

Abstract
Liver cancer is the fourth leading cause of cancer-related mortality and is distinguished by a relative paucity of chemotherapy options. It has been hypothesized that intratumor genetic heterogeneity may contribute to the high failure rate of chemotherapy. Here, we evaluated functional heterogeneity in a cohort of primary human liver cancer organoid lines. Each primary human liver cancer surgical specimen was used to generate multiple cancer organoid lines, obtained from distinct regions of the tumor. A total of 27 liver cancer lines were established and tested with 129 cancer drugs, generating 3,483 cell survival data points. We found a rich intratumor, functional (drug response) heterogeneity in our liver cancer patients. Furthermore, we established that the majority of drugs were either ineffective, or effective only in select organoid lines. In contrast, we found that a subset of drugs appeared pan-effective, displaying at least moderate activity in the majority of these cancer organoid lines. These drugs, which are FDA approved for indications other than liver cancers, deserve further consideration as either systemic or local therapeutics. Of note, molecular profiles, obtained for a reduced sample set, did not correlate with the drug response heterogeneity of liver cancer organoid lines. Taken together, these findings lay the foundation for in-depth studies of pan-effective drugs, as well as for functional personalized oncology approaches. Lastly, these functional studies demonstrate the utility of cancer organoid drug testing as part of a drug discovery pipeline.
Keywords: Hepatology, Oncology
Keywords: Drug screens, Drug therapy, Liver cancer
Cancer organoids define intra-tumor and inter-patient functional heterogeneity to identify pan-effective drugs in human primary liver cancers.
Introduction
Liver cancer has a global incidence of approximately 850,000 cases and represents the fourth leading cause of cancer-related mortality (1). Hepatocellular carcinoma (HCC) accounts for 80%–85% of all cases of primary liver cancer, while cholangiocarcinoma (CCA) accounts for most of the remaining of cases (2, 3). Survival is poor in both major types of primary liver cancer, with a 5-year survival rate as low as 15% (2, 3). The lack of effective therapeutics in liver cancers and the high failure rate of the vast majority of drugs to date led some investigators to hypothesize that HCC is “not druggable” (4). While multiple somatic alterations were identified in HCC, the vast majority of molecular/targeted agents tested to date have shown minimal if any significant clinical benefit (4). As an example, sorafenib, a poorly tolerated multikinase inhibitor approved for HCC, improves survival by only 3 months at the cost of severe side effects (5). Similar to clinical outcomes in HCC, and in spite of next-generation sequencing (NGS) efforts to elucidate molecular pathways in CCA (6), no targeted agent is part of the first-line therapy for CCA. A possible explanation for the lack of activity of targeted agents may be the presence of intratumor genetic heterogeneity (7). First recognized in primary renal carcinomas, genetic heterogeneity has now been reported in most solid tumors, including HCC and CCA (7–10). A hypothesized, but never demonstrated, the assumption is that intratumor genetic heterogeneity causes and perhaps could even predict drug response heterogeneity.
Functional diagnostics describes a category of techniques to enable experimental drug testing on live cancer cells obtained from a patient, with the goal of informing drug choice (8). While precision oncology is often defined based on NGS of cancer exomes, the incomplete understanding of the genotype-to-phenotype relationship limits the clinical value of a genome sequence on its own (11). Carefully characterized functional diagnostics could address this limitation. Initial efforts, however, have been limited by lack of standard study designs, assay formats, assay reproducibility and accuracy, the number of drugs used (typically small), and the type of drugs used (typically standard chemotherapeutics) (11). Patient-derived xenograft (PDX) models can also be utilized for cancer drug screening, either to characterize activity of new lead compounds, or in a personalized fashion to identify best chemotherapy for a specific patient (11). Unfortunately, liver cancers tend to display relatively low engraftment rates, rendering liver cancer PDX models impractical for functional diagnostics (12).
Next-generation functional diagnostics — including patient-derived organoids (PDOs) — have been recently developed (13–15). In contrast to prior studies, here we establish PDO lines from multiple distinct regions of each liver cancer. We test 129 cancer drugs on PDO lines from 5 patients. We show that liver cancers display intrapatient and interpatient functional heterogeneity. Most notably, our methodology identifies drugs that are pan-effective across all liver cancer lines and that are already FDA approved for other indications. Additionally, we estimate correlations in our data and implement principal component analysis (PCA) based on cancer exomes, transcriptomes, and drug response data for one of the patient samples. Finally, we compare our results to publicly available data repositories, including The Cancer Genome Atlas (TCGA), and find that gene expression of the PDO lines clusters with a subgroup of CCA patients identified in a recent study (16).
Results
Establishment of PDO lines from distinct regions of liver tumors.
Recently, PDOs were successfully established from primary liver cancers, and their histologic, genetic, and transcriptomic profiles mirrored the corresponding primary human tumors (17). We established PDOs from a human intrahepatic CCA. In line with our focus on biological replicates obtained from the same cancer tissue, we cut the resection piece into 20 tissue slices as shown in Figure 1A. PDOs were established according to published protocols (18). As shown in Figure 1B, we noted an epithelial organoid appearance of cystic growth starting on day 2 after plating. To identify the cellular source of the cystic PDO structure observed in white-light images, we chose an epithelial marker (EPCAM), the bile duct markers cytokeratin 19 and 7 (CK19 and CK7) (19), the mucin marker mucicarmin (20–22), as well as the stem cell markers LGR5 and SOX9. We found that both the primary tumor (Figure 1C) and the matched PDOs (Figure 1D) displayed similar staining for all markers tested (Figure 1, E and F). Similarly, 7 distinct regions from the same surgical specimen of a human HCC were utilized to establish 7 matched PDOs. We utilized markers for epithelial tissues (EPCAM), stemness (LGR5), liver origin (CK19), and hepatocyte-specific markers (AFP and HepPar1). As shown in Supplemental Figure 1 (supplemental material available online with this article; https://doi.org/10.1172/jci.insight.121490DS1), we confirmed that the PDO cultures displayed marker profiles similar to the original primary human tumors.
Figure 1. Establishment of multiregion organoid lines from a primary human liver cancer.

