

Abstract
Mutations in DEPDC5 and NPRL3 subunits of GATOR1, a modulator of mechanistic target of rapamycin (mTOR), are linked to malformations of cortical development (MCD). Brain specimens from these individuals reveal abnormal cortical lamination, altered cell morphology, and hyperphosphorylation of ribosomal S6 protein (PS6), a marker for mTOR activation. While numerous studies have examined GATOR1 subunit function in non-neuronal cell lines, few have directly assessed loss of GATOR 1 subunit function in neuronal cell types. We hypothesized that DEPDC5 or NPRL3 shRNA-mediated knockdown (DEPDC5/NPRL3 KD) leads to inappropriate functional activation of mTOR and mTOR-dependent alterations in neuronal morphology.
Neuronal size was determined in human specimens harboring DEPDC5 or NPRL3 mutations resected for epilepsy treatment. DEPDC5/NPRL3 KD effects on cell size, filopodial extension, subcellular mTOR complex 1 (mTORC1) localization, and mTORC1 activation during nutrient deprivation were assayed in mouse neuroblastoma cells (N2aC) and mouse subventricular zone derived neural progenitor cells (mNPCs). mTORC1-dependent effects of DEPDC5/NPRL3 KD were determined using the mTOR inhibitor rapamycin. Changes in mTOR subcellular localization and mTORC1 pathway activation following DEPDC5/NPRL3 KD were determined by examining the proximity of mTOR to the lysosomal surface during amino acid starvation.
Neurons exhibiting PS6 immunoreactivity (Ser 235/236) in human specimens were 1.5× larger than neurons in post-mortem control samples. DEPDC5/NPRL3 KD caused mTORC1, but not mTORC2, hyperactivation, soma enlargement, and increased filopodia in N2aC and mNPCs compared with wildtype cells. DEPDC5/NPRL3 KD led to inappropriate mTOR localization at the lysosome along with constitutive mTOR activation following amino acid deprivation. DEPDC5/NPRL3 KD effects on morphology and functional mTOR activation were reversed by rapamycin. mTOR-dependent effects of DEPDC5/NPRL3 KD on morphology and subcellular localization of mTOR in neurons suggests that loss-of-function in GATOR1 subunits may play a role in MCD formation during fetal brain development.
Keywords: GATOR1, focal cortical dysplasia, mTORopathy, epilepsy, neural progenitor cells, brain development
Introduction
Malformations of cortical development (MCDs) are common causes of epilepsy and intellectual disability (Desikan and Barkovich, 2016). Focal malformations (e.g., Focal Cortical Dysplasia; FCD) are a frequent cause of drug-resistant epilepsy in children and adults. FCD has been linked to either germline or somatic mutations in genes encoding protein components of the PI3K-AKT3-mTOR pathway occurring in neuroglial progenitor cells during brain development (Iffland and Crino, 2017). Appropriate mTOR signaling provides pivotal modulation of cell proliferation, migration, axon and dendrite outgrowth and cortical lamination during embryonic brain development and leads to the formation of the intact cerebral cortex (Switon et al., 2017).
A key regulatory node along the mTOR pathway is the GTPase-activating protein (GAP) activity towards Rags complex 1 (Bar-Peled et al., 2013). GATOR1 is a three-protein complex composed of Dep Domain Containing 5 (DEPDC5; 177 kDa), Nitrogen Permease Regulator-Like 2 (NPRL2; 44 kDa) and Nitrogen Permease Regulator-Like 3 (NPRL3; 64 kDa). Much of what is known about GATOR1 function has been defined in non-neural cell types. For example, GATOR1 prevents activation of the mTOR pathway during nutrient deprivation, e.g., when intracellular amino acid levels are low, by interfering with the ability of the mTOR complex 1 (mTORC1; a multi-protein complex containing mTOR and Raptor) to localize to the lysosomal surface (Bar-Peled et al., 2013). Localization of mTORC1 to the lysosome and subsequent interaction with Rag GTPases and Ragulator at the lysosomal surface is necessary to produce mTORC1 kinase activity (Bar-Peled et al., 2013). Recent studies have demonstrated that GATOR1 is the last checkpoint in a much wider amino acid-mTOR regulatory network that encompasses GATOR 2 (Chantranupong et al., 2014), sestrins (Parmigiani et al., 2014), CASTOR 1 (Saxton et al., 2016) and KICSTOR (Wolfson et al., 2017).
Loss-of-function mutations in DEPDC5 and NPRL3 have been associated with FCD (Baulac et al., 2015; Poduri, 2014; Scerri et al., 2015) in which histological examination of surgically resected brain specimens revealed morphologically abnormal PS6 immunoreactive neurons (Scerri, et al., 2015; Scheffer et al., 2014; Sim et al., 2016; Ricos et al., 2015). It is therefore believed that loss-of-function mutations in GATOR1 subunits cause constitutive mTORC1 activation leading to altered neuronal morphology, enhanced cell size, and abnormal cortical lamination (Marsan et al., 2016). Here, we demonstrate that DEPDC5/NPRL3 KD results in enhanced cell size, altered process outgrowth, and enhanced mTORC1 activation in mouse neural progenitor cells and human neuroblastoma cells. We show that DEPDC5/NPRL3 KD is associated with altered lysosomal localization of mTOR and aberrant responses to amino acid deprivation. We demonstrate that changes in cell morphology are mTOR-dependent and can be reversed by treatment with the mTOR inhibitor rapamycin.
Materials and Methods
Human Tissue Samples
Paraformaldehyde (PFA) fixed, paraffin embedded FCD tissue specimens were obtained following resective epilepsy surgery from patients with known DEPDC5 (n=2; Scerri et al., 2015) or NPRL3 mutations (n=2; Scerri et al., 2015; Sim et al., 2016). Histopathology was assessed by a neuropathologist using the ILAE Diagnostic Methods Commission guidelines (Blumcke et al., 2016) and specimens were classified as FCD type IIA (i.e., characterized by cytomegalic dysmorphic neurons and disorganized cortical lamination). Seven micron sections were probed with anti-PS6 antibodies (Ser 235/236; rabbit monoclonal; 1:1000; Cell Signaling, Danvers, MA). Age-matched specimens of frontal neocortex were obtained post-mortem (n=2) and analyzed in parallel with anti-MAP2 antibodies (Cell Signaling; rabbit monoclonal; 1:1000) and anti-PS6 (Ser 235/236; rabbit monoclonal; 1:1000; Cell Signaling, Danvers, MA). MAP2 staining was used to measure cell body size in control specimens as there was minimal PS6 observed in control. Size and morphology analysis of PS6 immunolabeled cells were performed using ImageQuant software and an automated tool for measuring cell area. In control specimens, measurements were only performed on MAP2+ layer V pyramidal neurons containing full and intact nuclei to ensure that the largest possible neurons were measured. Mean cell area ± standard error was determined using Student’s t-test (p<0.05). Measurements were performed blinded to mutation type. The Royal Children’s Hospital Human Research Ethics Committee and Lewis Katz School of Medicine at Temple University Institutional Review Board approved the study and specimens were obtained following informed consent.
Cell Culture
Three cell types were used for these experiments: Human Embryonic Kidney 293FT (HEK293FT) cells to provide a baseline readout for changes in mTOR pathway activation after knockdown (KD), amino acid deprivation and pharmacological manipulation; Neuro2a cells (N2aC) and subventricular zone (SVZ) derived mouse neural progenitor cells (mNPCs; J. Wolfe, Children’s Hospital of Philadelphia, Philadelphia, PA) to examine the effects of KD, amino acid depravation and pharmacological manipulation on neuronal progenitor cells – cell types integral to FCD formation during brain development.
mNPCs were cultured in complete medium consisting of Dulbecco’s modified Eagle’s medium (DMEM)/F12 (Invitrogen, Carlsbad, CA) supplemented with 1% N2 (Invitrogen), 1% fetal bovine serum (FBS) (Sigma-Aldrich, St. Louis, MO), 1% penicillin/streptomycin (Invitrogen), basic fibroblast growth factor (BFGF; 20 ng/ml) (Promega, Madison, WI), and heparin (5 μg/ml) (Sigma-Aldrich, St. Louis, MO). mNPCs were used to assess soma size, nuclear size, and filopodial outgrowth.
