsanou et al., 2014). Diets rich in n-3 polyunsaturated fatty acids, which are known to have anti-inflammatory properties (Trepanier et al., 2016) have beneficial effects on MIA-induced phenotypes when given to the mother during gestation and lactation or given to the offspring (Basil et al., 2018; Labrousse et al., 2018). Diets low in n-3 polyunsaturated fatty acids fed to the mother exacerbated LPS-induced cytokine elevations in the maternal plasma and fetal brain (Labrousse et al., 2018). These offspring exhibited spatial memory deficits while MIA offspring of mothers given a balanced diet in terms of polyunsaturated fatty acids did not have spatial memory deficits (Labrousse et al., 2018). Another group, which used Poly(I:C) to induce MIA, found that a diet rich in n-3 polyunsaturated fatty acids given to the offspring prevented MIA-induced PPI deficits and methylation changes in the hypothalamus (Basil et al., 2018; Li et al., 2015).
In addition to general anti-inflammatory drugs, some tested drugs target specific systems that are dysregulated in MIA offspring. To target the abnormalities in the GABAergic system, a benzodiazepine with partial selectivity for the α2, α3, and α5 subunits of the GABAA receptor was used. This treatment did not restore working memory or social interaction, but it did prevent MIA-induced hypersensitivity to amphetamine (Richetto et al., 2015). Treatment with the purinergic receptor inhibitor suramin normalized motor coordination and social preference, as well as the number of Purkinje cells in lobule VII of the cerebellum in MIA offspring (Naviaux et al., 2013). Antagonists for dopamine receptors D1R and D2R normalize PPI deficits in MIA offspring (Vuillermot et al., 2010). Co-administration of Poly (I:C) and parachlorophenylalanine, an inhibitor of tryptophan hydroxylase, prevented the MIA-induced reduction in serotonergic axon density (Goeden et al., 2016). Administration of 7,8-dihydroxyflavone, an agonist for the BDNF receptor tropomyosin receptor kinase B, during adolescence rescued MIA-induced impairments in PPI, novel object recognition, PV-immunoreactivity, and BDNF expression and signaling (Han et al., 2016), and normalized C1q expression (Han et al., 2017a). 7,8-dihydroxyflavone given to the mother during pregnancy and lactation also prevented cognitive deficits (Han et al., 2017b).
Supplements given to the mother during gestation can prevent the emergence of abnormalities in MIA offspring. When the mother’s diet was enriched with choline, mRNA levels of the nicotinic acetylcholine receptor alpha 7 subunit were normalized (Wu et al., 2015). Choline supplementation also restored normal marble burying, but did not prevent the PPI deficit in MIA offspring. Subcutaneous administration of the active form of vitamin D at the same time as Poly(I:C) normalized social approach and marble burying behavior in juvenile MIA offspring and amphetamine hypersensitivity in adult MIA offspring (Luan et al., 2018; Vuillermot et al., 2017). Co-administration of vitamin D also restored dorsoventral positioning of mature dopaminergic neurons in the fetal brain (Luan et al., 2018). The effect of vitamin D was not due to its anti-inflammatory properties as vitamin D did not block the elevation of cytokines in the maternal plasma and fetal brain (Vuillermot et al., 2017). Rather, vitamin D supplementation promotes expression of the transcription factor Nurr1 in mature dopaminergic neurons (Luan et al., 2018), suggesting an effect of vitamin D on epigenetic mechanisms. Using a similar approach with zinc resulted in a reduction in the number of GFAP positive cells, apoptotic cells, and TNF-α positive cells in the fetal brain (Chua et al., 2012) while dietary zinc supplementation throughout pregnancy improved novel object recognition in the offspring (Coyle et al., 2009). N-acetylcysteine in the drinking water of LPS-injected dams prevented synaptic plasticity deficits and improved working memory in MIA offspring (Lante et al., 2008). Water containing molecular hydrogen has been shown to be neuroprotective when given to either the mothers or the offspring (Imai et al., 2016; Imai et al., 2018; Nakano et al., 2015).
Dietary intervention during juvenile or adolescent stages can also prevent the MIA-induced phenotypes in the offspring. A diet containing glucoraphanin, a precursor to the antioxidant sulforaphane, rescues the deficit in PV immunoreactivity and impairments in novel object recognition in adult MIA offspring (Matsuura et al., 2018). In addition, a ketogenic diet given for 3–4 weeks increases sociability and decreases grooming behavior in male MIA offspring (Ruskin et al., 2017). D-serine in the drinking water during adolescence prevented cognitive impairments in adult MIA offspring (Fujita et al., 2016).
An interesting finding is that when pregnant dams received influenza vaccination 12 days prior to immune activation with LPS, the offspring did not exhibit many MIA-induced phenotypes including altered social behavior, anxiety- and depression-like behaviors, abnormal cortical lamination, and microglial activation (Wu et al., 2018). RNA sequencing results suggest that prenatal vaccination promotes expression of several genes involved in neuronal differentiation, which may underlie the abrogation of MIA-induced phenotypes in offspring of mothers that had been vaccinated (Wu et al., 2018).
A common theme linking many of these interventions is their potential to modulate the epigenome. Indeed, dietary supplementation with n-3 polyunsaturated fatty acids has already been shown to normalize PPI and DNA methylation in MIA offspring (Basil et al., 2018; Li et al., 2015). A number of other agents with therapeutic effects in MIA offspring have been shown elsewhere to regulate epigenetic mechanisms. For example, haloperidol, clozapine, and sulforaphane modulate DNA methylation (Dong et al., 2016; Kaufman-Szymczyk et al., 2015; Swathy and Banerjee, 2017). Haloperidol has also been shown to repress specific microRNAs that can also regulate DNA methylation (Swathy and Banerjee, 2017). Posttranslational modifications of histones can be regulated by risperidone, suramin, and sulforaphane (Kaufman-Szymczyk et al., 2015; Li et al., 2004; Villalba and Alcain, 2012). Whether these agents regulate the epigenome in MIA offspring remains an important direction for future study.
In summary, the efficacy of current antipsychotics in normalizing behaviors in MIA offspring suggests that the MIA model has predictive validity and can be used to test drugs for treating neurodevelopmental disorders for which maternal infection is a risk factor. In addition, anti-inflammatory drugs are promising candidates for new drugs. Thirdly, supplementation of a mother’s diet may help limit the adverse effects of maternal infection on neurodevelopment. Using agents to target epigenetic mechanisms is an important therapeutic strategy that remains to be addressed in the MIA model.
7. Conclusions
The developing brain is highly susceptible to environmental insults. Epidemiological data suggest that maternal infection during pregnancy increases the risk of neuropsychiatric disorders such as schizophrenia and autism in the offspring. The preclinical studies described here provide strong support for the association between prenatal immune insult and altered brain development that results in behavioral abnormalities. The mechanisms that link transient prenatal inflammation with delayed impairment of neuronal functions have begun to be investigated at the cellular and molecular levels spanning alterations in the immune molecules and cells, altered neuronal neurochemistry and function as well as epigenetic alterations. All of the major epigenetic mechanisms – histone modifications, DNA methylation, and microRNA expression – have been now shown to be associated with MIA. Moreover, we reviewed how these alterations have contributed to variations in phenotypes such as behavior through hypermethylation of DNA and altered histone methylation and acetylation; expression of components of glutamatergic, GABAergic, and serotonergic neurotransmission through hypoacetylation of histones, DNA hypermethylation, or hyperacetylation of histones, respectively; and expression of genes involved in neurodevelopment through reduced DNA methylation. Providing further support that epigenetic alterations are a key mechanism underlying MIA-induced phenotypes is the transgenerational aspect of MIA, whereby MIA induces behavioral alterations in the F1, F2, and F3 generations, suggesting an inheritable mechanism (such as epigenetics) is involved. In addition, the number of therapeutic interventions that may work through epigenetic modulation that have proven beneficial in MIA offspring strongly suggest that normalization of epigenetic mechanisms has the potential to alleviate a range of MIA-induced phenotypes. The current epigenetic studies offer a partial explanation of the mechanism involved in the production of the phenotypes observed in MIA offspring, but several questions remain. How does MIA induce a wide range of epigenetic alterations? Do these changes directly cause the diverse brain and behavioral phenotypes observed in MIA offspring? Can (and should) we effectively target specific epigenetic mechanisms for therapeutic purposes? In addition to these epigenetic-centered questions, in the future, impairments in specific brain circuits underlying specific behavioral impairments will be important to define. In addition, beyond the classical developmental disorders it is becoming increasingly evident that early insults could also be linked to neurodegenerative disorders such as Alzheimer’s disease, making this a question relevant to a larger population. Although studies investigating the interaction between genetic susceptibility and neurodevelopmental disorders have begun to emerge, similar studies with genetic risk factors for neurodegenerative disorders are still missing and will be a fruitful area of investigation. Finally, the animal models of MIA will continue to provide an important platform for preclinical testing of therapeutic reagents.
Figure 2: Brain anatomic changes in MIA offspring.
