

Abstract
Background:
Fragile X-associated tremor/ataxia syndrome (FXTAS) is a late-onset neurodegenerative disorder associated with premutation alleles of the FMR1 gene. Expansions of more than 200 CGG repeats give rise to fragile X syndrome, the most common inherited form of cognitive impairment. Fragile X-associated tremor/ataxia syndrome is characterized by cerebellar tremor and ataxia, and the presence of ubiquitin-positive inclusions in neurons and astrocytes. It has been previously suggested that fragile X-associated tremor/ataxia syndrome is associated with an inflammatory state based on signs of oxidative stress–mediated damage and iron deposition.
Objective:
Determine whether the pathology of fragile X-associated tremor/ataxia syndrome involves microglial activation and an inflammatory state.
Methods:
Using ionized calcium binding adaptor molecule 1 and cluster differentiation 68 antibodies to label microglia, we examined the number and state of activation of microglial cells in the putamen of 13 fragile X-associated tremor/ataxia syndrome and 9 control postmortem cases.
Results:
Nearly half of fragile X-associated tremor/ataxia syndrome cases (6 of 13) presented with dystrophic senescent microglial cells. In the remaining fragile X-associated tremor/ataxia syndrome cases (7 of 13), the number of microglial cells and their activation state were increased compared to controls.
Conclusions:
The presence of senescent microglial cells in half of fragile X-associated tremor/ataxia syndrome cases suggests that this indicator could be used, together with the presence of intranuclear inclusions and the presence of iron deposits, as a biomarker to aid in the postmortem diagnosis of fragile X-associated tremor/ataxia syndrome. An increased number and activation indicate that microglial cells play a role in the inflammatory state present in the fragile X-associated tremor/ataxia syndrome brain. Anti-inflammatory treatment of patients with fragile X-associated tremor/ataxia syndrome may be indicated to slow neurodegeneration.
Fragile X-associated tremor/ataxia syndrome (FXTAS) is a late-onset neurodegenerative disorder associated with premutation alleles (55–200 CGG repeats) of the fragile X mental retardation 1 (FMR1) gene; larger expansions (>200 CGG repeats, full mutation) give rise to fragile X syndrome (FXS), the most common inherited form of cognitive impairment. Carriers of premutation alleles are common in the general population, with an estimated frequency as high as 1 in 130 females and 1 in 250 males.1,2 However, only around 40% of male carriers and 14% of female carriers will eventually develop FXTAS.3,4 FXTAS is characterized by progressive action tremor, gait ataxia, cognitive decline, parkinsonism, neuropathy, and autonomic dysfunction.5 Central nervous system (CNS) pathology includes dystrophic white matter, intranuclear inclusions in neurons and astrocytes, and iron deposition.6–10 Although neurological symptoms of FXTAS have only been observed in adults, it is now clear that children who carry premutation alleles may also have forms of clinical involvement that include anxiety, attention deficit hyperactivity disorder, and autism spectrum disorders related to the premutation.9,11,12 Additionally, premutation CGG-repeat expansions in the mouse Fmr1 gene have been shown to alter embryonic neocortical development.13
Premutation carriers display a form of gene dysregulation that is quite distinct from the gene silencing observed with FXS. It is manifested by substantially increased levels of FMR1 mRNA and normal or moderately decreased levels of fragile X protein. The extent of this altered expression is a function of the size of the CGG-repeat expansion within the premutation range; larger CGG-repeat expansions are associated with higher levels of mRNA and lower levels of protein.14 Evidence from both human and animal studies implicates a direct toxic gain of function of the premutation CGG (pre-CGG) repeat-containing FMR1 mRNA.14–19 FMRpolyG peptides produced by out-of-frame transla-tion are observed in the nucleus20; however, their functional significance is not known.
FXTAS presents with high levels of oxidative stress in the brain and has been associated with an inflammatory state.21,22 We previously demonstrated that an alteration in iron transport into the brain is a characteristic feature of FXTAS and found substantial iron bound to hemosiderin in the parenchyma of the putamen in FXTAS.9,10,23 This iron deposition can also be detected as a symmetric hypointensity in the putamen and caudate in T2-weighted spin-echo magnetic resonance images in FXTAS patients.24,25 We also reported a deficit in proteins that transport and eliminate extra iron from cells (transferrin, ferroportin, and ceruloplasmin) and a concomitant increase in deposition of cellular iron in the choroid plexus.9,10 Iron is essential for cell metabolism; however, uncomplexed iron leads to oxidative stress and inflammation. The FXTAS proinflammatory state is also supported by an increase in oxidative/nitrative stress damage preceded by mitochondrial dysfunction in postmortem human brain samples.21 In addition, inflammation is supported by the lipid profile as well as the increase in oxidative stress–mediated damage to carbohydrates and proteins in serum of FXTAS patients.22 Fibroblasts from premutation individuals also show increased mitochondrial reactive oxygen species production accompanied by increased mtDNA deletions and increased biomarkers of lipid and protein oxidative–nitrative damage.26 Overall, these data indicate the presence of an inflammatory state in the FXTAS brain.
