

Abstract
pH is highly regulated in the mammalian central nervous systems. Neuronal calcium sensor-1 (NCS-1) can interact with numerous target proteins. Compared with C. elegans, evolution has avoided histidine residues at positions 102 and 83 in the human and Xenopus laevis NCS-1 protein, possibly to decrease conformational sensitivity to pH gradients in synaptic processes. We used all-atom molecular dynamics (MD) simulations to investigate the effects of amino acid substitutions between species on human NCS-1 by substituting Arg102 and Ser83 for histidine (R102H and S83H) at neutral and acidic pH. Our cumulative 5 μs simulations revealed that the R102H mutation slightly increases the structural flexibility of loop L2 and the R102Hprotonated mutation decrease protein stability. Community network analysis illustrates that the R102H and S83H mutations weaken the inter-domain and strengthen the intra-domain correlations. Secondary structure contents in the S83H and the S83Hprotonated mutants are similar to those in the wild type (WT), whereas the global structural stabilities and salt-bridge probabilities decrease. This study highlights conformational dynamics effects of R102H and S83H mutations respectively on NCS-1’s local structural flexibility and global stability, while the protonated histidine decreases the stability. Thus, histidines at positions 102 and 83 may not be compatible with NCS-1’s function whether in neutral or protonated states.
Keywords: Human neuronal calcium sensor 1 (NCS-1) protein, protonated histidine, conformational dynamics, dynamic community network analysis, salt bridge, PCA analysis, molecular dynamics simulations
1. INTRODUCTION
Neuronal calcium sensor (NCS) Ca2+-binding proteins are a conserved subfamily of the calmodulin superfamily. NCS proteins can interact with photoreceptor cells and regulate signal transduction in neurons.1 The NCS family consists of 15 members1b, 2 with multiple non-redundant functions.1a, 3 Their physiological functions are likely to be determined by their hydrophobic crevice which varies shape and size.1a, 4 Despite their physiological differences, they have a high sequence similarity. Structural studies showed that some NCS proteins such as NCS-1, recoverin, and GCAP1 have similar main chain topologies, while the species are distinguished by their high affinity for Ca2+.3
NCS-1, the first member of the NCS family,5 has been implicated in several physiological functions including regulation of synapse formation, neurite outgrowth,6 synaptic plasticity,7 regulation of neurotransmission, learning and memory,8 membrane trafficking,9 and voltage gated Ca2+ channels.10 These functions were recognized mainly from the observation that NCS-1 and orthologues can interact with a wide range of potential target proteins.1a Experiments by Hendricks et al5 and Strahl et al11 showed that NCS-1 was not functionally equivalent to naive yeast Frq1. However, Frequenin/NCS-1 protein sequences have been highly conserved throughout evolution. The sequences of known vertebrate and amphibian, vertebrate and invertebrate, human and Saccharomyces cerevisiae (S. cerevisiae) have sequence identify respectively around 100%, 75% and 60%.12 Arg102 and Ser83 are conserved in human and Xenopus NCS-1, but are histidine in C. elegans.
With these mutations, human and the other vertebrates (e.g. Rainbow trout) completely wiped out histidine residues in NCS-1 sequences. For C. elegans, histidine is important in extracellular alkalinity and pH sensing in GCY-14, while there is no evidence of NCS-1 involvement.13 However, pH in the mammalian central nervous systems is highly regulated.14 The pH value fluctuates across synaptic compartments and the effects of pH gradients on synaptic processes are well known.15 It is plausible to hypothesize that mutating out histidine may render the pH-independent conformational dynamics of NCS-1 in human and other vertebrates. Consequently, His residues at positions 102 and 83 may be not compatible with NCS-1’s function. Previously, it has been shown that protonation of conserved histidines may cause large conformation changes in the release factors.16 For the NCS-1 protein, the sensitivity of position 102 can be seen from previous studies of the R102Q mutation.17 Our recent MD simulations showed that substituting Arg102 for glutamine leads to a more occupied hydrophobic crevice by loop L3, which disables the relocation of the C-terminus.18 Several experimental and computational studies investigated the impact of mutations on the structural properties of proteins.18–19
In this study, we investigated the effects of the R102H and the S83H mutations on the structure of human NCS-1 protein at atomistic level. Two independent 500 ns MD simulations starting from the initial WT structure with explicit solvent were carried out for the WT, the R102H and the R102Hprotonated mutants and the S83H and the S83Hprotonated mutants. We found that the R102H mutation maintains the global conformational dynamics while significantly increasing the flexibility of loop L2. However, R102Hprotonated significantly decreases the structural stability of the protein. Previous studies found that loop L3 played a critical role in NCS-1 conformational dynamics,20 and our results reveal that R102H leads loop L3 to adopt a more extended state. The salt bridges in both R102H and the R102Hprotonated mutants are significantly affected, not only in the intra-domain but also inter-domain. Community network analysis shows that R102H and R102Hprotonated weaken the inter-domain and strengthen the intra-domain communication. On the other hand, the global stability and the salt-bridge probability are decreased by the S83H and the S83Hprotonated mutations, and the solvent accessible surface area of the hydrophobic residue located within the hydrophobic crevice (HC) is increased. The mutations significantly affect the free energy landscape of the protein. Our study reveals that R102H and the S83H affect the conformational dynamics of human NCS-1 in different ways. It is especially noteworthy that mutations mutated to protonated histidine decrease the structural stability, indicating strong pH dependence. Thus, histidine may be not compatible with NCS-1’s function.
2. MATERIALS AND METHODS
Human WT NCS-1 Protein and Its Mutants.
The solution NMR structure of the unmyristoylated calcium-bound human NCS-1 (PDB id: 2LCP) contains 20 models.20b The first minimum-energy model was utilized as the initial state for two independent simulations of WT NCS-1. The initial conformations of the R102H and the S83H mutants were modeled by taking the WT initial state as the starting point, substituting arginine 102 and serine 83 for neutral histidine respectively, followed by energy minimization. The initial conformations of the R102Hprotonated and the S83Hprotonated mutants were also modeled by taking the WT initial state as the starting point, substituting arginine 102 and serine 83 for protonated histidine respectively, followed by energy minimization. The N-terminus and the side chains of Lys and Arg were protonated (NH3+, Arg+, Lys+), and the C-terminus and the side chains of Glu and Asp were deprotonated (COO−, Glu−, Asp−).
Details of Molecular Dynamics Simulations.
MD simulations have been carried out using the GROMACS 4.5.3 package21 and the CHARMM27 force field with CMAP corrections,22 in accordance with a recent MD study by Bellucci et al.17a Two independent 500 ns MD simulations were performed for the WT, and the R102H and R102Hprotonated mutants and the S83H and S83Hprotonated mutants, using the first model with three calcium ions in the PDB structure as starting points.20b The parameters used for calcium ions were q = +2.0e, σ = 0.2436 nm, and ε = 0.5021 kJ/mol. The TIP3P water model was used. Additional Na+ and Cl− were added to the systems with a NaCl concentration of 0.1 M. The simulation box type is rhombic dodecahedron. In GROMACS, the simulation box is defined by three vectors a (d, 0, 0), b (0, d, 0), and c (d/2, d/2, √2d/2) and written as (d, d, √2d/2, d/2, d/2), where d is the edge length of the rhombic dodecahedron. In our simulations, the d value was 9.30 nm. Thus, the simulation boxes were described as (9.30, 9.30, 6.58, 4.65, 4.65) nm. Bond lengths within protein and water molecules were respectively constrained by the LINCS23 and SETTLE algorithms,24 The particle mesh Ewald (PME) method was used to calculate the electrostatic interaction with a real space cutoff of 1.0 nm, and the van der Waals interactions were calculated using a cutoff of 1.4 nm. The simulations were performed in isothermal−isobaric (NPT) ensemble using the periodic boundary condition. The solute and solvent were separately coupled to external temperature bath using the velocity rescaling method25 and pressure bath using the Parrinello−Rahman method.26 The temperature and pressure were maintained at 310 K and 1 bar using coupling constants of 0.1 and 1.0 ps, respectively.