N2aC (Sigma-Aldrich, St. Louis, MO) were cultured in complete medium consisting of DMEM/F12 (Invitrogen, Carlsbad, CA) supplemented with 10% FBS (Invitrogen, Carlsbad, CA) and 1% penicillin/streptomycin (Invitrogen, Carlsbad, CA). N2aC were differentiated in DMEM/F12 supplemented with 1% FBS, 1% penicillin/streptomycin and 20 mM retinoic acid replenished daily for 96hrs. HEK293FT (ATCC, Manassas, VA) cells were cultured in complete media consisting of DMEM supplemented with 10% FBS (Invitrogen, Carlsbad, CA), 1% Sodium pyruvate (Invitrogen, Carlsbad, CA), and 1% L-glutamine (Invitrogen, Carlsbad, CA). N2aC and HEK293FT cells were used to assess cell size by FACS and for functional (Western blot) studies. N2aC were used to determine the effects of DEPDC5/NPRL3 KD on localization of mTOR to the lysosomal surface during amino acid deprivation as well as functional studies. Differentiated N2aC were used to define the consequences of KD on morphology in neuronal cells. All cell cultures for all experiments were maintained in a humidified incubator at 37°C with 5% CO2.
Transfection procedures
Immediately prior to addition of transfection reagents, cells in culture were serum-starved by addition of fresh serum free media for the appropriate cell type. Serum free media was used for the duration of each experiment except where amino acid free conditions were required. Transfection for each cell type was performed using Lipofectamine LTX with Plus reagent (ThermoFisher Scientific, Waltham, MA) and 30 μg of shRNA-plasmid or overexpression-plasmid diluted in 300 μl Opti-MEM (Invitrogen, Carlsbad, CA). Lipofectamine LTX was specifically chosen to reduce lipofectamine cytotoxicity. In separate experiments, mouse DEPDC5 expression plasmid (MR21587, Myc-DDK tag), NPRL3 expression plasmid (MR208964; Myc-DDK tag), mouse DEPDC5 shRNA-plasmid (TG508165, tGFP) or NPRL3 shRNA containing plasmid (TG501301, tGFP) were transfected (all plasmids from Origene, Rockville, MD) into each cell type. Control transfections were performed using manufacturer provided tGFP-containing, scramble control plasmids (5′ GCACTACCAGAGCTAACTCAGATAGTACT 3′). All transfections were performed for 48 hours. Co-transfection experiments were performed by doubling the transfection reagents and using 30 μg of each plasmid. All transfection experiments were performed in duplicate.
Pharmacological and functional manipulation of transfected cells
To determine the mTOR-dependent effects of morphological and functional changes induced by DEPDC5/NPRL3 KD, rapamycin (50 nM, Cell Signaling Technologies, Danvers, MA), a p70 ribosomal S6 kinase (p70S6K) inhibitor (PF-4708671, 10 nM, Tocris Bioscience, Bristol, UK) or vehicle (dimethyl sulfoxide; DMSO, Sigma Aldrich, St. Louis, MO) was applied to the culture at the time of transfection and reapplied daily for 48hrs. Each experiment was performed in duplicate.
To define how amino acid free conditions affect mTOR activity after DEPDC5/NPRL3 KD, serum free media was removed from transfected cells (N2aC and HEK293FT) and washed once with amino acid free media (DMEM; US Biological, Salem MA; warmed to 37°C) to remove any remaining media. The cells were then incubated with amino acid free DMEM only or amino acid free DMEM with rapamycin (150 nM) for 90 minutes. Each experiment was performed in duplicate.
Immunocytochemistry
mNPCs, N2aC, and differentiated N2aC were fixed in 4% PFA at room temperature for 20 minutes and then permeabilized in phosphate-buffered saline (PBS) containing 0.3% Triton X-100 (ThermoFisher Scientific, Waltham, MA). Cells were blocked for 2 hours at room temperature (RT) in 5% normal serum from the species in which the secondary antibody was raised (Jackson ImmunoResearch, West Grove, PA). Cells were incubated in one of the following primary antibody in blocking solution containing 5% normal serum at 4°C overnight: F-actin (1:1000; Abcam, Cambridge, UK), mTOR (1:1000; Abcam, Cambridge, UK), Lysosome-associated membrane protein 2 (LAMP2; 1:1000; Thermofisher, Waltham, MA), anti-Myc-DDK (1:500; Origene, Rockville, MD), or microtubule associate protein 2 (MAP2; 1:1000; Abcam, Cambridge, UK). The secondary antibody, containing a flurochrome (Alexa Fluor 488, Alexa Fluor 594 or Alexa Fluor 647; all 1:100 dilution; Molecular Probes, Eugene, OR), was incubated with the cells for 2 hours at RT. Hoechst nuclear counterstain was used to visualize nuclei and was applied at a concentration of 1:100 for 20 min at RT (Molecular Probes, Eugene, OR).
Western blot analysis
Cells were lysed in RIPA lysis buffer (50 mM Tris HCl, pH 8.0; 150 mM NaCl; 1% NP-40; 0.5% sodium deoxycholate; and 0.1% SDS) with a protease (Sigma-Aldrich, St. Louis, MO) and phosphatase inhibitor (ThermoFisher Scientific, Waltham, MA) followed by brief sonication. Lysates were centrifuged at 4°C for 20 minutes at 14,000 g, and supernatants collected. Protein concentrations (μg/ml) were determined using the Bradford assay (Bradford, 1976). A sample mixture containing 30 μg of protein, Nupage Reducing Agent and Nupage Loading Buffer (Invitrogen, Carlsbad, CA) was denatured for 10 minutes at 90°C and loaded onto a Bolt BT Plus 4-12% gel (Invitrogen, Carlsbad, CA). After electrophoresis, proteins were transferred onto PVDF membranes (Immobilon; EMD millipore, Darmstadt, Germany) at 4°C. The membranes were blocked in Odyssey Blocking Buffer (Li-Cor, Lincoln, Nebraska) for 1 hour at RT. Membranes were probed overnight at 4°C in Odyssey Blocking Buffer with antibodies recognizing PS6 (Ser240/244; rabbit monoclonal; 1:1000; Cell Signaling, Danvers, MA), phospho-AKT (Ser473; rabbit monoclonal; 1:1000; Cell Signaling, Danvers, MA), or total S6 ribosomal protein (mouse monoclonal; 1:500; Cell Signaling, Danvers, MA). Odyssey secondary antibodies 800CW or 680RD were used to visualize bands on the blot (1:1000; Li-Cor, Lincoln, Nebraska). To ensure equal loading, antibodies recognizing GAPDH (rabbit monoclonal; 1:10,000, 2hrs, RT; Advanced Immunochemicals, Longbeach, CA) followed by Odyssey secondary (diluted 1:1,000, 2hrs, RT; Li-Cor, Lincoln, Nebraska) were used. Blots were developed using an Odyssey Clx scanner and scanned at 169 μm resolution with exposure for both 700 and 800 nm channels set at 3.5 (relative intensity, no units; Li-Cor, Lincoln, Nebraska).