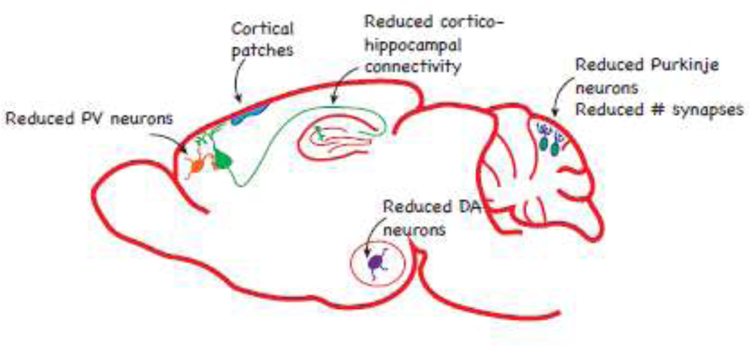
Brain alterations such as reduced brain volume (Fatemi et al., 2008; Patrich et al., 2016b; Piontkewitz et al., 2011; Short et al., 2010) is mediated by loss of neurons in several brain regions (Fatemi et al., 1999; Meyer et al., 2008c; Naviaux et al., 2013; Shi et al., 2009; Wischhof et al., 2015b; Zhang and van Praag, 2015). MIA also results in formation of disorganized cortical patches (Choi et al., 2016; Shin Yim et al., 2017).
Highlights:
Exposure to maternal immune activation (MIA) during specific windows of prenatal development can alter the normal trajectory of brain development.
MIA is associated with increased risk factor for various neurodevelopmental and neurodegenerative disorders.
MIA acts synergistically with genetic and environmental factors for neurodevelopmental disorders.
MIA results in altered immunological milieu for the developing brain.
MIA results in diverse alterations in neuron number, structure and function in different brain regions.
Global and specific epigenetic mechanisms have a potential to explain the diverse changes associated with MIA.
Abbreviations
- ASD
autism spectrum disorder
- GFAP
glial fibrillary acidic protein
- GSK3β
synthase kinase 3 beta
- IL
interleukin
- LPS
lipopolysaccharide
- MIA
maternal immune activation
- mEPSC
miniature excitatory postsynaptic current
- mIPSC
miniature inhibitory postsynaptic current
- mPFC
medial prefrontal cortex
- Poly (I:C)
polycytidylic acid
- PPI
prepulse inhibition
- PV
parvalbumin
- SZ
schizophrenia
- TNF-α
tumor necrosis factor-α
- USV
ultrasonic vocalizations
- VTA
ventral tegmental area
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Bibliography
- Aavani T, Rana SA, Hawkes R, Pittman QJ, 2015. Maternal immune activation produces cerebellar hyperplasia and alterations in motor and social behaviors in male and female mice. Cerebellum 14, 491–505. [DOI] [PubMed] [Google Scholar]
- Abazyan B, Nomura J, Kannan G, Ishizuka K, Tamashiro KL, Nucifora F, Pogorelov V, Ladenheim B, Yang C, Krasnova IN, Cadet JL, Pardo C, Mori S, Kamiya A, Vogel MW, Sawa A, Ross CA, Pletnikov MV, 2010. Prenatal interaction of mutant DISC1 and immune activation produces adult psychopathology. Biol Psychiatry 68, 1172–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allan AM, Liang X, Luo Y, Pak C, Li X, Szulwach KE, Chen D, Jin P, Zhao X, 2008. The loss of methyl-CpG binding protein 1 leads to autism-like behavioral deficits. Hum Mol Genet 17, 2047–2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonson AM, Balakrishnan B, Radlowski EC, Petr G, Johnson RW, 2018. Altered Hippocampal Gene Expression and Morphology in Fetal Piglets following Maternal Respiratory Viral Infection. Dev Neurosci 40, 104–119. [DOI] [PubMed] [Google Scholar]
- Antonson AM, Radlowski EC, Lawson MA, Rytych JL, Johnson RW, 2017. Maternal viral infection during pregnancy elicits anti-social behavior in neonatal piglet offspring independent of postnatal microglial cell activation. Brain Behav Immun 59, 300–312. [DOI] [PubMed] [Google Scholar]
- Arsenault D, St-Amour I, Cisbani G, Rousseau LS, Cicchetti F, 2014. The different effects of LPS and poly I:C prenatal immune challenges on the behavior, development and inflammatory responses in pregnant mice and their offspring. Brain Behav Immun 38, 77–90. [DOI] [PubMed] [Google Scholar]
- Ashdown H, Dumont Y, Ng M, Poole S, Boksa P, Luheshi GN, 2006. The role of cytokines in mediating effects of prenatal infection on the fetus: implications for schizophrenia. Mol Psychiatry 11, 47–55. [DOI] [PubMed] [Google Scholar]
- Atladottir HO, Thorsen P, Ostergaard L, Schendel DE, Lemcke S, Abdallah M, Parner ET, 2010. Maternal infection requiring hospitalization during pregnancy and autism spectrum disorders. J Autism Dev Disord 40, 1423–1430. [DOI] [PubMed] [Google Scholar]
- Babri S, Doosti MH, Salari AA, 2014. Strain-dependent effects of prenatal maternal immune activation on anxiety- and depression-like behaviors in offspring. Brain Behav Immun 37, 164–176. [DOI] [PubMed] [Google Scholar]
- Baharnoori M, Bhardwaj SK, Srivastava LK, 2012. Neonatal behavioral changes in rats with gestational exposure to lipopolysaccharide: a prenatal infection model for developmental neuropsychiatric disorders. Schizophr Bull 38, 444–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baharnoori M, Bhardwaj SK, Srivastava LK, 2013. Effect of maternal lipopolysaccharide administration on the development of dopaminergic receptors and transporter in the rat offspring. PLoS One 8, e54439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baharnoori M, Brake WG, Srivastava LK, 2009. Prenatal immune challenge induces developmental changes in the morphology of pyramidal neurons of the prefrontal cortex and hippocampus in rats. Schizophr Res 107, 99–109. [DOI] [PubMed] [Google Scholar]
- Bale TL, 2015. Epigenetic and transgenerational reprogramming of brain development. Nat Rev Neurosci 16, 332–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballendine SA, Greba Q, Dawicki W, Zhang X, Gordon JR, Howland JG, 2015. Behavioral alterations in rat offspring following maternal immune activation and ELR-CXC chemokine receptor antagonism during pregnancy: implications for neurodevelopmental psychiatric disorders. Prog Neuropsychopharmacol Biol Psychiatry 57, 155–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basil P, Li Q, Dempster EL, Mill J, Sham PC, Wong CC, McAlonan GM, 2014. Prenatal maternal immune activation causes epigenetic differences in adolescent mouse brain. Transl Psychiatry 4, e434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basil P, Li Q, Gui H, Hui TCK, Ling VHM, Wong CCY, Mill J, McAlonan GM, Sham PC, 2018. Prenatal immune activation alters the adult neural epigenome but can be partly stabilised by a n-3 polyunsaturated fatty acid diet. Transl Psychiatry 8, 125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batinic B, Santrac A, Divovic B, Timic T, Stankovic T, Obradovic A, Joksimovic S, Savic MM, 2016. Lipopolysaccharide exposure during late embryogenesis results in diminished locomotor activity and amphetamine response in females and spatial cognition impairment in males in adult, but not adolescent rat offspring. Behav Brain Res 299, 72–80. [DOI] [PubMed] [Google Scholar]
- Bauman MD, Iosif AM, Smith SE, Bregere C, Amaral DG, Patterson PH, 2014. Activation of the maternal immune system during pregnancy alters behavioral development of rhesus monkey offspring. Biol Psychiatry 75, 332–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger S, Ronovsky M, Horvath O, Berger A, Pollak DD, 2018. Impact of maternal immune activation on maternal care behavior, offspring emotionality and intergenerational transmission in C3H/He mice. Brain Behav Immun 70, 131–140. [DOI] [PubMed] [Google Scholar]
- Bilimoria PM, Stevens B, 2015. Microglia function during brain development: New insights from animal models. Brain Res 1617, 7–17. [DOI] [PubMed] [Google Scholar]
- Bitanihirwe BK, Peleg-Raibstein D, Mouttet F, Feldon J, Meyer U, 2010a. Late prenatal immune activation in mice leads to behavioral and neurochemical abnormalities relevant to the negative symptoms of schizophrenia. Neuropsychopharmacology 35, 2462–2478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bitanihirwe BK, Weber L, Feldon J, Meyer U, 2010b. Cognitive impairment following prenatal immune challenge in mice correlates with prefrontal cortical AKT1 deficiency. Int J Neuropsychopharmacol 13, 981–996. [DOI] [PubMed] [Google Scholar]
- Boksa P, Zhang Y, Nouel D, Wong A, Wong TP, 2016. Early Development of Parvalbumin-, Somatostatin-, and Cholecystokinin-Expressing Neurons in Rat Brain following Prenatal Immune Activation and Maternal Iron Deficiency. Dev Neurosci 38, 342–353. [DOI] [PubMed] [Google Scholar]
- Brown A, Derkits E, 2010. Prenatal Infection and Scizophrenia: A Review of Epidemiologic and Translational Studies. Am J Psychiatry 167, 261–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown AS, 2012. Epidemiologic studies of exposure to prenatal infection and risk of schizophrenia and autism. Dev Neurobiol 72, 1272–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bundo M, Toyoshima M, Okada Y, Akamatsu W, Ueda J, Nemoto-Miyauchi T, Sunaga F, Toritsuka M, Ikawa D, Kakita A, Kato M, Kasai K, Kishimoto T, Nawa H, Okano H, Yoshikawa T, Kato T, Iwamoto K, 2014. Increased l1 retrotransposition in the neuronal genome in schizophrenia. Neuron 81, 306–313. [DOI] [PubMed] [Google Scholar]
