

Abstract
Background
High-mobility group box 1 (HMGB1), a common extracellular damage associated molecular pattern molecule, is overexpressed in several solid tumors including pancreatic carcinoma. We previously observed that radiotherapy induced dying cells secrete HMGB1 and accelerate pancreatic carcinoma progression through an unclear mechanism.
Methods
Using the Millicell system as an in vitro co-culture model, we performed quantitative reverse transcriptase-polymerase chain reaction, western blot and sphere forming ability analyses to access the effect of dying-cell-derived HMGB1 on CD133+ cancer cell stemness in vitro and in vivo. Interactions between HMGB1 and Toll-like receptor 2(TLR2)/TLR4 were studied by co- immunoprecipitation. Western blot and short-hairpin RNA-based knockdown assays were conducted to detect HMGB1 and TLR2/TLR4 signaling activity.
Findings
Radiation-associated, dying-cell-derived HMGB1 maintained stemness and contributed to CD133+ cancer stem cell self-renewal in vitro and in vivo. In overexpressing and silencing experiments, we demonstrated that the process was activated by TLR2 receptor, whereas TLR4 antagonized HMGB1-TLR2 signaling. Wnt/β-catenin signaling supported the HMGB1-TLR2 mediated stemness of CD133+ cancer cells.
Interpretation
Our results show how irradiation-induced cell death might enhance the stemness of resident cancer cells, and indicate HMGB1-TLR2 signaling as a potential therapeutic target for preventing pancreatic cancer recurrence.
Keywords: HMGB1, Cancer stem cells, Radiotherapy, TLR2, TLR4
Graphical abstract
Highlights
-
•
Dying cells derived HMGB1 induces cancer cell stemness by radiotherapy.
-
•
TLR2 mediates paracrine HMGB1 to promote self-renewal of cancer stem cells, whereas TLR4 displays antagonistic effect.
-
•
Wnt/β-catenin signaling is involved in HMGB1-TLR2 mediated cancer stemness.
Research in context.
Evidence before this study
HMGB1, an extracellular damage-associated molecular pattern molecule, is overexpressed in several solid tumors including pancreatic carcinoma. Several recent studies implicated extracellular HMGB1 as being highly associated with cancer cell proliferation, resistance to cell death, angiogenesis, metastasis and invasion. Following anti-cancer therapy, dying cells are thought to secrete growth factors that stimulate the regrowth of neighboring resident cancer cells. In this study, we aimed to explore the role and regulation of dying-cell-derived HMGB1 on cross-talk between resident pancreatic cancer stem cells and the necrosis niche following radiotherapy.
Added value of this study
Extracellular HMGB1 is highly associated with each of the hallmarks of cancer. In this study, we investigated the role of dying-cell-derived HMGB1 on the stemness of CD133+ cells. TLR2 and TLR4 are the receptors responsible for HMGB1-mediated biological effects. Thus, we also determined the role of TLR2/TLR4 on maintaining CD133+ cell stemness and uncovering the underlying regulatory mechanism.
Implications of all the available evidence
We found that dying-cell-derived HMGB1 induced cancer cell stemness by radiotherapy. TLR2 mediated paracrine HMGB1 signaling to promote the self-renewal of cancer stem cells, whereas TLR4 displayed an antagonistic effect. Wnt/β-catenin signaling is involved in HMGB1-TLR2 mediated cancer cell stemness. Our results open the possibility to develop new effective strategies to prevent tumor repair in patients with residual disease. Combining radiotherapy and HMGB1 inhibitor, or targeting TLR2 induced cancer stem cell self-renewal may be considered for future therapeutic protocols.
Alt-text: Unlabelled Box
1. Introduction
Pancreatic carcinoma (PC) constitutes the fourth leading cause of cancer mortality [1]. Locally advanced PC is not suitable for curative surgery and is typically treated with either chemotherapy alone or chemotherapy combined with or radiotherapy [2]. The purpose of radiotherapy is to promote tumor cell death. However, mounting publications have demonstrated that dying tumor cells can induce the accelerated regrowth of the resident cancer cells and form a special microenvironment for tumor recurrence [[3], [4], [5]], making the role of radiotherapy in treating of PC controversial [6].
Cancer stem cells (CSCs) represent one plausible explanation for PC tumor radio-resistance. CSCs are undifferentiated cancer cells with unlimited ability for self-renew and differentiating into tumor cells to maintain the long-term tumorigenic potential [7,8]. Importantly, CSCs are more resistant to conventional cancer therapies, which may ultimately result in an enhanced invasive and migratory potential, making the cancer prone to metastatic disease progression [[9], [10], [11]]. CSCs are not only regulated by intrinsic factors, but are also mediated by conditions in their local microenvironments (niches), such as peri-vasculature, hypoxia and necrosis [12]. Although the necrosis niche (the intratumor microenvironment rich in dying cells, debris and their released soluble factors[13,14]) is essential for the maintenance and regeneration of CSCs, the mechanisms that enable CSCs to survive in such harsh conditions are still not understood.
Following radiotherapy, the necrosis niche is common in solid tumors. Soluble factors released from apoptotic cells into the necrosis niche are vital in mediating resident cancer cell repopulation and anti-apoptosis. The HMGB1, an extracellular damage associated molecular pattern (DAMP) molecule, is overexpressed in several solid tumors including pancreatic carcinoma [[15], [16], [17]]. HMGB1 can translocate from the nucleus to the extracellular space during cell death. As an extracellular signaling molecule, HMGB1 can bind and activate a variety of signaling transduction cell receptors, including (TLR-2, TLR-4, and TLR-9), receptor for advanced glycosylation end product (RAGE), and certain integrins that can act alone or in combination with other immune stimulants[18,19]. HMGB1 exhibits a dual role in tumor behavior [16,20,21]. As a protumor protein, extracellular HMGB1 is highly associated with cancer cell proliferation, resistance to cell death, resistance to therapy, invasion and metastasis [22]. It was recently reported that cancer-associated fibroblast derived HMGB1 maintained the stemness of breast CSCs [23]. This finding suggested that HMGB1 may modulate cancer invasion and metastasis by activating CSCs.
As radiotherapy can induce necrotic cell death, we are interested if extracellular HMGB1 mediates cross-talk between resident pancreatic CSCs and the necrosis niche following radiotherapy. CD133+ cancer cells, known for their abilities for self-renewal and giving rise to CD133 negative (CD133−) cells, were used as a CSC model in this study. We found that silencing HMGB1 in the feeder-irradiated cancer cells attenuated the sphere forming ability of CD133+ CSCs. We showed that HMGB1 affected cell stemness in a paracrine manner to that depended on TLR2 receptor, whereas TLR4 displayed certain antagonistic effect. Moreover, the Wnt/β-catenin signaling axis was involved in the process. Aberrant Wnt signaling has been tightly associated with regulating pancreatic cancer development and maintaining CSCs [24,25]. Taken together, our findings demonstrate that dying cells after radiotherapy induce and maintain the stemness of surviving CSCs by activating HMGB1-mediated paracrine signaling events, both in vitro and in vivo.
2. Materials and methods
2.1. Cell culture
Human pancreatic cancer cell lines (SW1990, Panc1, and AsPC1) were obtained from the Cell Bank of the China Academy of Sciences (Shanghai, China). Panc1 cells and SW1990 cells were cultured as previously described [28]. The AsPC1 cell line, derived from metastatic human pancreatic carcinoma, was maintained in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS), 100 U/mL penicillin and 100 U/mL streptomycin. The cells were maintained in a humidified atmosphere with 5% CO2 at 37 °C and passaged with 0.25% trypsin/EDTA every 3 days. The cells used in this study were passaged for <2 months after recovery.
2.2. In vitro and in vivo irradiation
Pancreatic cancer cells cultured in 6-cm dishes or Millicell inserts were irradiated at room temperature using an X-ray irradiator (Linear accelerator, Turebeam_STX, Varian, USA) with the indicated dose (4, 8, 10, or 12Gy). Corresponding controls were sham irradiated. Irradiated cells were immediately trypsinized and re-seeded for further use.
SW1990 cells (1 × 106) were injected subcutaneously into the right dorsal flanks of nude mice (n = 3) to establish the xenograft tumor model. When the tumor reached volume of 1 mm3, the nude mice were received 4 Gy of radiation every two days for three times. Five days following the last irradiation, the tumor tissues were dissected and dissolved into the single cell suspension. The Tumor Cell Isolation Kit(Shenzhen LabLit Co., Ltd., Shenzhen, China) was used to remove contaminating cells (mouse stroma cells and debris cells), according to the manufacturer's instructions. The percentage of CD133+ was determined by flow cytometry.
2.3. In vitro co-culture system
Segregated irradiated cancer cells and untreated cancer cells co-cultures were set as follows: 5 × 104 irradiated parental cancer cells or HMGB1− cancer cells were seeded on 0.4-μm inserts (Millicell) in Dulbecco's modified Eagle's medium (DMEM) with 10% FBS. After 12 h, the inserts were moved to 24-well plates containing indicated number untreated cancer cells/well in DMEM with 2% FBS. Different concentrations of rhHMGB1 (50, 100, 150, and 200 ng/mL) were added to the medium mentioned above in the inserts as a positive control. Empty inserts with the same medium were used as control.