FACS analysis
Live cells were trypsinized (0.25%), placed into 15 ml conical tubes and centrifuged at 1,200 RPM to create a cell pellet. Media was removed and cells were resuspended in DPBS with ethylenediaminetetraacetic acid (EDTA; 5 mM) and passed through a cell strainer into a conical glass tube to create a single cell suspension. Cells were sorted based on GFP fluorescence using a FACSAria II cell sorter (BD Bioscience, San Jose, CA; available in the Flow Cytometry Core at the Lewis Katz School of Medicine at Temple University, Philadelphia, PA). FloJo software (Ashland, Oregon) was used to measure forward scatter in populations of approximately 100,000 cells. Forward scatter is produced by diffraction of laser light around cells during FACS analysis and is proportional to cell diameter; the greater the degree of light scatter, the larger the diameter of the cell. Forward scatter analysis provides a rapid assay to assess cell size in a large number of cells based on marker protein expression, i.e., GFP.
mTOR-lysosomal colocalization analysis
mTOR activation requires localization of mTORC1 to the lysosomal surface. Therefore, we designed an approach to assess and quantitate colocalization of mTOR and lysosomal surface markers in N2aC after DEPDC5 or NPRL3 KD in amino acid free (AAF) or amino acid replete (complete) conditions. N2a Cells were transfected in serum free media as described above. After 48 hrs cells were incubated in AAF or complete media for 50 minutes and then PFA fixed. Cells were labeled with mTOR and LAMP2 (a lysosomal marker) antibodies. Using an upright fluorescent microscope (Leica, Wetzlar, Germany), GFP+ transfected cells were selected and a region of interest (ROI) was isolated from an individual cell. Fluorescence intensity of mTOR and LAMP2 was measured within the ROI using ImageJ (Schneider et al., 2012). A Pearson’s correlation test (Excel, Microsoft, Redmond, WA) was then performed to determine whether or not an increase in fluorescence intensity in mTOR correlated with an increase in fluorescence intensity in LAMP2 - the higher the correlation coefficient (R), the greater the degree of colocalization between mTOR and LAMP2 and thus, the greater the degree of mTORC1 activation. ROIs were measured for 10 N2aC in each group- DEPDC5 KD in AAF media, NPRL3 KD in AAF media, scramble control in AAF media, wildtype cells in AAF media and wildtype cells in complete media. An ANOVA was performed with the R values and a p< 0.05 was deemed statistically significant. To better visualize the degree of colocalization in digital images, each ROI was reconstructed into a 3D fluorescence intensity surface plot.
Quantitative and Statistical analysis of cell morphology data
Soma and nuclear diameter was measured in digital images of mNPCs in ImageJ (Schneider et al., 2012). Each measurement was taken using the longest dimension of each soma or nucleus in 50 total cells (25 cells in 2 replicates). Filopodial outgrowth was also measured in digital images of mNPCs in ImageJ by counting individual processes in each of 50 individual neurons (25 cells in 2 replicates). Only primary filopodia, defined as processes emanating directly off of the soma, were counted. In differentiated N2aC, MAP2+ processes emanating directly off of the soma were manually measured in digital images using ImageJ in 20 cells – 10 from each of 2 replicates. Cell size and filopodial measurements were obtained blinded to condition. Statistical analysis was performed in Origin (Northampton, MA) using ANOVA (p< 0.05 considered statistically significant).
Results
Increased cell size is observed in specimens from patients with DEPDC5 and NPRL3 mutations
Visual inspection of the 4 surgical tissue specimens confirmed the presence of cytomegalic dysmorphic neurons and highly disorganized lamination consistent with FCD type IIa histopathology. The NPRL3 specimens were from non-consanguineous first cousins (n=2) with drug-resistant focal epilepsy found to have a c1375_1376dupAC in NPRL3 (patients III3 and III4 in Sim et al., 2016). The DEPDC5 specimens were from non-consanguineous brothers (n=2) with drug-resistant focal epilepsy found to have a c.C1663T, p.Arg555* in DEPDC5 (patients III6 and III7 in Scerri et al., 2016). In total, 3000 PS6 labeled cells were counted in DEPDC5 (1500 cells) and NPRL3 (1500 cells) resected brain specimens, and 1500 cells were counted in control sections, visualized by MAP2. Mean maximal cell soma width of PS6 labeled cells was 1.6 times greater in the DEPDC5- and 1.4 times greater in NPRL3-associated specimens than in the control sections (112.3 and 90 microns, respectively, vs. 65.6 microns; p< 0.001; Fig. 1) suggesting effects of mTOR activation (as evidenced by PS6 immunoreactivity of these cell types) on cell size following loss-of-function mutations in either DEPDC5 or NPRL3.
Figure 1. Enhanced cell size in human DEPDC5- or NPRL3-associated brain tissue specimens.
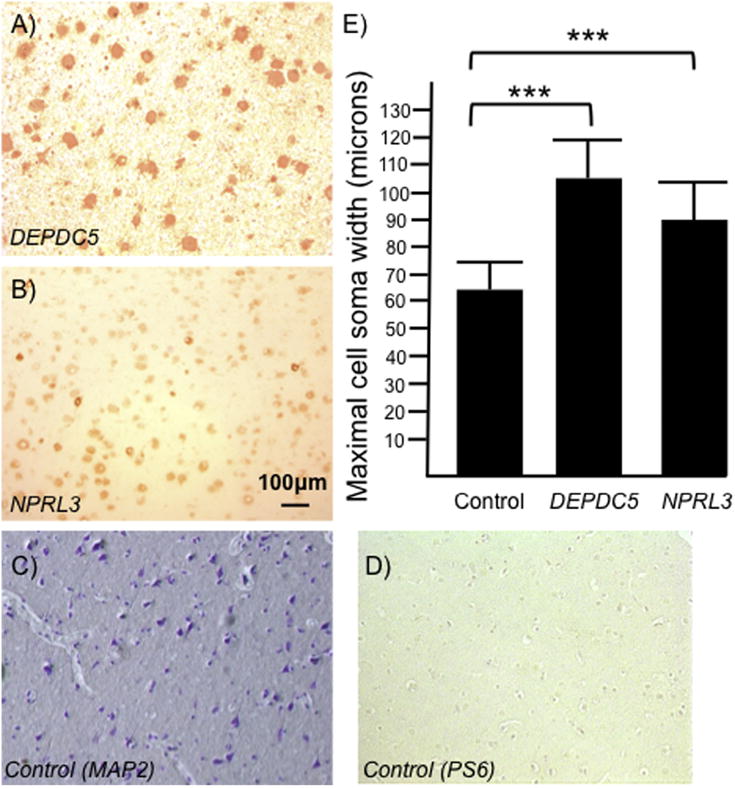
There is an increase in maximal cell soma width standard error of the mean (±SEM) in PS6 (Ser 235/236) immunoreactive neurons in resected specimens from individuals with DEPDC5 (A; n=2) and NPRL3 (B; n=2) mutations compared with age-matched post-mortem control layer V neurons (measured by MAP2 staining; C and D; n=2). Cells from the DEPDC5 specimen were 1.6 times larger than control (E; n=1500 cells per group; p < 0.001) and cells from the NPRL3 specimen were 1.4 times larger than control (E; 1500 cells per group, p < 0.001). Scale bar in (B) applies to all micrographs. *** = significantly different from control with a p < 0.001.
Knockdown of DEPDC5 or NPRL3 is achieved after shRNA transfection
Several commercially available antibodies directed against DEPDC5 or NPRL3 assayed in our lab did not yield clear or reliable results i.e., excessive non-specific bands detected by Western blot which prevented us from directly assessing the KD efficiency. We therefore utilized a recombinant expression system to indirectly measure DEPDC5 or NPRL3 through Myc-DDK tagged proteins. Expression constructs encoding full-length DEPDC5 or NPRL3 coupled to a Myc-DDK tag were co-transfected with the appropriate KD shRNA (Fig. 2) or co-transfected with a scramble control construct into HEK293FT cells. After co-transfection with shRNA, NPRL3-Myc-DDK or DEPDC5-Myc-DDK levels were decreased relative to the overexpression plasmid alone or the overexpression plasmid co-transfected with a scramble shRNA construct (Fig. 2). These findings demonstrate reliable knockdown with DEPDC5 or NPRL3 shRNA in vitro.
Figure 2. shRNA Knockdown of DEPDC5 or NPRL3.
