

Abstract
The ability to engineer natural proteins is pivotal to a future, pragmatic biology. CRISPR proteins have revolutionized genome modification, yet the CRISPR-Cas9 scaffold is not ideal for fusions or activation by cellular triggers. Here we show that topological rearrangement of Cas9 using circular permutation provides an advanced platform for RNA-guided genome modification and protection. Through systematic interrogation, we find that protein termini can be positioned adjacent to bound DNA, offering a straightforward mechanism for strategically fusing functional domains. Additionally, circular permutation enabled protease-sensing Cas9s (ProCas9s), a unique class of single molecule effectors possessing programmable inputs and outputs. ProCas9s can sense a wide range of proteases, and we demonstrate that ProCas9 can orchestrate a cellular response to pathogen-associated protease activity. Together, these results provide a toolkit of safer and more efficient genome modifying enzymes and molecular recorders for the advancement of precision genome engineering in research, agriculture, and biomedicine.
Graphical Abstract
In Brief
Programmable Cas9 variants with improved functionality are generated by protein engineering, including versions that work as protease sensors responding to viral infection.
Introduction
Type II CRISPR-Cas proteins, such as Cas9, are RNA-guided, DNA binding and cleaving enzymes that function as integral components of adaptive bacterial immune systems (Jinek et al., 2012). Due to its intuitive and robust function, Cas9 has been adapted as a programmable DNA double-strand break generation tool in vitro and in vivo (Cong et al., 2013; Fellmann et al., 2016; Jinek et al., 2012, 2013; Mali et al., 2013). Additionally, enzymatically deactivated Cas9 (dCas9) has been shown to function as a programmable DNA-binding protein which can be harnessed to site-specifically deliver additional protein domains such as chromatin modifiers, methyltransferases, nucleobase deaminases, and fluorescent markers (Chen et al., 2013; Gilbert et al., 2014; Guilinger et al., 2014; Hilton et al., 2015; Komor et al., 2016; Qi et al., 2013; Tsai et al., 2014a). As a consequence, Cas9 has revolutionized genome editing and functional interrogation of the genome. Despite this potential, a number of issues still hinder the broad utility of Cas9.
Foremost, Cas9’s always-on nature can lead to off-target genome editing due to prodigious activity (Richter et al., 2017). It is, relatedly, inherently difficult to generate cell or tissue specificity in an in vivo therapeutic delivery context unless the protein can be directly delivered to a defined compartment, such as the inner ear, eye, or brain (Kim et al., 2017; Staahl et al., 2017; Zuris et al., 2015). A lack of control on activity also limits the potential of using Cas9 in post-translational genetic circuits, such as a sensor that can respond to a defined cellular input or act as a molecular recorder of cellular events, e.g. (Roybal et al., 2016). Although there are now numerous strategies for controlling Cas9 activity via exogenous ligands and other inputs, the ability to control Cas9 activity via an endogenous signal would be highly desirable (Davis et al., 2015; Hemphill et al., 2015; Oakes et al., 2016; Richter et al., 2017).
Additionally, Cas9 did not evolve to function as a modular DNA-binding scaffold. Thus, its fusion to protein domains possessing additional activity has required elaborate optimization, such as long linkers or daisy-chained fusions (Chavez et al., 2015; Guilinger et al., 2014; Komor et al., 2016; Tanenbaum et al., 2014; Tsai et al., 2014a). Such tools may be greatly enhanced by the ability to fuse protein domains at a precise location in the Cas9 topology. Hence, developing an optimized Cas9 architecture for controlled nuclease activity and facilitating efficient construction of fusion proteins would expand and improve the future applications of this incredibly important enzyme.