(A) A cancer resection piece was cut into 20 tissue slices. The white-yellow appearance of the tissue in most of the slices was found to be fibrotic, intrahepatic cholangiocarcinoma. These tissue slices were utilized for histological, tissue banking, organoid establishment, and other analyses. (B) Bright-light images of PDOs established from CCA8. Cystic patient–derived organoid (PDO) structures were recognized starting on day 2 (D, day; P, passage). A representative cystic PDO was photographed from D1 to D10, at which point the culture was split. Representative images for this PDO culture are shown at P3 as well as P6. (C and D) Immunofluorescence staining of primary human cancer tissue and a matched PDO established from the same tissue slice for EPCAM (epithelial marker), CK19 (bile duct marker), and LGR5 and SOX9 (stem cell markers). DAPI was used to stain nuclei. (E and F) Primary human cancer tissue and a matched PDO established from the same tissue slice was used for hematoxylin and eosin staining as well as for immunohistochemistry (CK7 is a bile duct marker, and mucicarmine is a mucin marker). Scale bars: 200 μm (B) and 100 μm (C–F).
Drug testing and effect quantification in a PDO line.
Initial drug testing experiments were performed on one CCA PDO line, with a 9-drug panel, at a typical high-throughput drug screening concentration of 10 μM (23). For a positive control, we used 10% Triton X-100, while for a negative control we used DMSO at the same concentration (10 mM) used to dissolve the drugs. As shown in Figure 2, we observed that some drug treatments altered the shape of cystic PDO structures (from intact structures, to reduction of cyst size to their disappearance), as recently described (17). We tested cisplatin and gemcitabine, because they are first-line CCA therapeutics. We found that cisplatin had no effect on PDOs, while gemcitabine had a moderate effect (Figure 2 and Supplemental Figure 2). Please note that the viability measurements were verified in 3 different PDO lines (CCA8-5 in Figure 2A, CCA23-3 in Figure 2B, and CCA28-3 in Figure 2C, and Supplemental Figure 2). We then expanded the study to include National Cancer Institute (NCI) set VII, a 129 FDA-approved cancer drug library (https://wiki.nci.nih.gov/display/NCIDTPdata/Compound+Sets). In this drug library, we found that bortezomib, a first-generation proteasome inhibitor, exhibited excellent cancer inhibitory effects (Figure 2B). Next, we noted that combination therapies did not display additive effects on PDOs (Figure 2C). To better quantify the killing effect of this larger library of drugs, we employed CellTiter-Glo (an ATP-based cytotoxicity assay). The Z factor for the CellTiter-Glo assay was found to be 0.84 for the 9-drug panel screening, which is excellent for high-throughput drug screening (24). Supplemental Table 1 illustrates a typical color-coded viability readout (the drug indices — Supplemental Table 2). Note that the vast majority of drugs were ineffective, while 9 of the 129 drugs tested demonstrated over 95% killing activity. Using this method, we confirmed that the effect of combination therapy is similar to that of the more efficacious of the individual drugs (Supplemental Table 3). Of note, these assays reflect the direct, cell-intrinsic action of the drugs, and do not include complex in vivo interactions.
Figure 2. Low-throughput and high-throughput drug testing in cancer organoid lines and treatment efficacy determination, and summary of human primary liver cancer specimens, demographics, and laboratory derivation of sister PDO lines.

(A) A panel of 7 cancer drugs at a concentration of 10 μM, along with negative control (0.1% DMSO) and a positive control (10% Triton X-100), were used on a PDO line. The PDO treated with cisplatin appeared identical to the negative control, consistent with no drug efficacy. Gemcitabine stalls PDO growth, but does not kill cells. We verified that gemcitabine does not induce cell death through additional studies, as described in Supplemental Figure 2. (B) Cisplatin allows unrestricted growth of cancer PDOs — as demonstrated by continuously expanding cystic PDO structures — while gemcitabine had a cytostatic effect. The efficacy of bortezomib, a proteasome inhibitor, was validated in a time-course experiment. The cystic PDO structures were effectively prevented from maintaining their shape or expanding in size. (C) The most frequently used clinical drug combinations for CCA were used on a CCA PDO line. As shown, gemcitabine plus cisplatin had the same effect as gemcitabine alone. Similarly, in all 8 combinations tested, the overall efficacy was similar to that of the more efficacious of the 2 drugs used in combination. Scale bars: 200 μm (A) and 400 μm (B and C).
High-throughput drug screening in a large cohort of liver cancer PDO lines.
We collected 1 additional human primary HCC and 2 additional human primary CCA surgical specimens, for a total of 5 primary liver cancers, and established various numbers of PDOs (3–7) from each (Table 1). Next, all 27 liver cancer PDOs were treated with drugs. The experiment produced 27 viability readings (one per PDO) for each of the 129 drugs, for a total of 3,483 data points (Figure 3A and Supplemental Table 4). Thirteen drugs showed more than 90% killing across all 27 liver cancer PDO lines (red cells at the top of the heatmap in Figure 3A and to the far left in Figure 3B, which shows the mean and variance in drug response). Of these drugs, 4 (daunorubicin, ceritinib, doxorubicin, and mitomycin) had at least one PDO with suboptimal response. The remaining 9 drugs were pan-effective across the PDO lines (Table 2 shows the response to 7 pan-effective drugs). These 9 pan-effective drugs belong to 5 classes of antineoplastic agents (histone deacetylase [HDAC] inhibitors, proteasome inhibitors, DNA topoisomerase II inhibitors, protein translation inhibitors, and RNA synthesis inhibitors; Table 3). Only 2 out of these 9 pan-effective drugs had ever been tested as systemic chemotherapy in liver cancers (panobinostat and bortezomib, Table 3) and only 1 in CCA (bortezomib). One agent, idarubicin, has been tested as local, liver-directed therapy (transarterial chemoembolization [TACE]). In addition to screening at a concentration of 10 μM, we performed 10-fold dilutions for plicamycin and idarubicin. The concentration of drugs were 10 μM, 1 μM, 0.1 μM, 0.01 μM, and 0.001 μM. Based on these data, we calculated IC50 concentrations (Figure 3C). For plicamycin, except for CCA8-6, all PDO lines showed similar IC50 doses. For idarubicin, the IC50 range was much larger, from 2.5 nM in HCC 26-7 to 6.9 μM in 28-3. Intratumor responses were also variable; for example, for HCC26, the IC50 for 26-7 was 2.5 nM, while for 26-5 it was 0.9 μM. These results demonstrate inter- and intratumor heterogeneity in drug response.