- Buschert J, Sakalem ME, Saffari R, Hohoff C, Rothermundt M, Arolt V, Zhang W, Ambree O, 2016. Prenatal immune activation in mice blocks the effects of environmental enrichment on exploratory behavior and microglia density. Prog Neuropsychopharmacol Biol Psychiatry 67, 10–20. [DOI] [PubMed] [Google Scholar]
- Cai Z, Pan ZL, Pang Y, Evans OB, Rhodes PG, 2000. Cytokine induction in fetal rat brains and brain injury in neonatal rats after maternal lipopolysaccharide administration. Pediatr Res 47, 64–72. [DOI] [PubMed] [Google Scholar]
- Canetta S, Bolkan S, Padilla-Coreano N, Song LJ, Sahn R, Harrison NL, Gordon JA, Brown A, Kellendonk C, 2016. Maternal immune activation leads to selective functional deficits in offspring parvalbumin interneurons. Mol Psychiatry 21, 956–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan WY, Kohsaka S, Rezaie P, 2007. The origin and cell lineage of microglia: new concepts. Brain Res Rev 53, 344–354. [DOI] [PubMed] [Google Scholar]
- Cheray M, Joseph B, 2018. Epigenetics Control Microglia Plasticity. Front Cell Neurosci 12, 243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi GB, Yim YS, Wong H, Kim S, Kim H, Kim SV, Hoeffer CA, Littman DR, Huh JR, 2016. The maternal interleukin-17a pathway in mice promotes autism-like phenotypes in offspring. Science 351, 933–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chua JS, Cowley CJ, Manavis J, Rofe AM, Coyle P, 2012. Prenatal exposure to lipopolysaccharide results in neurodevelopmental damage that is ameliorated by zinc in mice. Brain Behav Immun 26, 326–336. [DOI] [PubMed] [Google Scholar]
- Clarke MC, Tanskanen A, Huttunen M, Whittaker JC, Cannon M, 2009. Evidence for an interaction between familial liability and prenatal exposure to infection in the causation of schizophrenia. Am J Psychiatry 166, 1025–1030. [DOI] [PubMed] [Google Scholar]
- Coiro P, Padmashri R, Suresh A, Spartz E, Pendyala G, Chou S, Jung Y, Meays B, Roy S, Gautam N, Alnouti Y, Li M, Dunaevsky A, 2015. Impaired synaptic development in a maternal immune activation mouse model of neurodevelopmental disorders. Brain Behav Immun 50, 249–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connor CM, Dincer A, Straubhaar J, Galler JR, Houston IB, Akbarian S, 2012. Maternal immune activation alters behavior in adult offspring, with subtle changes in the cortical transcriptome and epigenome. Schizophr Res 140, 175–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbin JG, Butt SJ, 2011. Developmental mechanisms for the generation of telencephalic interneurons. Dev Neurobiol 71, 710–732. [DOI] [PubMed] [Google Scholar]
- Corradini I, Focchi E, Rasile M, Morini R, Desiato G, Tomasoni R, Lizier M, Ghirardini E, Fesce R, Morone D, Barajon I, Antonucci F, Pozzi D, Matteoli M, 2018. Maternal Immune Activation Delays Excitatory-to-Inhibitory Gamma-Aminobutyric Acid Switch in Offspring. Biol Psychiatry 83, 680–691. [DOI] [PubMed] [Google Scholar]
- Cosenza-Nashat M, Zhao ML, Suh HS, Morgan J, Natividad R, Morgello S, Lee SC, 2009. Expression of the translocator protein of 18 kDa by microglia, macrophages and astrocytes based on immunohistochemical localization in abnormal human brain. Neuropathol Appl Neurobiol 35, 306–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyle P, Tran N, Fung JN, Summers BL, Rofe AM, 2009. Maternal dietary zinc supplementation prevents aberrant behaviour in an object recognition task in mice offspring exposed to LPS in early pregnancy. Behav Brain Res 197, 210–218. [DOI] [PubMed] [Google Scholar]
- Crum WR, Sawiak SJ, Chege W, Cooper JD, Williams SCR, Vernon AC, 2017. Evolution of structural abnormalities in the rat brain following in utero exposure to maternal immune activation: A longitudinal in vivo MRI study. Brain Behav Immun 63, 50–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Silveira VT, Medeiros DC, Ropke J, Guidine PA, Rezende GH, Moraes MF, Mendes EM, Macedo D, Moreira FA, de Oliveira AC, 2017. Effects of early or late prenatal immune activation in mice on behavioral and neuroanatomical abnormalities relevant to schizophrenia in the adulthood. Int J Dev Neurosci 58, 1–8. [DOI] [PubMed] [Google Scholar]
- Dantzer R, 2001. Cytokine-induced sickness behavior: mechanisms and implications. Annals of the New York Academy of Sciences 933, 222–234. [DOI] [PubMed] [Google Scholar]
- de Souza DF, Wartchow KM, Lunardi PS, Brolese G, Tortorelli LS, Batassini C, Biasibetti R, Goncalves CA, 2015. Changes in Astroglial Markers in a Maternal Immune Activation Model of Schizophrenia in Wistar Rats are Dependent on Sex. Front Cell Neurosci 9, 489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deane AR, Millar J, Bilkey DK, Ward RD, 2017. Maternal immune activation in rats produces temporal perception impairments in adult offspring analogous to those observed in schizophrenia. PLoS One 12, e0187719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deverman BE, Patterson PH, 2009. Cytokines and CNS development. Neuron 64, 61–78. [DOI] [PubMed] [Google Scholar]
- Dickerson DD, Restieaux AM, Bilkey DK, 2012. Clozapine administration ameliorates disrupted long-range synchrony in a neurodevelopmental animal model of schizophrenia. Schizophr Res 135, 112–115. [DOI] [PubMed] [Google Scholar]
- Dickerson DD, Wolff AR, Bilkey DK, 2010. Abnormal long-range neural synchrony in a maternal immune activation animal model of schizophrenia. J Neurosci 30, 12424–12431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donegan JJ, Boley AM, Lodge DJ, 2018. Embryonic stem cell transplants as a therapeutic strategy in a rodent model of autism. Neuropsychopharmacology [DOI] [PMC free article] [PubMed]
- Dong E, Tueting P, Matrisciano F, Grayson DR, Guidotti A, 2016. Behavioral and molecular neuroepigenetic alterations in prenatally stressed mice: relevance for the study of chromatin remodeling properties of antipsychotic drugs. Transl Psychiatry 6, e711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchatel RJ, Jobling P, Graham BA, Harms LR, Michie PT, Hodgson DM, Tooney PA, 2016. Increased white matter neuron density in a rat model of maternal immune activation - Implications for schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry 65, 118–126. [DOI] [PubMed] [Google Scholar]
- Eberharter A, Becker PB, 2002. Histone acetylation: a switch between repressive and permissive chromatin. Second in review series on chromatin dynamics. EMBO reports 3, 224–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehninger D, Sano Y, de Vries PJ, Dies K, Franz D, Geschwind DH, Kaur M, Lee YS, Li W, Lowe JK, Nakagawa JA, Sahin M, Smith K, Whittemore V, Silva AJ, 2012. Gestational immune activation and Tsc2 haploinsufficiency cooperate to disrupt fetal survival and may perturb social behavior in adult mice. Mol Psychiatry 17, 62–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmer BM, Estes ML, Barrow SL, McAllister AK, 2013. MHCI requires MEF2 transcription factors to negatively regulate synapse density during development and in disease. J Neurosci 33, 13791–13804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escobar M, Crouzin N, Cavalier M, Quentin J, Roussel J, Lante F, Batista-Novais AR, Cohen-Solal C, De Jesus Ferreira MC, Guiramand J, Barbanel G, Vignes M, 2011. Early, time-dependent disturbances of hippocampal synaptic transmission and plasticity after in utero immune challenge. Biol Psychiatry 70, 992–999. [DOI] [PubMed] [Google Scholar]
- Estes ML, McAllister AK, 2016. Maternal immune activation: Implications for neuropsychiatric disorders. Science 353, 772–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatemi SH, Emamian ES, Kist D, Sidwell RW, Nakajima K, Akhter P, Shier A, Sheikh S, Bailey K, 1999. Defective corticogenesis and reduction in Reelin immunoreactivity in cortex and hippocampus of prenatally infected neonatal mice. Mol Psychiatry 4, 145–154. [DOI] [PubMed] [Google Scholar]
- Fatemi SH, Pearce DA, Brooks AI, Sidwell RW, 2005. Prenatal viral infection in mouse causes differential expression of genes in brains of mouse progeny: a potential animal model for schizophrenia and autism. Synapse 57, 91–99. [DOI] [PubMed] [Google Scholar]
- Fatemi SH, Reutiman TJ, Folsom TD, Huang H, Oishi K, Mori S, Smee DF, Pearce DA, Winter C, Sohr R, Juckel G, 2008. Maternal infection leads to abnormal gene regulation and brain atrophy in mouse offspring: implications for genesis of neurodevelopmental disorders. Schizophr Res 99, 56–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez de Cossio L, Guzman A, van der Veldt S, Luheshi GN, 2017. Prenatal infection leads to ASD-like behavior and altered synaptic pruning in the mouse offspring. Brain Behav Immun 63, 88–98. [DOI] [PubMed] [Google Scholar]