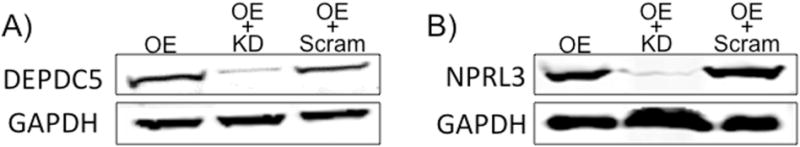
Knockdown of DEPDC5 (A) or NPRL3 (B) in HEK293FT cells was assayed following transfection of expression constructs encoding DEPDC5 or NPRL3 containing a Myc-DDK tag. The plasmids were transfected alone, co-transfected with the appropriate shRNA construct, or co-transfected with scramble control construct into HEK293FT cells. After co-transfection with shRNA, DEPDC5-Myc-DDK or NPRL3-Myc-DDK levels were decreased. Co-transfections of overexpression plasmids with scramble shRNA construct yielded no changes in Myc-DDK tagged DEPDC5 or NPRL3 protein levels compared to overexpression plasmid transfection alone. O.E.= overexpression, KD = knockdown, Scram = scramble control. Western blots performed in duplicate. Densitometry data are in Supplemental Fig. 3.
Mutations in DEPDC5 or NPRL3 have been associated with enhanced mTOR signaling activation as evidenced by increased phosphorylation of ribosomal S6 protein (PS6) in human brain specimens (Sim et al 2016). To prove this link, we hypothesized that DEPDC5/NPRL3 KD would lead to enhanced mTOR activation and increased PS6 levels in N2a and HEK293FT cells. DEPDC5/NPRL3 KD in both N2aC and HEK293FT cells resulted in an increase in PS6 levels compared to scramble control construct and wildtype cells (Fig. 3). DEPDC5 or NPRL3 shRNA transfected N2aC and HEK293FT cells treated with the mTOR inhibitor rapamycin exhibited decreased levels of PS6, thus indicating that DEPDC5/NPRL3 KD induced increases in PS6 levels are mTORC1-dependent. Levels of non-phosphorylated S6 protein were not altered by DEPDC5 or NPRL3 KD (Fig. 3C).
Figure 3. DEPDC5 or NPRL3 KD lead to enhanced PS6 levels but not changes in mTORC2 levels.
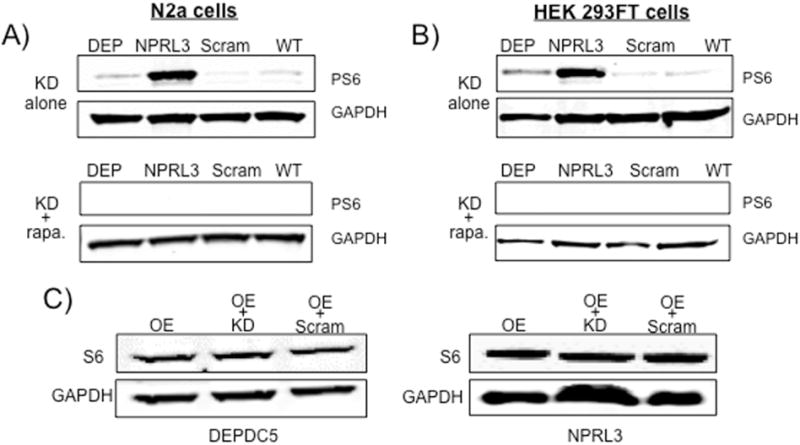
Increased phospho-S6 levels were observed in N2aC (A) and HEK293FT cells (B) after shRNA KD of DEPDC5 or NPRL3 compared to scramble control and wildtype cells. Application of rapamycin to cultured cells resulted in a decrease in PS6 levels (A and B). C) Overexpression or co-transfection of DEPDC5 or NPRL3 overexpression plasmid with shRNA or scramble control did not produce changes in total S6 levels. OE = overexpression; KD = knockdown, Scram = scramble control, DEP = DEPDC5, WT = Wildtype, rapa = rapamycin. Western blots performed in duplicate. Densitometry data are in Supplemental Fig. 4.
mTOR can also exist as another heteromeric complex, mTORC2, defined by protein binding to rictor. We assayed the mTORC2 substrate phospho-AKT (Ser473) in N2aC and HEK293FT cells, and found no effects, suggesting that DEPDC5 or NPRL3 KD does not alter mTORC2 activation (data not shown).
Increase in cell size and filopodia outgrowth is observed after DEPDC5 or NPRL3 KD in vitro
Enhanced soma size has been reported in surgically resected FCD specimens from patients with DEPDC5 or NPRL3 mutations (Baulac, 2016 and see Fig.1). We hypothesized that enhanced cell size is a direct and mTORC1-dependent effect of loss of either DEPDC5 or NPRL3 function. Thus, measurement of maximal soma size in mNPCs (digital images, p<0.001; n= 50 cells per group; 25 cells from each of 2 replicates; Fig. 4A, B) and FACS forward scatter analysis in N2aC and HEK293FT (≈100,000 GFP+ cells transfected with DEPDC5 or NPRL3 shRNA, scramble control shRNA construct; Fig. 4D), revealed that DEPDC5/NPRL3 KD led to an increased soma diameter compared to scramble control and wildtype cells.
Figure 4. DEPDC5 or NPRL3 KD result in morphological changes in N2aC and mNPCs.
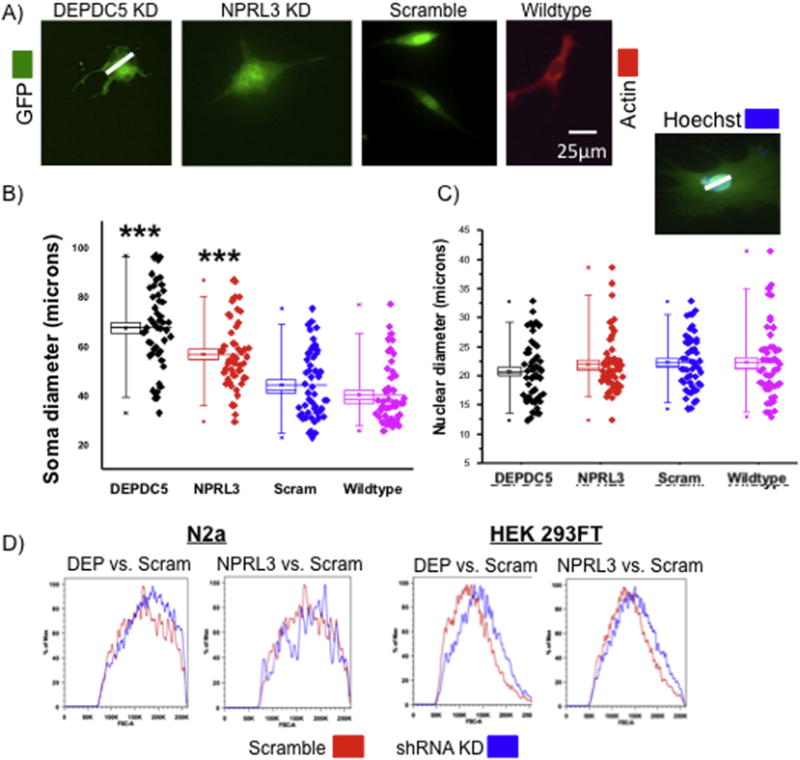
Representative images (A) of DEPDC5, NPRL3, and scramble control shRNA (white line in (A) depicts representative area of measurement) in mNPCs. (B) The soma diameter was significantly larger in DEPDC5 or NPRL3 shRNA transfected mNPCs than scramble control and wildtype cells (n= 50 per group- 25 cells from each of 2 replicates). (C) Nuclear diameter measurements taken from digital images of transfected mNPCs revealed no increase in nuclear size (inset above (C) represents an area of measurement; n=50 cells per group- 25 cells from each 2 replicates). (D) An increase in cell size was observed in N2aC and HEK 293FT after DEPDC5 or NPRL3 shRNA KD, compared to scramble control transfected cells, by FACS forward scatter analysis (n= 100,000 cells per group; red= scramble construct size, blue= size after KD). In (B) and (C), each box represents SEM with a mean line shown, whiskers reflect 5-95% confidence intervals. Scramble control transfected cells were not significantly larger than wildtype cells suggesting no effect of lipofectamine alone on cell size. DEP = DEPDC5, Scram = scramble control, GFP = green fluorescent protein, *** = significant difference between scramble and WT control with a p < 0.001.