2.4. Flow cytometry and fluorescent-activated cell sorting (FACS)
CD133 staining was performed as described previously [43]. Data were exported and graphed using FCS Express (DeNovo Software). To separate the CD133+ population by FACS, pancreatic cancer cells growing in SFM system were stained for CD133 expression. Cancer cells were incubated with trypsin–EDTA, dissociated and passed through a 40 μm sieve. Cells were pelleted by centrifugation at 500 ×g for 5 min at 4 °C, resuspended in 100 μL of monoclonal mouse anti-human CD133/PE antibody (1:50, catalog number:#130-110-962, Miltenyi Biotechnology, Germany), and incubated for 20 min at 4 °C. The sorting gates were established using cells stained with isotype-control PE-conjugated antibodies (BD Pharmingen). Sorted CD133+ cells were subjected to sphere-forming culture system for further use.
2.5. Quantitative real-time PCR
Total RNA was extracted from CD133+ cells, CD133− cells, HMGB1-knockdown cells and their respective parental cancer cells(with or without indicated treatment) using the RNeasy Kit (Qiagen). For mRNA analysis, cDNA was synthesized from 1 μg total RNA using the RevertAid RT Reverse Transcription Kit (Thermo Fisher Scientific). SYBR Green-based real-time PCR was subsequently performed in triplicate using the SYBR Green master mix (Thermo Fisher Scientific) on an Applied Biosystems StepOnePlus real-time PCR machine (Thermo Fisher Scientific). For analysis, the threshold cycle (Ct) values for each gene were normalized to those of GAPDH. The sequences of the primers used are shown in Table 1. (See Table 2, Table 3, Table 4.)
Table 1.
Primers used for quantitative real-time PCR.
| Name | Direction | Sequence (5′-3′) |
|---|---|---|
| HMGB1 | Forward | 5′-ACATCCACATACAGCCATTGC-3′ |
| Reverse | 5′-GGCAAGGATAGTGGTGTTGGA-3′ | |
| Oct4 | Forward | 5′-AAAGCAGAAACCCTCGT-3′ |
| Reverse | 5′-TCCAGGTTGCCTCTCAC-3′ | |
| Sox2 | Forward | 5′- CCCCTGGCATGGCTCTTGGC -3′ |
| Reverse | 5′- TCGGCGCCGGGGAGATACAT -3′ | |
| Nanog | Forward | 5′-GAGACAGAAATACCTCAGCC-3′ |
| Reverse | 5′-TCTGCGTCACACCATTG-3′ | |
| TLR2 | Forward | 5′-GCCTCTCCAAGGAAGAATCC-3′ |
| Reverse | 5′-TCCTGTTGTTGGACAGGTCA-3′ | |
| TLR4 | Forward | 5′- AAGCCGAAAGGTGATTGTTG -3′ |
| Reverse | 5′- CTGAGCAGGGTCTTCTCCAC -3′ | |
| LEF1 | Forward | 5′-AGGAACATCCCCACACTGAC-3′ |
| Reverse | 5′-AGGTCTTTTTGGCTCCTGCT-3′ | |
| Axin2 | Forward | 5′-CAAACTTTCGCCAACCGTGGTTG-3′ |
| Reverse | 5′-GGTGCAAAGACATAGCCAGAACC-3′ |
Table 2.
Sequences of shRNAs.
| Name | Sequence |
|---|---|
| HMGB1-shRNA1 | 5′-CCGGCCCAGATGCTTCAGTCAACTTCTCGAGAAGTTGACTGAAGCATCTGGGTTTTTG-3′ |
| HMGB1-shRNA2 | 5′-CCGGCCGTTATGAAAGAGAAATGAACTCGAGTTCATTTCTCTTTCATAACGGTTTTT-3′ |
| TLR2-shRNA1 | 5′-CCGGGCATCTGATAATGACAGAGTTCTCGAGAACTCTGTCATTATCAGATGCTTTTTG-3′ |
| TLR2-shRNA2 | 5′-CCGGGCACACGAATACACAGTGTAACTCGAGTTACACTGTGTATTCGTGTGCTTTTTG-3′ |
| TLR4-shRNA1 | 5′-CCGGCCAAGTAGTCTAGCTTTCTTACTCGAGTAAGAAAGCTAGACTACTTGGTTTTTG-3′ |
| TLR4-shRNA2 | 5′-CCGGCCCTGCTGGATGGTAAATCATCTCGAGATGATTTACCATCCAGCAGGGTTTTTG-3′ |
| β-catenin-shRNA1 | 5′-CCGGGCCATATTGCTCCAGGACAATCTCGAGATTGTCCTGGAGCAATATGGCTTTTTG-3′ |
| β-catenin-shRNA2 | 5′-CCGGCGAGGAGAGATCATCGACAATCTCGAGATTGTCGATGATCTCTCCTCGTTTTTG-3′ |
Table 3.
Tumorigenicity of CD133+ pancreatic cancer cells from mice treated with rhHMGB1 or a vehicle control.
| Injected cells | Treatment | Number of mice with tumors/total injected mice (% of mice with tumors) |
|||
|---|---|---|---|---|---|
| TLR2+ | TLR2− | TLR4+ | TLR4− | ||
| 1000 | PBS | 2/8(25) | 2/8(25) | 2/8(25) | 2/8(25) |
| rhHMGB1 | 4/8(50) | 2/8(25) | 2/8(25) | 4/8(50) | |
| (P = .608) | (P = 1.000) | (P = 1.000) | (P = .608) | ||
| 10,000 | PBS | 2/8(25) | 3/8(37.5) | 3/8(37.5) | 2/8(25) |
| rhHMGB1 | 6/8(75) | 2/8(25) | 3/8(37.5) | 6/8(75) | |
| (P = .132) | (P = 1.000) | (P = 1.000) | (P = .132) | ||
| 100,000 | PBS | 3/8(37.5) | 5/8(62.5) | 4/8(50) | 4/8(50) |
| rhHMGB1 | 6/8(75) | 4/8(50) | 3/8(37.5) | 6/8(75) | |
| (P = .315) | (P = 1.000) | (P = 1.000) | (P = .608) | ||
Table 4.
Tumorigenicity of TLR4+CD133+, TLR4−CD133+, TLR2+CD133+ and TLR2−CD133+ pancreatic cancer cells from mice treated with rhHMGB1.
| Treatment | Number of mice with tumors/total injected mice (% of mice with tumors) |
||||
|---|---|---|---|---|---|
| TLR2+ | TLR2− | TLR4+ | TLR4− | ||
| PBS | 7/24 (29.2) | 10/24 (41.7) | 9/24 (37.5) | 8/24 (33.3) | P = .890 |
| rhHMGB1 (150 ng/mL) | 16/24 (66.7) | 8/24 (33.3) | 8/24 (33.3) | 16/24 (66.7) | P = .014 |
| P = .020 | P = .766 | P = 1.000 | P = .042 | ||
2.6. Immunofluorescence analyses
Pancreatic cancer cells were fixed in 4% paraformaldehyde and blocked with 5% bovine serum albumin (BSA). Mouse monoclonal anti-HMGB1 (1:500, Abcam Company) was used as a primary antibody and detection was achieved using a Cy3-conjugated goat anti-mouse IgG (1:50, BA1032; Boster Biotech, Wuhan, China) and DAPI (Boster Biotech, Wuhan, China). All samples were examined under a laser confocal-scanning microscope. Image quantification was performed by assessing 20× high-power fields in a blinded manner.
2.7. In vitro sphere-forming assay
Pancreatic cancer cells were seeded at clonal densities into ultra-low adhesion plates (Corning, NY, USA) and suspended in DMEM/F12 with 20 ng/mL epidermal growth factor, 10 ng/mL basic fibroblast growth factor, NAAS (Thermo Fisher Scientific), 100 U/mL penicillin and 100 U/mL streptomycin for 2 weeks to allow sufficient time for spheres to form from single cells. The culture medium was replaced with fresh medium containing the indicated fresh reagents every day for 2 weeks. After 2 weeks, the number and size of spheres in each well were quantified.
2.8. Drug treatment
rhHMGB1 (HMG Biotech, Germany) was dissolved in distilled water to prepare a 1000 ng/mL stock solution. After the cells reached 80% confluency, they were incubated with rhHMGB1 (50, 100, 150, 200 ng/mL) for the indicated time and analyzed, as described below. Stevioside (a TLR2 antagonist, TOPSCIENCE, China) and TAK-242(a TLR4 antagonist, MedChemExpress, USA) were dissolved in dimethyl sulfoxide (DMSO). The cells were grown to 80% confluency, treated with 2 μM Stevioside or TAK-242 for 48 h and subjected to the following experiments.
2.9. RNAi and gene transfection
Pancreatic cancer cells RNAi and gene transfection were performed as previously described [28].
2.10. Gene transduction
The mammalian expression plasmids pCMV-Flag-TLR2 (PPL00524-2a) and pcDNA3.1-Myc-TLR4 (PPL00104-2a) were purchased from the Public Protein/Plasmid Library (PPL, Nanjing, China). Cells were transfected with the stated constructs according the manufacturer's instructions (Invitrogen, China).
2.11. Enzyme-linked immunosorbent assay (ELISA) analysis
To measure HMGB1 levels in the supernatants, ELISA was performed as previously described [28].