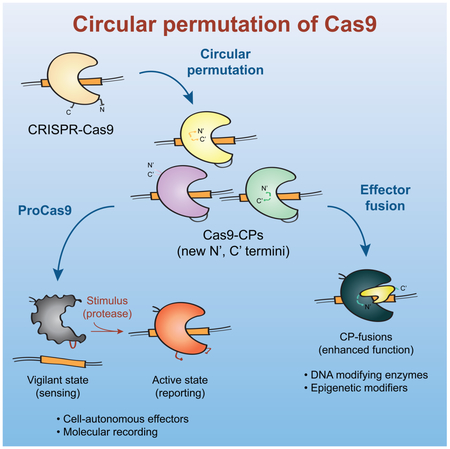
One unique route for creating novel and highly functional CRISPR architectures is by protein circular permutation (CP). CP is the topological rearrangement of a protein’s primary sequence - connecting its N- and C- terminus with a peptide linker, while concurrently splitting its sequence at a different position to create new, adjacent N- and C- termini (Figure 1A) (Yu and Lutz, 2011). Termini repositioning can change a protein’s behavior and naturally occurring or engineered circularly permuted proteins possess altered stability, substrate specificity, enzymatic rate, and novel quaternary structure (Beernink et al., 2001; Mehta et al., 2012; Qian and Lutz, 2005; Whitehead et al., 2009). The inherent requirement of linking the N- and C-termini has also been exploited to create ‘caged’ zymogen pro-enzymes in which a protease cleavage site is used as the linker sequence (Plainkum et al., 2003).
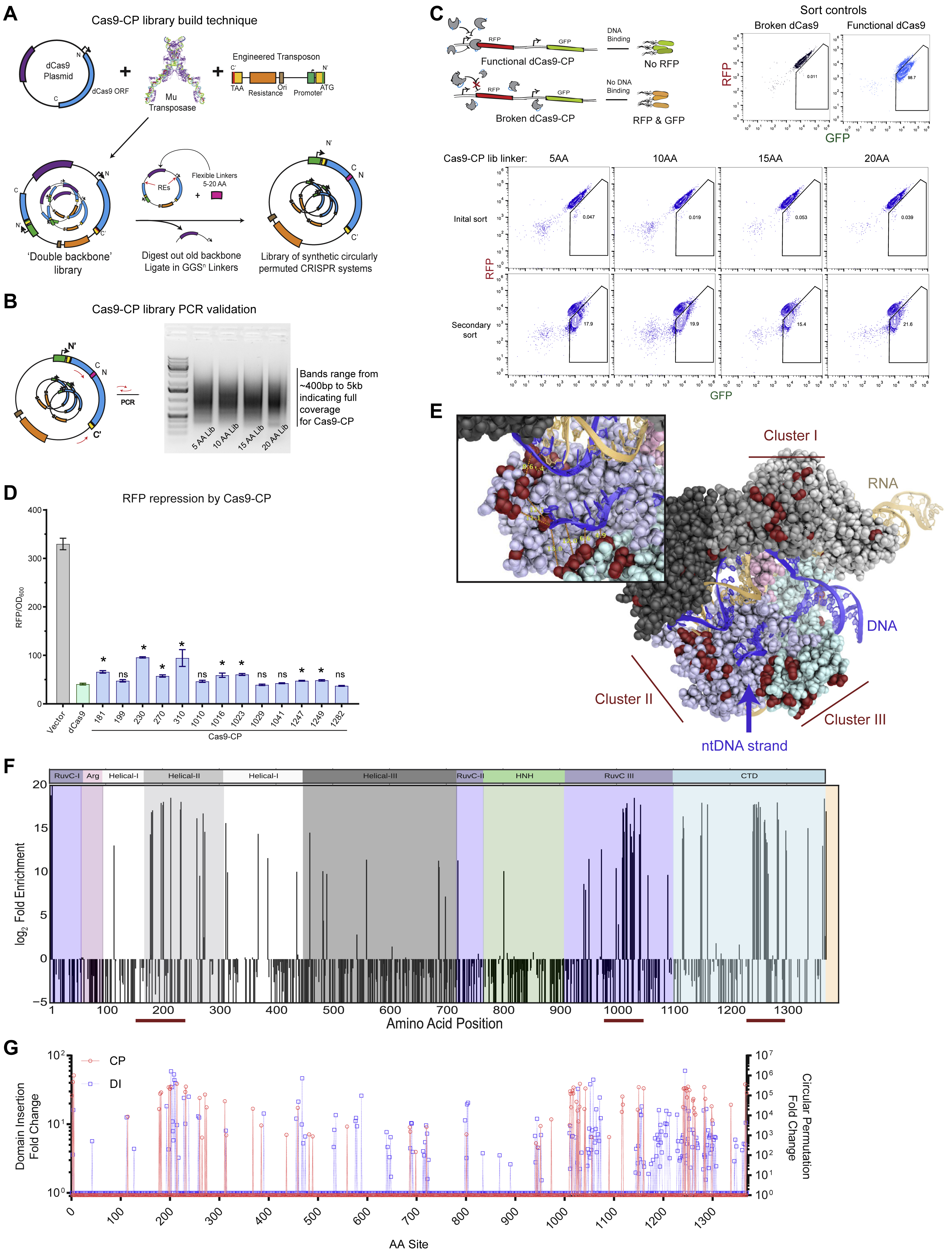
Figure 1. An unbiased Cas9 library screen identifies active circularly permuted Cas9 proteins.