Analysis Methods.
Analysis of the trajectories were performed using the tools implemented in the GROMACS 4.5.3 software package.27 The backbone RMSDs were calculated following structural alignment of the core structure (residues E11 to K174). The DSSP program was used to determine the secondary structure.28 Root-mean-square fluctuation (RMSF) was calculated for the core structure with respect to the MD generated average structure during the time scale of 400–500 ns. A salt bridge between a pair of oppositely charged residues was considered to be formed if the centroids of the side-chain charged groups in two oppositely charged residues lie within 0.4 nm of each other.29 The distances between the centroids of the side-chain charged groups were calculated to show the change of the salt bridges. The visual molecular dynamics (VMD 1.9.1) package was used for graphical structure analysis.30 Interaction networks of WT NCS-1 and of the R102H and the R102Hprotonated mutants were analyzed and displayed by Network View31 implemented in VMD. Principal component analysis (PCA) helps determine motions that contribute the most to the overall dynamics. A system of N atoms exists 3N-6 modes of possible internal fluctuations (the external rotation and translation of the system are described by six degrees of freedom). We focus on α-carbon atoms. The free energy landscape was constructed using the relation −RT*ln[H(x,y)], where H(x,y) was the histogram of two selected reaction coordinates, x and y (PC1 and PC2). We utilized the SWISS-MODEL32 website to select the functional conformations which have similar main chain topologies with human NCS-1. Because the conformations have different sequence lengths, we adopted the core structure of human NCS-1 as the longest homologous segment for the PCA analysis.
3. RESULTS AND DISCUSSION
NCS-1 is a member of the neuronal calcium sensor family and its solution structure of unmyristoilated calcium-bound state has been solved using NMR spectroscopy.20b Structural analyses have shown that its structure primarily consists of nine α-helices arranged in four EF-hands and three loops. Three of the four EF-hands (EF2, EF3, and EF4) can bind Ca2+ ions in corresponding sites. The conserved Cys/Pro substitution in the calcium binding motif in EF1 is unable to bind Ca2+.1c, 33 According to Heidarsson et al.,20b there are nine helices, helix 1 (H1) (residues E11-R18), H2 (E24-F34), H3 (A45-Q54), H4 (T62-F72), H5 (F82-S93), H6 (D98-Y108), H7 (R118-V132), H8 (E146-M155), and H9 (L166-K174) and the three loops, loop 1 (L1) (F56-P61), L2 (G133-P145), and L3 (D176-V190). The eight helices H2-H9 form a HC, of which helices H4, H5 and H6 form the floor of crevice, H3, H7 and H9 on the two sides form the long “walls”, and helices H2 and H8 close the HC at the opposite edges. In the absence of a binding partner, loop L3 occupies the HC as a ligand mimic and turns the HC into a part-occupied state. The protein consists of two domains which are connected by the hinge loop R94-L97: N-domain consisting of residues M1-S93 and C-domain consisting of residues D98-V190.20b
The R102H and the R102Hprotonated Mutants Increase the Flexibility of Loop L2, while R102Hprotonated Significantly Decreases the Structural Stability of the WT.
Prior to probing the structural features of the R102H and the R102Hprotonated mutants of human NCS-1 protein, we examined the conformational dynamics of the WT NCS-1 by performing two independent 500 ns MD stimulations at 310 K (WT-MD1, WT-MD2).
Figure 1 presents the time evolution of the backbone RMSDs of the core structure (residue E11-K174) of WT NCS-1 with respect to the initial state in the two different MD trajectories. It can be seen from Figure 1a that the backbone RMSDs of the core structure of the WT increase quickly in the first 50 ns, then change gradually, and stabilize separately around 0.33 and 0.31 nm after t = 250 ns for WT-MD1 and WT-MD2.
Figure 1.
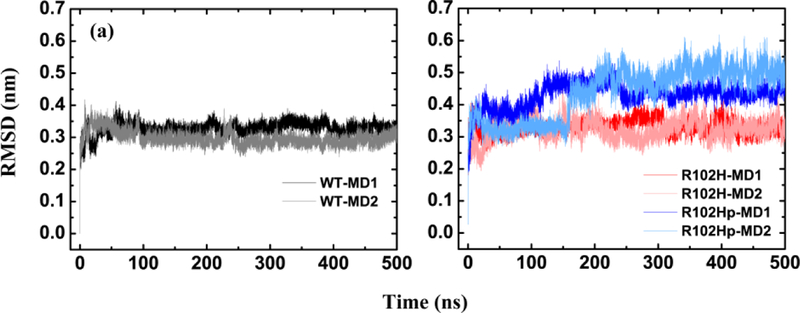
Time evolution of the backbone RMSDs include the core structure (E11 to K174) for WT NCS-1 (black and gray curve), its R102H mutant NCS-1 (red and pink curve) and its R102Hprotonated mutant (blue and light blue curve) in two different MD trajectories (WT-MD1, WT-MD2 for the WT NCS-1, R102H-MD1, R102H-MD2 for the R102H mutant and R102Hp-MD1, R102Hp-MD2 for the R102Hprotonated mutant).
We then probed the structural changes of the R102H and the R102Hprotonated mutants from the initial state by two MD simulations respectively. The backbone RMSDs of the core structure for the R102H and the R102Hprotonated mutants with respect to the initial state of the WT are plotted in Figure 1b. It can be seen that the core structure of R102H mutant stabilizes around 0.33 nm for both MD runs, which are similar to those of the WT. However, the R102Hprotonated mutant largely increases the backbone RMSD values during the last 200 ns and the trajectories are stabilized around 0.45 (run2) and 0.50 nm (run1). The results indicate that the R102Hprotonated mutation significantly decreases the structural stability of the protein while it is maintained for the R102H mutation at neutral pH. The stability of WT and the mutants was also explored by calculating the percentage of helical structure. It can be seen from Figure S1 that it remains around 52% and 51% for the R102Hprotonated mutant, similar with those of the WT and R102H mutant during the simulations. The results reveal similar global conformational dynamics for the R102H and R102Hprotonated mutants and the WT. This is also seen from the secondary structure profiles (Figure S2) of the R102H and the R102Hprotonated mutants which are mostly maintained in the MD trajectories, indicating the larger RMSD values of the R102Hprotonated mutant might originate from conformational rearrangement of the tertiary structure.