2.12. Co-IP assay
SW1990 cells (5 × 106/10-cm dish) were plated in 6-cm dishes and at a density of 1 × 105 cells/well to achieve a confluence of 70–80% overnight. Later, the cells were transfected using the Lipofectamine 2000 reagent (Invitrogen, Mississauga, Ontario, Canada) and 4–6 μg of plasmid DNA per dish. rhHMGB1 (150 ng/mL) was added 24 h later. Cells were dislodged from the dish by flushing with cold PBS, collected by centrifugation, and lysed in ice-cold buffer (50 mM Tris-HCl at pH 7.4, 20% glycerol, 1 mM EDTA, 150 mM NaCl, 0.5% Triton X-100, 0.02% SDS, 1 mM dithiothreitol, 2 mM phenylmethylsulfonyl fluoride, 1 μg/mL aprotinin, 10 μg/mL pepstatin, and 1 μg/mL leupeptin). After 5 min, the final concentration wad adjusted to 400 nM with 5 M NaCl. After another 5 min on ice, an equal volume of ice-cold water was added and thoroughly mixed before immediate centrifugation in a microfuge (12,000 rpm, 10 min). Supernatants were collected and further used for IP. Lysates prepared from 60 mm dish were mixed with monoclonal antibodies (per IP: anti-Flag [Sigma], 2 μg; anti-HMGB1 [Abcam], 0.6–1.0 μg; anti-TLR2 [CST], 0.6–1.0 μg; 5–6 h at 4 °C while rocking on a nutator). During the final hour, 15 μL of protein G-Sepharose beads (settled volume) was added to each tube (beads were pre-blocked overnight with 10% BSA in PBS). The beads were washed three times with 1:1 diluted lysis buffer; proteins were eluted with Laemmli loading buffer and analyzed by SDS-PAGE and immunoblotting.
2.13. Western blot analysis
Protein concentrations were determined by bicinchoninic acid (BCA) method. Western blotting assay was performed as described previously [43]. Antibody against TLR4 was purchased from Abcam Company (Cambridge, USA). Antibodies against TLR2, GAPDH, Oct4, Sox2 and Nanog were purchased from Santa Cruz Biotechnology (USA). Antibodies against HMGB1, CD133, Sox2, CD44, c-myc, GSK3β, p-GSK3β, β-catenin, Myc, and Flag were obtained from Cell Signaling Technology, Inc. (USA). Secondary antibody (either anti-rabbit or anti-mouse) were purchased from Boster Biotechnology Company (China).
2.14. Xenograft tumor models
Animal studies were approved by the Committee on the Use of Live Animals for Teaching and Research of Jiangsu University. Four-week-old female BALB/c nude mice (purchased from The Compare Medicine Center, Yangzhou University, China) were maintained under standard conditions according to the institutional guidelines.
Sorted CD133+ SW1990 cancer cells were co-cultured with irradiated parental cancer cells (HMGB1+ cells), rhHMGB1(150 ng/mL), irradiated HMGB1− cells (HMGB1 shRNA1 and HMGB1 shRNA2), or empty medium(control group) for 5 days. Then, 1 × 106 treated cancer cells were implanted subcutaneously into the right dorsal flanks of nude mice. The tumor growth speed and volume were monitored every two days until the end point at day 27.
TLR2-overexpressing CD133+ SW1990 cells (TLR2+), TLR2 silenced CD133+ SW1990 (TLR2−), TLR4-overexpressing CD133+ SW1990 cells (TLR4+), and control CD133+ SW1990 cells (TLR4−) were treated with rhHMGB1(150 ng/mL) or PBS for 5 days. For in vivo tumor-initiation assays, the above cultured cancer cells were transplanted subcutaneously with 1 × 103, 1 × 104, or 1 × 105 cells/mouse into the right subcutaneous flank of nude mice (four mice per group). Mice were monitored every day until the end point on day 34, when palpable tumors were taken as a positive. The mice were euthanized, and excised fresh tumor tissues were measured.
2.15. Patient selection
TCGA data (https://tcga-data.nci.nih.gov/tcga; TCGA BRCA exp. HiSeqV2 PANCAN-2014-05-02) including those from 183 pancreatic carcinoma patient specimens were utilized to further analyze the relationship between HMGB1, TLR2, TLR4, CD133, CD44, Oct4, Nanog, and β-catenin. High and low groups were defined as above and below the mean respectively.
2.16. Statistical analysis
All data are presented as the mean ± standard error of the mean (SEM). Linear regression and F testing were used to determine correlation between CD133, CD44, Oct4, Nanog, and β-catenin expression and HMGB1, or HMGB1 receptor (TLR4 and TLR2) protein levels in pancreatic cancer. Two-tailed Mann–Whitney U tests were used to compare the statistical differences between the treatment groups in vivo. Fisher exact test was used to compare the tumorigenesis. The significances of differences between groups were analyzed using Student's t-tests, one-way analysis of variance (ANOVA) or two-way ANOVA. P < .05 was considered to reflect a statistically significant difference. All the experiments were repeated at least three times.
3. Results
3.1. Irradiation enriched the stemness of survival cancer cells
To examine whether radiotherapy of pancreatic cancer enriches for CSCs, pancreatic cancer cells (SW1990, Panc1, and AsPC1 cells) were irradiated with clinically relevant doses (4, 8, and 10 Gy) in vitro[26,27]. Flow cytometry was used to analysis temporal changes in CD133+ cancer cells at 24 h, 48 h, and 72 h post-irradiation, which showed that irradiation increased the percentage of CD133+ cells in all three pancreatic cancer cell lines with a peak time of 48 h (Fig. 1a and Fig. S1a). The three cell lines responded differently to radiotherapy, as 7–10-fold induction in SW1990 cells, 4–7-fold induction in Panc1 cells, and 2–4-fold induction in AsPC1 cells were observed at 48 h post-irradiation (Fig. 1a). Western blot and qRT-PCR data also showed increased protein- and mRNA-expression levels of CD133, Oct4, Sox2 and Nanog upon radiation, with peak signals measured at 48 h post 8 Gy irradiation (Fig.1b and c). The extreme limiting dilution assay is the gold standard assay for determining CSCs in vivo. SW1990 (1 × 104) cells with or without irradiation (8 Gy) were injected into the right flank subcutaneously of the nude mouse, respectively. Irradiation cancer cells (8 Gy) displayed higher tumorigenicity than the control group (Fig. S1b). Thus, both the in vitro and in vivo results demonstrated that irradiation enriched the CSC population.
Fig. 1.

X-ray irradiation enriched the stemness of surviving cancer cells in vitro and in vivo. (a) Established pancreatic cancer cell lines (SW1990, Panc1, and AsPC1) were exposed to various doses (0, 4, 8, and 10 Gy) X-ray irradiation and analyzed for CD133+ cancer cells by flow cytometry at 0, 24, 48, 72 h post treatment. (b) Western blot analysis of the protein expression levels of stem cell related markers (CD133, Oct4, Sox2, and Nanog) in pancreatic cancer cell lines (SW1990, Panc1, and AsPC1) at 48 h post-treated with various doses (0, 4, 8, or 10 Gy) X-ray irradiation. GAPDH expression was detected as a loading control. (c) qRT-PCR analysis of stem cell-related markers (CD133, Oct4, Sox2, and Nanog) in pancreatic cancer cell lines (SW1990, Panc1, and AsPC1) at 48 h post-treated with various doses (0, 4, 8, or 10 Gy) X-ray irradiation. GAPDH was detected as a loading control. (d) Schematic representation of the in vivo irradiation protocol. SW1990 cancer cells were used to establish subcutaneous tumors. When the tumors reached a volume of 1mm3, the nude mice received 4 Gy X-ray irradiation every 2 d for 6 d. Fresh tumor tissues were harvested 5 d after the last irradiation. (e) Fresh tumor tissues were digested into single cell suspension and the percentages of CD133+ were accessed by flow cytometry. Experiments were repeated three times, and the data are expressed as the mean ± SEM. *P < .05, **P < .01, ***P < .001. Statistical analysis was performed using Student's t-test (c, e right).
Fig. S1.

Irradiation-enriched CSCs. (a) The gating strategy used for analyzing CD133+ cancer cells by flow cytometry. Established pancreatic cancer cell lines (SW1990, Panc1, and AsPC1) were exposed to various doses (0, 4, 8, of 10 Gy) of X-ray irradiation for 0, 48, or 72 h. (b) Extreme limiting-dilution assays were performed to assess the subcutaneous tumorigenic ability of SW1990 cells treated with or without irradiation (8 Gy) in nude mice at 30 days post-injection. Experiments were repeated three times, and the data are expressed as the mean ± SEM (a). *P < .05, **P < .01, ***P < .001.
To exclude the impact of two dimensional culturing conditions and to further confirm the relationship between irradiation and cancer stemness in vivo, a subcutaneous xenograft tumor model was established by injecting 1 × 106 SW1990 cancer cells into the right flank of each nude mouse. Tumor-bearing nude mice received X-ray irradiation when the tumor reached 1 mm3. Five days following the last irradiation, the freshly harvested tumor tissues were immediately dissected for FACS analysis (Fig. 1d). The results showed that X-ray irradiation markedly increased the percentage of CD133+ cells (4.82 ± 0.7% at 0Gy vs. 17.68 ± 1.63% at 4Gy, Fig. 1e), suggesting that irradiation can enrich the CD133+ cancer cells and the stemness of survival cancer cells in vivo.