(A) Overview of circular permutation and library generation for Cas9.
(B) Enrichment values of the unbiased screen as determined by flow cytometry and colony forming units. Error bars indicate standard deviation in all panels.
(C) Deep sequencing read averages for the Pre- and Post- Cas9-CP library members, demonstrating a strong clustering of highly enriched library members with internal (within 4 AA of the N- and C-termini) and empirically validated controls. The dotted line highlights an approximate boundary that represents >100-fold enrichment in the screen.
(D) Model of new Cas9-CP termini (in red) based on PDB ID: 5F9R with domains colored according to the sequence bar (below). New termini are mapped onto the AA sequence bar.
(E) Endpoint values for dCas9-CP 12 hr E. coli CRISPRi DNA binding and RFP repression system compared with WT dCas9 and a protein expression vector control in triplicate (error bars are s.d., * = p<0.05, ns = not significant, t-test).
(F) CFU/mL readings in an E. coli genomic cleavage assay read out by cell death compared with a protein expression vector control, WT dCas9 and WT Cas9 (n = 3, error bars are s.d., * = p<0.05, ns = not significant, t-test).
(G) Cleavage efficiency of a genomic reporter in mammalian cells in triplicate (described in Figures S2B and S2C), observed via indel formation and GFP reporter disruption. hCas9 is human codon optimized Cas9; bCas9 indicates bacterial codon-based Cas9 constructs (error bars are s.d., * = p<0.05, ns = not significant, t-test).
Here, we demonstrate how circular permutation can be used to re-engineer the molecular sequence of Cas9 in order to both better control its activity and create a more optimal DNA binding scaffold for fusion proteins. By coupling systematic library creation with high-throughput fitness assays and deep sequencing, we define the set of possible Cas9 circular permutants (collectively, Cas9-CPs). Our analysis shows that Cas9 is highly malleable to circular permutation, and several regions of the protein - notably the Helical-II, RuvC, and C-terminal Domain (CTD) - possess hotspots that can be opened at numerous positions to generate a diversity of Cas9-CPs. We further show that engineering of the linker sequence with site-specific protease sequences, derived from a variety of pathogenic plant and human viruses, yields ‘caged’ pro-enzyme Cas9 variants that can be activated by proteolytic cleavage. This modular approach is generalizable and the proteins, which we term ProCas9s, are capable of sensing and stably recording or responding to the presence of numerous, distinct families of viral proteases - including those from Flaviviruses - in both E. coli and various mammalian cell types. In total, this work establishes an architecture for generating Cas9s that can be activated by cell-, tissue-, or pathway-specific endogenous signals, and provides a resource of Cas9-CPs to simplify and optimize the process of constructing Cas9-fusion proteins for precision genome modification.
Results
Circular permutation of Cas9
To investigate the topological malleability of Streptococcus pyogenes Cas9 (hereafter Cas9), we generated a random transposon insertion library in vitro by adapting an engineered transposon from Silberg and colleagues (Jones et al., 2016) to contain a plasmid backbone, inducible promoter, and stop codon (Figure S1A and Table S1). As the original N- and C- termini of Cas9 are 40 to 60 Å apart (Anders et al., 2014), the requirements for Cas9 circular permutation are not known. We therefore permuted dCas9 using a series of linkers (GGS repeats, varying from five to 20 AA) between the original N- and C- termini, providing increasing steric freedom (Figure S1A). Transposition of the engineered cassette and pooled molecular cloning yielded high insertional diversity for all libraries, as indicated by the length distributions of polymerase chain reaction (PCR) amplicons (S1B). Deep sequencing of the 20 AA linker library further demonstrated that ~1 out of every 2 AA in Cas9 were observed transposition sites in the original pool, for a total of 661 circular permutant (CP) variants in the library (Table S2).