Figure 2 shows the Cα-RMSF curves of the WT and R102H and R102Hprotonated mutants. Overall, similar fluctuations can be seen in the R102H and the R102Hprotonated mutants and the WT except for few regions. However, not only loop L2 in the R102H mutant possesses larger fluctuations than other parts of the R102H mutant; R102Hprotonated mutant loop L1 also shows a larger RMSF value. The results indicate that the mutations increase the local flexibility especially in loop L2, while the R102Hprotonated mutation plays a particular role in the local conformational dynamics of loop L1. Figure S2 shows that the secondary structure and the overall folding of the WT and the R102H and the R102Hprotonated mutants remain mostly unchanged. Therefore, the increased flexibilities of loop L1 and loop L2 suggest that the tertiary structure might undergo certain conformational rearrangements with time while the secondary structures are largely maintained.
Figure 2.
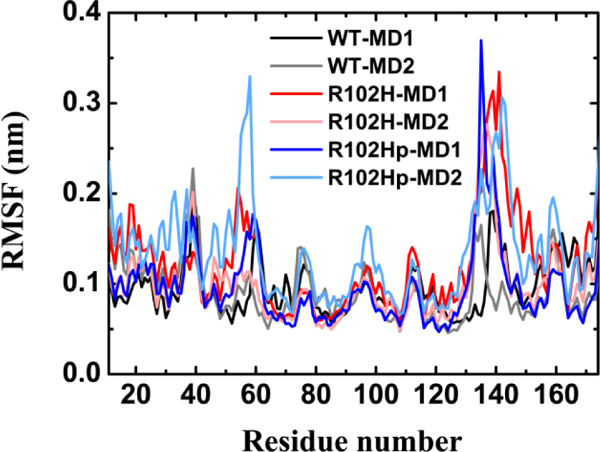
Cα-RMSF of core structure for WT NCS-1, the R102H mutant and the R102Hprotonated mutant in the two independent MD runs. Cα-RMSF values were calculated using the last 100 ns data of each MD trajectory for all systems.
The R102H Mutation Induces Loop L3 to Adopt a More Extended State.
Previous studies demonstrated that L3 plays a critical role in the structure stability of NCS-1.17b, 20a To examine whether the R102H or the R102Hprotonated mutation influence the conformational dynamics of loop L3, we calculated the Cα-Cα distance between D176 and V190 (the starting and ending residues of loop L3) in both WT and mutants. The probability distributions of the distance between D176 and V190 in the two MD trajectories respectively for WT and mutants are plotted in the Figure 3. It can be seen that the distance probability distribution peaks are located at 1.9 and 2.6 nm and 1.8 and 2.3 nm in the two WT trajectories, while the peaks are located at 3.2 and 3.0 nm in the two R102H mutant trajectories. The data show that loop L3 in the WT is mainly in a collapsed state, while it adopts a more extended state in the R102H mutant.
Figure 3.
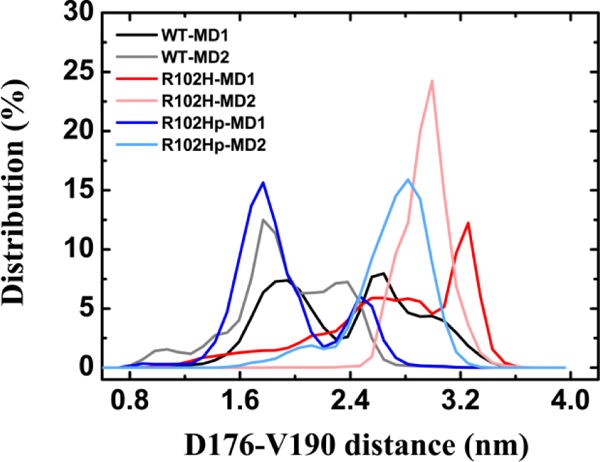
Probability distributions of the Cα-Cα distance between the starting and ending residues of loop L3 (D176 and V190) for WT NCS-1, the R102H and the R102Hprotonated mutants in the two different MD runs. The D176−V190 distances are calculated for the 0–500 ns of each MD trajectory for all systems.
The above results are similar to our recent study18 where the NCS-1R102Q mutation may extend loop L3, in agreement with a hypothesis by Bellucci et al. that a mutation that consists of replacing Arg102 with an uncharged residue could affect the mobility of H9 and L3.17a It is noteworthy that this hypothesis was based on their finding that the stabilization of inter- versus intra-helix salt bridges involving D98 relies on the possibility of D98 interacting with R102 or K174.17a However, it can be seen from Figure 4 that R102Hprotonated diminished the possibility of D98 interacting with R102 or K174. The change of salt bridges can provide an explanation for the phenomenon that loop L3 is mainly in a collapsed state in the R102Hprotonated mutant, indicating that the mobility of loop L3 is largely maintained.
Figure 4.
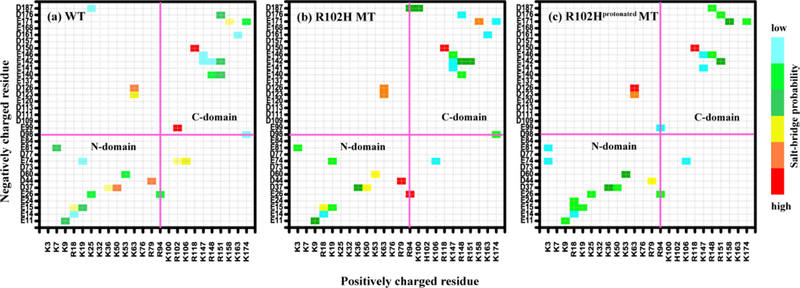
The R102H mutation and the R102Hprotonated mutations shift the salt bridge network of WT NCS-1. The salt-bridge probability maps are given in (a) for the WT NCS-1, in (b) for the R102H mutant and in (c) for R102Hprotonated mutant.
The R102H Mutation Affects the Salt Bridge Network Significantly.
Salt bridges play an important role in stabilizing the structures of proteins,34 whether in buried35 or solvent-exposed36 locations. A salt bridge is considered to be formed if the centroids of the side-chain charged groups lie within 0.4 nm of each other.29 NCS-1 is a highly charged protein. To detect the effect of the R102H and the R102Hprotonated mutations on salt the bridges, we calculated their formation probabilities by positively and negatively charged residues in the WT and the mutants. The salt-bridge probability maps are given in Figure 4 for WT NCS-1, the R102H and the R102Hprotonated mutants. It is clear that the R102H mutation significantly changes the overall salt bridge network. Some salt bridges disappear, such as E99-R102, E74-R102, E140-R151, K7-E81 and K25-D187, while K100-D187 and E141-K147 are newly formed. The most affected intra-domain salt bridges include K3-E81, K7-E81, K19-E74, K25-E26, E26-R94, K36-D37, K53-D60 in the N-domain and D98-K174, E99-R102, K100-D187, E140-R151, E141-K147, E142-R148, E146-K147, R151-D176 in the C-domain. Among these, E26-R94, K63-D123, E74-R102, D98-K174, E99-R102 and K100-D187 are also reported in the recent MD studies by Bellucci et al.17a and by us.18, 20a, 37
Both R102H and R102Hprotonated mutations remove the inter-domain salt bridges K25-D187 and E74-R102 and reduce the probability of E74-K106. The significant change of the population of these inter-domain salt bridges would influence the interaction between the N-domain and the C-domain, which may affect protein dynamics and ligand selectivity. However, the inter-domain weakened interactions could be partly compensated by the increased probability of K63-D123 and K63-D126 salt bridge to maintain a well-folded structure in the R102H and R102Hprotonated mutants, respectively.