3.2. Dying cells released HMGB1 following irradiation
Following radiotherapy, many inflammatory factors are released from dying cells, which may drive cancer cell stemness. We previously reported that HMGB1 was released from irradiation-induced dying cells in a time dependent manner [28]. We detected basal HMGB1 expression in the pancreatic cancer cell lines SW1990, Panc1, and AsPC1, with highest HMGB1 expression (both in term of mRNA and protein levels) in Panc1 cells, and lowest in Aspc1 cells (Fig. 2a and b). By performing CCK8 and Annexin V-FITC/PI assays, we accessed the cell death induced by various X-ray doses (0, 4, 8, or 10 Gy) at 48 h post-irradiation and found that cell death occurred in a dose-dependent manner (Fig. S2a and b). Our preliminary experiments showed that 12 Gy X-ray irradiation was lethal as it induced cell death in 50% of the pancreatic cancer cells. Thus, we chose dosed of 4, 8, 10 Gy to evaluate the role of HMGB1 in activating CD133+ CSCs during irradiation. The results showed that irradiation elevated HMGB1 protein expression, with peak induced by 8 Gy irradiation (Fig. 2c). Since HMGB1 needs to translocate from the nucleus into extracellular compartments to function as a pro-inflammatory mediator, we then treated pancreatic cancer cells with 8 Gy irradiation and re-cultured cells for 0, 6, 12, 24, 36, 48, 60, or 72 h. Subsequently, we checked HMGB1 protein expression in the irradiated cancer cells and in the cell culture supernatants. Irradiation induced HMGB1 expression in pancreatic cancer cells (Fig. 2d), and HMGB1 was gradually released into the cultured supernatant (with a peak period between 36 h to 60 h) in all three cell lines (Fig. 2e). HMGB1 translocation from the nucleus into the cytoplasm or extracellular compartment of pancreatic cancer cells was also visualized by immunofluorescence (Fig. 2f), which further confirmed that irradiated dying cancer cells released HMGB1 into the extracellular space in vitro.
Fig. 2.

HMGB1 was released from dying cells in vitro. (a, b) Western blot and qRT-PCR analysis of the baseline protein and mRNA levels of HMGB1 in pancreatic cancer cell lines (SW1990, Panc1, and AsPC1). GAPDH expression was detected as loading control. (c) Western blot analysis of the protein expression levels of HMGB1 in pancreatic cancer cells lines (SW1990, Panc1, and AsPC1) at 24 h post-irradiation (0, 4, 8, or 10 Gy). GAPDH was detected as a loading control. (d) Western blot analysis of HMGB1 protein-expression levels in irradiated (8 Gy) cancer cells (SW1990, Panc1, and AsPC1) at 0, 6, 12, 24, 36, 48, or 72 h post-treatment. GAPDH was detected as a loading control. (e) ELISA-based analysis of the HMGB1concentration in the culture supernatants of 8-Gy-irradiated cancer cells (SW1990, Panc1, and AsPC1) at 0, 6, 12, 24, 36, 48, 72 h post-treatment. (f) Immunofluorescence assays were performed to access the expression level and location of HMGB1 in pancreatic cancer cell lines (SW1990, Panc1, and AsPC1) at 48 h post-irradiation (8 or 12 Gy). Red: HMGB1; Blue: nucleus. Scale bar, 50 μm. Experiments were repeated three times, and the data are expressed as the mean ± SEM. *P < .05, **P < .01, ***P < .001. Statistical analysis was performed using Student's t-test (b, c right).
Fig. S2.

Irradiation dose induced cell death. (a) CCK8 testing was performed to assess the viability of pancreatic cancer cells (SW1990, Panc1, and AsPC1) at 48 h post-treatment with various doses of X-rays (0, 4, 8, or 10 Gy). (b) Annexin V-FITC/PI staining was used to assess pancreatic cancer cell (SW1990, Panc1, and AsPC1) death at 48 h post-treatment with various doses of X-rays (0, 4, 8, or 10 Gy). Experiments were repeated three times, and the data are expressed as the mean ± SEM. Statistical analysis was performed using Student's t-test (a).
3.3. Dying cell-derived HMGB1 regulated CD133+ cancer cell stemness
To elucidate the functional role of irradiation induced extracellular HMGB1 in the self-renewal and proliferation of CD133+ CSCs, two stable HMGB1-knockdown cell clones (HMGB1 shRNA1 and shRNA2) were established, and high silencing efficiency (80% silencing, due to the expression of short hairpin RNAs[shRNAs]) was confirmed by western blotting (Fig. 3a). These cells were then irradiated, and the cultured supernatant was collected 48 h after treatment. HMGB1 levels in the supernatant were downregulated by 75–85% when HMGB1 expression was inhibited (Fig. 3b). We then used supernatants from irradiated parental Ctrl shRNA cancer cells (HMGB1+) and HMGB1 shRNA1 cancer cells (HMGB1-) to further determine their potential to induce sphere formation and expansion of the sorted CD133+ cancer cells. Different concentrations of recombinant human HMGB1 (rhHMGB1; 50, 100, 150, and 200 ng/mL) were used as dose controls and we found that increasing the rhHMGB1 concentration drove enlargement of the sphere, without increasing the total number of spheres (Fig. 3c and d). HMGB1+ supernatants promoted sphere sizes to a level comparable level to 200 ng/mL rhHMGB1, whereas HMGB1- supernatants had no effect on inducing sphere formation either in terms of size or numbers (Fig. 3d). Consistently, qRT-PCR and western blot analysis showed that, similar to our findings after treatment with 150 ng/mL rhHMGB1, HMGB1+ supernatants showed enhanced mRNA and protein expression levels of Oct4, Sox2 and Nanog in CD133+ cancer cells, whereas HMGB1- supernatants showed a very mild effect on inducing the expression of these genes (Fig. 3e and f). These data further supported the functional role of irradiation-induced extracellular HMGB1 production in up-regulating cancer cell stemness. Treating the bulk SW1990 cancer cells with 150 ng/mL of rhHMGB1, we found that rhHMGB1 can also enlarge CD133+ cancer cells among bulk cells (Fig. S3a).
Fig. 3.

Dying cell-derived HMGB1 regulated CD133+ cancer cells stemness. (a) Western blot analysis of shRNA-mediated knockdown of HMGB1 protein expression in pancreatic cancer cell lines. GAPDH expression was detected as a loading control. (b) ELISA-based analysis of HMGB1 concentrations in the culture supernatant of HMGB1-knockdown cancer cell. (c, d) The number and size of spheres formed from CD133+ cancer cells were measured following a 14-d co-cultured with the following agents: i) lethally irradiated HMGB1+ cancer cells; ii) HMGB1− cancer cells; iii) shControl + rhHMGB1(150 ng/mL); iv) shHMGB1 + rhHMGB1(150 ng/mL); v) various concentrations of rhHMGB1(50, 100, 150, and 200 ng/mL). (e, f) Western blot and qRT-PCR analysis of mRNA- and protein- expression levels of stem cell markers (Oct4, Sox2, and Nanog) in CD133+ cancer cells after a 14-d co-cultured with the following agents: i) lethally irradiated HMGB1+ cancer cells; ii) HMGB1− cancer cells; iii) rhHMGB1 (150 ng/mL). GAPDH was detected as a loading control. Scale bar, 1000 μm. Experiments were repeated three times, and the data are expressed as the mean ± SEM. *P < .05, **P < .01, ***P < .001. Statistical analysis was performed using Student's t-test (a right, b, d, e, f right).
Fig. S3.

Role of rhHMGB1 in bulk pancreatic cancer cell stemness and proliferation. (a) Flow cytometric analysis of the percentage of CD133+ cancer cells among bulk cells at 72 h post-treatment with 150 ng/mL rhHMGB1. (b) CCK8 testing was performed to assess the ability of CD133+ cancer cells to proliferate when co-cultured with the following agents: i) PBS; ii) irradiated HMGB1+ cells; iii) irradiated HMGB1− cells (HMGB1-shRNA1); iv) rhHMGB1 (150 ng/mL) for 0, 2, 4, or 6 d. (c) Flow cytometric analysis of cell cycle progression in CD133+ cancer cells at 48 h post-treatment with 150 ng/mL rhHMGB1. (d) Colony-forming assay analysis of the colony forming ability at 48 h post-treatment with 150 ng/mL rhHMGB1. Experiments were repeated three times, and the data are expressed as the mean ± SEM.
To further confirm the role of HMGB1 in CD133+ cell proliferation and cell cycle progression, we performed CCK8 and flow cytometry assays. The results showed that treated with rhHMGB1 significantly promoted CD133+ cell proliferation and induced the cells in S phase (Fig. S3b and c). To confirm the effect of HMGB1 on anoikis, primary pancreatic cancer cells were grown as monolayer cultures and treated with vehicle or rhHMGB1 (150 ng/mL). Forty-eight hours later, these cells were examined in vitro colony-forming assays without rhHMGB1. Prior rhHMGB1-treated cells formed more colonies (Fig. S3d), demonstrating that HMGB1 induced expansion of cells with self-renewal properties without the involvement of apoptosis.