CP libraries, constructed around dCas9, were screened for function in an E. coli-based repression (i.e. CRISPRi) assay targeting the expression of either red or green fluorescent protein (RFP/GFP) (Qi et al., 2013; Oakes et al., 2014, 2016). In brief, dCas9-CP libraries were targeted to repress RFP expression while GFP was used as a control for cell viability. Functional dCas9-CP library members were isolated through a sequential double sorting procedure that enriched functional clones 100-10,000-fold (Figures 1B, 1C, S1A-C, and Table S2). A subset of isolated clones was plated for each of the libraries (i.e. 5, 10, 15 and 20 AA linkers) and sequenced. For the 5 and 10 AA linker library, only a minimal number of CPs around the original termini was observed. However, the 15 and 20 AA linker libraries yielded a number of CP variants and isolated clones were found to be highly functional in bacterial CRISPRi assays (Figures 1E, S1D, and Table 1).
Table 1. Key Cas9 circular permutants.
Nomenclature and local sequence of select Cas9 circular permutants (Cas9-CPs). The superscript in the name indicates the original amino acid (AA) in Streptococcus pyogenes Cas9 that now serves as the new N-terminus.
| Name | Domain at CP site | Original seq at CP site | New start site (AA) |
|---|---|---|---|
| Cas9-CP181 | Helical-II | …PDNSD∣VDKLF… | 181 |
| Cas9-CP199 | Helical-II | …QLFEE∣NPINA… | 199 |
| Cas9-CP230 | Helical-II | …LIAQL∣PGEKK… | 230 |
| Cas9-CP270 | Helical-II | …QLSKD∣TYDDD… | 270 |
| Cas9-CP310 | Helical-II | …ILRVN∣TEITK… | 310 |
| Cas9-CP1010 | RuvC-III | …ESEFV∣YGDYK… | 1010 |
| Cas9-CP1016 | RuvC-III | …GDYKV∣YDVRK… | 1016 |
| Cas9-CP1023 | RuvC-III | …VRKMI∣AKSEQ… | 1023 |
| Cas9-CP1029 | RuvC-III | …KSEQE∣IGKAT… | 1029 |
| Cas9-CP1041 | RuvC-III | …YFFYS∣NIMNF… | 1041 |
| Cas9-CP1247 | CTD | …YEKLK∣GSPED… | 1247 |
| Cas9-CP1249 | CTD | …KLKGS∣PEDNE… | 1249 |
| Cas9-CP1282 | CTD | …SKRVI∣LADAN… | 1282 |
Since the majority of functional clones were found in the 20 AA linker library, we proceeded to deep sequence this library, to generate an enrichment profile of permutation across Cas9. We identified 77 sites that were highly enriched (>100 fold) following the double sorting procedure (Figures 1C and SIF). Notably, all confirmed hits (Figure 1E) and internal controls fell within this group. Mapping the observed sites onto the protein sequence (Figure 1D) revealed three hotspots of CPs (all numbering based on Streptococcus pyogenes Cas9 protein sequence): in the Helical-II (AA 178-314), in the RuvC-III (AA 940-1150) and in the CTD (AA 1240-1299) domains (Figures 1D and S1E-G). These hotspots qualitatively correspond with those we have previously identified for Cas9 domain insertion (Oakes et al., 2016), indicating that the underlying structural and biochemical constraints may be similar (Figure S1G). Intriguingly, among the newly discovered termini, a number are in direct contact (< 5 Å) with the non-target strand, yielding Cas9-CPs containing ideal fusion points for protein domains to modify the isolated single-strand that heretofore required long linkers to gain such access (i.e. base editors) (Figure S1E) (Gaudelli et al., 2017; Guilinger et al., 2014; Komor et al., 2016; Tsai et al., 2014a).
The isolated Cas9-CPs were next tested for their cleavage activity relative to wild-type (WT) Cas9. Briefly, two variants from each of the three hotspots (specifically, CP sites 199, 230, 1010, 1029, 1249, and 1282) were constructed with a 20 AA linker between the original N- and C-termini and recoded with functional nuclease active sites (Table 1). Testing of these constructs for genomic cleavage and killing activity in E. coli demonstrated that all possessed similar activity as WT Cas9 (Figure 1F). To assess how well these findings extrapolate to mammalian systems, we established a rapid human genome editing reporter assay with a quantitative fluorescence-based readout of target disruption activity and editing efficiency (Figures S2A-C; Methods). When compared relative to WT Cas9 in this assay, our Cas9-CPs showed surprisingly high genome editing efficiency (Figure 1G). While we observed more variation than in the E. coli based experiments, four tested CP variants (CP199, CP1029, CP1249, CP1282) showed 80% or more of WT activity. Overall, these results demonstrate that Cas9 can be circularly permuted to create novel proteins that may maintain wild-type like levels of DNA binding and cleavage activity.