Table 1. Summary of human primary liver cancer specimens, demographics, and laboratory derivation of sister PDO lines.

Figure 3. The identification of drugs that are pan-effective in a large cohort of 27 primary liver cancer PDO lines.

(A) Color-coded heatmap of drug killing effects for 129 drugs (y axis; the indices of 129 drugs are in Supplemental Table 4) over 27 primary liver cancer PDO lines (x axis). The color scale is shown at the bottom of the figure, from blue (100% cell viability) to red (0% cell viability). The heatmap is organized with pan-effective drugs at the top of the figure. Color-coded values are the mean of 3 technical replicates. (B) The median cell viability across each of the 27 PDO lines was calculated for each drug and displayed in a box-and-whisker plot along with the variance. Thirteen drugs (to the far left in this panel) were found to induce an average cell viability across the 27 lines of less than 10%. (C) IC50 calculation for 2 of the pan-effective drugs, idarubicin and plicamycin, and for 22 organoids out of 27 from the 5 patients. The upper subpanel is the log-scale IC50 for 2 patients, CCA23 and HCC26; the lower subpanel shows the IC50 drug dose for each line from the 5 patients. Drug screening was performed in triplicate.
Table 2. Seven pan-effective drugs tested across all 27 primary liver cancer PDO lines.

Table 3. Clinical trial information for 7 pan-effective drugs.

Interpatient functional heterogeneity — the identification of interpatient-divergent drugs.
Several drugs were effective in only a subgroup of PDO lines. For example, we noted that dasatinib, previously reported as efficacious in a set of liver cancer organoids (17), was only moderately effective (survival of less than 15%) in only 3 of 27 liver cancer lines (Supplemental Figure 5). To investigate interpatient-divergent drugs, we averaged the drug effects across all PDOs established from each cancer. This mean cell viability per patient is an estimate of how the tumor, as a whole, would respond to a certain drug. We demonstrate these data using a heatmap, with the order of cancers and the order of drugs determined by unsupervised hierarchical clustering (Figure 4A and Supplemental Table 6). The vast majority of drugs had poor overall effect (blue rows). However, some drugs had variable interpatient effects (Supplemental Table 7). Note that while some interpatient-divergent drugs are efficacious in just one cancer (such as ceritinib and valrubicin in HCC25), others are effective in a majority of cancers, but not in all of them (such as ponatinib or sorafenib).
Figure 4. The identification of drugs with interpatient and intrapatient divergent effects.

(A) Heatmap of drug response for each of the 5 cancers (x axis) across 129 drugs (y axis). The mean cell viability measurement for each patient-drug pair is shown. Drug testing was performed at a screening concentration of 10 μM. Index values at the right refer to individual drugs (described in Supplemental Table 4). The red color represents good killing effect (low cell viability after drug treatment) and blue represents poor killing effect (high cell viability after drug testing). The cluster of drugs represented in red at the top of the figure includes the 7 pan-effective drugs described in Figure 3. (B) Heatmap similar to that in A but showing the standard deviation of cell viability for each patient-drug combination (see drug order in Supplemental Table 4). Here, blue signifies low intracancer standard deviation and red signifies high intracancer standard deviation of drug effect.
The identification of intratumor-divergent drugs.
A number of drugs were selectively effective against a subset of PDOs derived from individual patients and therefore displayed intrapatient drug response heterogeneity. We calculated the variation of drug response of PDO lines derived from individual patients represented by the deviation from mean drug effect (Figure 4B and Supplemental Table 6). Targeted drugs, such as tyrosine kinase inhibitors (TKIs; crizotinib, sorafenib, and others) were well represented as intratumor-divergent drugs. Next, we focused on patient CCA8 and performed in-depth analysis of all 6 PDO lines derived from this tumor. Several drugs displayed uniform killing effects over the 6 PDOs, while others had substantially divergent effects. Most drugs had a similar effect over the 6 PDOs, as evinced by the low standard deviation (blue color in Figure 4B). Of note, drugs that displayed low standard deviation in terms of effects on cancer cells are either equally effective (such as ixazomib and carfilzomib), or equally ineffective (such as thalidomide and gefitinib, Supplemental Table 8). The drugs displayed in red in Figure 4B have large variation (Supplemental Table 9). For example, belinostat demonstrated moderate activity (25% survival) in 1 of the 6 PDO lines and generally poor activity (mean 54% survival) in the other 5. Dasatinib, previously reported as efficacious in a liver cancer organoid drug testing study (17), was efficacious in 3 of the 6 (mean 14% survival), displayed moderate activity in 2 of the 6 (mean 25% survival), and poor activity in 1 of the 6 PDO (59% survival). Gemcitabine showed moderate activity in 1 of the 6 (24% survival) and poor activity in 5 of the 6 (mean 69% survival) PDO lines. Some drugs, such as ceritinib, demonstrated a sharp difference between high activity in 3 of the 6 (mean 1% survival) and almost no activity (mean 80% survival) in the remaining 3 of the 6 PDOs. These drugs presumably have organoid-dependent targets rather than nonspecific cytotoxic effects.
We used variance component analysis to quantify the importance of within-tumor versus between-tumor factors responsible for drug response heterogeneity (see Methods section). We found that 73% of the variance in the whole drug screen is attributed to between-tumor variation and 27% to within-tumor variation. Globally ineffective drugs, described as those with greater than 80% mean viability (n = 69), have a low overall variance, and only 7% of it is explained by the between-tumor variation. For the TKIs, we estimated that 80% of the variance comes from between-tumor variability and 20% from within-tumor variability. A summary of the variance component analysis can be found in Table 4.
Table 4. Variance component analysis.