3.4. TLR2 and TLR4 mediated HMGB1-induced stemness of CD133+ cancer cell in different manners
To study the signaling pathways in which irradiation-induced HMGB1 mediate cancer cell stemness, we first examined the total mRNA- and protein-expression patterns of two common HMGB1 receptors (TLR-2 and TLR-4) in pancreatic cancer cells. The results suggest that among the three pancreatic cancer cell lines, SW1990 cells expressed the highest levels of TLR2 and the lowest levels of TLR4, whereas AsPC1 cells displayed the highest level levels of TLR4 and the lowest levels of TLR2 (Fig. 4a). Furthermore, compared to CD133− cells, CD133+ cancer cells expressed higher levels of TLR2 protein and mRNA, whereas TLR4 expression levels were reduced in CD133+ cancer cells (Fig. 4a), indicating that TLR2 and TLR4 serve different roles in cancer cell stemness. Immunoprecipitation assays with SW1990 cell lystates showed TLR2 and TLR4 formed a complex alone, or together with HMGB1 (Fig. 4b).
Fig. 4.

HMGB1 maintained and enhanced the stemness of CD133+ cancer cells in TLR2-dependent manner. (a) qRT-PCR and western blot analysis of the baseline protein levels of TLR2 and TLR4 in total cancer cells and isolated CD133+ cancer cells from pancreatic cancer cell lines (SW1990, Panc1, and AsPC1). GAPDH expression was detected as a loading control. (b)Co-IP analysis of the binding ability between HMGB1 and TLR2 or TLR4 in CD133+ SW1990 cancer cells. (c) TLR4+ CD133+ SW1990, TLR4+ CD133+ AsPC1, TLR2+ CD133+ SW1990 and TLR2+ CD133+ AsPC1 cancer cells were treated with 150 ng/mL rhHMGB1, and western blot analysis was performed to examine the expression level of Oct4, Sox2, and Nanog at 72 h post-treatment. GAPDH was detected as a loading control. (d) Sphere sizes were measured for TLR4+ CD133+ SW1990, TLR4+ CD133+ AsPC1, TLR2+ CD133+ SW1990 and TLR2+ CD133+ AsPC1 at 14 d following rhHMGB1 treatment. (e) TLR4− CD133+ SW1990, TLR4− CD133+ AsPC1, TLR2− CD133+ SW1990, and TLR2− CD133+ AsPC1 cancer cells were treated with 150 ng/mL rhHMGB1, and western blot analysis was performed to study the expression levels of Oct4, Sox2, and Nanog at 72 h post-treatment. GAPDH was detected as a loading control. (f) Sphere sizes were measured for TLR4− CD133+ SW1990, TLR4− CD133+ AsPC1, TLR2− CD133+ SW1990 and TLR2− CD133+ AsPC1 cells at 14 d after treatment with 150 ng/mL rhHMGB1. (g) Western blot analysis of the expression levels of Oct4, Sox2, and Nanog in TLR4+ CD133+ SW1990 cells treated with the following agents: i) DMSO; ii) rhHMGB1(150 ng/mL) + DMSO; iii) rhHMGB1(150 ng/mL) + Stevioside; iv) rhHMGB1(150 ng/mL) + TAK-242. GAPDH was detected as a loading control. Scale bar, 1000 μm. Experiments were repeated three times, and the data are expressed as the mean ± SEM. *P < .05, **P < .01, ***P < .001. Statistical analysis was performed using Student's t-test (d right, e right, f down, g right).
To further assess the role of TLR2 and TLR4 in HMGB1 regulated stemness, we over-expressed TLR2 or TLR4 in CD133+ SW1990 and AsPC1 cancer cells (TLR2+ CD133+ SW1990, TLR4+ CD133+ SW1990, TLR2+ CD133+ AsPC1, and TLR4+ CD133+ AsPC1) and then treated the cells with rhHMGB1 (150 ng/mL). Interestingly, increased Oct4, Sox2 and Nanog protein expression and sphere size occurred in TLR2+ CD133+ SW1990 and TLR2+ CD133+ AsPC1 cells in the presence of rhHMGB1 (Fig. 4c and d). In contrast, cancer cell stemness was inhibited in TLR4 over-expressing cells (TLR4+ CD133+ AsPC1 and TLR4+ CD133+ SW1990; Fig. 4c and d), indicating the opposite roles of TLR2 and TLR4 in HMGB1-induced cancer cell stemness. To test this hypothesis, by transfecting specific shRNA oligos, we transiently silenced TLR2 and TLR4 expression in CD133+ SW1990 (TLR2− CD133+ SW1990) and AsPC1 (TLR4− CD133+ AsPC1) cells. FACS analysis also showed that overexpression or siRNA did not affect the percentage of CD133+ cancer cell without HMGB1 treatment (Fig. S4a). As expected, TLR2 inhibition downregulated cancer cell stemness, whereas silencing TLR4 slightly upregulated the stem cell markers Nanog and Sox2, especially in AsPC1 cells in the presence of rhHMGB1 (Fig. 4e and f). We also treated TLR4+ CD133+ SW1990 cells separately with Stevioside (a TLR2 antagonist) and TAK-242 (a TLR4 antagonist). Compared with the control group (rhHMGB1 + DMSO), we observed decreased protein expression of stemness related-markers in the presence of rhHMGB1 and Stevioside, whereas increased expression of these stemness related-markers was observed in the presence of rhHMGB1 and TAK-242(Fig. 4 g). We further confirmed the role of TLR2 and TLR4 expression in radiotherapy resistance following HMGB1 treatment. Compared to the control groups, TLR4+ CD133+ SW1990 cells and TLR2+ CD133+ SW1990 cells showed enhanced radiotherapy resistance in the presence of HMGB1, whereas TLR2−CD133+ SW1990 and TLR4− CD133+ SW1990 cells showed reduced radiotherapy resistance (Fig. S4b and c).
Fig. S4.

Effect of over-expression or silencing TLR2 or TLR4 on bulk cancer cell stemness and radiotherapy resistance. (a) FACS analysis of the percentage of CD133+ cancer cells in TLR2-overexpressing or TLR2 siRNA-transfected bulk cells. (b) CCK8 testing was performed to assess the viability of CD133+ SW1990 cancer cells overexpressing TLR2 or transfected with TLR2 siRNA, followed by treatment with 150 ng/mL rhHMGB1. (c) CCK8 testing was performed to assess the viability of CD133+ SW1990 cancer cells overexpressing TLR4 or transfected with TLR4 siRNA, followed by treatment with 150 ng/mL rhHMGB1. Experiments were repeated three times, and the data are expressed as the mean ± SEM. Statistical analysis was performed using Student's t-test (b, c).
These results confirmed HMGB1 was capable of maintaining and enhancing CD133+ pancreatic cancer cell stemness through the TLR2 receptor, which further induced radiotherapy resistance. Moreover, TLR4 receptor displayed certain antagonistic effect on the HMGB1–TLR2 axis induced stemness effect.
3.5. Wnt/β-catenin pathway was involved in HMGB1–TLR2 axis mediated stemness of CD133+ cells
As the Wnt/β-catenin is an important regulator of CSCs, we hypothesized that it may function downstream of HMGB1/TLR2 signaling to maintain and enhance the stemness of CD133+ cancer cells. Sorted CD133+ SW1990 cells were con-cultured with supernatants from irradiated parental Ctrl shRNA cancer cells (HMGB1+), HMGB1 shRNA1 cancer cells (HMGB1−), rhHMGB1 (150 ng/mL) and mock medium (Control) in the Millicell system for 48 h. We found elevated expression of Wnt-signaling markers (p-GSK3β and β-catenin), signaling components downstream of β-catenin (CD44 and c-myc), and Wnt target gene transcripts (LEF1 and Axin2), in the HMGB1+ and rhHMGB1-treated groups, but not in the HMGB1− group (Fig. 5a and b). Silencing TLR2 in SW1990 cells (TLR2 shRNA2) substantially inhibited Wnt signaling in the presence of rhHMGB1, but silencing TLR4 in SW1990 cells (TLR4 shRNA2) slightly enhanced Wnt signals (Fig. 5a), suggesting that irradiation-induced extracellular HMGB1 activated Wnt signaling via the TLR2 receptor. Silencing β-catenin in SW1990 cells (β-catenin shRNA1 and shRNA2) reversed the rhHMGB1-induced expression of Oct4, Sox2 and Nanog, as well as the sphere size (Fig. 5c and d), further suggesting that the HMGB1–TLR2 axis maintains and enhances the stemness of CD133+ cancer cells by activating Wnt/β-catenin signaling.
Fig. 5.

The Wnt/β-catenin pathway was involved in the HMGB1/TLR2-mediated stemness of CD133+ cells. (a) Western blot analysis of the expression levels of p-GSK3β, GSK3β, β-catenin, CD44, and c-myc in CD133+ SW1990 cells co-cultured with irradiated HMGB1+cells, irradiated HMGB1− cells, rhHMGB1(150 ng/mL), rhHMGB1 + TLR2 shRNA, and rhHMGB1 + TLR4 shRNA. GAPDH expression was detected as a loading control. (b) qRT-PCR analysis of LEF1 and Axin2 mRNA levels in CD133+ SW1990 cells co-cultured with irradiated HMGB1+cells, irradiated HMGB1− cells, rhHMGB1(150 ng/mL), or empty medium. GAPDH was detected as a loading control. (c) Western blot analyses of the levels of Oct4, Sox2, and Nanog in β-catenin knockdown CD133+ SW1990 cells treated with 150 ng/mL rhHMGB1. (d) The sphere size of β-catenin knockdown CD133+ SW1990 cells at 14 d post-treatment with 150 ng/mL rhHMGB1. Scale bar, 1000 μm. Experiments were repeated three times, and the data are expressed as the mean ± SEM. *P < .05, **P < .01, ***P < .001. Statistical analysis was performed using Student's t-test (a right, b, d right).