Microglia-mediated neuroinflammation is a critical contributor to the pathogenesis of neurodegenerative diseases and has been implicated in numerous diseases and conditions, such as hypoxia, stroke, epilepsy, and neuropathic pain.27 Microglial morphology varies throughout the brain depending upon brain region, age, and presence or absence of immune challenge or injury. In healthy cortical gray matter, microglia typically have a small soma and multiple thin, ramified processes that radiate from the soma. In white matter, microglial cells orientate along fiber tracts and have an elongated soma. Under nonpathological conditions, microglial cells are highly motile and have a continuous extension and retraction of long and thin radial processes.28 In response to injury or pathology, microglial cells exhibit a series of molecular and morphological changes that include a distinct activated appearance with a rounded “amoeboid” soma and a few thick processes. Activated microglial cells phagocytose cellular debris that can be detected within their cytoplasm.28–31 These morphological changes provide a tool to assess the activation status of microglia.32 Microglial cells in brain tissue from a small number of subjects diagnosed with Alzheimer’s disease (AD) display a distinct morphology that is characterized as senescent. Senescent microglia appear dystrophic and show diverse structural deformities that are increasingly prevalent with aging, including cytoplasmic processes that are twisted, beaded, gnarled, and fragmented.33
We examined the number and state of activation of microglia in the putamen of FXTAS and control postmortem patients. We found that almost half of the cases of FXTAS presented with dystrophic senescent microglial cells and that the presence of senescent microglia was correlated with the number of CGG repeats and high levels of iron accumulation in the putamen. Cases that lacked senescent microglia presented with an increased number of microglial cells that exhibited an increased state of activation compared to controls. This pattern seems to be similar in other brain regions of FXTAS. These data suggest that microglial cells play a role in the inflammatory state that is present in the FXTAS brain. It is not clear whether microglial activation initiates neurodegeneration, or instead whether microglial activation is a response to neurodegeneration. However, the results we present suggest that anti-inflammatory treatment for patients with FXTAS may be indicated to counteract neurodegeneration.
Materials and Methods
Sample Collection
Samples from 13 FXTAS postmortem human subjects and 9 postmortem control subjects were obtained from the FXTAS brain repository at the University of California, Davis (Sacramento, CA), School of Medicine (Table 1). Control tissue was obtained from the Pathology Department at the University of California, Davis, Health System. Tissue specimens were obtained through consented autopsies with institutional review board approval. FXTAS patients had symptoms for many years before death and were clinically diagnosed based on the presence of intention tremor, cerebellar ataxia, parkinsonism, memory and executive function deficits, and autonomic dysfunction, which were confirmed with the postmortem presence of intranuclear ubiquitin inclusions in brain cells. Control tissue was obtained from subjects who did not have any significant neurological history, including encephalitis, epilepsy, demyelinating disease, dementia, or concurrent neurodegenerative disease.
TABLE 1.
Information for the control and FXTAS cases included in this study.
| Case ID | A Diagnosis | CGG Repeats | Age | Sex | PMI (Hours) | Cause of Death |
|---|---|---|---|---|---|---|
| 1 | Control | N/A | 53 | W | 63 | Cor pulmonale |
| 2 | Control | N/A | 68 | W | 44 | Breast cancer |
| 3 | Control | N/A | 54 | M | 64 | Pulmonary fungal infection |
| 4 | Control | N/A | 60 | M | 68 | Adenocarcinoma |
| 5 | Control | N/A | 82 | W | >72 | Subdural hematoma |
| 6 | Control | N/A | 57 | M | 23 | NK |
| 7 | Control | N/A | 66 | M | NK | NK |
| 8 | Control | N/A | 61 | M | 37 | NK |
| 9 | Control | N/A | 44 | M | 24 | NK |
| 10 | FXTAS | 65 | 87 | M | NK | NK |
| 11 | FXTAS | 73 | 80 | W | 5 | Natural causes, stopped eating |
| 12 | FXTAS | 100 | 68 | M | 87 | Cardiac arrest |
| 13 | FXTAS | 75 | 70 | M | 2.5 | NK |
| 14 | FXTAS | 113 | 78 | M | NK | Natural cause |
| 15 | FXTAS | 97 | 58 | M | 14 | Natural causes, life support eliminated |
| 16 | FXTAS | 67 | 82 | M | NK | NK |
| 17 | FXTAS | 74 | 78 | M | 24 | Cardiomyopathy |
| 18 | FXTAS | 96 | 69 | M | 6.5 | Cardiac failure |
| 19 | FXTAS | 146 | 72 | M | 13.5 | Myelodysplasia |
| 20 | FXTAS | 75 | 52 | W | NK | Multiple sclerosis |
| 21 | FXTAS | 66 | 85 | M | 62 | NK |
| 22 | FXTAS | 86 | 85 | M | >72 | Cardiorespiratory arrest |
Information includes number of CGG repeats, age, sex, PMI (postmortem interval), and cause of death. Abbreviation: N/A, not applicable; NK, not knwon.