We next assessed whether DEPDC5 or NPRL3 KD led to nuclear enlargement in mNPCs. Maximal nuclear diameter was assessed by manual visual count under fluorescence microscopy (Hoechst nuclear counterstain, n= 50 cells per group; 25 cells from each of 2 replicates). There was no change in nuclear diameter identified following DEPDC5/NPRL3 KD compared to control or wildtype cells (Fig. 4C), suggesting that cell size enlargement reflected cytoplasmic expansion alone.
SVZ-derived mNPCs are undifferentiated and therefore have no defined axons or dendrites. However, these cells do extend short filopodia which are precursors to neurites when the cell differentiates. We tested the hypothesis that DEPDC5/NPRL3 KD leads to altered filopodial outgrowth in mNPCs compared with scramble control or wildtype cells. We used filamentous actin (F-actin) staining to define the number of primary filopodia emanating directly from the soma (Sainath and Gallo, 2015). After KD, mNPCs displayed a statistically significant increase in the number of filopodia extending from the soma versus scramble control or wildtype mNPCs (n=50; 25 cells from each of 2 replicates, p< 0.01, Fig. 5A, B). DEPDC5 or NPRL3 shRNA transfected mNPCs averaged approximately two filopodia per cell while scramble and wildtype averaged only 1.6 and 1.2 filopodia, respectively (Fig. 5B).
Figure 5. DEPDC5 or NPRL3 KD lead to enhanced filopodial number.
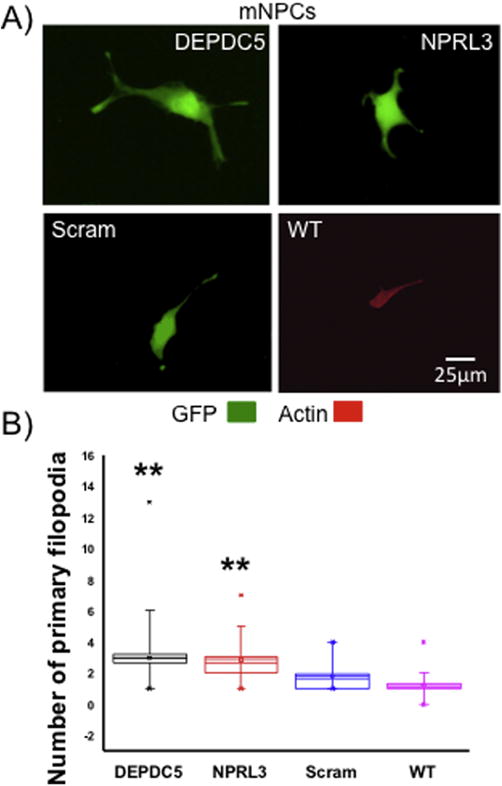
DEPDC5 or NPRL3 shRNA transfected mNPCs had higher numbers of filopodia (defined by actin labeling) than scramble control or wildtype mNPCs (n= 50 per group- 25 cells from each of 2 replicates; A and B). Filopodia outgrowth in scramble control and wildtype mNPCs was not significantly different. In (B), each box represents SEM with a mean line shown, whiskers reflect 5-95% confidence intervals. Scram= scramble control, GFP= green fluorescent proteins, WT= wildtype; ** = significant difference between scramble and WT control with a p < 0.01.
We hypothesized that the morphological and signaling changes resulting from DEPDC5 or NPRL3 KD were mTORC1-dependent and therefore would be prevented by application of rapamycin in vitro. Each cell type was transfected and treated daily with rapamycin, starting at the time of transfection, for 48 hrs and then imaged. Following DEPDC5/NPRL3 KD, rapamycin treated mNPCs displayed a reduction in soma diameter compared with vehicle treated cells (Fig. 6A,B; n=50, 25 cells from each of 2 replicates; see statistics in Table 1). Increased filopodia outgrowth following DEPDC5/NPRL3 KD was blocked by rapamycin (Fig. 6C and Table 1). In N2aC and HEK293FT cells, forward scatter FACS analysis (≈100,000 cells per group) showed a decrease in cell size following rapamycin treatment (Fig. 7 A,B).
Figure 6. Soma size and process outgrowth are mTOR-dependent.
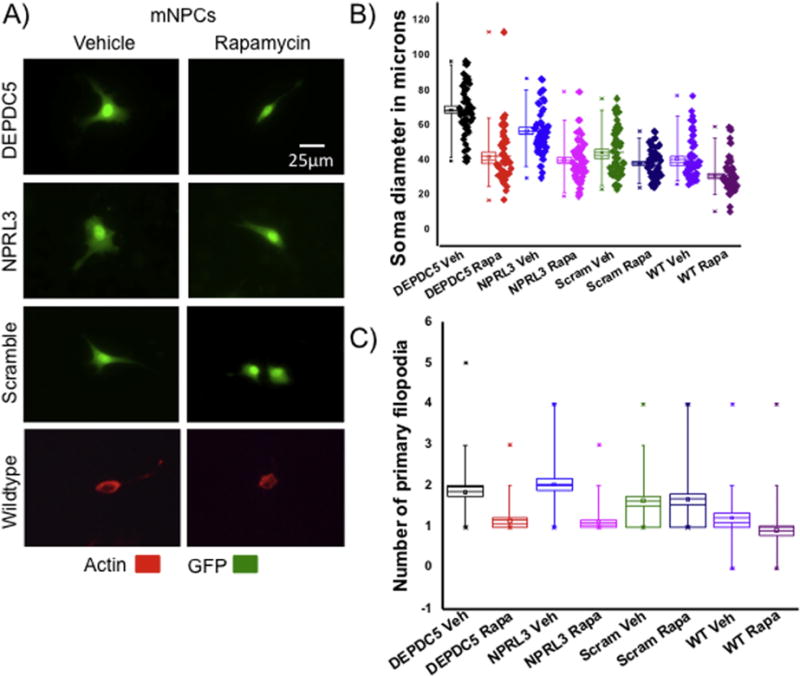
After rapamycin treatment (50nM, 48hrs), a decrease in soma diameter (B) and number of filopodia (C) was observed following DEPDC5/NPRL3 KD in mNPCs compared to scramble and wildtype mNPCs treated with vehicle (DMSO; representative images in A). Statistical significance of morphological changes using ANOVA can be found in Table 1 (n = 50 cells per group– 25 cells from each of 2 replicates). In (B) and (C), each box represents SEM with a mean line shown, whiskers reflect 5-95% confidence intervals. WT= wildtype cells, GFP= green fluorescent protein, veh = vehicle, rapa = rapamycin.
Table 1.
Statistical data for mNPCs treated with rapamycin versus vehicle treated cells.
| Group 1 | vs. Group 2 | p value: soma size | p value: # primary filopodia |
|---|---|---|---|
| DEPDC5 vehicle | DEPDC5 w/rapa | p < 0.001 | p < 0.001 |
| DEPDC5 vehicle | Scram vehicle | p < 0.001 | p < 0.05 |
| NPRL3 vehicle | NPRL3 w/rapa | p < 0.001 | p < 0.01 |
| NPRL3 vehicle | Scram vehicle | p < 0.001 | p < 0.01 |
| Scram vehicle | Scram w/rapa | p < 0.01 | Not Sig. |
| WT vehicle | WT w/rapa | p < 0.01 | p = 0.05 |
| Scram vehicle | WT vehicle | Not Sig. | Not Sig. |
Figure 7. Soma size is mTOR-dependent.
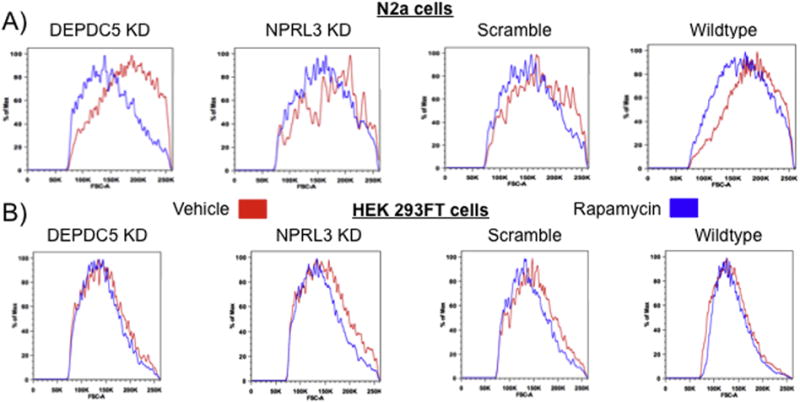
FACS forward-scatter analysis of N2aC (A) and HEK293FT cells (B) demonstrate a decrease in soma size after rapamycin treatment (50 nM; 48hrs; blue) following DEPDC5 or NPRL3 KD, and in scramble and wildtype cells versus vehicle control (DMSO; red).