A recent optical tweezer experiment38 studied the folding mechanism of human NCS-1 protein. According to Heidarsson et al., the complete folding of the C-domain (containing helices H6, H7, H8, and H9) was crucial for the subsequent folding of the N-domain (containing helices H1, H2, H3, H4, and H5), revealing inter-domain dependence. The results of our simulations of mutation-affected inter-domain correlation support this experimental finding.38
As can be seen in Figure 4, three of most affected salt bridges in the R102H mutant (E140-R151, E141-K147 and E142-R148) involve three loop L2 residues E140, E141 and E142. The R102H mutation removes the population of E140-R151 salt bridge and increases the populations of E141-K147 and E142-R148. As residue E140 is located closer to the middle of loop L2 than E141 and E142, the disruption of the E140-R151 salt bridge would release the motion of loop L2. The salt bridges that involve loop L2 residues are more affected in the R102Hprotonated mutant. The R102Hprotonated mutation removes the population of E140-R148, E140-R151, E142-K147 and E142-R148. The above analysis of the salt bridges in all systems provides an explanation for the increased flexibility of loop L2 in both the R102H and R102Hprotonated mutants (see Figure 2).
Figure 4 also reveals another significant change in the three salt bridges that involve two loop L3 residues D176 (the first residue of loop L3) and D187 (the C-terminal residue of loop L3) in the R102H mutant. Residue pairs forming the three salt bridges are D151 in H8 and D176 in loop L3, K100 in H6 and D187 in loop L3, and R94 in the hinge loop and D187 in loop L3. The R102H mutation decreases the probability of the R151-D176 and increases the probabilities of the K100-D187 and R94-D187 salt bridges. The results show that the R102H mutation weakens the intersegment interaction between the N-terminal of loop L3 and the H8 and strengthens the attraction between the C-terminal of loop L3 and the segments H6 and hinge-loop, which may facilitate an extended loop L3 conformation. The data illustrate that the mobility of loop L3 could be affected by the R102H mutation, providing an explanation for loop L3 adopting a more extended state.
The R102H and the R102Hprotonated Mutations Weaken the Inter-Domain and Strengthen the Intra-Domain communication
The dynamic effects of the R102H and the R102Hprotonated mutations on the inter-domain interactions of the NCS-1 were also studied using MD-based community network analysis.39 In Figure 5 nodes center on the Cα atoms, and those pairs of residues (nodes) interacting at least 75% of the time (within 4.5 Å) throughout the simulation time were defined as edges. The time averaged connectivity of the nodes is used to identify the disjoint subnetwork called community.40 Nodes connecting within the same community have stronger connectivity, and those connecting to nodes outside the community have weaker connections. The strength of the dynamic correlation between two nodes can be detected by the thickness of the edges.
Figure 5.
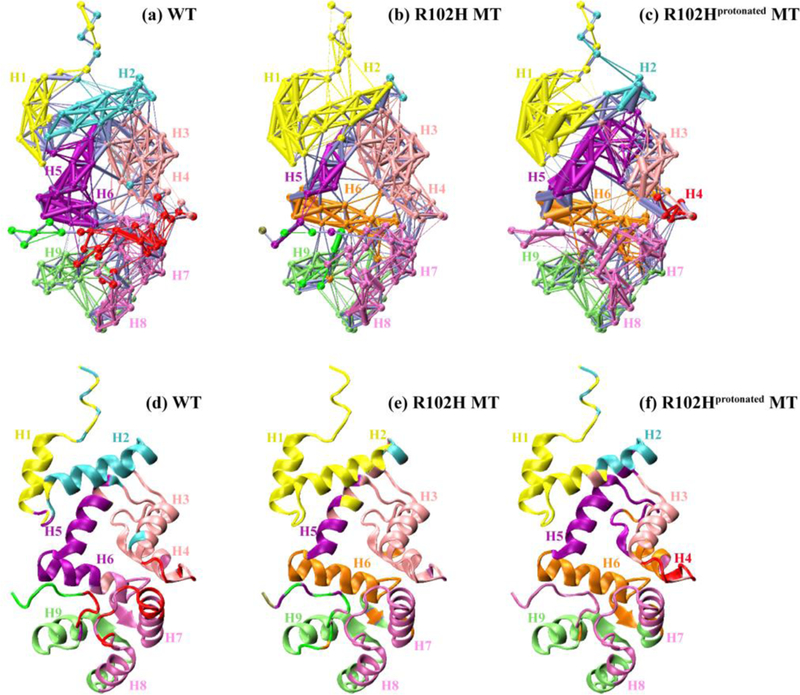
Community networks formed in the WT NCS-1 (a), the R102H mutant (b) and the R102Hprotonated mutant (c) with edge widths corresponding to their weights based on MD simulations. Each community has its own color, superimposed on the initial MD simulation structure. The corresponding cartoon representations at t = 0 ns are shown in (d), (e) and (f).
The differences between the community networks of the WT and mutants can be seen in Figure 5. The Girvan-Newman algorithm splits the network respectively into 8, 9 and 8 communities for the WT, the R102H and the R102Hprotonated mutants. The intra-community connections are denser in mutants than those of WT, mainly between helices H1 and H2. We probed the inter-domain coupling connected by helices H5 and H6. Figure 5a shows that the two helices belong to one community (purple) and the connections are dense in the WT, illustrating the strong connections between the N- and C-domain. In contrast, the community that consists of helices H5 and H6 is split into two communities in the R102H and the R102Hprotonated mutants, implying that the inter-domain cross-talk could be weakened by the mutations. The data show that even though the secondary structure contents of mutants are largely maintained (Figure S2), the mutations alter the residue-residue connections and the dynamic communities. The results illustrate that the mutations weaken the inter-domain and strengthen the intra-domain correlation, in accordance with our previous work on the Δ 176-190 truncated NCS-1.20a
The S83H and the S83Hprotonated Mutations Also Affect the Conformational Dynamics − albeit differently.
The backbone RMSDs of the core structure of the S83H and the S83Hprotonated mutants were probed by MD. As Figure 6 shows, the backbone RMSDs of S83H mutant stabilize around 0.44 and 0.42 nm and are significantly larger than those of the WT, indicating that the S83H mutation decreases the stability of the protein. However, the backbone RMSDs of the core structure of the S83Hprotonated mutant stabilize around 0.35 nm during the last 200 ns, which are between the values of the WT and the S83H mutant.
Figure 6.
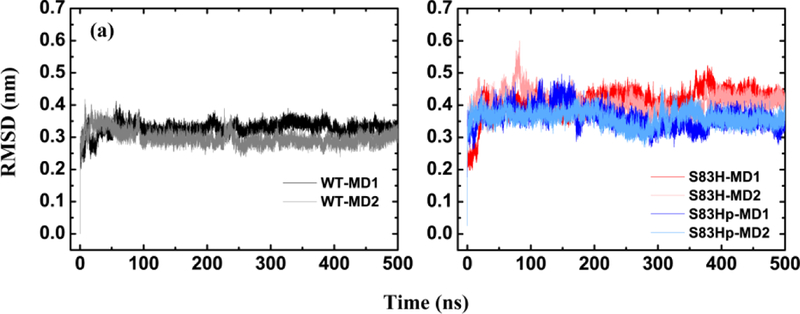
Time evolution of the backbone RMSDs include the core structure (E11 to K174) for WT NCS-1 (black and gray curve), its S83H mutant NCS-1 (red and pink curve) and its S83Hprotonated mutant (blue and light blue curve) in two different MD trajectories (WT-MD1, WT-MD2 for the WT NCS-1, S83H-MD1, S83H-MD2 for the S83H mutant and S83Hp-MD1, S83Hp-MD2 for the S83Hprotonated mutant).