CGG Sizing
Genomic DNA was isolated from brain tissue (100 mg) using standard procedures (Trizol; Invitrogen, Carlsbad, CA). CGG repeat allele size was determined using PCR and Southern blot analysis as previously described.34,35
Immunostaining
A block of tissue (2 × 2 × 0.25 μm2) containing mid-putamen for each case was immersed in 30% sucrose (Thermo Fisher Scientific, Waltham, MA) and embedded in OCT (Thermo Fisher Scientific). Blocks were cut into 12-μm sections using a cryostat. We used the primary monoclonal rabbit anti-Iba (ionized calcium binding adaptor molecule; 1:500; Wako Chemicals USA, Inc., Richmond, VA) and rabbit anti-CD68 (1:200; Abcam, Cambridge, MA) antibodies, secondary antibody donkey antirabbit conjugated with biotin (1:150; The Jackson Laboratory, Bar Harbor, ME), amplified with avidin-biotin complex (Vector Laboratories, Burlingame, CA), and developed with 3,3’-diaminobenzidine (Vector Laboratories). Tissue was dehydrated and defatted, mounted on glass slides, and coverslipped with dibutylphthalate polystyrene xylene.
Perl Method
Tissue sections were submerged in a 1:1 ratio of 20% hydrochloric acid (Thermo Fisher Scientific) and 10% potassium ferrocyanide (Thermo Fisher Scientific) solution for 20 minutes at room temperature. Slides were washed with water, dehydrated with ethanol, and cleared with xylene before being coverslipped with Permount (Thermo Fisher Scientific).
Microglial Activation Paradigm
We classified microglial cells into six morphological states of activation from nonactivated state (1) to a fully activated state (6).31,36,37 Stage 1: Microglia cell has processes that are ramified and spread out, with a small soma. Stage 2: The soma has increased in size to approximately 1.5 to 2 times the soma diameter of a stage 1 cell. The cell processes have started to retreat, and the branches next to the cell soma are thickened. Stage 3: The cell soma diameter is enlarged to 2 to 3 times the soma diameter of stage 1. All cell processes have retracted and thickened. Stage 4: The soma diameter is 3 to 4 times larger than the stage 1 soma. All thin cell processes have completely retracted, and only the thick cell branches remain. Stage 5: Soma diameter is 5 times larger than the soma diameter in stage 1. The thick processes are replaced by thin processes oriented in the direction of cell movement. All branches are gone. Stage 6: transformation from microglia to macrophage. We referred to senescence stages 1 to 3 as early senescence and 4 to 6 as advance senescence.
Cell Quantification and Statistical Analysis
Cell quantification was performed in a Nikon Eclipse E200 microscope with Stereo Investigator software (MBF Bioscience, Williston, VT). A 300-μm2 area of the putamen was analyzed in three slides from each case. The average data collected from the three slices were used for analysis. Values were compared between FXTAS and control subjects using t-test analysis. A P value of 0.05 was used for statistical significance. To account for family-wise error, Bonferroni’s correction was applied using a P value of 0.01. After this correction, the effect of the CGG repetition number in the presence or absence of senescence was not significant.
Results
We previously reported increased iron deposition in the brain of FXTAS cases, with the most pronounced deposits being found in the putamen. To determine whether inflammatory activation is present in FXTAS, we gathered brain tissue from the putamen of the 13 FXTAS and 9 control cases. Age of FXTAS subjects ranged from 52 to 87 years, with an average of 74.1 years. Control subjects ranged from age 44 to 82 years with an average of 60.5 years. FXTAS brains had an average postmortem interval (PMI) of 31.8 hours, whereas control brains had an average PMI of 49.3 hours. The number of CGG repeats in the FXTAS group was between 65 and 146, with an average of 87.1 repeats (Table 1).
Over One Third of FXTAS Cases Had Dystrophic Senescent Microglia
We performed immunostaining with an antibody against ionized calcium binding adaptor molecule 1 (Iba1), a marker of microglial cells and macrophages, in the putamen of all cases included in the study. Our analysis of Iba1-stained tissue revealed that 6 FXTAS cases (6 of 13; 46%) presented with a notably distinct pattern of staining when compared to controls (Fig. 1). Iba1 immunostaining in these cases labeled cells and cellular processes that were beaded and punctate, similar to the pattern of Iba1 staining in dystrophic or senescent microglia in human tissue from some cases of AD.38–40 None of the control cases had senescent microglial cells. Dystrophic microglia in FXTAS cases had distinct morphologies that corresponded to a state of senescence. Some cases had recognizable Iba1+ microglial cells that exhibited spheroidal swellings of their processes, as well as beading and fragmentation (Fig. 1A–C). We referred to these as “early senescent” microglia (stages of senescence 1–3; see Materials and Methods). Other cases had microglia with very fragmented darkly stained punctate processes and isolated rounded soma/nuclei (Fig. 1D,F), as previously described.40 We referred to these as “advanced senescent” microglia (stages of senescence 4–5). In early senescence cases, senescent microglial cells were present in some regions of the putamen, but not others; however once the senescence process was more advance, all microglial cells in the putamen showed the fragmented morphology typical of senescence. The senescent pattern of staining was not present in control cases.
FIG. 1.