PDO drug response data compared to publicly available data repositories.
Genomics of Drug Response Data in Cancer (GDSC), and the Cancer Cell Line Encyclopedia (CCLE), are large-scale, publicly available data repositories with drug response data for various cell lines. The IC50 and area under the curve (AUC) measurements from these 2 data repositories have been shown to be in good agreement (25). Here, we compared the drug response data for the 27 PDOs to the CCA and HCC cell lines in the GDSC and CCLE. From CCLE, we obtained the data for all of the available CCA and HCC cell lines (n = 20) that were tested using drugs in our screen (11 drugs) (26). A list of the cell lines and drugs can be found in Supplemental Table 10. The highest dose that the drugs in the CCLE data set were tested at is 8 μM. Recall that our drug response data are for 10 μM. The cell viability has been calculated in the CCLE using treatment with DMSO as negative control and treatment with MG132 as positive control. The correlation matrix for the drug response of the cell lines (Supplemental Figure 4A) and the visualization of the first 2 principle components (Supplemental Figure 4B) highlight the drug response discrepancy. For the most part, the PDOs and the cell lines form distinct clusters. Interestingly, the drug response of C3A (an HCC cell line) clusters together with the drug response of the PDOs (Supplemental Figure 4A) using a minimal variance clustering method. Specifically, the pan-effective drug panobinostat had a good killing effect on the cell lines and PDOs (mean 5% survival). Meanwhile, the intertumor-divergent drug sorafenib had poor killing effect on the cell lines (mean 76% survival). Similarly, from GDSC, we obtained available data for CCA and HCC cell lines (n = 16) that were tested with drugs in our data set (29 unique drugs) (27). A list of the cell lines and drugs can be found in Supplemental Table 10. The drugs olaparib and afatinib have 2 different drug identification numbers with distinct drug response data in the GDSC; both drug responses were included in our analysis. The drugs in the GDSC are tested at various concentrations; we used the highest available drug dose (which in these data ranges from 0.5 to 16 μM). To obtain a cell viability measurement we used the mean intensity from experiments with blank wells (wells with no drugs and no cells) as positive control and the mean intensity of experiments with cell lines treated with DMSO as negative control. The correlation matrix for the drug response of the cell lines (Supplemental Figure 5A) showed that 2 of the PDOs, CCA8-6 and HCC25-1, cluster with the HCC cell lines JHH-1, JHH-2, and SNU-182. Additionally, the visualization of the first 2 principle components (Supplemental Figure 5B) showed that the cell lines TGBC1TKB (a biliary tract cancer cell line) and SNU-449 (an HCC cell line) cluster with the PDOs (using the k-means algorithm, with k = 3). The cell lines (n = 6) in the top cluster on the x axis in Figure 5A had a poor correlation to the drug response of the PDOs. Furthermore, the PDO HCC25-2 does not cluster with the remaining PDOs. On the individual-drug level, the intratumor-divergent drug doxorubicin had a good killing effect on the majority of PDOs and cell lines (mean 14% survival). Lastly, the interpatient-divergent drug belinostat (mean 43% survival) had a good killing effect on the cell lines (mean 11% survival).
Figure 5. Comparing gene expression to drug response in patient CCA8.

Heatmaps generated for patient CCA8 using hierarchical clustering agglomeration (minimal variance method) for (A) the cell viability data (with the 129 drugs on the y axis and the color bar representing the survival fraction), and (B) the gene expression data for the 100 genes with the greatest variance in the expression level (shown here as the log2 of the fragments per kilobase of transcript per million reads [FPKM], with the color bar representing the standard score that shows if the expression is above [yellow] or below [blue] the mean). In both data sets, the following 2 PDO pairs clustered together: CCA8-7 and CCA8-11, and CCA8-5 and CCA8-10.
Exome and RNA sequencing.
In order to understand whether molecular correlates could predict drug response, we performed whole-exome sequencing (WES) as well as RNA sequencing on the 6 distinct PDO lines from patient CCA8. Targeted regions were sequenced with an average depth of 141× coverage, and 98% of the exome sequenced to at least 20× depth (Supplemental Table 11). While understudied in human CCA, genetic intratumor heterogeneity has been recognized in other human cancers, such as renal cell carcinoma and HCC (7, 8, 10). We found that 2 of the 6 PDOs (CCA8-7 and CCA8-11) displayed a frameshift mutation in fibroblast growth factor receptor 1 (FGFR1). The other 4 did not display any mutations in FGFR1. These findings predict that CCA8-7 and CCA8-11 would respond to ponatinib, a multikinase inhibitor, while the other 4 PDOs that do not display this mutation would not respond to ponatinib. Indeed, at a screening concentration of 10 μM, CCA8-7 and CCA8-11 were killed by ponatinib (Figure 6). These findings suggest that mutational profiles are able to predict response to targeted therapeutics. Nonetheless, at decreasing concentrations of ponatinib, CCA8-7 and CCA8-11 lose response (Figure 6). The calculated IC50 for CCA8-7 was 4.22 μM and for CCA8-11 it was 7.78 μM (Supplemental Figure 3). The published maximum serum concentration (Cmax) from the FDA for ponatinib, however, is 0.137 μM. The implication is that while CCA8-7 and CCA8-11 respond to ponatinib at a high screening concentration of 10 μM (as predicted by WES), there would be no response in vivo at Cmax concentrations.
Figure 6. Response to ponatinib in CCA8 organoid lines.

At a concentration of 10 μM (first row from the top), ponatinib was effective in CCA8-7 and CCA8-11 (note the destruction of the cystic structure), and ineffective in the other 3 lines. The response of CCA8-7 and CCA8-11 was lost at 1 μM and lower concentrations. Scale bar: 400 μm. Please see the text and Supplemental Figure 3 for IC50 curves and calculations.
Out of the known targets of the TKIs in our screen, only the FGFR1 mutation described above was found in the 6 PDO lines. Additional mutations reported in CCA (28, 29) were found in our patient data. All PDOs have KMT2C and PTCHD3 mutations, CCA8-10 has FMN2 and USP2 mutations, PDOs CCA8-6 and CCA8-10 have ARID1B mutations, CCA8-10 and CCA8-11 have RTK mutations, and CCA8-5, CCA8-9, CCA8-10, and CCA8-11 have HDAC5 mutations. These mutations do not have known therapeutic targets.