We calculated the percentage of helical structure for these three systems. As shown in Figure 7, the S83H mutation leads to a slight decrease, indicating that S83H mutation decreases the structural stability. It can be seen that the average numbers of helical percentage in the WT are around 0.54, while those of the S83Hprotonated mutation are 0.52 and 0.54 for the two runs. It is possible that a short α-helix appeared in loop L3 (see Figure S2j), suggesting that the S83Hprotonated mutation also decreases the structural stability of NCS-1.
Figure 7.
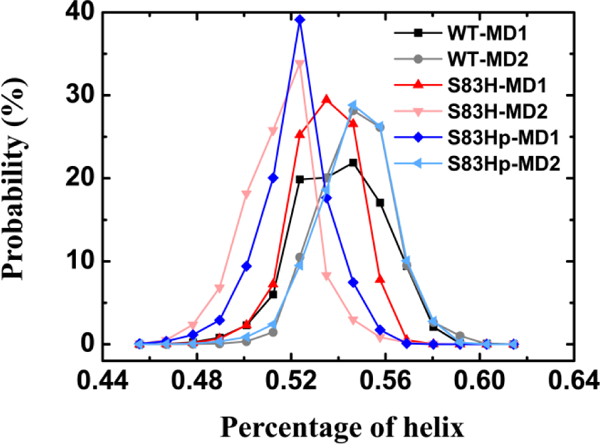
Probability distribution of percentage of helix in the MD trajectories during 200–500 ns for the WT NCS-1, the S83H and the S83Hprotonated mutant systems.
The Cα-RMSF curves of the WT, the S83H and the S83Hprotonated mutants were plotted in Figure S3. The Cα-RMSF curves of the S83H mutant overlap significantly those of the WT. The results indicate that the S83H mutation does not change the protein flexibility. However, the S83Hprotonated mutation shows a larger fluctuation than the WT at positions 130–135, indicating the mutation increase the flexibility of the N-terminal of loop L2.
We probed the size of the hydrophobic crevice (HC) by calculating the solvent accessible surface area (SASA) of the hydrophobic residue located within the HC (SASA_HC). The probability distribution function (PDF) of SASA_HC was plotted in Figure 8. The SASA_HC PDF peaks are centered at 15.3 and 14.9 nm2 respectively for the WT-MD1 and WT-MD2. The peaks are centered at 16.1 and 17.9 nm2 for the S83H mutant and 16.0 and 16.9 nm2 for the S83Hprotonated mutant. The data show that both the S83H and the S83Hprotonated mutations can induce the hydrophobic crevice to adopt a more expanded state.
Figure 8.
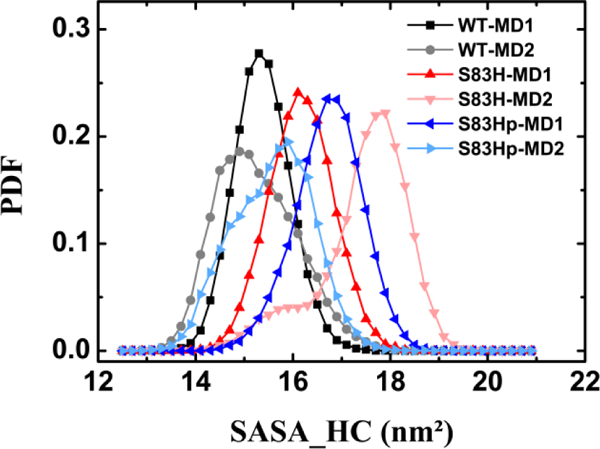
Probability distribution function (PDF) of solvent accessible surface area (SASA) for the hydrophobic crevice (HC) formed by the hydrophobic residue located inside the HC for the WT NCS-1, the S83H and the S83Hprotonated mutants.
We plotted the salt bridge network in Figure S4 to probe the effect of the S83H and the S83Hprotonated mutations on the salt bridges. It can be seen that the salt-bridge probability reduces in both mutants. The affected salt bridges were K7-E81 and K19-E74 in the N-domain and E99-R102, E140-R148, E142-R151, R118-D150 and E147-K147 in the C-domain. The E142-K147 and E142-R148 salt bridges in the C-domain were also affected by S83Hprotonated. The disruption of inter-domain salt bridges is also visualized in Figure S4, including E74-R102, K63-D123, K63-D126 and K25-D187 salt bridges. Our simulations demonstrate that the mutations not only affect the intra-domain salt-bridge network but also alter the inter-domain salt bridges. The results reveal the inter-domain correlation, explaining the decreased structural stability of both mutants (Figure 6).
The Mutations Alter the Dynamics Overlap with Other NCS family proteins.
We utilized the SWISS-MODEL32 website to select functional conformations. NCS-1, recoverin, and GCAP1 not only are both the members of the NCS family,1a but also have similar main chain topologies.41 We selected functional conformations that originated from the structures of these proteins (Table 1). We adopted the core structure of human NCS-1 as the longest homologous segment for the PCA analysis, and all sequences are converted to the human NCS-1 sequence.
Table 1.
Selected structures have similar main chain topologies with the human NCS-1. All of structures below are found in homo sapiens.
| Number | PDB ID | Protein Description | Method |
|---|---|---|---|
| 1 | 2LCP | Solution structure of human NCS-1 | NMR |
| 2 | 1G8I | Crystal structure of human NCS-1 | X-RAY |
| 3 | 5AFP | Crystal structure of Rattus norvegicus NCS-1 | X-RAY |
| 4 | 5AER | Crystal structure of Rattus norvegicus NCS-1 | X-RAY |
| 5 | 4GUK | Crystal structure of human NCS-1 | X-RAY |
| 6 | 1FPW | Solution structure of Yeast frequenin | NMR |
| 7 | 2JU0 | Solution structure of Yeast frequenin | NMR |
| 8 | 2I94 | Solution structure of Bos taurus recoverin | NMR |
| 9 | 4YI8 | Crystal structure of Bos taurus recoverin | X-RAY |
| 10 | 1OMV | Crystal structure of Bos taurus recoverin | X-RAY |
| 11 | 4M2O | Crystal structure of Bos taurus recoverin | X-RAY |
| 12 | 2D8N | Crystal structure of human recoverin | X-RAY |
| 13 | 2R2I | Crystal structure of Gallus gallus GCAP1 | X-RAY |
The free energy landscape was projected onto two reaction coordinates: PC1 and PC2. Analysis shows that the top two PCs capture more than 40 % of the accumulated variance and the first four PCs covered more than 60 % (Figure S5). The free energy landscape is plotted in Figure 9. The WT landscape is compacted and there are two dominant potential wells at (1, −0.5) and (−1.5, 0). Structure 1 (PDB ID: 2LCP) in the MD simulations is right between the two, located at the center of the plot (Figure S6). For the R102H mutant, there are also two dominant potential wells respectively at (1, −0.5) and (−1.5, 0). However, the landscape is dispersed. One basin in both systems corresponds to NCS-1 (1–7) and recoverin (8–12), and the second to GCAP1 (13). As for the R102Hprotonated mutant, the landscape is becoming more dispersed. Comparing with the WT, the two basins in the R102Hprotonated mutant respectively moved to left and right, to a larger extent than the R102H mutant. The distribution of the energy surface suggests that both the R102H and the Ri02Hprotonated mutations alter the motions of WT NCS-1, but there also exist some similar motion modes. S83H and the S83Hprotonated mutations also change the free energy landscape. The two potential wells in the S83H mutant landscape are located at (−1, 0) and (4, 0). Contrasting the WT, the R102H and the R102Hprotonated mutants, the potential well near the center position at (1, −0.5) vanished in the S83H mutant landscape and only second at (−1.5, 0) captures fewer structures. With the S83Hprotonated mutation, however, the landscape is compacted and the basin that located at (1, −1) captures more features than the S83H mutant. The difference in the energy surfaces indicates that the S83H mutation changes the WT motion modes more than the S83Hprotonated mutation, and diminishes similar motion modes in the other NCS proteins.