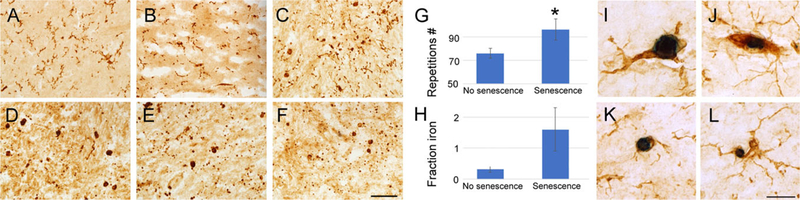
Senescent microglial cells are present in half of the cases of FXTAS. Dystrophic microglia with distinct morphologies corresponding to early and advanced states of senescence. (A–C) Early senescent microglial have spheroidal swellings of their processes, beading, and fragmentation. (D–F) Advance senescent microglia have very fragmented processes with punctate morphology and isolated rounded soma/nuclei. (G) Presence of senescent microglia was correlated with the number of CGG repeats in FXTAS. (H) Cases of FXTAS with senescent microglia accumulated more iron in the putamen; however, this was not statistically significant. (I–L) Microglial cells (Iba1, brown) contain high amounts of iron bound to hemosiderin (dark blue, Perl method) in the putamen in FXTAS. Scale bar in (A–D): 100 μm; in (I–L): 25 μm. [Color figure can be viewed at wileyonlinelibrary.com]
We found no correlation between the presence or absence of senescent microglia with age or with PMI (age, average [AVG] nonsenescence = 77.0 years, AVG senescence = 70.8 years; P = 0.3; PMI, AVG nonsenescence = 46.3 hours, AVG senescence = 24.5 hours; P = 0.07). However, there was a correlation between the presence or absence of senescent microglia and number of CGG repeats (repeat no.: AVG nonsenescence = 76.0, AVG senescence = 96.2; P = 0.05). We next compared the presence or absence of senescent microglia with the amount of iron accumulation in the same region of the putamen. An increased amount of iron in the putamen of these cases was reported by our team in a previous publication.9 We compared previously reported levels of iron and the presence of senescence and found that cases with senescent microglia accumulated an average of 5 times more iron in the putamen that those without senescent microglia. However, this correlation was not statistically significant (fractional area of putamen parenchyma occupied by iron deposits in AVG nonsenescence = 0.3, AVG senescence = 1.5; P = 0.1; Fig. 1). We next tested whether senescent microglial cells expressed the activated microglial/macrophage marker, CD68, and found that they do, suggesting that a state of activation may precede the process of senescence (Fig. 2O). The remaining FXTAS cases (7) did not have apparent senescent microglial cells. We excluded the 6 cases classified as senescent from further analysis.
FIG. 2.
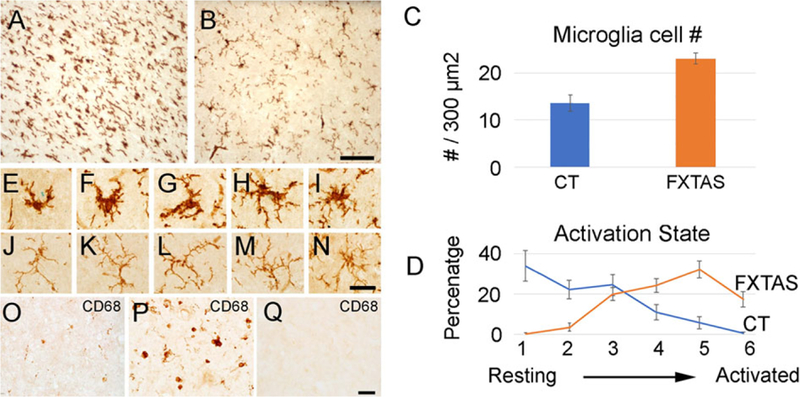
The number and state of activation of microglial cells is increased in FXTAS. (A) Representative images of Iba1+ microglial cells in the putamen in FXTAS (A) and control cases (B). (C) Microglial cell number is increased in FXTAS cases when compared to control cases. (D) The pattern of activation between the FXTAS and control cases was inverted, and microglial cells in FXTAS were more activated than in control cases. Blue: control; orange: FXTAS. (E–I) Activated microglial cells in the putamen of FXTAS cases. (J–N) Resting microglial cells in the putamen of control cases. (O–Q) CD68+ in senescent microglial cells (O), activated microglial cells (P), and control tissue (Q). Scale bar in (A,B): 100 μm; in (E–Q): 25 μm. [Color figure can be viewed at wileyonlinelibrary.com]
The Number of Microglial Cells Was Increased, and Microglial Activation Was Elevated in FXTAS
We quantified the number of Iba+ microglial cells in the putamen (300 μm2), and found that the number of microglia was higher in FXTAS than in control patients (control [CT], 13.6 ± 1.8; FXTAS, 23.1 ± 1.1; P < 0.001; Fig. 2C). Sections of brain tissue from controls showed Iba1+ microglial cells that were largely “resting,” characterized by thin, finely branched cellular processes radiating from small cell bodies. In contrast, brain tissue from FXTAS patients had a striking number of activated microglia with large cell bodies and thick, short processes. Based on morphological criteria, we classified microglial cells into six activation states from nonactivated state (1) to a fully activated state (6).31,36,37 We determined that activation status of microglial cells in FXTAS and control cases differed, and that microglial cells in FXTAS were significantly more activated than in control cases (Fig. 2D). An average of 80.9% of microglial cells in control cases were classified in the lower states of activation (states 1–3), and only 17.2% were classified in the higher states of activation (states 4–6). In contrast, 23.9% of microglial cells in FXTAS cases were classified in the lower states of activation (1–3), whereas 74.1% of cells were in the highest states of activation (4–6; Fig. 2D). These data demonstrate a significant change in morphological activation of microglia cells in the putamen of FXTAS cases and suggest a role for microglial cells in the neuropathological disease process.