3.6. Knockout of HMGB1 and/or TLR2 inhibited CD133+ cancer cell tumorigenesis and stemness in vivo
To validate our in vitro observation that irradiated dying cells can produce extracellular HMGB1 and regulates cancer cell stemness, we performed the limiting dilution tumor initiation assay using sorted CD133+ SW1990 cells in the nude mice. Similar to previous experiments, CD133+ SW1990 cells were cultured in supernatants from irradiated parental Ctrl shRNA cancer cells (HMGB1+), HMGB1 shRNA1 cancer cells (HMGB1−-1), HMGB1 shRNA2 cancer cells (HMGB1−-2), rhHMGB1 (150 ng/mL) and mock medium (Control) for 5 days before 1 × 105 cancer cells were implanted subcutaneously into the nude mice. Consistent with in vitro results, tumors from HMGB1+ and rhHMGB1 groups grew faster and larger than the other groups (Fig. 6a and b).
Fig. 6.

Knockout of HMGB1 and/or TLR2 inhibited CD133+ cancer cell tumorigenesis and stemnss in vivo. (a, b) CD133+ SW1990 cancer cells co-cultured with irradiated HMGB1+ cells, irradiated HMGB1− cells (HMGB1-shRNA1, HMGB1-shRNA2), rhHMGB1(150 ng/mL) or empty medium for 5 d and then implanted subcutaneously into the right dorsal flanks of nude mice. The tumor volume (a) and tumor-growth rate (b) were measured at 27 d post-injection. (c, d) TLR4+CD133+, TLR4−CD133+, TLR2+CD133+ and TLR2−CD133+ SW1990 cancer cells treated with or without rhHMGB1(150 ng/mL), with limiting dilution tumor-initiation ability studied with eight mice per group. The tumor volume (a) and growth-growth rate (b) were measured at 34 d post injection. Experiments were repeated three times, and the data are expressed as the mean ± SEM. *P < .05, **P < .01, ***P < .001. Statistical analysis was performed using Student's t-test. Statistical analysis was determined by Fisher exact test (b, d).
In addition, TLR2-overexpressing CD133+ SW1990 cells (TLR2+), TLR2 silencing CD133+ SW1990 (TLR2−), TLR4-overexpressing CD133+ SW1990 cells (TLR4+), and the control CD133+ SW1990 cells (TLR4−) were treated rhHMGB1(150 ng/mL) or phosphate-buffered saline (PBS) for 5 days, and then the cells were implanted subcutaneously into nude mice at 1 × 103, 1 × 104, or 1 × 105 cells/mouse. TLR2+ CD133+ and TLR4− CD133+ cells treated with rhHMGB1 showed significantly higher tumorigenicity than the other groups. As few as 1 × 103 TLR2+ CD133+ (4/8) cells treated with rhHMGB1 developed into tumors. At least 1 × 105 cancer cells were necessary to consistently generate a tumor in the rhHMGB1-treated TLR4+ CD133+ (3/8) and TLR2− CD133+ (4/8) groups, and rhHMGB1 treated TLR2+ CD133+ and TLR4− CD133+ cancer cells groups exhibited higher tumor-growth rates and larger tumor volumes (Fig. 6C, Table 3 and Table 4), supporting the possibility that the HMGB1-TLR2 axis enhanced the stemness of cancer cells in vivo. TLR4 negatively regulated cancer cell stemness in vivo, especially in the presence of rhHMGB1, which is also in agreement with our in vitro observation.
3.7. The expression of CSC markers correlated with levels of HMGB1, TLR2, and β-catenin in human pancreatic carcinoma
CD133 and CD44 are believed to be markers of CSCs in pancreatic carcinoma. Using information from The Cancer Genome Atlas (TCGA) database (https://genome-cancer.ucsc.edu, n = 183), correlations between the expression of HMGB1, TLR2, β-catenin and CSC related markers (Oct4, Nanog, CD133, and CD44) were evaluated. Using TCGA protein array data, we found that the expression of Oct4, Nanog, CD133, CD44, and β-catenin showed a linear relationship with TLR2 in a heat-map, whereas HMGB1 and TLR4 showed no significant correlation (Fig. 7a).
Fig. 7.

Stem cell-related markers expression correlated with HMGB1, TLR2, and TLR4 levels in human pancreatic carcinoma. (a) Analysis of the relationships between HMGB1 expression and CD133, CD44, Oct4, Nanog, and β-catenin expression in human pancreatic cancer tissues from the TCGA database. (b) Analysis of the relationships between TLR2 expression and CD133, CD44, Oct4, Nanog, and β-catenin expression in human pancreatic cancer tissues from the TCGA database. (c) Analysis of the relationships between TLR4 expression and CD133, CD44, Oct4, Nanog, and β-catenin expression in human pancreatic cancer tissues from the TCGA database. The results are presented in a heat map: n = 183. The data are presented as fold changes in cancer specimens as compared to matched normal tissues. Statistical analysis was performed using Pearson's correlation analysis (a right, b right, c right).
4. Discussion
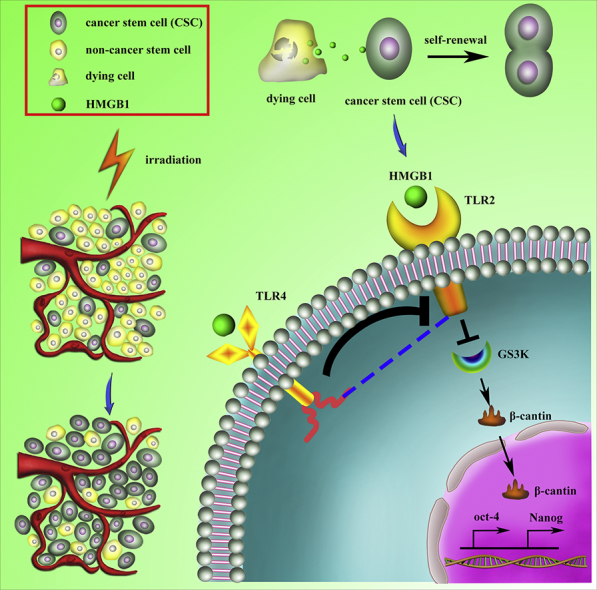
In the present study, we demonstrated that X-ray irradiation-induced cell death promoted self-renewal in vitro and enhanced the tumorigenicity of CD133+ pancreatic CSCs in vivo. We also found that cell death-induced HMGB1 binding with TLR2 in CD133+ cancer cells enhanced their self-renewal ability, whereas TLR4 displayed an antagonistic effect. Furthermore, the HMGB1/TLR2/Wnt/β-catenin pathway promotes self-renewal of CD133+ cancer cells, which enriches for a radiotherapy-resistant CSC population (Fig. 8); therefore, blocking this pathway may inhibit both enrichment of the CD133+ stem cell population and tumor recurrence. Similar results were also found in hepatocellular carcinoma, glioblastoma, and breast cancer cell lines (Fig. S5).
Fig. 8.

Schematic representation of how radiotherapy-induced cell death may activate paracrine HMGB1-TLR2/4 signaling and regulate the stemness of resident CSCs. Radiotherapy induced the cancer cells death which enriched for CSCs. HMGB1 is released by dying cells and binds specific receptors (TLR2 and TLR4) expressed on CSCs. HMGB1/TLR2 stimulates Wnt/β-catenin in a paracrine manner, which promotes the self-renewal of CSCs, while HMGB1/TLR4 abrogates this effect.
Fig. S5.

Irradiation-induced dying cell-derived HMGB1 enriched cancer stemness in other types of tumors. (a) Flow cytometric analysis of the percentage of CD133+ cancer cells among HepG2 cancer cells at 48 h post-irradiation with 0, 4, 8, or 10 Gy X-rays. (b) Western blot analysis of HMGB1 protein expression in culture supernatants at 48 h post-irradiation (8 Gy) in HepG2, U87, and MCF-7 cancer cells. GAPDH expression was detected as a loading control. (c) Western blot analysis of Oct4 and Nanog protein expression in HepG2, U87, and MCF-7 cells treated with 150 ng/mL rhHMGB1. GAPDH was detected as a loading control. Experiments were repeated three times, and the data are expressed as the mean ± SEM.
Similar to normal tissue repair, tumor recurrence following radiotherapy resulted from therapy-resistant CSCs. CSCs are responsible for tumor therapy resistance, metastasis, relapse, and the real threat to tumor eradication [8,29]. Our results confirmed that irradiation increases the number of CSCs in pancreatic cancer cell lines. The increased number may be ascribed to: i) selective killing of differentiated cancer cells, leading to a relative increase in the CSC population; ii) self-renewal and proliferation of CSCs, leading to an absolute increase in the CSC population; and iii) the reprogramming of differentiated cancer cells to acquire stem cell traits. Our in vitro study confirmed the role of an HMGB1-enriched microenvironment following irradiation in inducing self-renewal of CSCs. Based on our study, the increased fractions of CD133+ cells were distinct among the three pancreatic cancer cell lines. Two possibilities can explain this difference. First, each cell line contains a different level of basal CD133+ fraction. According to the former reports, AsPC-1 and SW1990 contain a high proportion of the CD133+ subpopulation [30], which may explain why these cell lines had a more dramatic increase in the CSC population after irradiation. The second possibility is that the response to stimulation from the located environment signals varied. We observed stem-related gene- and protein expression-level changes in a dose-independent manner. Indeed, irradiation induced significantly higher expression of stemness-related genes and proteins. Irradiation stress can induce the ectopic overexpression of stemness related transcription factors. Based on previous research, we speculate that this may be ascribed to the baseline expression levels of transcription factors in cancer cells and the gene copy increased derive threshold [31].