Figure 9.
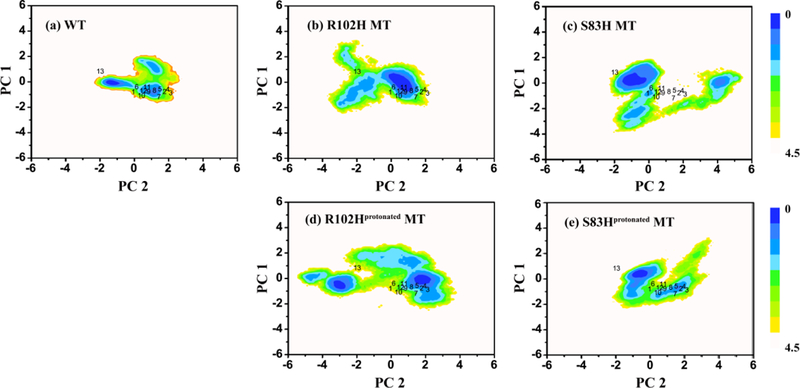
Free energy surface (in kcal/mol) of the WT (a), the R102H mutant (b), the S83H mutant (c), the R102Hprotonated mutant (d), the S83Hprotonated mutant (e) as a function of PC1 and PC2 with PDB-obtained crystallographic structures mapped on.
4. CONCLUSIONS
To probe the effects of the amino acid substitutions between species on the structural properties of NCS-1, we investigated the influence of the R102H and S83H mutations on the conformational dynamics at the atomic level by performing ten independent 500 ns MD simulations. The R102H mutant maintains the secondary structure content. The global stabilities of the WT and the R102H mutant are similar. However, the R102H mutation significantly increases the local flexibility of loop L2. Loop L3 prefers to adopt a collapsed state in WT, and a relatively extended state in the R102H mutant. Both R102H and the R102Hprotonated mutations alter the salt-bridge probabilities, especially at the interdomain, which may influence the domain-domain interaction. Analysis of the community networks illustrates that the R102H and R102Hprotonated mutations weaken the inter-domain and strengthen the intra-domain communication.
We also tested other mutants for the neutral Ser83 to protonated histidine substitution. The S83H and the S83Hprotonated mutations influence the structural stability, and decrease the salt-bridge probability. All mutations change the free energy landscape of the WT human NCS-1, indicating that the motion modes are affected. Similar motion modes in the other NCS proteins diminished. Overall, our study provides a detailed atomistic picture of amino acid substitutions affecting the conformational dynamics in different ways. The local flexibility and global stability of protein are changed by the R102H and the S83H mutations respectively. While histidine is not compatible with NCS-1’s functional dynamics no matter at neutral or protonated states, protonated histidine residues at acidic pH decrease the structural stability substantially. Our results indicated that human NCS-1 protein avoids histidine to decrease its pH sensitivity.
Supplementary Material
ACKNOWLEDGEMENTS
Simulations were performed at the High Performance Computing Server (PowerEdge T710) of Shanghai University of Sport. This project has been funded in whole or in part with Federal funds from the National Cancer Institute, National Institutes of Health, under contract number HHSN261200800001E. This research was supported (in part) by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
Footnotes
ASSOCIATED CONTENT
Supporting Information
Four figures present the time evolution of percentage of helix for WT NCS-1 and its R102H and R102Hprotonated mutants, the secondary structure profiles for WT NCS-1 and its mutants, the root-mean-square fluctuation (RMSF) was calculated for core structure with respect to the MD generated average structure during the time scale of 400–500 ns for WT NCS-1 and its S83H and S83Hprotonated mutants, the salt bridge probability maps for WT NCS-1 and its S83H and S83Hprotonated mutants, the accumulation of variance of variance that captured from the PCs of the all systems’ motion modes, the free energy landscape of all systems with 2LCP mapped on.
Notes
The authors declare no competing financial interest.
REFERENCES
- 1.(a)Burgoyne RD; Haynes LP, Understanding the physiological roles of the neuronal calcium sensor proteins. Molecular Brain 2012, 5; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Weiss JL; Hui H; Burgoyne RD, Neuronal Calcium Sensor-1 Regulation of Calcium Channels, Secretion, and Neuronal Outgrowth. Cellular and Molecular Neurobiology 2010, 30 (8), 1283–1292; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Bourne Y; Dannenberg J; Pollmann V; Marchot P; Pongs O, Immunocytochemical localization and crystal structure of human frequenin (neuronal calcium sensor 1). Journal of Biological Chemistry 2001, 276 (15), 11949–11955. [DOI] [PubMed] [Google Scholar]
- 2.Reyes-Bermudez A; Miller DJ; Sprungala S, The Neuronal Calcium Sensor Protein Acrocalcin: A Potential Target of Calmodulin Regulation during Development in the Coral Acropora millepora. Plos One 2012, 7 (12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Burgoyne RD, Neuronal calcium sensor proteins: generating diversity in neuronal Ca2+ signalling. Nature Reviews Neuroscience 2007, 8 (3), 182–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ames JBLS, Molecular structure and target recognition of neuronal calcium sensor proteins. Biochim Biophys Acta 2012, 1820(8), 1205–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hendricks KB; Wang BQ; Schnieders EA; Thorner J, Yeast homologue of neuronal frequenin is a regulator of phosphatidylinositol-4-OH kinase. Nature Cell Biology 1999, 1 (4), 234–241. [DOI] [PubMed] [Google Scholar]
- 6.(a) Iketani MIC; Nakamura F; Jeromin A; Mikoshiba KG,Y; Takei K, Regulation of neurite outgrowth mediated by neuronal calcium sensor-1 and inositol 1,4,5-trisphosphate receptor in nerve growth cones. Neuroscience 2009, 161, 743–752; [DOI] [PubMed] [Google Scholar]; (b) Romero-Pozuelo J; Dason JS; Atwood HL; Ferrus A, Chronic and acute alterations in the functional levels of Frequenins 1 and 2 reveal their roles in synaptic transmission and axon terminal morphology. European Journal of Neuroscience 2007, 26 (9), 2428–2443. [DOI] [PubMed] [Google Scholar]
- 7.(a) Sippy T; Cruz-Martin A; Jeromin A; Schweizer FE, Acute changes in short-term plasticity at synapses with elevated levels of neuronal calcium sensor-1. Nature Neuroscience 2003, 6 (10), 1031–1038; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Jo J; Heon S; Kim MJ; Son GH; Park Y; Henley JM; Weiss JL; Sheng M; Collingridge GL; Cho K, METABOTROPIC GLUTAMATE RECEPTOR-MEDIATED LTD INVOLVES TWO INTERACTING CA(2+) SENSORS, NCS-1 AND PICK1. European Psychiatry 2009, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Saab BJ; Georgiou J; Nath A:,Lee FJS; Wang M; Michalon A; Liu F; Mansuy IM; Roder JC, NCS-1 in the Dentate Gyrus Promotes Exploration, Synaptic Plasticity, and Rapid Acquisition of Spatial Memory. Neuron 2009, 63 (5), 643–656; [DOI] [PubMed] [Google Scholar]; (b) Gomez M; De Castro E; Guarin E; Sasakura H; Kuhara A; Mori I; Bartfai T; Bargmann CI; Nef P, Ca2+ signaling via the neuronal calcium sensor-1 regulates associative learning and memory in C-elegans. Neuron 2001, 30 (1), 241–248. [DOI] [PubMed] [Google Scholar]