To further verify the increase of microglial activation in FXTAS, we stained the putamen with an antibody against CD68, a marker for activated microglia cells. We quantified the number of CD68+ cells (300 μm2), and found that, compared to controls, FXTAS putamen had an increase in cells expressing CD68 (CT, 2.7 ± 10.9; FXTAS, 14.4 ± 2.6; P < 0.004; Fig. 2P,Q).
These data, together, indicate that the majority of microglial cells were activated in FXTAS.
Changes in Microglial Activation Present in the Putamen Seem to Affect Most Brain Regions
We stained other brain regions of 2 FXTAS cases with microglial cell activation, and our qualitative analysis indicated that microglial number and activation status were also increased in the cerebral cortex (frontal, parietal, occipital, and temporal), hippocampus, amygdala, thalamus, midbrain, hindbrain, and cerebellum (Fig. 3). These results suggest that increased microglial activation may be widespread throughout the CNS in FXTAS.
FIG. 3.
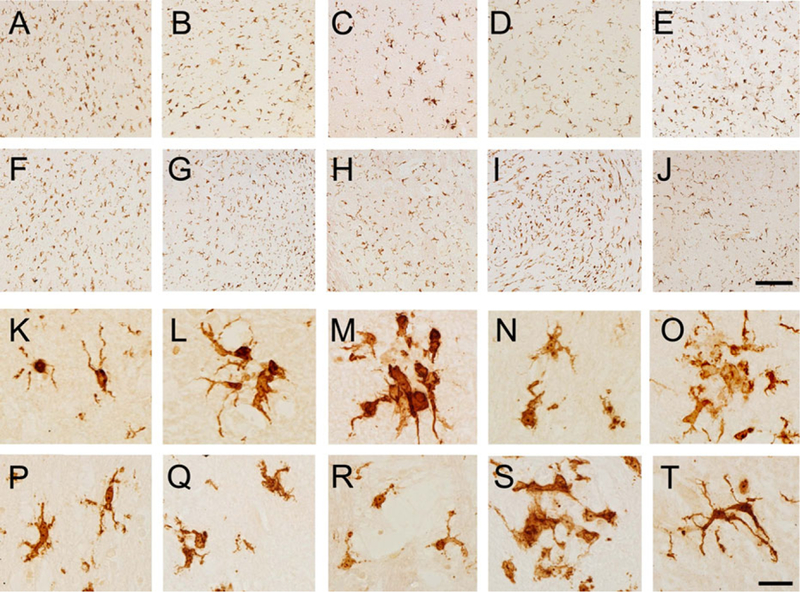
The increase in the number and state of activation described in the putamen seems to be present in other cortical and subcortical brain areas. (A,K) Frontal cortex; (B,L) temporal cortex; (C,M) parietal cortex; (D,N) occipital cortex; (E,O) hippocampus; (F,P) thalamus; (G,Q) midbrain; (H,R) pons; (I,S) medulla; (JT) cerebellum. Scale bar (A–J): 250 μm; (K–T): 25 μm. [Color figure can be viewed at wileyonlinelibrary.com]
Discussion
FXTAS presents with signs of oxidative stress damage that have been associated previously with an inflammatory state. We previously reported that there is a large increase in iron deposition in the brain of FXTAS patients, with the highest increase in the putamen.9,10,23 These findings may be related to an increase in free radicals and oxidative stress that occurs in FXTAS.21,22 To investigate an inflammatory state in FXTAS and a potential role for microglial cells in inflammation, we analyzed the nature of microglial cells in FXTAS. Because we previously found that increased iron deposition is most notable in the putamen in FXTAS, we analyzed the number and state of activation of microglial cells in the putamen of 13 FXTAS cases.
Dysmorphic Senescent Microglial Cells Are a Pathological Trait of FXTAS
We found that almost half of the FXTAS cases analyzed (6 of 13) presented with dysmorphic microglial cells in the putamen. These cells were characterized by a beaded, punctate, and fractured morphology. Dysmorphic morphology of microglia in neuropathology was first described in the 1930s.41 However, since then there have not been many reports about this phenotypic trait of microglia in neuropathological brain tissue. Most of the work on this topic has been performed by the Streit group.38,40,42 Streit and colleagues demonstrated that the degree of microglial dysmorphism is linked to age and senescence. With aging, an increasing proportion of microglial cells display abnormal morphological features, such as shortened, gnarled, beaded, or fragmented cytoplasmic processes, as well as loss of fine ramifications and formation of spheroidal swellings. These changes are designated collectively as microglial dystrophy.40 Depending on how far senescent degeneration has progressed, fragmentation of the microglial cytoplasm (cytorrhexis) may be partial or it may involve the entire cytoplasm leaving multiple fragments that in some cases still delineate the cells’ contours, or in the most advanced case of disintegration are widely scattered about.42 They also showed that senescent microglial cells are present in some diseases; specifically, they described dysmorphic senescent microglial cells in a small number of cases of Down syndrome with AD and dementia with Lewy bodies.38,39,43–46 The altered morphology of senescent microglia is a reflection of abnormal cellular function. Our finding of microglial dysmorphism in FXTAS could reflect aging, but the dysmorphism we observed in FXTAS did not correlate with age. In addition, the extent of dysmorphic microglia in FXTAS went beyond that described in the aging brain. We found that the cellular processes of senescent microglial cells in FXTAS were almost completely degraded and appeared as individual puncta. Microglial soma/nuclei were disconnected from processes and appeared as round or oval isolated structures. Presence of senescent microglia was not correlated with the state of preservation of the tissue, given that the PMI was much lower in cases with no senescence than in those with senescence. Moreover, previous reports have shown that the dystrophic appearance of microglial cells is not affected by PMI.43 However, we found that senescence was significantly correlated with the number of CGG repeats in FXTAS, pointing to a threshold in severity of disease at which the process of microglial senescence takes place.