- Fortier ME, Luheshi GN, Boksa P, 2007. Effects of prenatal infection on prepulse inhibition in the rat depend on the nature of the infectious agent and the stage of pregnancy. Behav Brain Res 181, 270–277. [DOI] [PubMed] [Google Scholar]
- Fujita Y, Ishima T, Hashimoto K, 2016. Supplementation with D-serine prevents the onset of cognitive deficits in adult offspring after maternal immune activation. Sci Rep 6, 37261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garay PA, Hsiao EY, Patterson PH, McAllister AK, 2013. Maternal immune activation causes age- and region-specific changes in brain cytokines in offspring throughout development. Brain Behav Immun 31, 54–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garay PA, McAllister AK, 2010. Novel roles for immune molecules in neural development: implications for neurodevelopmental disorders. Front Synaptic Neurosci 2, 136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garbett KA, Hsiao EY, Kalman S, Patterson PH, Mirnics K, 2012. Effects of maternal immune activation on gene expression patterns in the fetal brain. Transl Psychiatry 2, e98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giovanoli S, Engler H, Engler A, Richetto J, Feldon J, Riva MA, Schedlowski M, Meyer U, 2016a. Preventive effects of minocycline in a neurodevelopmental two-hit model with relevance to schizophrenia. Transl Psychiatry 6, e772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giovanoli S, Engler H, Engler A, Richetto J, Voget M, Willi R, Winter C, Riva MA, Mortensen PB, Feldon J, Schedlowski M, Meyer U, 2013. Stress in puberty unmasks latent neuropathological consequences of prenatal immune activation in mice. Science 339, 1095–1099. [DOI] [PubMed] [Google Scholar]
- Giovanoli S, Notter T, Richetto J, Labouesse MA, Vuillermot S, Riva MA, Meyer U, 2015. Late prenatal immune activation causes hippocampal deficits in the absence of persistent inflammation across aging. J Neuroinflammation 12, 221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giovanoli S, Weber-Stadlbauer U, Schedlowski M, Meyer U, Engler H, 2016b. Prenatal immune activation causes hippocampal synaptic deficits in the absence of overt microglia anomalies. Brain Behav Immun 55, 25–38. [DOI] [PubMed] [Google Scholar]
- Goeden N, Velasquez J, Arnold KA, Chan Y, Lund BT, Anderson GM, Bonnin A, 2016. Maternal Inflammation Disrupts Fetal Neurodevelopment via Increased Placental Output of Serotonin to the Fetal Brain. J Neurosci 36, 6041–6049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan JS, Xie H, Ding X, 2015. The role of epigenetic regulation in learning and memory. Exp Neurol 268, 30–36. [DOI] [PubMed] [Google Scholar]
- Hadar R, Dong L, Del-Valle-Anton L, Guneykaya D, Voget M, Edemann-Callesen H, Schweibold R, Djodari-Irani A, Goetz T, Ewing S, Kettenmann H, Wolf SA, Winter C, 2017. Deep brain stimulation during early adolescence prevents microglial alterations in a model of maternal immune activation. Brain Behav Immun 63, 71–80. [DOI] [PubMed] [Google Scholar]
- Hagberg H, Gressens P, Mallard C, 2012. Inflammation during fetal and neonatal life: implications for neurologic and neuropsychiatric disease in children and adults. Ann Neurol 71, 444–457. [DOI] [PubMed] [Google Scholar]
- Han M, Zhang JC, Hashimoto K, 2017a. Increased Levels of C1q in the Prefrontal Cortex of Adult Offspring after Maternal Immune Activation: Prevention by 7,8-Dihydroxyflavone. Clin Psychopharmacol Neurosci 15, 64–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han M, Zhang JC, Huang XF, Hashimoto K, 2017b. Intake of 7,8-dihydroxyflavone from pregnancy to weaning prevents cognitive deficits in adult offspring after maternal immune activation. Eur Arch Psychiatry Clin Neurosci 267, 479–483. [DOI] [PubMed] [Google Scholar]
- Han M, Zhang JC, Yao W, Yang C, Ishima T, Ren Q, Ma M, Dong C, Huang XF, Hashimoto K, 2016. Intake of 7,8-Dihydroxyflavone During Juvenile and Adolescent Stages Prevents Onset of Psychosis in Adult Offspring After Maternal Immune Activation. Sci Rep 6, 36087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao LY, Hao XQ, Li SH, Li XH, 2010. Prenatal exposure to lipopolysaccharide results in cognitive deficits in age-increasing offspring rats. Neuroscience 166, 763–770. [DOI] [PubMed] [Google Scholar]
- Harvey L, Boksa P, 2012. A stereological comparison of GAD67 and reelin expression in the hippocampal stratum oriens of offspring from two mouse models of maternal inflammation during pregnancy. Neuropharmacology 62, 1767–1776. [DOI] [PubMed] [Google Scholar]
- Hava G, Vered L, Yael M, Mordechai H, Mahoud H, 2006. Alterations in behavior in adult offspring mice following maternal inflammation during pregnancy. Dev Psychobiol 48, 162–168. [DOI] [PubMed] [Google Scholar]
- Hodge DR, Cho E, Copeland TD, Guszczynski T, Yang E, Seth AK, Farrar WL, 2007. IL-6 enhances the nuclear translocation of DNA cytosine-5-methyltransferase 1 (DNMT1) via phosphorylation of the nuclear localization sequence by the AKT kinase. Cancer genomics & proteomics 4, 387–398. [PubMed] [Google Scholar]
- Hodge DR, Xiao W, Clausen PA, Heidecker G, Szyf M, Farrar WL, 2001. Interleukin-6 regulation of the human DNA methyltransferase (HDNMT) gene in human erythroleukemia cells. J Biol Chem 276, 39508–39511. [DOI] [PubMed] [Google Scholar]
- Hollins SL, Zavitsanou K, Walker FR, Cairns MJ, 2014. Alteration of imprinted Dlk1-Dio3 miRNA cluster expression in the entorhinal cortex induced by maternal immune activation and adolescent cannabinoid exposure. Transl Psychiatry 4, e452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao EY, McBride SW, Chow J, Mazmanian SK, Patterson PH, 2012. Modeling an autism risk factor in mice leads to permanent immune dysregulation. Proc Natl Acad Sci U S A 109, 12776–12781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao EY, Patterson PH, 2011. Activation of the maternal immune system induces endocrine changes in the placenta via IL-6. Brain Behav Immun 25, 604–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsueh PT, Lin HH, Wang HH, Liu CL, Ni WF, Liu JK, Chang HH, Sun DS, Chen YS, Chen YL, 2018. Immune imbalance of global gene expression, and cytokine, chemokine and selectin levels in the brains of offspring with social deficits via maternal immune activation. Genes, brain, and behavior 17, e12479. [DOI] [PubMed] [Google Scholar]
- Imai K, Kotani T, Tsuda H, Mano Y, Nakano T, Ushida T, Li H, Miki R, Sumigama S, Iwase A, Hirakawa A, Ohno K, Toyokuni S, Takeuchi H, Mizuno T, Suzumura A, Kikkawa F, 2016. Neuroprotective potential of molecular hydrogen against perinatal brain injury via suppression of activated microglia. Free Radic Biol Med 91, 154–163. [DOI] [PubMed] [Google Scholar]
- Imai K, Kotani T, Tsuda H, Nakano T, Ushida T, Iwase A, Nagai T, Toyokuni S, Suzumura A, Kikkawa F, 2018. Administration of molecular hydrogen during pregnancy improves behavioral abnormalities of offspring in a maternal immune activation model. Sci Rep 8, 9221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inestrosa NC, Varela-Nallar L, 2015. Wnt signalling in neuronal differentiation and development. Cell Tissue Res 359, 215–223. [DOI] [PubMed] [Google Scholar]
- Ito HT, Smith SE, Hsiao E, Patterson PH, 2010. Maternal immune activation alters nonspatial information processing in the hippocampus of the adult offspring. Brain Behav Immun 24, 930–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang HS, Shin WJ, Lee JE, Do JT, 2017. CpG and Non-CpG Methylation in Epigenetic Gene Regulation and Brain Function. Genes (Basel) 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong HK, Ji K, Min K, Joe EH, 2013. Brain inflammation and microglia: facts and misconceptions. Exp Neurobiol 22, 59–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang HY, Xu LL, Shao L, Xia RM, Yu ZH, Ling ZX, Yang F, Deng M, Ruan B, 2016. Maternal infection during pregnancy and risk of autism spectrum disorders: A systematic review and meta-analysis. Brain Behav Immun 58, 165–172. [DOI] [PubMed] [Google Scholar]
- Jones PA, 2012. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet 13, 484–492. [DOI] [PubMed] [Google Scholar]
- Juckel G, Manitz MP, Brune M, Friebe A, Heneka MT, Wolf RJ, 2011. Microglial activation in a neuroinflammational animal model of schizophrenia--a pilot study. Schizophr Res 131, 96–100. [DOI] [PubMed] [Google Scholar]
- Kaufman-Szymczyk A, Majewski G, Lubecka-Pietruszewska K, Fabianowska-Majewska K, 2015. The Role of Sulforaphane in Epigenetic Mechanisms, Including Interdependence between Histone Modification and DNA Methylation. International journal of molecular sciences 16, 29732–29743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessler MS, Bosch OJ, Bunck M, Landgraf R, Neumann ID, 2011. Maternal care differs in mice bred for high vs. low trait anxiety: impact of brain vasopressin and cross-fostering. Soc Neurosci 6, 156–168. [DOI] [PubMed] [Google Scholar]