- 9.Haynes LP; Fitzgerald DJ; Wareing B; O’Callaghan DW; Morgan A; Burgoyne RD, Analysis of the interacting partners of the neuronal calcium-binding proteins L-CaBP1, hippocalcin, NCS-1 and neurocalcin delta. Proteomics 2006, 6 (6), 1822–1832. [DOI] [PubMed] [Google Scholar]
- 10.(a) Weiss JL; Archer DA; Burgoyne RD, Neuronal Ca2+ sensor-1/frequenin autocrine pathway regulating Ca2+ bovine adrenal chromaffin cells. Journal of Biological Chemistry 2000, 275 (51), 40082–40087; [DOI] [PubMed] [Google Scholar]; (b) Tsujimoto T; Jeromin A; Saitoh N; Roder JC; Takahashi T, Neuronal calcium sensor 1 and activity-dependent facilitation of P/Q-type calcium currents at presynaptic nerve terminals. Science 2002, 295 (5563), 2276–2279; [DOI] [PubMed] [Google Scholar]; (c) Dason JS; Romero-Pozuelo J; Marin L; Iyengar BG; Klose MK; Ferrus A; Atwood HL, Frequenin/NCS-1 and the Ca2+-channel alpha(1)-subunit co-regulate synaptic transmission and nerve-terminal growth. Journal of Cell Science 2009, 122 (22), 4109–4121. [DOI] [PubMed] [Google Scholar]
- 11.Strahl TGB,; Dannenberg J;Thorner J; Pongs O, Molecular Structure and Target Recognition of Neuronal Calcium Sensor Proteins. J.Biol.Chem. 2003, 278, 49589. [DOI] [PubMed] [Google Scholar]
- 12.Philiippov PP, Neuronal calcium sensor proteins. Nova Science Publishers, Inc; 2006, 5. [Google Scholar]
- 13.Murayama T; Takayama J; Fujiwara M; Maruyama IN, Environmental alkalinity sensing mediated by the transmembrane guanylyl cyclase GCY-14 in C. elegans. Current biology : CB 2013, 23 (11), 1007–12. [DOI] [PubMed] [Google Scholar]
- 14.Obara M; Szeliga M; Albrecht J, Regulation of pH in the mammalian central nervous system under normal and pathological conditions: facts and hypotheses. Neurochemistry international 2008, 52 (6), 905–19. [DOI] [PubMed] [Google Scholar]
- 15.Sinning A; Hubner CA, Minireview: pH and synaptic transmission. FEBS letters 2013, 587 (13), 1923–8. [DOI] [PubMed] [Google Scholar]
- 16.Ma B; Nussinov R, Release factors eRF1 and RF2: a universal mechanism controls the large conformational changes. J Biol Chem 2004, 279 (51), 53875–85. [DOI] [PubMed] [Google Scholar]
- 17.(a) Bellucci L; Corni S; Di Felice R; Paci E, The Structure of Neuronal Calcium Sensor-1 in Solution Revealed by Molecular Dynamics Simulations. Plos One 2013, 8 (9); [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Handley MTW; Lian LY; Haynes LP; Burgoyne RD, Structural and Functional Deficits in a Neuronal Calcium Sensor-1 Mutant Identified in a Case of Autistic Spectrum Disorder. Plos One 2010, 5 (5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhu Y Z. WY; Luo Y; Zou Y; Ma BY; Zhang QW, R102Q mutation shifts the salt-bridge network and reduces the structural flexibility of human neuronal calcium sensor-1 protein..J.Phys. Chem. B 2014, 118, 13112–13122. [DOI] [PubMed] [Google Scholar]
- 19.(a) Zou Y; Sun YX; Zhu YZ; Ma BY; Nussinov R; Zhang QW, Critical Nucleus Structure and Aggregation Mechanism of the C-terminal Fragment of Copper-Zinc Superoxide Dismutase Protein. Acs Chemical Neuroscience 2016, 7 (3), 286–296; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ma B; Nussinov R, Stabilities and conformations of Alzheimer’s beta -amyloid peptide oligomers (Abeta 16–22, Abeta 16–35, and Abeta 10–35): Sequence effects. Proc Natl Acad Sci U S A 2002, 99 (22), 14126–31; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Nussinov R, Allosteric modulators can restore function in an amino acid neurotransmitter receptor by slightly altering intra-molecular communication pathways. British Journal of Pharmacology 2012, 165 (7), 2110–2112; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Miller Y; Ma B; Nussinov R, Polymorphism of Alzheimer’s Abeta17–42 (p3) oligomers: the importance of the turn location and its conformation. Biophys J 2009, 97 (4), 1168–77; [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Zheng J; van de Veerdonk FL; Crossland KL; Smeekens SP; Chan CM; Al Shehri T; Abinun M; Gennery AR; Mann J; Lendrem DW; Netea MG; Rowan AD; Lilic D, Gain-of-function STAT1 mutations impair STAT3 activity in patients with chronic mucocutaneous candidiasis (CMC). European journal of immunology 2015, 45 (10), 2834–46; [DOI] [PubMed] [Google Scholar]; (f) Ma B; Nussinov R, The stability of monomeric intermediates controls amyloid formation: Abeta25–35 and its N27Q mutant. Biophys J 2006, 90 (10), 3365–74; [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Nussinov R; Muratcioglu S; Tsai CJ; Jang H; Gursoy A; Keskin O, The Key Role of Calmodulin in KRAS-Driven Adenocarcinomas. Molecular cancer research : MCR 2015, 13 (9), 1265–73; [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Miller Y; Ma B; Nussinov R, The unique Alzheimer’s beta-amyloid triangular fibril has a cavity along the fibril axis under physiological conditions. J Am Chem Soc 2011, 133 (8), 2742–8; [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Zheng J; Wang H; Yao J; Zou X, More antitumor efficacy of the PI3K inhibitor GDC-0941 in breast cancer with PIK3CA mutation or HER2 amplification status in vitro. Die Pharmazie 2014, 69 (1), 38–42. [PubMed] [Google Scholar]
- 20.(a) Zhu YZ; Yang S; Qi RX; Zou Y; Ma BY; Nussinov R; Zhang QW, Effects of the C-Terminal Tail on the Conformational Dynamics of Human Neuronal Calcium Sensor-1 Protein. Journal of Physical Chemistry B 2015, 119 (44), 14236–14244; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Heidarsson PO; Bjerrum-Bohr IJ; Jensen GA; Pongs O; Finn BE; Poulsen FM; Kragelund BB, The C-Terminal Tail of Human Neuronal Calcium Sensor 1 Regulates the Conformational Stability of the Ca2+-Activated State. Journal of Molecular Biology 2012, 417 (1–2), 51–64. [DOI] [PubMed] [Google Scholar]