Dystrophic microglial cells would be expected in neurodegenerative disorders characterized by excessive iron accumulation, as is the case in FXTAS, and have been described previously in AD.43–45 Because iron promotes oxidative damage, iron storage increases susceptibility to cell degeneration. Microglial cells are especially susceptible, because they participate in the maintenance of brain iron homeostasis through storage of excess iron.47,48 Accordingly, senescent microglial cells have been previously found in AD; however, this finding is not very common.46
Microglial cells, in contrast to other neural types such as neurons, have the capacity to proliferate in the adult brain. This process occurs because of microglial state of activation. It has been proposed that upon a given number of mitoses, microglial cells cease proliferating and become senescent. Under specific disease conditions that produce constant activation, the proliferative capacity of microglial cells may accelerate, and senescence could occur earlier in life.49 In FXTAS, we found that half of the cases presented with senescent microglia, the highest proportion ever reported for a neurodegenerative disease. Senescence of microglial cells, and therefore their lack of function and death, would be expected to alter proper function of the brain. This would potentially be associated with a process of neurodegeneration. Accordingly, presence of senescent microglial cells in half of FXTAS cases could be used, as is the presence of intranuclear inclusion and presence of iron, as a biomarker for postmortem diagnosis of FXTAS.
The Number and State of Activation of Microglial Cells Is Increased in FXTAS
Cases of FXTAS that were not senescent had a generalized increase in number and state of activation of microglial cells. Microglial cell activation takes place in response to injury and degeneration and is accompanied by changes in immune profile, morphology, proliferation, migration, and inflammatory cytokine production. The increased number of microglial cells in FXTAS may indicate that microglial proliferation has taken place, and therefore it is an indication of a process of microglial activation. Accordingly, we found an increase in the apparent morphological state of activation of microglial cells and expression of the microglial activation marker CD68. Proinflammatory cytokines released by activated microglia can exacerbate inflammation inducing neuronal degeneration, which could accelerate the progression of FXTAS. Both inflammation and reduced function of senescent microglial cells may together contribute to the pathological and functional decline in FXTAS.
Microglial Senescence May Be Preceded by a Prolonged Period of Microglial Activation in FXTAS
Senescence and activation of microglial cells may be independent processes; however, senescence may be the natural progression after a prolonged state of activation in microglia. Microglial senescence is not present in aged rodents because of the short life of these animals, but it can be achieved by inducing a severe neuropathological state; for example, through administration of the G-series nerve agent, soman,50 or in the SOD1G93A transgenic rat, an animal model of motor neuron disease, after infection with bacillus bacteria.51 In this rat model, microglial cytorrhexis occurs after prolonged microglial activation, supporting the idea that long-lasting microglial activation leads to immune exhaustion and microglial burnout.51 This is also supported by FXTAS cases with early microglial senescence in some brain areas and activated microglia in others. In addition, the senescent microglial cells, or the debris remnant of them, expressed CD68, a marker for activated microglia.
Conclusion
Senescence and activation characterize the pathology of FXTAS. Both inflammation and reduced microglial function are probably contributors to the pathological and functional decline in FXTAS. However, it is not clear whether microglial senescence and activation play a role in initiation of neurodegeneration and/or progression. Our data suggest that anti-inflammatory treatment of patients with FXTAS may be indicated to slow neurodegeneration.
Acknowledgments
Funding agencies: NIMH 2R01MH094681, NINDS R011NS107131, and Shriners Hospital for Children funding to VMC, NICHD GPHD036071 to RH, NIGMS GM113929 to PH, and NIMH R01MH101188–01 to SCN.
Footnotes
Relevant conflicts of interest/financial disclosures: Nothing to report.
Full financial disclosures and author roles may be found in the online version of this article.