- Kim S, Kim H, Yim YS, Ha S, Atarashi K, Tan TG, Longman RS, Honda K, Littman DR, Choi GB, Huh JR, 2017. Maternal gut bacteria promote neurodevelopmental abnormalities in mouse offspring. Nature 549, 528–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirsten TB, Bernardi MM, 2017. Prenatal lipopolysaccharide induces hypothalamic dopaminergic hypoactivity and autistic-like behaviors: Repetitive self-grooming and stereotypies. Behav Brain Res 331, 25–29. [DOI] [PubMed] [Google Scholar]
- Kirsten TB, Taricano M, Florio JC, Palermo-Neto J, Bernardi MM, 2010. Prenatal lipopolysaccharide reduces motor activity after an immune challenge in adult male offspring. Behav Brain Res 211, 77–82. [DOI] [PubMed] [Google Scholar]
- Knuesel I, Chicha L, Britschgi M, Schobel SA, Bodmer M, Hellings JA, Toovey S, Prinssen EP, 2014. Maternal immune activation and abnormal brain development across CNS disorders. Nat Rev Neurol 10, 643–660. [DOI] [PubMed] [Google Scholar]
- Konefal SC, Stellwagen D, 2017. Tumour necrosis factor-mediated homeostatic synaptic plasticity in behavioural models: testing a role in maternal immune activation. Philos Trans R Soc Lond B Biol Sci 372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kundakovic M, Jaric I, 2017. The Epigenetic Link between Prenatal Adverse Environments and Neurodevelopmental Disorders. Genes (Basel) 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labouesse MA, Dong E, Grayson DR, Guidotti A, Meyer U, 2015. Maternal immune activation induces GAD1 and GAD2 promoter remodeling in the offspring prefrontal cortex. Epigenetics 10, 1143–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labrousse VF, Leyrolle Q, Amadieu C, Aubert A, Sere A, Coutureau E, Gregoire S, Bretillon L, Pallet V, Gressens P, Joffre C, Nadjar A, Laye S, 2018. Dietary omega-3 deficiency exacerbates inflammation and reveals spatial memory deficits in mice exposed to lipopolysaccharide during gestation. Brain Behav Immun 73, 427–440. [DOI] [PubMed] [Google Scholar]
- Lante F, Meunier J, Guiramand J, De Jesus Ferreira MC, Cambonie G, Aimar R, Cohen-Solal C, Maurice T, Vignes M, Barbanel G, 2008. Late N-acetylcysteine treatment prevents the deficits induced in the offspring of dams exposed to an immune stress during gestation. Hippocampus 18, 602–609. [DOI] [PubMed] [Google Scholar]
- Larson SJ, Dunn AJ, 2001. Behavioral effects of cytokines. Brain Behav Immun 15, 371–387. [DOI] [PubMed] [Google Scholar]
- Laskaris LE, Di Biase MA, Everall I, Chana G, Christopoulos A, Skafidas E, Cropley VL, Pantelis C, 2016. Microglial activation and progressive brain changes in schizophrenia. British journal of pharmacology 173, 666–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leger M, Quiedeville A, Bouet V, Haelewyn B, Boulouard M, Schumann-Bard P, Freret T, 2013. Object recognition test in mice. Nat Protoc 8, 2531–2537. [DOI] [PubMed] [Google Scholar]
- Li J, Guo Y, Schroeder FA, Youngs RM, Schmidt TW, Ferris C, Konradi C, Akbarian S, 2004. Dopamine D2-like antagonists induce chromatin remodeling in striatal neurons through cyclic AMP-protein kinase A and NMDA receptor signaling. J Neurochem 90, 1117–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Leung YO, Zhou I, Ho LC, Kong W, Basil P, Wei R, Lam S, Zhang X, Law AC, Chua SE, Sham PC, Wu EX, McAlonan GM, 2015. Dietary supplementation with n-3 fatty acids from weaning limits brain biochemistry and behavioural changes elicited by prenatal exposure to maternal inflammation in the mouse model. Transl Psychiatry 5, e641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li WY, Chang YC, Lee LJ, Lee LJ, 2014. Prenatal infection affects the neuronal architecture and cognitive function in adult mice. Dev Neurosci 36, 359–370. [DOI] [PubMed] [Google Scholar]
- Li Y, Missig G, Finger BC, Landino SM, Alexander AJ, Mokler EL, Robbins JO, Manasian Y, Kim W, Kim KS, McDougle CJ, Carlezon WA Jr., Bolshakov VY, 2018. Maternal and Early Postnatal Immune Activation Produce Dissociable Effects on Neurotransmission in mPFC-Amygdala Circuits. J Neurosci 38, 3358–3372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipina TV, Zai C, Hlousek D, Roder JC, Wong AH, 2013. Maternal immune activation during gestation interacts with Disc1 point mutation to exacerbate schizophrenia-related behaviors in mice. J Neurosci 33, 7654–7666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombardo MV, Moon HM, Su J, Palmer TD, Courchesne E, Pramparo T, 2018. Maternal immune activation dysregulation of the fetal brain transcriptome and relevance to the pathophysiology of autism spectrum disorder. Mol Psychiatry 23, 1001–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luan W, Hammond LA, Vuillermot S, Meyer U, Eyles DW, 2018. Maternal Vitamin D Prevents Abnormal Dopaminergic Development and Function in a Mouse Model of Prenatal Immune Activation. Sci Rep 8, 9741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luoni A, Richetto J, Longo L, Riva MA, 2017. Chronic lurasidone treatment normalizes GABAergic marker alterations in the dorsal hippocampus of mice exposed to prenatal immune activation. Eur Neuropsychopharmacol 27, 170–179. [DOI] [PubMed] [Google Scholar]
- MacDowell KS, Munarriz-Cuezva E, Caso JR, Madrigal JL, Zabala A, Meana JJ, Garcia-Bueno B, Leza JC, 2017. Paliperidone reverts Toll-like receptor 3 signaling pathway activation and cognitive deficits in a maternal immune activation mouse model of schizophrenia. Neuropharmacology 116, 196–207. [DOI] [PubMed] [Google Scholar]
- Machado CJ, Whitaker AM, Smith SE, Patterson PH, Bauman MD, 2015. Maternal immune activation in nonhuman primates alters social attention in juvenile offspring. Biol Psychiatry 77, 823–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malkova NV, Yu CZ, Hsiao EY, Moore MJ, Patterson PH, 2012. Maternal immune activation yields offspring displaying mouse versions of the three core symptoms of autism. Brain Behav Immun 26, 607–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuura A, Ishima T, Fujita Y, Iwayama Y, Hasegawa S, Kawahara-Miki R, Maekawa M, Toyoshima M, Ushida Y, Suganuma H, Kida S, Yoshikawa T, Iyo M, Hashimoto K, 2018. Dietary glucoraphanin prevents the onset of psychosis in the adult offspring after maternal immune activation. Sci Rep 8, 2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattei D, Djodari-Irani A, Hadar R, Pelz A, de Cossio LF, Goetz T, Matyash M, Kettenmann H, Winter C, Wolf SA, 2014. Minocycline rescues decrease in neurogenesis, increase in microglia cytokines and deficits in sensorimotor gating in an animal model of schizophrenia. Brain Behav Immun 38, 175–184. [DOI] [PubMed] [Google Scholar]
- Mattei D, Ivanov A, Ferrai C, Jordan P, Guneykaya D, Buonfiglioli A, Schaafsma W, Przanowski P, Deuther-Conrad W, Brust P, Hesse S, Patt M, Sabri O, Ross TL, Eggen BJL, Boddeke E, Kaminska B, Beule D, Pombo A, Kettenmann H, Wolf SA, 2017. Maternal immune activation results in complex microglial transcriptome signature in the adult offspring that is reversed by minocycline treatment. Transl Psychiatry 7, e1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meehan C, Harms L, Frost JD, Barreto R, Todd J, Schall U, Shannon Weickert C, Zavitsanou K, Michie PT, Hodgson DM, 2017. Effects of immune activation during early or late gestation on schizophrenia-related behaviour in adult rat offspring. Brain Behav Immun 63, 8–20. [DOI] [PubMed] [Google Scholar]
- Mehler MF, 2008. Epigenetic principles and mechanisms underlying nervous system functions in health and disease. Prog Neurobiol 86, 305–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer U, 2014. Prenatal poly(i:C) exposure and other developmental immune activation models in rodent systems. Biol Psychiatry 75, 307–315. [DOI] [PubMed] [Google Scholar]
- Meyer U, Feldon J, 2012. To poly(I:C) or not to poly(I:C): advancing preclinical schizophrenia research through the use of prenatal immune activation models. Neuropharmacology 62, 1308–1321. [DOI] [PubMed] [Google Scholar]
- Meyer U, Feldon J, Schedlowski M, Yee BK, 2005. Towards an immuno-precipitated neurodevelopmental animal model of schizophrenia. Neurosci Biobehav Rev 29, 913–947. [DOI] [PubMed] [Google Scholar]
- Meyer U, Feldon J, Schedlowski M, Yee BK, 2006a. Immunological stress at the maternal-foetal interface: a link between neurodevelopment and adult psychopathology. Brain Behav Immun 20, 378–388. [DOI] [PubMed] [Google Scholar]