Upon differentiation with retinoic acid, N2aC display MAP2 positive (MAP2+) processes and assume the characteristic morphology of neurons. Therefore we tested whether DEPDC5/NPRL3 KD could affect morphology in differentiated neuronal cells. After KD, N2aC neurons showed an increase in soma size and number of MAP2+ processes compared to scramble control and wildtype cells. These results indicate that DEPDC5/NPRL3 KD produces morphological changes in neural progenitor cells and differentiated neurons. Both of these morphological changes were reversible with rapamycin (Supplemental Fig. 1). In order to confirm that the morphological changes observed above were due directly to rapamycin-mediated reduction of mTORC1 phosphorylation of p70S6K (which phosphorylates S6), a direct inhibitor of p70S6K (PF-4708671; 10nM, 48 hrs), was used (Pearce et al., 2010; Srivastava et al., 2016). After PF-4708671 application to mNPCs, a decrease in soma size and filopodia outgrowth was observed (Supplemental Fig. 2; n=50 per group, 25 cells from each of 2 replicates) similar to treatment with rapamycin.
DEPDC5 or NPRL3 KD alters subcellular localization of mTOR in neural progenitor cells
mTORC1 is required to move from the cytoplasm to the lysosomal membrane in order to function properly (Sancak et al., 2010), a process that is modulated by intracellular amino acid levels and guided by GATOR1 and GATOR 2 complexes. In non-neural cell lines, the GATOR1 complex prevents localization of mTORC1 to the lysosomal membrane in the setting of low cellular levels of amino acids, thereby preventing mTOR action under conditions of nutrient deprivation. Indeed in HEK293T cells, DEPDC5 shRNA KD leads to enhanced localization of mTOR on the lysosomal membrane even when amino acids are absent (Bar-Peled, 2013). Thus, we hypothesized that DEPDC5/NPRL3 KD would lead to increased colocalization between mTOR and the lysosomal membrane in neural cell lines. DEPDC5/NPRL3 transfected, scramble transfected and wildtype N2aC were incubated in AAF or complete media for 50 minutes and then PFA fixed. Cells were dually fluorescently labeled with anti-mTOR and anti-LAMP2 antibodies and the fluorescence intensity of these proteins was quantified and assessed with a Pearson’s correlation test. We determined whether the highest fluorescence intensity in mTOR was correlated with the highest fluorescence intensity in LAMP2. Areas of pink, colocalization between mTOR (blue) and LAMP2 (red), represent the highest degree of colocalization; whereas areas of red (LAMP2) represent low degrees of colocalization. N2aC (n=10 per group) transfected with DEPDC5 or NPRL3 shRNA displayed a significantly increased colocalization between mTOR and LAMP2 compared to scramble control and wildtype cells (Fig. 8A). After DEPDC5/NPRL3 KD an increased average R value was observed (0.89; Fig. 8B; p< 0.01) compared to scramble control and wildtype cells (0.57 and 0.63, respectively; 3D ROI reconstruction, Fig. 8A, B). Wildtype N2aC in complete media displayed a similar correlation coefficient to DEPDC5/NPRL3 KD cells in AAF media (p< 0.01). These results demonstrate that loss of DEPDC5 or NPRL3 leads to inappropriate lysosomal localization of mTOR in neural cells in the absence of amino acids.
Figure 8. DEPDC5 or NPRL3 KD promote constitutive changes in subcellular mTOR localization and enhanced PS6 levels during amino acid deprivation.
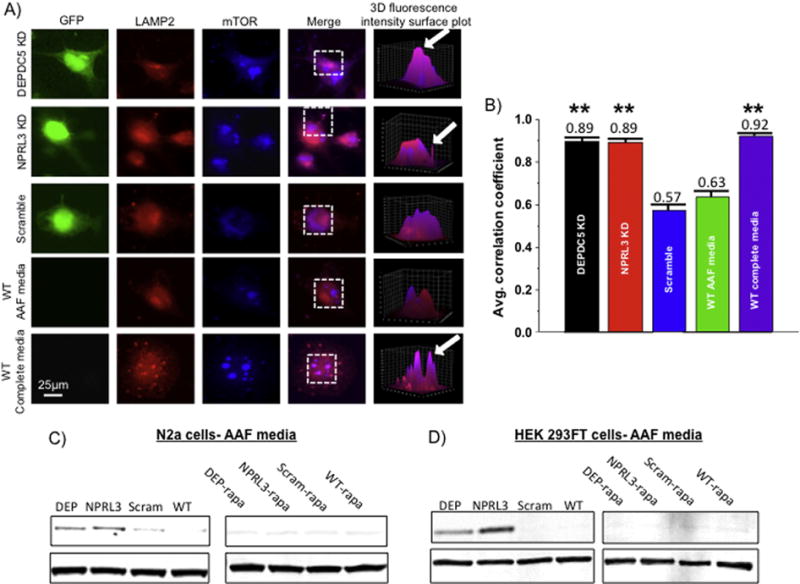
Colocalization analysis was performed on DEPDC5 KD, NPRL3 KD, scramble control, and wildtype N2aC in amino acid free (AAF) or amino acid replete (complete) media (A). A fluorescence intensity surface plot was generated (see 3D reconstruction) for each region of interest (dashed boxes). Areas of pink represent colocalization of mTOR (blue) and the lysosomal membrane protein 2 (LAMP2, red). To quantify fluorescence, a Pearson’s correlation coefficient (R) was calculated for each cell and then averaged across all cells in a group (n= 10 per group). Statistical significance was determined using ANOVA. mTOR and LAMP2 were colocalized after DEPDC5 or NPRL3 KD and incubation in AAF media (B). Reduced colocalization was observed in scramble control and wildtype N2aC after incubation in AAF media (B). High degrees of colocalization between mTOR and LAMP2 were observed in wildtype N2aC incubated in complete media (B). In AAF media, DEPDC5 and NPRL3 KD in both N2aC (C) and HEK293FT cells (D) resulted in elevated levels of PS6 while scramble control and wildtype cells display little or no S6 phosphorylation. Rapamycin application after KD and amino acid starvation dramatically reduced PS6 levels. Western blots performed in duplicate. Densitometry data are in Supplemental Fig. 5. LAMP2= lysosome associated membrane protein 2, mTOR= mechanistic target of rapamycin, GFP= green fluorescent protein, AAF= amino acid free. Area of 3D reconstruction (ROI) is noted by the dashed boxes in the ‘merge’ column. ** = significant difference between scramble and WT control with a p < 0.01.
Inappropriate mTOR activation during nutrient starvation following DEPDC5 or NPRL3 KD
GATOR1 is known to inhibit mTOR signaling as a means of cellular conservancy during amino acid starvation in non-neural cell types (Bar-Peled et al., 2013; Wei and Lilly, 2014). We thus hypothesized that DEPDC5/NPRL3 KD would lead to inappropriate mTOR activation despite amino acid starvation in N2aC. Following DEPDC5 or NPRL3 shRNA transfection, N2aC were fully amino acid starved for 90 minutes in AAF media. Western assay revealed enhanced S6 phosphorylation (Ser240/244) compared to scramble control and wildtype cells (Fig. 8C). Treatment of these cells with rapamycin during amino acid starvation reversed the effects on mTORC1 signaling and we observed decreased levels of PS6 compared to untreated, amino acid starved cells (Fig. 8). These data were corroborated in HEK 293FT cells after 90 min of amino acid starvation in AAF media followed by rapamycin treatment (Fig. 8D). These data demonstrate inappropriate activation of mTORC1 in the setting of nutrient deprivation in neural cells and reversal with rapamycin.