Comparison of gene expression and functional response in patient CCA8.
A univariate linear regression used to test for pairwise significant correlation (with Bonferroni’s correction for multiple testing of genes and drugs) did not reveal any significant correlations between individual coding somatic DNA variants or expression levels of individual genes, and response profiles for each drug, across the 6 PDOs. A heatmap of the drug response for the 6 PDO lines from patient CCA8 with hierarchical clustering is illustrated in Figure 5A. The results show that the following regions cluster: CCA8-7 and CCA8-11, CCA8-5 and CCA8-10, CCA8-6 and CCA8-9. A heatmap of the gene expression data for the same PDO lines is in Figure 5B. Interestingly, for these data, the regions CCA8-7 and CCA8-11, CCA8-5 and CCA8-10 also cluster together. These data suggest that gene expression could indicate which PDO lines would respond in similar ways to drug treatments. Additionally, we performed PCA of the drug response, gene expression, and somatic mutations, with results presented in the supplemental material.
Finally, we compared the gene expression data from the 6 PDOs from patient CCA8 to TCGA. TCGA has 36 patient samples for CCA (classified as TCGA-CHOL), which were in part analyzed in a recent study (16). As in Farshidfar et al. (16), we excluded from our analysis 541 genes that are associated with normal liver function. Figure 7 shows that the gene expression (fragments per kilobase of transcript per million reads, FPKM) of the mean of the 6 PDOs for patient CCA8 cluster with 8 of the TCGA samples. Notably, this cluster is similar (including 4 out of 5 patients) to the one identified as group II, a group enriched in extrahepatic or perihilar CCA (16).
Figure 7. The gene expression from patient CCA8 clusters with a subset of patients from TCGA.

Heatmap of the gene expression (the log2 of the FPKM on the y axis with the color bar representing the deviation from the mean, Z score, as in Figure 5B) for the cholangiocarcinoma sample set from TCGA (n = 36) compared with the mean gene expression of the 6 PDOs from patient CCA8. The hierarchical clustering agglomeration (using Pearson’s correlation and the minimal variance method) was based on the 1,000 genes with the greatest variance after excluding 541 normal liver genes as was done in Farshidfar et al. (16). Four main sample clusters were observed. The gene expression for patient CCA8 clusters with 8 of the samples from the TCGA.
Discussion
Intratumor genetic heterogeneity has been demonstrated in several cancers (7–10). To our knowledge, our study is the first to report intratumor drug response heterogeneity in any solid human cancer. We have shown that the 129 cancer drugs tested on 27 primary liver cancer organoid lines can be broadly classified into 4 classes (Supplemental Table 12). A small group of drugs were found to be pan-effective. Although these analyses reflect cell-intrinsic sensitivity to drugs and therefore may not reflect complex in vivo interactions, the identification of these class I type of drugs may constitute the first step in a rational strategy of drug selection, repurposing, or discovery. We also found a number of drugs that displayed intrapatient functional divergence (class II) (Figure 4B). This functional heterogeneity within patients supports the use of PDO lines derived from distinct regions of the cancer. Furthermore, we found a group of drugs that displayed significant interpatient divergence (class III). These drugs appeared effective in 1 or more human cancers, but not in all (Figure 4A and Supplemental Tables 7 and 12). Interestingly, 2 of the top interpatient-divergent drugs (sorafenib and gemcitabine) are in clinical use for CCA and HCC. The fact that some patients appear to respond, while others do not, suggests a possible role for organoid-based functional testing in informing drug choice for liver cancer patients. Lastly, we found that most drugs were generally ineffective (class IV). One could speculate that treatment with these drugs would result in selection for tumor regions corresponding to drug-resistant sublines. Of note, TKIs (such as lapatinib and axitinib, Supplemental Table 12) were well represented in this class of drugs.
The development of new oncology drugs typically starts with preclinical models (30). This approach, pioneered at the NCI in the 1950s, aimed to test new drug leads on in vitro cancer models. After 1985, the growing realization that human solid cancers are molecularly heterogeneous led to the development and utilization of 60 human cell lines for drug testing in oncology (the NCI-60) (30). Here, we present evidence of functional heterogeneity, even within the same cancer. For example, we found that dasatinib, recently reported as effective in a liver cancer organoid study (17), was effective (survival of less than 15%) in only 3 out of 27 PDO lines. In comparing a subset of the drug response of the 27 PDOs in this study to publicly available cell line drug response data (Supplemental Figures 4 and 5), we found a vast diversity in their responses (the exception being the pan-effective drug panobinostat that was represented in the CCLE data repository). These findings suggest an explanation for why drug candidates with promising preclinical results obtained in several cell lines or cancer PDO lines could nevertheless fail in clinical practice. By extension, the current study along with other published studies argue that cancer organoid lines established from large cohorts of patients may become a valuable resource for go/no-go decisions early in the typically expensive cancer drug development pipelines (31). In addition, the organoid platform may serve as part of a drug repurposing strategy. Drug repurposing strategies are well known in diverse areas of medicine, including oncology (32, 33). Repurposing is attractive because of the anticipated reduction in costs and acceleration of development timelines (31).
The analysis of the gene expression data set for patient CCA8 with the publicly available data from TCGA revealed that it was representative of a previously identified subgroup of CCA patients (Figure 7) (34). Furthermore, our analysis using hierarchical clustering agglomeration (Figure 5) indicates that gene expression may be suggestive of drug response for patient CCA8. However, our data set did not enable the identification of correlations between molecular and functional heterogeneity. It is formally possible that a larger number of PDOs for which all 3 sets of data (exome, transcriptome, and drug response) are collected may allow correlative analyses. It should be noted that, in a clinical setting, for technical and cost reasons only a limited number of PDOs can be generated from an individual patient cancer with the purpose of informing treatment. Therefore, the constraint imposed by limited sample size is unlikely to be overcome in a patient-specific clinical scenario. Alternatively, it is possible that numerous factors on the tumor, individual, and population levels contribute to complex biology underlying a cancer’s susceptibility to various anticancer agents, resulting in a mathematically impossible predictive task. The implications are that cancer behaviors (including when treated with drugs) are still incompletely understood and that our current genetic and transcriptomic strategies require further development and analysis to allow accurate predictions.