- 21.Hess B; Kutzner C; van der Spoel D; Lindahl E, GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. Journal of Chemical Theory and Computation 2008, 4 (3), 435–447. [DOI] [PubMed] [Google Scholar]
- 22.Bjelkmar P; Larsson P; Cuendet MA; Hess B; Lindahl E, Implementation of the CHARMM Force Field in GROMACS: Analysis of Protein Stability Effects from Correction Maps, Virtual Interaction Sites, and Water Models. Journal of Chemical Theory and Computation 2010, 6 (2), 459–466. [DOI] [PubMed] [Google Scholar]
- 23.Hess B; Bekker H; Berendsen HJC; Fraaije J, LINCS: A linear constraint solver for molecular simulations. Journal of Computational Chemistry 1997, 18 (12), 1463–1472. [Google Scholar]
- 24.Miyamoto S; Kollman PA, SETTLE - AN ANALYTICAL VERSION OF THE SHAKE AND RATTLE ALGORITHM FOR RIGID WATER MODELS. Journal of Computational Chemistry 1992, 13 (8), 952–962. [Google Scholar]
- 25.Bussi G; Donadio D; Parrinello M, COMP 8-Canonical sampling through velocity rescaling. Abstracts of Papers of the American Chemical Society 2007, 234. [DOI] [PubMed] [Google Scholar]
- 26.Parrinello MRA,, POLYMORPHIC TRANSITIONS IN SINGLE-CRYSTALS - A NEW MOLECULAR-DYNAMICS METHOD. Journal of Applied Physics 1981, 52 (12), 7182–7190. [Google Scholar]
- 27.Hess B, GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. Abstracts of Papers of the American Chemical Society 2009, 237. [DOI] [PubMed] [Google Scholar]
- 28.Kabsch WSC,, DICTIONARY OF PROTEIN SECONDARY STRUCTURE - PATTERN RECOGNITION OF HYDROGEN-BONDED AND GEOMETRICAL FEATURES. Biopolymers 1983, 22 (12), 2577–2637. [DOI] [PubMed] [Google Scholar]
- 29.Ma BY; Kumar S; Tsai CJ; Nussinov R, Folding funnels and binding mechanisms. Protein Engineering 1999, 12 (9), 713–720. [DOI] [PubMed] [Google Scholar]
- 30.Humphrey WD,A; Schulten K, VMD: visual molecular dynamics. J. Mol. Graphics 1996, 14, 33–38. [DOI] [PubMed] [Google Scholar]
- 31.Eargle J; Luthey-Schulten Z, NetworkView: 3D display and analysis of protein center dot RNA interaction networks. Bioinformatics 2012, 28 (22), 3000–3001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.(a) Arnold K; Bordoli L; Kopp J; Schwede T, The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics 2006, 22 (2), 195–201; [DOI] [PubMed] [Google Scholar]; (b) Biasini M; Bienert S; Waterhouse A; Arnold K; Studer G; Schmidt T; Kiefer F; Gallo Cassarino T.; Bertoni M; Bordoli L; Schwede T, SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic acids research 2014, 42, W252–8; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Guex N; Peitsch MC; Schwede T, Automated comparative protein structure modeling with SWISS-MODEL and Swiss-PdbViewer: a historical perspective. Electrophoresis 2009, 30 Suppl 1, S162–73; [DOI] [PubMed] [Google Scholar]; (d) Kiefer F; Arnold K; Kunzli M; Bordoli L; Schwede T, The SWISS-MODEL Repository and associated resources. Nucleic acids research 2009, 37, D387–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Burgoyne RD, The neuronal calcium-sensor proteins. Biochimica Et Biophysica Acta-Molecular Cell Research 2004, 1742 (1–3), 59–68. [DOI] [PubMed] [Google Scholar]
- 34.(a) Kumar S; Tsai CJ; Ma BY; Nussinov R, Contribution of salt bridges toward protein thermostability. Journal of Biomolecular Structure & Dynamics 2000, 79–85; [DOI] [PubMed] [Google Scholar]; (b) Kumar S; Ma BY; Tsai CJ; Nussinov R, Electrostatic strengths of salt bridges in thermophilic and mesophilic glutamate dehydrogenase monomers. Proteins-Structure Function and Genetics 2000, 38 (4), 368–383. [DOI] [PubMed] [Google Scholar]
- 35.(a) Waldburger CD; Schildbach JF; Sauer RT, ARE BURIED SALT BRIDGES IMPORTANT FOR PROTEIN STABILITY AND CONFORMATIONAL SPECIFICITY. Nature Structural Biology 1995, 2 (2), 122–128; [DOI] [PubMed] [Google Scholar]; (b) Tissot AC; Vuilleumier S; Fersht AR, Importance of two buried salt bridges in the stability and folding pathway of barnase. Biochemistry 1996, 35 (21), 6786–6794. [DOI] [PubMed] [Google Scholar]
- 36.(a) Makhatadze GI; Loladze VV; Ermolenko DN; Chen XF; Thomas ST, Contribution of surface salt bridges to protein stability: Guidelines for protein engineering. Journal of Molecular Biology 2003, 327 (5), 1135–1148; [DOI] [PubMed] [Google Scholar]; (b) Ibarra-Molero B; Zitzewitz JA; Matthews CR, Salt-bridges can stabilize but do not accelerate the folding of the homodimeric coiled-coil peptide GCN4-p1. Journal of Molecular Biology 2004, 336 (5), 989–996; [DOI] [PubMed] [Google Scholar]; (c) Williams DV; Byrne A; Stewart J; Andersen NH, Optimal Salt Bridge for Trp-Cage Stabilization. Biochemistry 2011, 50 (7), 1143–1152; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Andersson HS; Figueredo SM; Haugaard-Kedstrom LM; Bengtsson E; Daly NL; Qu XQ; Craik DJ; Ouellette AJ; Rosengren KJ, The alpha-defensin salt-bridge induces backbone stability to facilitate folding and confer proteolytic resistance. Amino Acids 2012, 43 (4), 1471–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhu Y; Ma B; Qi R; Nussinov R; Zhang Q, Temperature-Dependent Conformational Properties of Human Neuronal Calcium Sensor-1 Protein Revealed by All-Atom Simulations. The journal of physical chemistry. B 2016, 120 (14), 3551–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Heidarsson PO; Otazo MR; Bellucci L; Mossa A; Imparato A; Paci E; Corni S; Di Felice R; Kragelund BB; Cecconi C, Single-Molecule Folding Mechanism of an EF-Hand Neuronal Calcium Sensor. Structure 2013, 21 (10), 1812–1821. [DOI] [PubMed] [Google Scholar]
- 39.Sethi A; Eargle J; Black AA; Luthey-Schulten Z, Dynamical networks in tRNA: protein complexes. Proceedings of the National Academy of Sciences of the United States of America 2009, 106 (16), 6620–6625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Girvan M; Newman MEJ, Community structure in social and biological networks. Proceedings of the National Academy of Sciences of the United States of America 2002, 99 (12), 7821–7826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ames JB; Lim S, Molecular structure and target recognition of neuronal calcium sensor proteins. Biochim Biophys Acta 2012, 1820 (8), 1205–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.