References
- 1.Kogan CS, Turk J, Hagerman RJ, Cornish KM. Impact of the Fragile X mental retardation 1 (FMR1) gene premutation on neuropsychiatric functioning in adult males without fragile X-associated Tremor/Ataxia syndrome: a controlled study. Am J Med Genet B Neuropsychiatr Genet 2008;147B:859–872. [DOI] [PubMed] [Google Scholar]
- 2.Tassone F, Greco CM, Hunsaker MR, et al. Neuropathological, clinical and molecular pathology in female fragile X premutation carriers with and without FXTAS. Genes Brain Behav 2012;11:577–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Coffey SM, Cook K, Tartaglia N, et al. Expanded clinical phenotype of women with the FMR1 premutation. Am J Med Genet A 2008; 146A:1009–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rodriguez-Revenga L, Madrigal I, Pagonabarraga J, et al. Penetrance of FMR1 premutation associated pathologies in fragile X syndrome families. Eur J Hum Genet 2009;17:1359–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hagerman P Fragile X-associated tremor/ataxia syndrome (FXTAS): pathology and mechanisms. Acta Neuropathol 2013;126:1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Greco CM, Berman RF, Martin RM, et al. Neuropathology of fragile X-associated tremor/ataxia syndrome (FXTAS). Brain 2006;129:243–255. [DOI] [PubMed] [Google Scholar]
- 7.Greco CM, Hagerman RJ, Tassone F, et al. Neuronal intranuclear inclusions in a new cerebellar tremor/ataxia syndrome among fragile X carriers. Brain 2002;125:1760–1771. [DOI] [PubMed] [Google Scholar]
- 8.Greco CM, Tassone F, Garcia-Arocena D, et al. Clinical and neuropathologic findings in a woman with the FMR1 premutation and multiple sclerosis. Arch Neurol 2008;65:1114–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ariza J, Rogers H, Hartvigsen A, et al. Iron accumulation and dysregulation in the putamen in fragile X-associated tremor/ataxia syndrome. Mov Disord 2017;32:585–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ariza J, Steward C, Rueckert F, et al. Dysregulated iron metabolism in the choroid plexus in fragile X-associated tremor/ataxia syndrome. Brain Res 2015;1598:88–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Farzin F, Perry H, Hessl D, et al. Autism spectrum disorders and attention-deficit/hyperactivity disorder in boys with the fragile X premutation. J Dev Behav Pediatr 2006;27(2 Suppl):S137–S144. [DOI] [PubMed] [Google Scholar]
- 12.Chonchaiya W, Au J, Schneider A, et al. Increased prevalence of seizures in boys who were probands with the FMR1 premutation and comorbid autism spectrum disorder. Hum Genet 2012;131:581–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cunningham CL, Martinez Cerdeno V, Navarro Porras E, et al. Premutation CGG-repeat expansion of the Fmr1 gene impairs mouse neocortical development. Hum Mol Genet 2011;20:64–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tassone F, Hagerman RJ, Taylor AK, et al. Elevated levels of FMR1 mRNA in carrier males: a new mechanism of involvement in the fragile-X syndrome. Am J Hum Genet 2000;66:6–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Willemsen R, Hoogeveen-Westerveld M, Reis S, et al. The FMR1 CGG repeat mouse displays ubiquitin-positive intranuclear neuronal inclusions; implications for the cerebellar tremor/ataxia syndrome. Hum Mol Genet 2003;12:949–959. [DOI] [PubMed] [Google Scholar]
- 16.Brouwer JR, Mientjes EJ, Bakker CE, et al. Elevated Fmr1 mRNA levels and reduced protein expression in a mouse model with an unmethylated Fragile X full mutation. Exp Cell Res 2007;313:244–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sellier C, Rau F, Liu Y, et al. Sam68 sequestration and partial loss of function are associated with splicing alterations in FXTAS patients. EMBO J 2010;29:1248–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tassone F, Hagerman RJ, Garcia-Arocena D, Khandjian EW, Greco CM, Hagerman PJ. Intranuclear inclusions in neural cells with premutation alleles in fragile X associated tremor/ataxia syndrome. J Med Genet 2004;41:e43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hashem V, Galloway JN, Mori M, et al. Ectopic expression of CGG containing mRNA is neurotoxic in mammals. Hum Mol Genet 2009;18:2443–2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Todd PK, Oh SY, Krans A, et al. CGG repeat-associated translation mediates neurodegeneration in Fragile X tremor ataxia syndrome. Neuron 2013;78:440–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ross-Inta C, Omanska-Klusek A, Wong S, et al. Evidence of mitochondrial dysfunction in fragile X-associated tremor/ataxia syndrome. Biochem J 2010;429:545–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Giulivi C, Napoli E, Tassone F, Halmai J, Hagerman R. Plasma metabolic profile delineates roles for neurodegeneration, proinflammatory damage and mitochondrial dysfunction in the FMR1 premutation. Biochem J 2016;473:3871–3888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rogers H, Ariza J, Monterrubio A, Hagerman P, Martinez-Cerdeno V. Cerebellar mild iron accumulation in a subset of FMR1 premutation carriers with FXTAS. Cerebellum 2016;15:641–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Scaglione C, Ginestroni A, Vella A, et al. MRI and SPECT of midbrain and striatal degeneration in fragile X-associated tremor/ataxia syndrome. J Neurol 2008;255:144–146. [DOI] [PubMed] [Google Scholar]