Finally, these data support the concept of multiple cancer biopsy protocols to ensure that functional intratumor heterogeneity is well represented in a PDO-driven platform. For example, as shown in Figure 6, depending on the site of the biopsy, the patient would have been diagnosed (or not) as carrying an FGFR1 mutation. However, in terms of predicting response to the pan-effective drugs discovered here, no known genetic alteration (mutational or otherwise) predicts the response of human cancers to proteasome inhibitors (35, 36). Furthermore, early transcriptomic biomarkers of sensitivity to proteasome inhibitors had not yet been clinically validated (35–37). In addition, HDAC inhibitors also appear to be pan-effective, which is mechanistically plausible given that chromatin-modifying genes were found in one of our earlier studies as top mutated genes in CCA (38). Similar to proteasome inhibitors, HDAC inhibitors also lack genetic tests that can predict response. In addition, RNA sequencing and WES have not been useful to predict response to microtubule inhibitors, DNA topoisomerase 2 inhibitors, or the “Others” class, since the mechanism of action for these drugs is generally not DNA mutation specific. In an effort to address these obstacles, we developed this multiple region biopsy protocol and combined it with a PDO drug testing platform with the aim of informing chemotherapy choices in a patient-individualized manner. Further studies will elucidate the correct balance between drug pan-effectiveness and utility of personalized drug testing for select group of patients.
Methods
Human tissue.
Fresh human HCC or CCA tissue was obtained from the Johns Hopkins Hospital (JHH), under an IRB-approved informed consent. A diagnosis of HCC or CCA was performed by JHH pathologists, in accord with current standards in the field.
Establishment of PDOs from human HCC and CCA.
Resection liver tissue (HCC or CCA) obtained from patients was minced into small pieces measuring approximately 9 mm3. Tissue was then rinsed 3 times with DMEM supplemented with 1% FBS at 4°C in 50-ml Falcon tubes. The tissue was then dissociated with collagenase (2 mg/ml, Sigma-Aldrich) supplemented with DNAse 1 (0.1 mg/ml in DMEM, Sigma-Aldrich) at 37°C. Digestion was stopped by adding cold DMEM supplemented with 10% FBS. Cells were filtered, washed by centrifugation, and then counted. After mixing with growth factor–reduced Matrigel (Corning, catalog 356231), cells were seeded into 24-well plates. After the Matrigel solidified within 15 minutes, warm organoid culture medium was added.
PDO medium.
Medium reported previously for normal liver organoids and liver cancer organoids was adapted for use in the current study (13, 18). The PDO medium is Advanced DMEM/F12 supplemented with 1× HEPES, 1× GlutaMax (Invitrogen), 1× primocin (InvivoGen), 1× B27 Supplement (Gibco), 1× N-2 Supplement (Gibco), 30% Wnt-conditioned medium, 20% Rspondin-conditioned medium, 0.5 μM A83-01 (Tocris), 0.05 μg/ml EGF (Sigma-Aldrich), 0.1 μg/ml FGF-10, 0.03 μg/ml HGF (Peprotech), 12.5 μM FSK (LC Laboratories), 0.01 μM gastrin (Sigma-Aldrich), 0.1 μg/ml noggin (Peprotech), 1.25 mM N-acetylcysteine (Sigma-Aldrich), and 12.5 mM nicotinamide (Sigma-Aldrich). For initial establishment of the PDOs, we used 50% (vol/vol) Wnt-conditioned medium and 30% (vol/vol) Rspondin-conditioned medium in the first week, 10 μM Y27632 (LC Laboratories) was added at the PDO establishment and passage step.
WES and analysis.
Total DNA was extracted from PDOs with Qiagen DNA kits (catalog 69504). We used 1.5 μg DNA for WES with an Agilent V5 capture probe set and sequenced using an Illumina HiSeq 2000 platform. The sequences of each sample were generated as 90- to 100-bp paired-end reads, and for each sample, approximately 10 Gb of unique sequence was generated. Standard quality control procedures were applied. Tumor-normal pair read alignment and variant identification, using SAMtools and GATK, were performed using standard parameters. A somatic variant was considered present in a tissue sample if it was present in at least 10% of reads. Using somatic coding (missense, nonsense, and canonical splice site) variations only, we performed phylogenetic tree estimation, using the maximum parsimony method with default settings in MEGA7 (39).
High-throughput drug screening of CCA PDOs.
A panel of 129 anticancer compounds (the NCI-Approved Oncology Drugs Set VII) was utilized. PDOs were plated in 96-well or 384-well plates and cultured for 3 days. Next, media were changed with drug-containing media. The initial screen was performed at a concentration of 10 μM. After 4 days, cell viability was determined in each well with CellTiter-Glo (catalog G7572, Promega) following the manufacturer’s instructions. Luminescent signal was measured with a plate reader (PerkinElmer EnVision Plate Reader). Drugs that induced less than 50% viability were chosen for future studies. IC50 experiments were performed at 10 μM, 1 μM, 100 nM, 10 nM, and 1 nM. All drug screens were performed with experimental triplicates.
Variance component analysis.
Variance component models describe the distributions of random variables in structured populations where subgroups have distinct means but share a common variance. These models provide a framework for hypothesis testing and provide unbiased estimates of variances. Most commonly, populations refer to individuals and the structure arises from families. Here, the population refers to organoids from a distinct tumor region treated with a specific drug, and the structure arises because subsets of organoids are from a single patient. The observation of organoids i from patient t is denoted yti. The variance component model considers nested hypotheses for yti, stated in terms of hypotheses H0 and H1 equivalent to models M0 (the null model assumes equal means and equal variance) and M1 (the alternative model assumes the variance is shared):
Equation 1.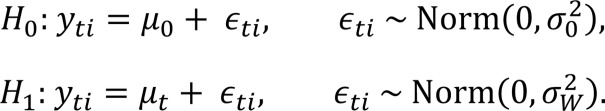
The null hypothesis, M0, specifies that survival is independently, identically distributed for each organoid, with a mean μ0 and variance shared across all tumors. The alternative hypothesis, M1, introduces a tumor-dependent mean, μt. The tumors continue to share the same within-tumor variance, . Testing H0 versus H1 is accomplished using 1-way analysis of variance (ANOVA).