Discussion
We demonstrate that DEPDC5 and NPRL3 knockdown causes abnormal cellular morphology in two murine neural cell lines and that these morphological changes are mTOR pathway-dependent. We show that modulation of mTOR signaling in response to nutrient deprivation is disrupted following DEPDC5 or NPRL3 KD and that reduced DEPDC5 or NPRL3 leads to altered subcellular localization of mTOR at the lysosome. These effects on cell structure and signaling provide mechanisms by which DEPDC5 and NPRL3 mutations in developing human brain lead to abnormal cytoarchitecture observed in FCD (Baulac et al., 2015) and provide groundwork for clinical studies examining the efficacy of mTOR inhibitors in DEPDC5- or NPRL3-associated FCD.
DEPDC5- and NPRL3-associated focal malformations are rare causes of epilepsy that encompass a broad spectrum of epilepsy syndromes (Poduri, 2014), many defined by dysmorphic neurons displaying enhanced mTOR activation. We show that neurons labeled with PS6 antibodies are enlarged in tissues resected from patients with DEPDC5 or NPRL3 mutations and model this experimentally by DEPDC5 or NPRL3 KD in vitro. We demonstrate that mTOR inhibition or inhibition of the downstream p70S6 kinase can prevent cell soma enlargement in vitro. Indeed, an increase in PS6 levels and cytomegaly was seen following germline DEPDC5 knockout in the rat (Marsan et al., 2016). It is interesting to note that while soma size was increased after KD, the size of the nucleus was unchanged. These results suggest a specific effect on cytoplasmic rather than nuclear volume.
We also find mTOR-dependent increases in filopodia and dendritic outgrowth in mNPCs and differentiated N2aC following DEPDC5 or NPRL3 KD suggesting an early developmental effect on cell polarity and process outgrowth. These data fit well with previous work demonstrating that Tsc1 and Tsc2, upstream regulators of mTOR activation in response to growth factor stimulation, also regulate process outgrowth (Choi et al., 2008). For example, Tsc1 or Tsc2 knockout in vivo and in vitro leads to disrupted axon outgrowth with the formation of supernumerary axons in both hippocampal and cortical neurons. Conversely, filopodial outgrowth may play an important role in early neuronal migration. Abnormal filopodia outgrowth following DEPDC5 or NPRL3 KD may provide a partial explanation for abnormal laminar neuronal positioning caused by DEPDC5 or NPRL3 mutations in human FCD. For example, Tsc2 knockout mouse embryonic fibroblasts demonstrate altered migration (Goncharova et al., 2014) and mTOR pathway hyperactivation caused by Tsc2 knockout in mice promotes activation of the Dab-Reelin pathway (Moon et al., 2015) – a pathway integral to neuronal migration. Finally, knockout or KD of select mTOR genes including Lkb1 (Asada et al., 2007), Strada (Orlova et al., 2010), or Rheb (Lafourcade et al., 2013) leads to altered motility and migration of neurons both in vitro and in vivo suggesting that the mTOR pathway is a critical modulator of cell motility during brain development.
Amino acid concentrations in the developing brain may be dynamic and thus contribute to mTOR regulation via GATOR1. In the setting of DEPDC5 or NPRL3 mutations, constitutive mTOR activation in the setting of normally fluctuating amino acid levels could have deleterious effects on neuronal function. Indeed, NPRL2 and NPRL3 KD in Drosophila oocytes coupled with amino acid starvation resulted in apoptosis (Wei and Lilly, 2014). We show that while DEPDC5/NPRL3 KD results in increased localization of mTOR to the lysosome during amino acid starvation, the functional consequences of this can be prevented with rapamycin application.
DEPDC5 and NPRL3 play important roles in mTOR modulation in response to ambient amino acid levels and that further investigation of how amino acid levels fluctuate during cortical development seems warranted. An additional component of the cellular amino acid regulatory cascade, KICSTOR, was recently identified (Wolfson et al., 2017). As KICSTOR is responsible for guiding the GATOR1 complex to the lysosomal surface and KICSTOR subunits are associated with neurological disease (Pajusalu et al., 2015; Peng et al., 2017), further studies are required to elucidate its role in FCD and how it may affect neuronal morphology and function These studies may provide novel avenues of treatment for mTOR-associated cortical malformations by manipulating dietary amino acids. In addition, recent evidence suggests a link between NPRL2, another GATOR1 subunit, and FCD (Ricos et al., 2015) although whether NPRL2 plays a similar role in mTORC1 activation in neurons is unknown.
Of note, we did not observe a statistical difference between “scram” and “scram rapa” in our filopodia number counts (Table 1, Fig. 6). All cells were transfected under serum free conditions and therefore recruitment of mTOR to the lysosomal surface in non-KD cells is attenuated. Thus, rapamycin application produced a very small, non-significant, decrease in filopodia number. This is in contrast to DEPDC5 and NPRL3 KD under the same conditions where filopodia number is significantly increased. Subsequent rapamycin application produces significant changes in filopodia number.
We provide a conclusive link between impaired GATOR1 subunit function and mTOR pathway hyperactivity in neurons and neuronal progenitor cells. These data provide the groundwork for future studies examining how mTOR inhibitors, such as rapamycin, can be used to reduce, correct or prevent the deleterious consequences of focal malformations of cortical development.
Supplementary Material
Highlights.
Mutations in two GATOR1 subunits, DEPDC5 and NPRL3 have been linked with MCD associated with epilepsy.
We demonstrate that DEPDC5 or NPRL3 KD alters cell size, polarity, and morphology, as well as subcellular localization of mTOR in vitro in neuronal progenitor cells.
The morphological consequences of DEPDC5 or NPRL3 KD are mTORC1 dependent and preventable by the mTOR inhibitor rapamycin.
GATOR1 may provide a novel target for treatment of a number of epilepsy-associated MCD.