- Meyer U, Knuesel I, Nyffeler M, Feldon J, 2010a. Chronic clozapine treatment improves prenatal infection-induced working memory deficits without influencing adult hippocampal neurogenesis. Psychopharmacology (Berl) 208, 531–543. [DOI] [PubMed] [Google Scholar]
- Meyer U, Murray PJ, Urwyler A, Yee BK, Schedlowski M, Feldon J, 2008a. Adult behavioral and pharmacological dysfunctions following disruption of the fetal brain balance between pro-inflammatory and IL-10-mediated anti-inflammatory signaling. Mol Psychiatry 13, 208–221. [DOI] [PubMed] [Google Scholar]
- Meyer U, Nyffeler M, Engler A, Urwyler A, Schedlowski M, Knuesel I, Yee BK, Feldon J, 2006b. The time of prenatal immune challenge determines the specificity of inflammation-mediated brain and behavioral pathology. J Neurosci 26, 4752–4762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer U, Nyffeler M, Schwendener S, Knuesel I, Yee BK, Feldon J, 2008b. Relative prenatal and postnatal maternal contributions to schizophrenia-related neurochemical dysfunction after in utero immune challenge. Neuropsychopharmacology 33, 441–456. [DOI] [PubMed] [Google Scholar]
- Meyer U, Nyffeler M, Yee BK, Knuesel I, Feldon J, 2008c. Adult brain and behavioral pathological markers of prenatal immune challenge during early/middle and late fetal development in mice. Brain Behav Immun 22, 469–486. [DOI] [PubMed] [Google Scholar]
- Meyer U, Schwendener S, Feldon J, Yee BK, 2006c. Prenatal and postnatal maternal contributions in the infection model of schizophrenia. Exp Brain Res 173, 243–257. [DOI] [PubMed] [Google Scholar]
- Meyer U, Spoerri E, Yee BK, Schwarz MJ, Feldon J, 2010b. Evaluating early preventive antipsychotic and antidepressant drug treatment in an infection-based neurodevelopmental mouse model of schizophrenia. Schizophr Bull 36, 607–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer U, Yee BK, Feldon J, 2007. The neurodevelopmental impact of prenatal infections at different times of pregnancy: the earlier the worse? Neuroscientist 13, 241–256. [DOI] [PubMed] [Google Scholar]
- Miale IL, Sidman RL, 1961. An autoradiographic analysis of histogenesis in the mouse cerebellum. Exp Neurol 4, 277–296. [DOI] [PubMed] [Google Scholar]
- Missault S, Van den Eynde K, Vanden Berghe W, Fransen E, Weeren A, Timmermans JP, Kumar-Singh S, Dedeurwaerdere S, 2014. The risk for behavioural deficits is determined by the maternal immune response to prenatal immune challenge in a neurodevelopmental model. Brain Behav Immun 42, 138–146. [DOI] [PubMed] [Google Scholar]
- Money KM, Barke TL, Serezani A, Gannon M, Garbett KA, Aronoff DM, Mirnics K, 2017. Gestational diabetes exacerbates maternal immune activation effects in the developing brain. Mol Psychiatry [DOI] [PMC free article] [PubMed]
- Morais LH, Felice D, Golubeva AV, Moloney G, Dinan TG, Cryan JF, 2018. Strain differences in the susceptibility to the gut-brain axis and neurobehavioural alterations induced by maternal immune activation in mice. Behav Pharmacol 29, 181–198. [DOI] [PubMed] [Google Scholar]
- Mueller FS, Polesel M, Richetto J, Meyer U, Weber-Stadlbauer U, 2018. Mouse models of maternal immune activation: Mind your caging system! Brain Behav Immun 73, 643–660. [DOI] [PubMed] [Google Scholar]
- Muotri AR, Gage FH, 2006. Generation of neuronal variability and complexity. Nature 441, 1087–1093. [DOI] [PubMed] [Google Scholar]
- Muotri AR, Marchetto MC, Coufal NG, Oefner R, Yeo G, Nakashima K, Gage FH, 2010. L1 retrotransposition in neurons is modulated by MeCP2. Nature 468, 443–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray BG, Davies DA, Molder JJ, Howland JG, 2017. Maternal immune activation during pregnancy in rats impairs working memory capacity of the offspring. Neurobiol Learn Mem 141, 150–156. [DOI] [PubMed] [Google Scholar]
- Nakano T, Kotani T, Mano Y, Tsuda H, Imai K, Ushida T, Li H, Miki R, Sumigama S, Sato Y, Iwase A, Hirakawa A, Asai M, Toyokuni S, Kikkawa F, 2015. Maternal molecular hydrogen administration on lipopolysaccharide-induced mouse fetal brain injury. Journal of clinical biochemistry and nutrition 57, 178–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naviaux RK, Zolkipli Z, Wang L, Nakayama T, Naviaux JC, Le TP, Schuchbauer MA, Rogac M, Tang Q, Dugan LL, Powell SB, 2013. Antipurinergic therapy corrects the autism-like features in the poly(IC) mouse model. PLoS One 8, e57380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson ED, Kavalali ET, Monteggia LM, 2008. Activity-dependent suppression of miniature neurotransmission through the regulation of DNA methylation. J Neurosci 28, 395–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nouel D, Burt M, Zhang Y, Harvey L, Boksa P, 2012. Prenatal exposure to bacterial endotoxin reduces the number of GAD67- and reelin-immunoreactive neurons in the hippocampus of rat offspring. Eur Neuropsychopharmacol 22, 300–307. [DOI] [PubMed] [Google Scholar]
- O’Leary C, Desbonnet L, Clarke N, Petit E, Tighe O, Lai D, Harvey R, Waddington JL, O’Tuathaigh C, 2014. Phenotypic effects of maternal immune activation and early postnatal milieu in mice mutant for the schizophrenia risk gene neuregulin-1. Neuroscience 277, 294–305. [DOI] [PubMed] [Google Scholar]
- O’Loughlin E, Pakan JMP, Yilmazer-Hanke D, McDermott KW, 2017. Acute in utero exposure to lipopolysaccharide induces inflammation in the pre- and postnatal brain and alters the glial cytoarchitecture in the developing amygdala. J Neuroinflammation 14, 212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh-Nishi A, Obayashi S, Sugihara I, Minamimoto T, Suhara T, 2010. Maternal immune activation by polyriboinosinic-polyribocytidilic acid injection produces synaptic dysfunction but not neuronal loss in the hippocampus of juvenile rat offspring. Brain Res 1363, 170–179. [DOI] [PubMed] [Google Scholar]
- Oskvig DB, Elkahloun AG, Johnson KR, Phillips TM, Herkenham M, 2012. Maternal immune activation by LPS selectively alters specific gene expression profiles of interneuron migration and oxidative stress in the fetus without triggering a fetal immune response. Brain Behav Immun 26, 623–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozawa K, Hashimoto K, Kishimoto T, Shimizu E, Ishikura H, Iyo M, 2006. Immune activation during pregnancy in mice leads to dopaminergic hyperfunction and cognitive impairment in the offspring: a neurodevelopmental animal model of schizophrenia. Biol Psychiatry 59, 546–554. [DOI] [PubMed] [Google Scholar]
- Pang Y, Rodts-Palenik S, Cai Z, Bennett WA, Rhodes PG, 2005. Suppression of glial activation is involved in the protection of IL-10 on maternal E. coli induced neonatal white matter injury. Brain Res Dev Brain Res 157, 141–149. [DOI] [PubMed] [Google Scholar]
- Patrich E, Piontkewitz Y, Peretz A, Weiner I, Attali B, 2016a. Maternal immune activation produces neonatal excitability defects in offspring hippocampal neurons from pregnant rats treated with poly I:C. Sci Rep 6, 19106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patrich E, Piontkewitz Y, Peretz A, Weiner I, Attali B, 2016b. Maturation- and sex-sensitive depression of hippocampal excitatory transmission in a rat schizophrenia model. Brain Behav Immun 51, 240–251. [DOI] [PubMed] [Google Scholar]
- Patterson PH, 2009. Immune involvement in schizophrenia and autism: etiology, pathology and animal models. Behav Brain Res 204, 313–321. [DOI] [PubMed] [Google Scholar]
- Paylor JW, Lins BR, Greba Q, Moen N, de Moraes RS, Howland JG, Winship IR, 2016. Developmental disruption of perineuronal nets in the medial prefrontal cortex after maternal immune activation. Sci Rep 6, 37580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pendyala G, Chou S, Jung Y, Coiro P, Spartz E, Padmashri R, Li M, Dunaevsky A, 2017. Maternal Immune Activation Causes Behavioral Impairments and Altered Cerebellar Cytokine and Synaptic Protein Expression. Neuropsychopharmacology 42, 1435–1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piontkewitz Y, Arad M, Weiner I, 2011. Risperidone administered during asymptomatic period of adolescence prevents the emergence of brain structural pathology and behavioral abnormalities in an animal model of schizophrenia. Schizophr Bull 37, 1257–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piontkewitz Y, Bernstein HG, Dobrowolny H, Bogerts B, Weiner I, Keilhoff G, 2012. Effects of risperidone treatment in adolescence on hippocampal neurogenesis, parvalbumin expression, and vascularization following prenatal immune activation in rats. Brain Behav Immun 26, 353–363. [DOI] [PubMed] [Google Scholar]