Statistics.
Using linear regression, we found no significant correlation between drug response and gene expression at a family-wise error rate of 0.05 (equivalent to a single-test P value of 0.05 divided by the number of gene-drug combinations, 1.9 × 10–8).
We quantified the proportion of the variance in the drug response data that was attributed to intrapatient versus interpatient variability (see the previous Methods section Variance component analysis). We examined the cell survival data using both arithmetic and log scales. We concluded that the errors in the data are likely constant from sample to sample and thus we chose to use the arithmetic scale in our analysis. The hypotheses testing was carried using 1-way ANOVA. If the null hypothesis was rejected, the variance was decomposed into intra- and interpatient variance (see Table 4).
The gene expression from patient CCA8, shown in Figure 5B and Figure 7, is presented using the standard score, Z score, which shows how many standard deviations from the mean the data are. To minimize the outlier effect, we used log2 of the FPKM data.
All clustering agglomeration was conducted with Pearson’s correlation coefficients using the minimal variance method for hierarchical clustering, which minimizes the total within-cluster variance (also known as Ward’s method).
Study approval.
Collection of the specimens for this study was approved by the IRB of the Johns Hopkins University School of Medicine.
Author contributions
Study concept and design (FMS, LL), acquisition of data (LL, HK, KH, MJW, JH, BP, AMC, CLW, TMP, GG, EM, FMS), analysis and interpretation of data (LL, HK, KH, GG, AJE, JSB, FMS), drafting of the manuscript (LL, HK, KH, JSB, FMS), critical revision of the manuscript (AJE, JSB, FMS), statistical analysis (HK, JSB, FMS).
Supplementary Material
Acknowledgments
Support for this project was received through grants to FMS from the NIH (R01CA190040, R01EB017742), and from Maryland Innovation Initiative (TEDCO, Project 0118-008); as well as grants from the NIH/NCI to AJE and JSB (U01CA217846) and to AJE (U01CA221007, U54CA2101732, and 3P30CA006973); and from the Jayne Koskinas Ted Giovanis Foundation for Health and Policy and the Breast Cancer Research Foundation to AJE and JSB. The Jayne Koskinas Ted Giovanis Foundation for Health and Policy and the Breast Cancer Research Foundation are private foundations committed to critical funding of cancer research. The opinions, findings, conclusions or recommendations expressed in this material are those of the author(s) and not necessarily those of the Jayne Koskinas Ted Giovanis Foundation for Health and Policy or the Breast Cancer Research Foundation, or their respective directors, officers, or staffs.
Version 1. 01/24/2019
Electronic publication
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
License: Copyright 2019, American Society for Clinical Investigation.
Reference information: JCI Insight. 2019;4(2):e121490. https://doi.org/10.1172/jci.insight.121490.
Contributor Information
Ling Li, Email: lli42@jhmi.edu.
Hildur Knutsdottir, Email: hildurknuts@gmail.com.
Jin He, Email: jhe11@jhmi.edu.
Benjamin Philosophe, Email: bphilosophe@jhmi.edu.
Gabriel Ghiaur, Email: gghiaur1@jhmi.edu.
Esteban Mezey, Email: emezey@jhmi.edu.
Joel S. Bader, Email: joel.bader@jhu.edu.
References
- 1.Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 2.Singal AG, El-Serag HB. Hepatocellular carcinoma from epidemiology to prevention: Translating knowledge into practice. Clin Gastroenterol Hepatol. 2015;13(12):2140–2151. doi: 10.1016/j.cgh.2015.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Valle J, et al. Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. N Engl J Med. 2010;362(14):1273–1281. doi: 10.1056/NEJMoa0908721. [DOI] [PubMed] [Google Scholar]
- 4.Llovet JM, et al. Hepatocellular carcinoma. Nat Rev Dis Primers. 2016;2:16018. doi: 10.1038/nrdp.2016.18. [DOI] [PubMed] [Google Scholar]
- 5.Llovet JM, Villanueva A, Lachenmayer A, Finn RS. Advances in targeted therapies for hepatocellular carcinoma in the genomic era. Nat Rev Clin Oncol. 2015;12(8):436. doi: 10.1038/nrclinonc.2015.121. [DOI] [PubMed] [Google Scholar]
- 6.Jiao Y, et al. Exome sequencing identifies frequent inactivating mutations in BAP1, ARID1A and PBRM1 in intrahepatic cholangiocarcinomas. Nat Genet. 2013;45(12):1470–1473. doi: 10.1038/ng.2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gerlinger M, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366(10):883–892. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Friemel J, et al. Intratumor heterogeneity in hepatocellular carcinoma. Clin Cancer Res. 2015;21(8):1951–1961. doi: 10.1158/1078-0432.CCR-14-0122. [DOI] [PubMed] [Google Scholar]
- 9.Gao Q, et al. Cell culture system for analysis of genetic heterogeneity within hepatocellular carcinomas and response to pharmacologic agents. Gastroenterology. 2017;152(1):232–242.e4. doi: 10.1053/j.gastro.2016.09.008. [DOI] [PubMed] [Google Scholar]
- 10.Xue R, et al. Variable intra-tumor genomic heterogeneity of multiple lesions in patients with hepatocellular carcinoma. Gastroenterology. 2016;150(4):998–1008. doi: 10.1053/j.gastro.2015.12.033. [DOI] [PubMed] [Google Scholar]