Acknowledgments
This work was supported by a postdoctoral fellowship award from the Shriners Hospitals for Children to PHI; Melbourne Children’s Clinician Scientist Fellowship to RJL; NHMRC Career Development Fellowship [GNT1032364], the Victorian Government’s Operational Infrastructure Support Program and Australian Government NHMRC IRIISS to PJL; National Institutes of Health [R01NS082343 and R01NS099452] and Citizens United for Research in Epilepsy to PBC.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest: None
References
- Asada N, Sanada K, Fukada Y. LKB1 regulates neuronal migration and neuronal differentiation in the developing neocortex through centrosomal positioning. J Neurosci. 2007;27:11769–11775. doi: 10.1523/JNEUROSCI.1938-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar-Peled L, Chantranupong L, Cherniack AD, Chen WW, Ottina KA, Grabiner BC, Spear ED, Carter SL, Meyerson M, Sabatini DM. A Tumor suppressor complex with GAP activity for the Rag GTPases that signal amino acid sufficiency to mTORC1. Science. 2013;340:1100–1106. doi: 10.1126/science.1232044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baulac S. mTOR signaling pathway genes in focal epilepsies. Prog Brain Res. 2016;226:61–79. doi: 10.1016/bs.pbr.2016.04.013. [DOI] [PubMed] [Google Scholar]
- Baulac S, Ishida S, Marsan E, Miquel C, Biraben A, Nguyen DK, Nordli D, Cossette P, Nguyen S, Lambrecq V, Vlaicu M, Daniau M, Bielle F, Andermann E, Andermann F, Leguern E, Chassoux F, Picard F. Familial focal epilepsy with focal cortical dysplasia due to DEPDC5 mutations. Ann Neurol. 2015;77:675–683. doi: 10.1002/ana.24368. [DOI] [PubMed] [Google Scholar]
- Blumcke I, Aronica E, Miyata H, Sarnat HB, Thom M, Roessler K, Rydenhag B, Jehi L, Krsek P, Wiebe S, Spreafico R. International recommendation for a comprehensive neuropathologic workup of epilepsy surgery brain tissue: A consensus Task Force report from the ILAE Commission on Diagnostic Methods. Epilepsia. 2016;57:348–358. doi: 10.1111/epi.13319. [DOI] [PubMed] [Google Scholar]
- Chantranupong L, Wolfson RL, Orozco JM, Saxton RA, Scaria SM, Bar-Peled L, Spooner E, Isasa M, Gygi SP, Sabatini DM. The Sestrins interact with GATOR2 to negatively regulate the amino-acid-sensing pathway upstream of mTORC1. Cell Rep. 2014;9:1–8. doi: 10.1016/j.celrep.2014.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi YJ, Di NA, Kramvis I, Meikle L, Kwiatkowski DJ, Sahin M, He X. Tuberous sclerosis complex proteins control axon formation. Genes Dev. 2008;22:2485–2495. doi: 10.1101/gad.1685008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desikan RS, Barkovich AJ. Malformations of cortical development. Ann Neurol. 2016 doi: 10.1002/ana.24793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goncharova EA, James ML, Kudryashova TV, Goncharov DA, Krymskaya VP. Tumor suppressors TSC1 and TSC2 differentially modulate actin cytoskeleton and motility of mouse embryonic fibroblasts. PLoS One. 2014;9:e111476. doi: 10.1371/journal.pone.0111476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iffland PH, Crino PB. Focal Cortical Dysplasia: Gene Mutations, Cell Signaling, and Therapeutic Implications. Annu Rev Pathol. 2017;12:547–571. doi: 10.1146/annurev-pathol-052016-100138. [DOI] [PubMed] [Google Scholar]
- Lafourcade CA, Lin TV, Feliciano DM, Zhang L, Hsieh LS, Bordey A. Rheb activation in subventricular zone progenitors leads to heterotopia, ectopic neuronal differentiation, and rapamycin-sensitive olfactory micronodules and dendrite hypertrophy of newborn neurons. J Neurosci. 2013;33:2419–2431. doi: 10.1523/JNEUROSCI.1840-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsan E, Ishida S, Schramm A, Weckhuysen S, Muraca G, Lecas S, Liang N, Treins C, Pende M, Roussel D, Le Van QM, Mashimo T, Kaneko T, Yamamoto T, Sakuma T, Mahon S, Miles R, Leguern E, Charpier S, Baulac S. Depdc5 knockout rat: A novel model of mTORopathy. Neurobiol Dis. 2016;89:180–189. doi: 10.1016/j.nbd.2016.02.010. [DOI] [PubMed] [Google Scholar]
- Moon UY, Park JY, Park R, Cho JY, Hughes LJ, McKenna J, III, Goetzl L, Cho SH, Crino PB, Gambello MJ, Kim S. Impaired Reelin-Dab1 Signaling Contributes to Neuronal Migration Deficits of Tuberous Sclerosis Complex. Cell Rep. 2015;12:965–978. doi: 10.1016/j.celrep.2015.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlova KA, Parker WE, Heuer GG, Tsai V, Yoon J, Baybis M, Fenning RS, Strauss K, Crino PB. STRADalpha deficiency results in aberrant mTORC1 signaling during corticogenesis in humans and mice. J Clin Invest. 2010;120:1591–1602. doi: 10.1172/JCI41592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pajusalu S, Reimand T, Ounap K. Novel homozygous mutation in KPTN gene causing a familial intellectual disability-macrocephaly syndrome. Am J Med Genet A. 2015;167A:1913–1915. doi: 10.1002/ajmg.a.37105. [DOI] [PubMed] [Google Scholar]
- Parmigiani A, Nourbakhsh A, Ding B, Wang W, Kim YC, Akopiants K, Guan KL, Karin M, Budanov AV. Sestrins inhibit mTORC1 kinase activation through the GATOR complex. Cell Rep. 2014;9:1281–1291. doi: 10.1016/j.celrep.2014.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce LR, Alton GR, Richter DT, Kath JC, Lingardo L, Chapman J, Hwang C, Alessi DR. Characterization of PF-4708671, a novel and highly specific inhibitor of p70 ribosomal S6 kinase (S6K1) Biochem J. 2010;431:245–255. doi: 10.1042/BJ20101024. [DOI] [PubMed] [Google Scholar]
- Peng M, Yin N, Li MO. SZT2 dictates GATOR control of mTORC1 signalling. Nature. 2017;543:433–437. doi: 10.1038/nature21378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poduri A. DEPDC5 does it all: shared genetics for diverse epilepsy syndromes. Ann Neurol. 2014;75:631–633. doi: 10.1002/ana.24160. [DOI] [PubMed] [Google Scholar]
- Ricos MG, et al. Mutations in the mTOR pathway regulators NPRL2 and NPRL3 cause focal epilepsy. Ann Neurol. 2015 doi: 10.1002/ana.24547. [DOI] [PubMed] [Google Scholar]
- Sainath R, Gallo G. Cytoskeletal and signaling mechanisms of neurite formation. Cell Tissue Res. 2015;359:267–278. doi: 10.1007/s00441-014-1955-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxton RA, Chantranupong L, Knockenhauer KE, Schwartz TU, Sabatini DM. Mechanism of arginine sensing by CASTOR1 upstream of mTORC1. Nature. 2016;536:229–233. doi: 10.1038/nature19079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scerri T, Riseley JR, Gillies G, Pope K, Burgess R, Mandelstam SA, Dibbens L, Chow CW, Maixner W, Harvey AS, Jackson GD, Amor DJ, Delatycki MB, Crino PB, Berkovic SF, Scheffer IE, Bahlo M, Lockhart PJ, Leventer RJ. Familial cortical dysplasia type IIA caused by a germline mutation in DEPDC5. Ann Clin Transl Neurol. 2015;2:575–580. doi: 10.1002/acn3.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheffer IE, Heron SE, Regan BM, Mandelstam S, Crompton DE, Hodgson BL, Licchetta L, Provini F, Bisulli F, Vadlamudi L, Gecz J, Connelly A, Tinuper P, Ricos MG, Berkovic SF, Dibbens LM. Mutations in mammalian target of rapamycin regulator DEPDC5 cause focal epilepsy with brain malformations. Ann Neurol. 2014;75:782–787. doi: 10.1002/ana.24126. [DOI] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sim JC, Scerri T, Fanjul-Fernandez M, Riseley JR, Gillies G, Pope K, van RH, Heng JI, Mandelstam SA, McGillivray G, MacGregor D, Kannan L, Maixner W, Harvey AS, Amor DJ, Delatycki MB, Crino PB, Bahlo M, Lockhart PJ, Leventer RJ. Familial cortical dysplasia caused by mutation in the mammalian target of rapamycin regulator NPRL3. Ann Neurol. 2016;79:132–137. doi: 10.1002/ana.24502. [DOI] [PubMed] [Google Scholar]
- Srivastava IN, Shperdheja J, Baybis M, Ferguson T, Crino PB. mTOR pathway inhibition prevents neuroinflammation and neuronal death in a mouse model of cerebral palsy. Neurobiol Dis. 2016;85:144–154. doi: 10.1016/j.nbd.2015.10.001. [DOI] [PubMed] [Google Scholar]
- Switon K, Kotulska K, Janusz-Kaminska A, Zmorzynska J, Jaworski J. Molecular neurobiology of mTOR. Neuroscience. 2017;341:112–153. doi: 10.1016/j.neuroscience.2016.11.017. [DOI] [PubMed] [Google Scholar]
- Wei Y, Lilly MA. The TORC1 inhibitors Nprl2 and Nprl3 mediate an adaptive response to amino-acid starvation in Drosophila. Cell Death Differ. 2014;21:1460–1468. doi: 10.1038/cdd.2014.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfson RL, Chantranupong L, Wyant GA, Gu X, Orozco JM, Shen K, Condon KJ, Petri S, Kedir J, Scaria SM, Abu-Remaileh M, Frankel WN, Sabatini DM. KICSTOR recruits GATOR1 to the lysosome and is necessary for nutrients to regulate mTORC1. Nature. 2017;543:438–442. doi: 10.1038/nature21423. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.