- 25.Wang JY, Hessl D, Schneider A, Tassone F, Hagerman RJ, Rivera SM. Fragile X-associated tremor/ataxia syndrome: influence of the FMR1 gene on motor fiber tracts in males with normal and premutation alleles. JAMA Neurol 2013;70:1022–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Song G, Napoli E, Wong S, et al. Altered redox mitochondrial biology in the neurodegenerative disorder fragile X-tremor/ataxia syndrome: use of antioxidants in precision medicine. Mol Med 2016;22:548–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Heneka MT, Carson MJ, El Khoury J, et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol 2015;14:388–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Davalos D, Grutzendler J, Yang G, et al. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci 2005;8:752–758. [DOI] [PubMed] [Google Scholar]
- 29.Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005;308:1314–1318. [DOI] [PubMed] [Google Scholar]
- 30.Cunningham CL, Martinez-Cerdeno V, Noctor SC. Microglia regulate the number of neural precursor cells in the developing cerebral cortex. J Neurosci 2013;33:4216–4233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cunningham CL, Martinez-Cerdeno V, Noctor SC. Microglia regulate the number of neural precursor cells in the developing cerebral cortex. J Neurosci 2013;33:4216–4233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kozlowski C, Weimer RM. An automated method to quantify microglia morphology and application to monitor activation state longitudinally in vivo. PLoS One 2012;7:e31814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Smith AM, Dragunow M. The human side of microglia. Trends Neurosci 2014;37:125–135. [DOI] [PubMed] [Google Scholar]
- 34.Tassone F, Pan R, Amiri K, Taylor AK, Hagerman PJ. A rapid polymerase chain reaction-based screening method for identification of all expanded alleles of the fragile X (FMR1) gene in newborn and high-risk populations. J Mol Diagn 2008;10:43–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Filipovic-Sadic S, Sah S, Chen L, et al. A novel FMR1 PCR method for the routine detection of low abundance expanded alleles and full mutations in fragile X syndrome. Clin Chem 2010;56:399–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cunningham CL, Martinez-Cerdeno V, Noctor SC. Diversity of neural precursor cell types in the prenatal macaque cerebral cortex exists largely within the astroglial cell lineage. PLoS One 2013;8:e63848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jonas RA, Yuan TF, Liang YX, Jonas JB, Tay DK, Ellis-Behnke RG. The spider effect: morphological and orienting classification of microglia in response to stimuli in vivo. PLoS One 2012;7:e30763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Streit WJ, Xue QS. Microglia in dementia with Lewy bodies. Brain Behav Immun 2016;55:191–201. [DOI] [PubMed] [Google Scholar]
- 39.Bachstetter AD, Van Eldik LJ, Schmitt FA, et al. Disease-related microglia heterogeneity in the hippocampus of Alzheimer’s disease, dementia with Lewy bodies, and hippocampal sclerosis of aging. Acta Neuropathol Commun 2015;3:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Streit WJ, Xue QS, Tischer J, Bechmann I. Microglial pathology. Acta Neuropathol Commun 2014;2:142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ferraro A The origin and formation of senile plaques. Arch Neurol Psychiatry 1931;25:1042–1062. [Google Scholar]
- 42.Streit WJ, Xue QS. Human CNS immune senescence and neurodegeneration. Curr Opin Immunol 2014;29:93–96. [DOI] [PubMed] [Google Scholar]
- 43.Lopes KO, Sparks DL, Streit WJ. Microglial dystrophy in the aged and Alzheimer’s disease brain is associated with ferritin immunore-activity. Glia 2008;56:1048–1060. [DOI] [PubMed] [Google Scholar]
- 44.Streit WJ. Microglia and Alzheimer’s disease pathogenesis. J Neurosci Res 2004;77:1–8. [DOI] [PubMed] [Google Scholar]
- 45.Streit WJ, Braak H, Xue QS, Bechmann I. Dystrophic (senescent) rather than activated microglial cells are associated with tau pathology and likely precede neurodegeneration in Alzheimer’s disease. Acta Neuropathol 2009;118:475–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xue QS, Streit WJ. Microglial pathology in Down syndrome. Acta Neuropathol 2011;122:455–466. [DOI] [PubMed] [Google Scholar]
- 47.Cheepsunthorn P, Palmer C, Connor JR. Cellular distribution of ferritin subunits in postnatal rat brain. J Comp Neurol 1998;400:73–86. [PubMed] [Google Scholar]
- 48.Han J, Day JR, Connor JR, Beard JL. H and L ferritin subunit mRNA expression differs in brains of control and iron-deficient rats. J Nutr 2002;132:2769–2774. [DOI] [PubMed] [Google Scholar]
- 49.Interpreting sampling results. In: StereoInvestigator 8 User Guide Williston, VT: MicroBrightField; 2008:9. [Google Scholar]
- 50.Johnson EA, Dao TL, Guignet MA, Geddes CE, Koemeter-Cox AI, Kan RK. Increased expression of the chemokines CXCL1 and MIP-1alpha by resident brain cells precedes neutrophil infiltration in the brain following prolonged soman-induced status epilepticus in rats. J Neuroinflammation 2011;8:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fendrick SE, Xue QS, Streit WJ. Formation of multinucleated giant cells and microglial degeneration in rats expressing a mutant Cu/Zn superoxide dismutase gene. J Neuroinflammation 2007;4:9. [DOI] [PMC free article] [PubMed] [Google Scholar]