- Ploeger A, Raijmakers ME, van der Maas HL, Galis F, 2010. The association between autism and errors in early embryogenesis: what is the causal mechanism? Biol Psychiatry 67, 602–607. [DOI] [PubMed] [Google Scholar]
- Poo MM, 2001. Neurotrophins as synaptic modulators. Nat Rev Neurosci 2, 24–32. [DOI] [PubMed] [Google Scholar]
- Pratt L, Ni L, Ponzio NM, Jonakait GM, 2013. Maternal inflammation promotes fetal microglial activation and increased cholinergic expression in the fetal basal forebrain: role of interleukin-6. Pediatr Res 74, 393–401. [DOI] [PubMed] [Google Scholar]
- Rahman T, Zavitsanou K, Purves-Tyson T, Harms LR, Meehan C, Schall U, Todd J, Hodgson DM, Michie PT, Weickert CS, 2017. Effects of Immune Activation during Early or Late Gestation on N-Methyl-d-Aspartate Receptor Measures in Adult Rat Offspring. Front Psychiatry 8, 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapoport JL, Giedd JN, Gogtay N, 2012. Neurodevelopmental model of schizophrenia: update 2012. Mol Psychiatry 17, 1228–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reisinger S, Khan D, Kong E, Berger A, Pollak A, Pollak DD, 2015. The poly(I:C)-induced maternal immune activation model in preclinical neuropsychiatric drug discovery. Pharmacol Ther 149, 213–226. [DOI] [PubMed] [Google Scholar]
- Reisinger SN, Kong E, Khan D, Schulz S, Ronovsky M, Berger S, Horvath O, Cabatic M, Berger A, Pollak DD, 2016. Maternal immune activation epigenetically regulates hippocampal serotonin transporter levels. Neurobiol Stress 4, 34–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richetto J, Calabrese F, Riva MA, Meyer U, 2014. Prenatal immune activation induces maturation-dependent alterations in the prefrontal GABAergic transcriptome. Schizophr Bull 40, 351–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richetto J, Chesters R, Cattaneo A, Labouesse MA, Gutierrez AMC, Wood TC, Luoni A, Meyer U, Vernon A, Riva MA, 2017a. Genome-Wide Transcriptional Profiling and Structural Magnetic Resonance Imaging in the Maternal Immune Activation Model of Neurodevelopmental Disorders. Cereb Cortex 27, 3397–3413. [DOI] [PubMed] [Google Scholar]
- Richetto J, Labouesse MA, Poe MM, Cook JM, Grace AA, Riva MA, Meyer U, 2015. Behavioral effects of the benzodiazepine-positive allosteric modulator SH-053–2’F-S-CH(3) in an immune-mediated neurodevelopmental disruption model. Int J Neuropsychopharmacol 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richetto J, Massart R, Weber-Stadlbauer U, Szyf M, Riva MA, Meyer U, 2017b. Genome-wide DNA Methylation Changes in a Mouse Model of Infection-Mediated Neurodevelopmental Disorders. Biol Psychiatry 81, 265–276. [DOI] [PubMed] [Google Scholar]
- Richtand NM, Ahlbrand R, Horn P, Stanford K, Bronson SL, McNamara RK, 2011. Effects of risperidone and paliperidone pre-treatment on locomotor response following prenatal immune activation. J Psychiatr Res 45, 1194–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richtand NM, Ahlbrand R, Horn P, Tambyraja R, Grainger M, Bronson SL, McNamara RK, 2012. Fluoxetine and aripiprazole treatment following prenatal immune activation exert longstanding effects on rat locomotor response. Physiol Behav 106, 171–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson SA, Skinner RJ, Care AS, 2006. Essential Role for IL-10 in Resistance to Lipopolysaccharide-Induced Preterm Labor in Mice. The Journal of Immunology 177, 4888–4896. [DOI] [PubMed] [Google Scholar]
- Ronovsky M, Berger S, Zambon A, Reisinger SN, Horvath O, Pollak A, Lindtner C, Berger A, Pollak DD, 2017. Maternal immune activation transgenerationally modulates maternal care and offspring depression-like behavior. Brain Behav Immun 63, 127–136. [DOI] [PubMed] [Google Scholar]
- Rose DR, Careaga M, Van de Water J, McAllister K, Bauman MD, Ashwood P, 2017. Long-term altered immune responses following fetal priming in a non-human primate model of maternal immune activation. Brain Behav Immun 63, 60–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roumier A, Pascual O, Bechade C, Wakselman S, Poncer JC, Real E, Triller A, Bessis A, 2008. Prenatal activation of microglia induces delayed impairment of glutamatergic synaptic function. PLoS One 3, e2595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousset CI, Chalon S, Cantagrel S, Bodard S, Andres C, Gressens P, Saliba E, 2006. Maternal exposure to LPS induces hypomyelination in the internal capsule and programmed cell death in the deep gray matter in newborn rats. Pediatr Res 59, 428–433. [DOI] [PubMed] [Google Scholar]
- Ruskin DN, Murphy MI, Slade SL, Masino SA, 2017. Ketogenic diet improves behaviors in a maternal immune activation model of autism spectrum disorder. PLoS One 12, e0171643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuelsson AM, Jennische E, Hansson HA, Holmang A, 2006. Prenatal exposure to interleukin-6 results in inflammatory neurodegeneration in hippocampus with NMDA/GABA(A) dysregulation and impaired spatial learning. Am J Physiol Regul Integr Comp Physiol 290, R1345–1356. [DOI] [PubMed] [Google Scholar]
- Sando R 3rd, Gounko N, Pieraut S, Liao L, Yates J 3rd, Maximov A, 2012. HDAC4 governs a transcriptional program essential for synaptic plasticity and memory. Cell 151, 821–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savanthrapadian S, Wolff AR, Logan BJ, Eckert MJ, Bilkey DK, Abraham WC, 2013. Enhanced hippocampal neuronal excitability and LTP persistence associated with reduced behavioral flexibility in the maternal immune activation model of schizophrenia. Hippocampus 23, 1395–1409. [DOI] [PubMed] [Google Scholar]
- Scattoni ML, Crawley J, Ricceri L, 2009. Ultrasonic vocalizations: a tool for behavioural phenotyping of mouse models of neurodevelopmental disorders. Neurosci Biobehav Rev 33, 508–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaafsma W, Basterra LB, Jacobs S, Brouwer N, Meerlo P, Schaafsma A, Boddeke E, Eggen BJL, 2017. Maternal inflammation induces immune activation of fetal microglia and leads to disrupted microglia immune responses, behavior, and learning performance in adulthood. Neurobiol Dis 106, 291–300. [DOI] [PubMed] [Google Scholar]
- Schwartzer JJ, Careaga M, Onore CE, Rushakoff JA, Berman RF, Ashwood P, 2013. Maternal immune activation and strain specific interactions in the development of autism-like behaviors in mice. Transl Psychiatry 3, e240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi L, Smith SE, Malkova N, Tse D, Su Y, Patterson PH, 2009. Activation of the maternal immune system alters cerebellar development in the offspring. Brain Behav Immun 23, 116–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin Yim Y, Park A, Berrios J, Lafourcade M, Pascual LM, Soares N, Yeon Kim J, Kim S, Kim H, Waisman A, Littman DR, Wickersham IR, Harnett MT, Huh JR, Choi GB, 2017. Reversing behavioural abnormalities in mice exposed to maternal inflammation. Nature 549, 482–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Short SJ, Lubach GR, Karasin AI, Olsen CW, Styner M, Knickmeyer RC, Gilmore JH, Coe CL, 2010. Maternal influenza infection during pregnancy impacts postnatal brain development in the rhesus monkey. Biol Psychiatry 67, 965–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrestha S, Offer SM, 2016. Epigenetic Regulations of GABAergic Neurotransmission: Relevance for Neurological Disorders and Epigenetic Therapy. Medical Epigenetics 4, 1–19. [Google Scholar]
- Silverman JL, Yang M, Lord C, Crawley JN, 2010. Behavioural phenotyping assays for mouse models of autism. Nat Rev Neurosci 11, 490–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SE, Li J, Garbett K, Mirnics K, Patterson PH, 2007. Maternal immune activation alters fetal brain development through interleukin-6. J Neurosci 27, 10695–10702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SEP, Hsiao E, Patterson PH 2010. Activation of the Maternal Immune System as a Risk Factor for Neuropsychiatric Disorders. In: Maternal Influences on Fetal Neurodevelopment: Clinical and Research Aspects pp. 97–115. Eds. Zimmerman AW, Connors SL. Springer New York: New York, NY. [Google Scholar]
- Smolders S, Smolders SM, Swinnen N, Gartner A, Rigo JM, Legendre P, Brone B, 2015. Maternal immune activation evoked by polyinosinic:polycytidylic acid does not evoke microglial cell activation in the embryo. Front Cell Neurosci 9, 301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadler F, Kolb G, Rubusch L, Baker SP, Jones EG, Akbarian S, 2005. Histone methylation at gene promoters is associated with developmental regulation and region-specific expression of ionotropic and metabotropic glutamate receptors in human brain. J Neurochem 94, 324–336. [DOI] [PubMed] [Google Scholar]