To maintain stemness and the self-renewal ability, CSCs are also tightly regulated by specific tumor microenvironments (also called niches) [32]. Either cell–cell communication or extracellular protein–cell interactions in the niches regulate the self-renewal of CSCs and, thus, tumorigenicity in the primary and secondary sites [12]. As a common DAMP molecule, HMGB1 can be passively released from dying or stressed cells, including necrotic cells following radiotherapy [33]. The effects of extracellular HMGB1 on tumor biology are complicated and sometimes contradictory. Some reports demonstrated that extracellular HMGB1 may favor cancer cell proliferation and inhibit apoptosis through its growth factor capability [33,34]. Other reports provided evidence that extracellular HMGB1 can induce cancer cell death through promoting genome instability or limiting the Warburg effect [35,36]. Conti et al. reported autocrine HMGB1 from breast cancer cells and CSCs could sustain CSC renewal [37]. Another study showed that autophagic cancer associated fibroblasts (CAFs) released HMGB1 and could promote stemness and tumorigenicity of luminal breast cancer [23]. According to our in vitro results, HMGB1 in the supernatant was progressively up-regulated at 36 h, and then maintained at a high level for 72 h after radiotherapy. The dramatic increase of the CD133+ population started at 48 h after radiotherapy. These results suggest that HMGB1 functioned as an important regulator of CD133+ cancer cell stemness and self-renewal. Thus, extracellular HMGB1 can function in a paracrine or autocrine manner to affect the stemness of cancer cells. The role of HMGB1 in regulating cancer cell biology may also depend on its oxidation/redox status. We also confirmed that reduce state of extracellular HMGB1 was important for maintaining the stemness of CD133+ cancer cells, while the oxidized state of extracellular HMGB1 promoted the differentiation of CD133+ cancer cells (data not shown).
By interacting with several cell surface receptors (including RAGE, TLRs, and CD24), extracellular HMGB1 induces various functional responses. It was reported that TLRs play an important role in the pathophysiology of pancreatic cancer [38]. Our findings also suggest that TLR2 and TLR4 are the major receptor responsible for HMGB1-mediated effects on pancreatic cancer cell stemness and self-renewal. Previous reports showed that TLR2 maintained CSC self-renewal in ovarian and breast cancer, while TLR4 suppressed CSC self-renewal in glioblastoma[37,39,40]. Our results demonstrate for the first time the opposite roles of TLR2 and TLR4 in pancreatic CSC self-renewal. Based on our in vitro results, the basal expression levels of TLR2 and TLR4 varied between different pancreatic cancer cell lines. Interestingly, their responses to HMGB1 mediated stemness effects were also different. TLR2 inhibition significantly reduced the formation of mammospheres and the expression of stem cell markers, while inhibition TLR4 had the opposite consequences. These results suggested that TLR2 is a positive regulator that mediates HMGB1 induced stemness, while TLR4 acts as a negative mediator, although the molecular mechanism between TLR4–TLR2 crosstalk was not well elucidated in this study. It was reported that TLR2 and TLR4 can induce each other's expression in acute lung injury and that their crosstalk serves as an important amplifier of inflammation [41]. Ovarian cancer research showed that TLR2 enhanced the self-renewal of MyD88+ EOC cells, while TLR4 induced MyD88+ EOC cells proliferation [39,42]. We speculate that crosstalk between TLR2 and TLR4 determines the proper balance of CSCs and differentiated cells in HMGB1-mediating tumor recurrence. HMGB1-TLR2 signaling induces CSC self-renewal and therefore maintenance/expansion of the stem cell pool. In contrast, HMGB1-TLR4 signaling promotes the differentiation of CSCs and further gives rise to heterogeneous daughter cells. Similar to tissue repair upon injury, the balance between self-renewal and differentiation of CSCs is critical for promote tumor repair and recurrence following radiotherapy. The exact crosstalk mechanism between TLR2 and TLR4 in pancreatic cancer cells need to be investigated further and at a deeper level.
In the present study, we identified the HMGB1-TLR2/TLR4-Wnt/β-catenin signaling pathway as being initiated by irradiated cancer cells, and our data implicated a specific cancer cell subpopulation, CD133+ CSCs, in mediating tumor recurrence following radiotherapy. Our results open the possibility for developing new effective strategies to prevent tumor repair in patients with residual disease. Combining radiotherapy and an HMGB1 inhibitor or targeting TLR2-induced CSC self-renewal may be considered for future therapeutic protocols.
Funding sources
This study was supported by grants from the National Natural Science Foundation of China (grant number 81502663), the Social Development Foundation of Jiangsu Province (grant number BE2018691), Young Medical Talents of Jiangsu (grant numbers QNRC2016831, QNRC2016833, and QNRC2016839), the Jinshan Talent Project of Zhenjiang City, and the Social Development Foundation of Zhenjiang City (grant numbers SH2017027 and SH2018063, SH2018031).
Roles of the funding sources
The corresponding author confirms that he had full access to all the data in the study and had final responsibility for the decision to submit for publication.
Declaration of interests
The authors declare no conflict of interests.
Author contributions
Haitao Zhu, Aihua Gong, and Dongqing Wang served as corresponding authors and organized the study. Xuelian Chen, Lirong Zhang, and Lian Song performed the experiments. Xiaojie Cai and Yanfang Liu analyzed the data. Xin Fan collected the clinical tumor tissue samples. Tao You performed the radiotherapy. Fang Cheng drafted the manuscript. All authors read and approved of the final manuscript.
The following are the supplementary data related to this article.
References
- 1.Peran I., Madhavan S., Byers S.W., McCoy M.D. Curation of the pancreatic ductal adenocarcinoma subset of the cancer genome atlas is essential for accurate conclusions about survival-related molecular mechanisms. Clin Cancer Res. 2018;24(16):3813–3819. doi: 10.1158/1078-0432.CCR-18-0290. [DOI] [PubMed] [Google Scholar]
- 2.Fokas E., O'Neill E., Gordon-Weeks A., Mukherjee S., McKenna W.G., Muschel R.J. Pancreatic ductal adenocarcinoma: From genetics to biology to radiobiology to oncoimmunology and all the way back to the clinic. Biochim Biophys Acta. 2015;1855(1):61–82. doi: 10.1016/j.bbcan.2014.12.001. [DOI] [PubMed] [Google Scholar]
- 3.Huang Q., Li F., Liu X., Li W., Shi W., Liu F.F. Caspase 3-mediated stimulation of tumor cell repopulation during cancer radiotherapy. Nat Med. 2011;17(7):860–866. doi: 10.1038/nm.2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu X., He Y., Li F., Huang Q., Kato T.A., Hall R.P. Redefining the roles of apoptotic factors in carcinogenesis. Mol Cell Oncol. 2016;3(3) doi: 10.1080/23723556.2015.1054550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Feng X., Yu Y., He S., Cheng J., Gong Y., Zhang Z. Dying glioma cells establish a proangiogenic microenvironment through a caspase 3 dependent mechanism. Cancer Lett. 2017;385:12–20. doi: 10.1016/j.canlet.2016.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lauffer D.C., Kuhn P.A., Kueng M., Thalmann S.U., Risse G., Tercier P.A. Pancreatic Cancer: feasibility and outcome after radiochemotherapy with high dose external radiotherapy for non-resected and R1 resected patients. Cureus. 2018;10(5):e2713. doi: 10.7759/cureus.2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Batlle E., Clevers H. Cancer stem cells revisited. Nat Med. 2017;23(10):1124–1134. doi: 10.1038/nm.4409. [DOI] [PubMed] [Google Scholar]
- 8.Subramaniam D., Kaushik G., Dandawate P., Anant S. Targeting cancer stem cells for chemoprevention of pancreatic cancer. Curr Med Chem. 2018;25(22):2585–2594. doi: 10.2174/0929867324666170127095832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang W.J., Wu S.P., Liu J.B., Shi Y.S., Huang X., Zhang Q.B. MYC regulation of CHK1 and CHK2 promotes radioresistance in a stem cell-like population of nasopharyngeal carcinoma cells. Cancer Res. 2013;73(3):1219–1231. doi: 10.1158/0008-5472.CAN-12-1408. [DOI] [PubMed] [Google Scholar]
- 10.Ahmed S.U., Carruthers R., Gilmour L., Yildirim S., Watts C., Chalmers A.J. Selective inhibition of parallel DNA damage response pathways optimizes radiosensitization of glioblastoma stem-like cells. Cancer Res. 2015;75(20):4416–4428. doi: 10.1158/0008-5472.CAN-14-3790. [DOI] [PubMed] [Google Scholar]