- 11.Friedman AA, Letai A, Fisher DE, Flaherty KT. Precision medicine for cancer with next-generation functional diagnostics. Nat Rev Cancer. 2015;15(12):747–756. doi: 10.1038/nrc4015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cavalloni G, et al. Establishment of a patient-derived intrahepatic cholangiocarcinoma xenograft model with KRAS mutation. BMC Cancer. 2016;16:90. doi: 10.1186/s12885-016-2136-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Broutier L, et al. Human primary liver cancer-derived organoid cultures for disease modeling and drug screening. Nat Med. 2017;23(12):1424–1435. doi: 10.1038/nm.4438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gao D, et al. Organoid cultures derived from patients with advanced prostate cancer. Cell. 2014;159(1):176–187. doi: 10.1016/j.cell.2014.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.van de Wetering M, et al. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell. 2015;161(4):933–945. doi: 10.1016/j.cell.2015.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Farshidfar F, et al. Integrative genomic analysis of cholangiocarcinoma identifies distinct IDH-mutant molecular profiles. Cell Rep. 2017;19(13):2878–2880. doi: 10.1016/j.celrep.2017.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Broutier L, et al. Human primary liver cancer-derived organoid cultures for disease modeling and drug screening. Nat Med. 2017;23(12):1424–1435. doi: 10.1038/nm.4438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huch M, et al. Long-term culture of genome-stable bipotent stem cells from adult human liver. Cell. 2015;160(1–2):299–312. doi: 10.1016/j.cell.2014.11.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jain R, Fischer S, Serra S, Chetty R. The use of cytokeratin 19 (CK19) immunohistochemistry in lesions of the pancreas, gastrointestinal tract, and liver. Appl Immunohistochem Mol Morphol. 2010;18(1):9–15. doi: 10.1097/PAI.0b013e3181ad36ea. [DOI] [PubMed] [Google Scholar]
- 20.Chu P, Wu E, Weiss LM. Cytokeratin 7 and cytokeratin 20 expression in epithelial neoplasms: a survey of 435 cases. Mod Pathol. 2000;13(9):962–972. doi: 10.1038/modpathol.3880175. [DOI] [PubMed] [Google Scholar]
- 21.Onoe S, et al. Clinicopathological significance of mucin production in patients with papillary cholangiocarcinoma. World J Surg. 2015;39(5):1177–1184. doi: 10.1007/s00268-014-2923-2. [DOI] [PubMed] [Google Scholar]
- 22.Fraveto A, et al. Sensitivity of human intrahepatic cholangiocarcinoma subtypes to chemotherapeutics and molecular targeted agents: A study on primary cell cultures. PLoS ONE. 2015;10(11):e0142124. doi: 10.1371/journal.pone.0142124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hughes JP, Rees S, Kalindjian SB, Philpott KL. Principles of early drug discovery. Br J Pharmacol. 2011;162(6):1239–1249. doi: 10.1111/j.1476-5381.2010.01127.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang JH, Chung TD, Oldenburg KR. A Simple statistical parameter for use in evaluation and validation of high throughput screening assays. J Biomol Screen. 1999;4(2):67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 25.Bonora M, et al. Molecular mechanisms of cell death: central implication of ATP synthase in mitochondrial permeability transition. Oncogene. 2015;34(12):1608. doi: 10.1038/onc.2014.462. [DOI] [PubMed] [Google Scholar]
- 26.Barretina J, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483(7391):603–607. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang W, et al. Genomics of Drug Sensitivity in Cancer (GDSC): a resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 2013;41(Database issue):D955–D961. doi: 10.1093/nar/gks1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hashimoto T, et al. ABO mismatched living related donor liver transplantation. Transplant Proc. 1996;28(3):1217–1219. [PubMed] [Google Scholar]
- 29.Sasaki M, Sato Y, Nakanuma Y. Mutational landscape of combined hepatocellular carcinoma and cholangiocarcinoma, and its clinicopathological significance. Histopathology. 2017;70(3):423–434. doi: 10.1111/his.13084. [DOI] [PubMed] [Google Scholar]
- 30.Chabner BA. NCI-60 cell line screening: A radical departure in its time. J Natl Cancer Inst. 2016;108(5):djv388. doi: 10.1093/jnci/djv388. [DOI] [PubMed] [Google Scholar]
- 31.Bertolini F, Sukhatme VP, Bouche G. Drug repurposing in oncology--patient and health systems opportunities. Nat Rev Clin Oncol. 2015;12(12):732–742. doi: 10.1038/nrclinonc.2015.169. [DOI] [PubMed] [Google Scholar]
- 32.Ashburn TT, Thor KB. Drug repositioning: identifying and developing new uses for existing drugs. Nat Rev Drug Discov. 2004;3(8):673–683. doi: 10.1038/nrd1468. [DOI] [PubMed] [Google Scholar]
- 33.Pantziarka P, Bouche G, Meheus L, Sukhatme V, Sukhatme VP, Vikas P. The Repurposing Drugs in Oncology (ReDO) Project. Ecancermedicalscience. 2014;8:442. doi: 10.3332/ecancer.2014.442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Farshidfar F, et al. Integrative genomic analysis of cholangiocarcinoma identifies distinct IDH-mutant molecular profiles. Cell Rep. 2017;18(11):2780–2794. doi: 10.1016/j.celrep.2017.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Leung-Hagesteijn C, et al. Xbp1s-negative tumor B cells and pre-plasmablasts mediate therapeutic proteasome inhibitor resistance in multiple myeloma. Cancer Cell. 2013;24(3):289–304. doi: 10.1016/j.ccr.2013.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Orlowski RZ. Why proteasome inhibitors cannot ERADicate multiple myeloma. Cancer Cell. 2013;24(3):275–277. doi: 10.1016/j.ccr.2013.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mitra AK, et al. A gene expression signature distinguishes innate response and resistance to proteasome inhibitors in multiple myeloma. Blood Cancer J. 2017;7(6):e581. doi: 10.1038/bcj.2017.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jiao Y, et al. Exome sequencing identifies frequent inactivating mutations in BAP1, ARID1A and PBRM1 in intrahepatic cholangiocarcinomas. Nat Genet. 2013;45(12):1470–1473. doi: 10.1038/ng.2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kumar S, Stecher G, Tamura K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for bigger datasets. Mol Biol Evol. 2016;33(7):1870–1874. doi: 10.1093/molbev/msw054. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.