- Stiles J, Jernigan TL, 2010. The basics of brain development. Neuropsychol Rev 20, 327–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun E, Shi Y, 2015. MicroRNAs: Small molecules with big roles in neurodevelopment and diseases. Exp Neurol 268, 46–53. [DOI] [PubMed] [Google Scholar]
- Sun H, Kennedy PJ, Nestler EJ, 2013. Epigenetics of the depressed brain: role of histone acetylation and methylation. Neuropsychopharmacology 38, 124–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swathy B, Banerjee M, 2017. Haloperidol induces pharmacoepigenetic response by modulating miRNA expression, global DNA methylation and expression profiles of methylation maintenance genes and genes involved in neurotransmission in neuronal cells. PLoS One 12, e0184209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang B, Jia H, Kast RJ, Thomas EA, 2013. Epigenetic changes at gene promoters in response to immune activation in utero. Brain Behav Immun 30, 168–175. [DOI] [PubMed] [Google Scholar]
- Teixeira CM, Kron MM, Masachs N, Zhang H, Lagace DC, Martinez A, Reillo I, Duan X, Bosch C, Pujadas L, Brunso L, Song H, Eisch AJ, Borrell V, Howell BW, Parent JM, Soriano E, 2012. Cell-autonomous inactivation of the reelin pathway impairs adult neurogenesis in the hippocampus. J Neurosci 32, 12051–12065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trakhtenberg EF, Goldberg JL, 2012. Epigenetic regulation of axon and dendrite growth. Front Mol Neurosci 5, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trepanier MO, Hopperton KE, Orr SK, Bazinet RP, 2016. N-3 polyunsaturated fatty acids in animal models with neuroinflammation: An update. Eur J Pharmacol 785, 187–206. [DOI] [PubMed] [Google Scholar]
- Van den Eynde K, Missault S, Fransen E, Raeymaekers L, Willems R, Drinkenburg W, Timmermans JP, Kumar-Singh S, Dedeurwaerdere S, 2014. Hypolocomotive behaviour associated with increased microglia in a prenatal immune activation model with relevance to schizophrenia. Behav Brain Res 258, 179–186. [DOI] [PubMed] [Google Scholar]
- Villalba JM, Alcain FJ, 2012. Sirtuin activators and inhibitors. BioFactors (Oxford, England) 38, 349–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuillermot S, Feldon J, Meyer U, 2011. Nurr1 is not essential for the development of prepulse inhibition deficits induced by prenatal immune activation. Brain Behav Immun 25, 1316–1321. [DOI] [PubMed] [Google Scholar]
- Vuillermot S, Joodmardi E, Perlmann T, Ogren SO, Feldon J, Meyer U, 2012. Prenatal immune activation interacts with genetic Nurr1 deficiency in the development of attentional impairments. J Neurosci 32, 436–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuillermot S, Luan W, Meyer U, Eyles D, 2017. Vitamin D treatment during pregnancy prevents autism-related phenotypes in a mouse model of maternal immune activation. Mol Autism 8, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuillermot S, Weber L, Feldon J, Meyer U, 2010. A longitudinal examination of the neurodevelopmental impact of prenatal immune activation in mice reveals primary defects in dopaminergic development relevant to schizophrenia. J Neurosci 30, 1270–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber-Stadlbauer U, Richetto J, Labouesse MA, Bohacek J, Mansuy IM, Meyer U, 2017. Transgenerational transmission and modification of pathological traits induced by prenatal immune activation. Mol Psychiatry 22, 102–112. [DOI] [PubMed] [Google Scholar]
- Weir RK, Forghany R, Smith SE, Patterson PH, McAllister AK, Schumann CM, Bauman MD, 2015. Preliminary evidence of neuropathology in nonhuman primates prenatally exposed to maternal immune activation. Brain Behav Immun 48, 139–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willi R, Harmeier A, Giovanoli S, Meyer U, 2013. Altered GSK3beta signaling in an infection-based mouse model of developmental neuropsychiatric disease. Neuropharmacology 73, 56–65. [DOI] [PubMed] [Google Scholar]
- Winter C, Djodari-Irani A, Sohr R, Morgenstern R, Feldon J, Juckel G, Meyer U, 2009. Prenatal immune activation leads to multiple changes in basal neurotransmitter levels in the adult brain: implications for brain disorders of neurodevelopmental origin such as schizophrenia. Int J Neuropsychopharmacol 12, 513–524. [DOI] [PubMed] [Google Scholar]
- Wischhof L, Irrsack E, Dietz F, Koch M, 2015a. Maternal lipopolysaccharide treatment differentially affects 5-HT(2A) and mGlu2/3 receptor function in the adult male and female rat offspring. Neuropharmacology 97, 275–288. [DOI] [PubMed] [Google Scholar]
- Wischhof L, Irrsack E, Osorio C, Koch M, 2015b. Prenatal LPS-exposure--a neurodevelopmental rat model of schizophrenia--differentially affects cognitive functions, myelination and parvalbumin expression in male and female offspring. Prog Neuropsychopharmacol Biol Psychiatry 57, 17–30. [DOI] [PubMed] [Google Scholar]
- Wolff AR, Bilkey DK, 2015. Prenatal immune activation alters hippocampal place cell firing characteristics in adult animals. Brain Behav Immun 48, 232–243. [DOI] [PubMed] [Google Scholar]
- Wu WL, Adams CE, Stevens KE, Chow KH, Freedman R, Patterson PH, 2015. The interaction between maternal immune activation and alpha 7 nicotinic acetylcholine receptor in regulating behaviors in the offspring. Brain Behav Immun 46, 192–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu WL, Hsiao EY, Yan Z, Mazmanian SK, Patterson PH, 2017. The placental interleukin-6 signaling controls fetal brain development and behavior. Brain Behav Immun 62, 11–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Qi F, Song D, He Z, Zuo Z, Yang Y, Liu Q, Hu S, Wang X, Zheng X, Yang J, Yuan Q, Zou J, Guo K, Yao Z, 2018. Prenatal influenza vaccination rescues impairments of social behavior and lamination in a mouse model of autism. J Neuroinflammation 15, 228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zavitsanou K, Lim CK, Purves-Tyson T, Karl T, Kassiou M, Banister SD, Guillemin GJ, Weickert CS, 2014. Effect of maternal immune activation on the kynurenine pathway in preadolescent rat offspring and on MK801-induced hyperlocomotion in adulthood: amelioration by COX-2 inhibition. Brain Behav Immun 41, 173–181. [DOI] [PubMed] [Google Scholar]
- Zerbo O, Iosif AM, Walker C, Ozonoff S, Hansen RL, Hertz-Picciotto I, 2013. Is maternal influenza or fever during pregnancy associated with autism or developmental delays? Results from the CHARGE (CHildhood Autism Risks from Genetics and Environment) study. J Autism Dev Disord 43, 25–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerbo O, Qian Y, Yoshida C, Grether JK, Van de Water J, Croen LA, 2015. Maternal Infection During Pregnancy and Autism Spectrum Disorders. J Autism Dev Disord 45, 4015–4025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Jing Y, Zhang H, Bilkey DK, Liu P, 2018. Maternal immune activation altered microglial immunoreactivity in the brain of postnatal day 2 rat offspring. Synapse, e22072. [DOI] [PubMed]
- Zhang Y, Cazakoff BN, Thai CA, Howland JG, 2012. Prenatal exposure to a viral mimetic alters behavioural flexibility in male, but not female, rats. Neuropharmacology 62, 1299–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, van Praag H, 2015. Maternal immune activation differentially impacts mature and adult-born hippocampal neurons in male mice. Brain Behav Immun 45, 60–70. [DOI] [PubMed] [Google Scholar]
- Zhu F, Zheng Y, Liu Y, Zhang X, Zhao J, 2014. Minocycline alleviates behavioral deficits and inhibits microglial activation in the offspring of pregnant mice after administration of polyriboinosinic-polyribocytidilic acid. Psychiatry Res 219, 680–686. [DOI] [PubMed] [Google Scholar]
- Zibetti C, Adamo A, Binda C, Forneris F, Toffolo E, Verpelli C, Ginelli E, Mattevi A, Sala C, Battaglioli E, 2010. Alternative splicing of the histone demethylase LSD1/KDM1 contributes to the modulation of neurite morphogenesis in the mammalian nervous system. J Neurosci 30, 2521–2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zijlstra GJ, Ten Hacken NH, Hoffmann RF, van Oosterhout AJ, Heijink IH, 2012. Interleukin-17A induces glucocorticoid insensitivity in human bronchial epithelial cells. The European respiratory journal 39, 439–445. [DOI] [PubMed] [Google Scholar]
- Zuckerman L, Rehavi M, Nachman R, Weiner I, 2003. Immune activation during pregnancy in rats leads to a postpubertal emergence of disrupted latent inhibition, dopaminergic hyperfunction, and altered limbic morphology in the offspring: a novel neurodevelopmental model of schizophrenia. Neuropsychopharmacology 28, 1778–1789. [DOI] [PubMed] [Google Scholar]