- 11.Diehn M., Cho R.W., Lobo N.A., Kalisky T., Dorie M.J., Kulp A.N. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature. 2009;458(7239):780–783. doi: 10.1038/nature07733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Filatova A., Acker T., Garvalov B.K. The cancer stem cell niche(s): the crosstalk between glioma stem cells and their microenvironment. Biochim Biophys Acta. 2013;1830(2):2496–2508. doi: 10.1016/j.bbagen.2012.10.008. [DOI] [PubMed] [Google Scholar]
- 13.Sundar S.J., Hsieh J.K., Manjila S., Lathia J.D., Sloan A. The role of cancer stem cells in glioblastoma. Neurosurg Focus. 2014;37(6):E6. doi: 10.3171/2014.9.FOCUS14494. [DOI] [PubMed] [Google Scholar]
- 14.Ishii A., Kimura T., Sadahiro H., Kawano H., Takubo K., Suzuki M. Histological characterization of the tumorigenic “peri-necrotic niche” harboring quiescent stem-like tumor cells in glioblastoma. PLoS One. 2016;11(1) doi: 10.1371/journal.pone.0147366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jube S., Rivera Z.S., Bianchi M.E., Powers A., Wang E., Pagano I. Cancer cell secretion of the DAMP protein HMGB1 supports progression in malignant mesothelioma. Cancer Res. 2012;72(13):3290–3301. doi: 10.1158/0008-5472.CAN-11-3481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cebrian M.J., Bauden M., Andersson R., Holdenrieder S., Ansari D. Paradoxical role of HMGB1 in pancreatic cancer: tumor suppressor or tumor promoter? Anticancer Res. 2016;36(9):4381–4389. doi: 10.21873/anticanres.10981. [DOI] [PubMed] [Google Scholar]
- 17.Huang B.F., Tzeng H.E., Chen P.C., Wang C.Q., Su C.M., Wang Y. HMGB1 genetic polymorphisms are biomarkers for the development and progression of breast cancer. Int J Med Sci. 2018;15(6):580–586. doi: 10.7150/ijms.23462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kang R., Chen R., Zhang Q., Hou W., Wu S., Cao L. HMGB1 in health and disease. Mol Aspects Med. 2014;40:1–116. doi: 10.1016/j.mam.2014.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Livesey K.M., Kang R., Vernon P., Buchser W., Loughran P., Watkins S.C. p53/HMGB1 complexes regulate autophagy and apoptosis. Cancer Res. 2012;72(8):1996–2005. doi: 10.1158/0008-5472.CAN-11-2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tang D., Kang R., Zeh H.J., 3rd, Lotze M.T. High-mobility group box 1 and cancer. Biochim Biophys Acta. 2010;1799(1–2):131–140. doi: 10.1016/j.bbagrm.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.LI G, Liang X, Kaus J, Basse P, Zeh H, Lotze M. Natural killer (NK) cell high-mobility group box 1 (HMGB1) is required for anti-tumor function (TUM2P.895). J Immunol 2014;192(1 Suppl):71.19–71.19.
- 22.Kang R., Zhang Q., Zeh H.J., 3rd, Lotze M.T., Tang D. HMGB1 in cancer: good, bad, or both? Clin Cancer Res. 2013;19(15):4046–4057. doi: 10.1158/1078-0432.CCR-13-0495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhao X.L., Lin Y., Jiang J., Tang Z., Yang S., Lu L. High-mobility group box 1 released by autophagic cancer-associated fibroblasts maintains the stemness of luminal breast cancer cells. J Pathol. 2017;243(3):376–389. doi: 10.1002/path.4958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhan T., Rindtorff N., Boutros M. Wnt signaling in cancer. Oncogene. 2017;36(11):1461–1473. doi: 10.1038/onc.2016.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jiang X., Hao H.X., Growney J.D., Woolfenden S., Bottiglio C., Ng N. Inactivating mutations of RNF43 confer Wnt dependency in pancreatic ductal adenocarcinoma. Proc Natl Acad Sci U S A. 2013;110(31):12649–12654. doi: 10.1073/pnas.1307218110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Golden E.B., Frances D., Pellicciotta I., Demaria S., Helen Barcellos-Hoff M., Formenti S.C. Radiation fosters dose-dependent and chemotherapy-induced immunogenic cell death. Oncoimmunology. 2014;3 doi: 10.4161/onci.28518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee S.Y., Jeong E.K., Ju M.K., Jeon H.M., Kim M.Y., Kim C.H. Induction of metastasis, cancer stem cell phenotype, and oncogenic metabolism in cancer cells by ionizing radiation. Mol Cancer. 2017;16(1):10. doi: 10.1186/s12943-016-0577-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen X., Zhang L., Jiang Y., Song L., Liu Y., Cheng F. Radiotherapy-induced cell death activates paracrine HMGB1-TLR2 signaling and accelerates pancreatic carcinoma metastasis. J Exp Clin Cancer Res. 2018;37(1):77. doi: 10.1186/s13046-018-0726-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Krause M., Dubrovska A., Linge A., Baumann M. Cancer stem cells: Radioresistance, prediction of radiotherapy outcome and specific targets for combined treatments. Adv Drug Deliv Rev. 2017;109:63–73. doi: 10.1016/j.addr.2016.02.002. [DOI] [PubMed] [Google Scholar]
- 30.Satoh K., Hamada S., Shimosegawa T. Involvement of epithelial to mesenchymal transition in the development of pancreatic ductal adenocarcinoma. J Gastroenterol. 2015;50(2):140–146. doi: 10.1007/s00535-014-0997-0. [DOI] [PubMed] [Google Scholar]
- 31.Lagadec C., Vlashi E., Della Donna L., Dekmezian C., Pajonk F. Radiation-induced reprogramming of breast cancer cells. Stem Cells. 2012;30(5):833–844. doi: 10.1002/stem.1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Peitzsch C., Kurth I., Kunz-Schughart L., Baumann M., Dubrovska A. Discovery of the cancer stem cell related determinants of radioresistance. Radiother Oncol. 2013;108(3):378–387. doi: 10.1016/j.radonc.2013.06.003. [DOI] [PubMed] [Google Scholar]
- 33.Zhang Z., Wang M., Zhou L., Feng X., Cheng J., Yu Y. Increased HMGB1 and cleaved caspase-3 stimulate the proliferation of tumor cells and are correlated with the poor prognosis in colorectal cancer. J Exp Clin Cancer Res. 2015;34:51. doi: 10.1186/s13046-015-0166-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang K., Anumanthan G., Scheaffer S., Cornelius L.A. HMGB1/RAGE mediates UVB-induced secretory inflammatory response and resistance to apoptosis in human melanocytes. J Invest Dermatol. 2019;139(1):202–212. doi: 10.1016/j.jid.2018.05.035. (pii: S0022-202X(18)32348-0) [DOI] [PubMed] [Google Scholar]
- 35.Gdynia G., Sauer S.W., Kopitz J., Fuchs D., Duglova K., Ruppert T. The HMGB1 protein induces a metabolic type of tumor cell death by blocking aerobic respiration. Nat Commun. 2016;7 doi: 10.1038/ncomms10764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tang D., Loze M.T., Zeh H.J., Kang R. The redox protein HMGB1 regulates cell death and survival in cancer treatment. Autophagy. 2010;6(8):1181–1183. doi: 10.4161/auto.6.8.13367. [DOI] [PubMed] [Google Scholar]
- 37.Conti L., Lanzardo S., Arigoni M., Antonazzo R., Radaelli E., Cantarella D. The noninflammatory role of high mobility group box 1/Toll-like receptor 2 axis in the self-renewal of mammary cancer stem cells. FASEB J. 2013;27(12):4731–4744. doi: 10.1096/fj.13-230201. [DOI] [PubMed] [Google Scholar]
- 38.Vaz J., Andersson R. Intervention on toll-like receptors in pancreatic cancer. World J Gastroenterol. 2014;20(19):5808–5817. doi: 10.3748/wjg.v20.i19.5808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chefetz I., Alvero A.B., Holmberg J.C., Lebowitz N., Craveiro V., Yang-Hartwich Y. TLR2 enhances ovarian cancer stem cell self-renewal and promotes tumor repair and recurrence. Cell Cycle. 2013;12(3):511–521. doi: 10.4161/cc.23406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Alvarado A.G., Thiagarajan P.S., Mulkearns-Hubert E.E., Silver D.J., Hale J.S., Alban T.J. Glioblastoma cancer stem cells evade innate immune suppression of self-renewal through reduced TLR4 expression. Cell Stem Cell. 2017;20(4) doi: 10.1016/j.stem.2016.12.001. (450–61 e4) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fan J. TLR cross-talk mechanism of hemorrhagic shock-primed pulmonary neutrophil infiltration. Open Crit Care Med J. 2010;2:1–8. doi: 10.2174/1874828700902010001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang A.C., Ma Y.B., Wu F.X., Ma Z.F., Liu N.F., Gao R. TLR4 induces tumor growth and inhibits paclitaxel activity in MyD88-positive human ovarian carcinoma in vitro. Oncol Lett. 2014;7(3):871–877. doi: 10.3892/ol.2013.1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang L., Wang D., Li Y., Liu Y., Xie X., Wu Y. CCL21/CCR7 axis contributed to CD133+ pancreatic cancer stem-like cell metastasis via EMT and Erk/NF-kappaB pathway. PLoS One. 2016;11(8) doi: 10.1371/journal.pone.0158529. [DOI] [PMC free article] [PubMed] [Google Scholar]