Cas9-CP activity can be regulated by proteolytic cleavage
Characterization of the libraries described above revealed that circular permutation is highly sensitive to the linker length connecting the original N- and C-terminus. PCR analysis of pooled libraries (Figure S3A) indicated that a linker length of 5 AA or 10 AA was not sufficient to generate Cas9-CP diversity. Conversely, libraries of 15 or 20 AA linkers (Figure S3A) qualitatively possessed extensive permutable diversity. We therefore tested the importance of linker length on confirmed sites identified above (Figure 1E). The same six Cas9-CPs (i.e. Cas9-CP199 through Cas9-CP1282) were cloned with linkers (GGS repeats) from 5 to 30 amino acids and tested for repression of GFP in an E. coli based CRISPRi assay (Figure 2A). In agreement with the pooled libraries, we found that all Cas9-CPs with linkers of 5 and 10 AA in length were markedly disrupted in activity, while those with longer linkers were active. Notably, activity did not increase with linker length beyond 15 AA (Figure 2A).
Figure 2. Linker length can be utilized to control Cas9-CP activity.

(A) Endpoint analysis of an E. coli CRISPRi based GFP repression assay run in triplicate using Cas9- CPs identified as functional with 20 AA linkers, evaluated with GGSn linkers of length 5, 10, 15, 20, 25 and 30 AA. Error bars indicate standard deviation in all panels.
(B) Schematic describing the rationale behind using a Cas9-CP with a short AA linker as a ‘caged’ molecule.
(C) Endpoint analysis of an E. coli CRISPRi-based GFP expression time course with all six Cas9-CPs containing a 7 AA TEV linker (ENLYFQ/S) in the presence of a functional TEV protease (TEV, blue) compared with deactivated TEV protease with the catalytic triad mutant C151A (dTEV, gray) (n = 3, error bars are s.d., * = p<0.05, ns = not significant, t-test).
(D) Schematic and western blot against the Flag epitope on the C terminus of the CP-TEVs after the endpoint measurement (Figure 2C). Expected kDa indicates the predicted band size if cleavages occurs at the TEV site in the CP linker region.
The sensitivity of CPs to linker length led us to hypothesize that Cas9-CPs could be made into ‘caged’ variants that could switch from an inactive form to an active one upon post-translational modification (Figure 2B). It has previously been observed that circularly permuted proteins can be sensitive to the length of the linker between their old N- and C-termini (Yu and Lutz, 2011). This requirement has been exploited to create zymogen pro-enzymes by replacing the linker with a site-specific protease sequence, such that proteolytic cleavage converts a short linker into an effectively infinite linker with concomitant turn-on in protein activity. Although potentially useful for applications in biosensing (e.g. pathogen or cancer detection) all existing sensors were constructed around either ribonuclease A (Johnson et al., 2006; Plainkum et al., 2003) or barnase (Butler et al., 2009) and possess limited in vivo potential due to their inherent nonspecific, toxic activity.
To test the possibility of turning Cas9-CPs into activatable switches using a well-studied protease, we engineered the six representative CP variants with the 7 AA cleavage site (ENLYFQ/S) of the Tobacco Etch Virus (TEV) Nuclear Inclusion antigen (NIa) protease as the linker sequence (Seon Han et al., 2013). We found that this 7 AA linker was able to fully disrupt Cas9-CP activity in our E. coli CRISPRi GFP repression assay (Figure 2C and S3B). Upon addition of a fully active TEV protease, activity was restored to a varying degree in all six Cas9-CPTEV constructs. Notably, Cas9-CP199 switched from completely off to fully on (Figure 2C) and performed consistently over a 20-hour time course (Figure S3C). This switch behaved well across the population in single cell assays and did not activate when a TEV catalytic triad mutant, C151A, was expressed (dTEV) (Figure S3D). Finally, to verify if TEV is cleaving Cas9-CPs at the CP linker, we recovered cells from the endpoint of the CRISPRi assay (Figure 2C) for western blot analysis against a 2x Flag-tag cloned onto the C-terminus of the protein. As expected, when an active TEV protease was present, products were observed corresponding to the size of the C-terminal circularly permuted fragment (Figure 2D).
ProCas9s exemplify a general strategy for regulating caged Cas9s with site-specific proteases
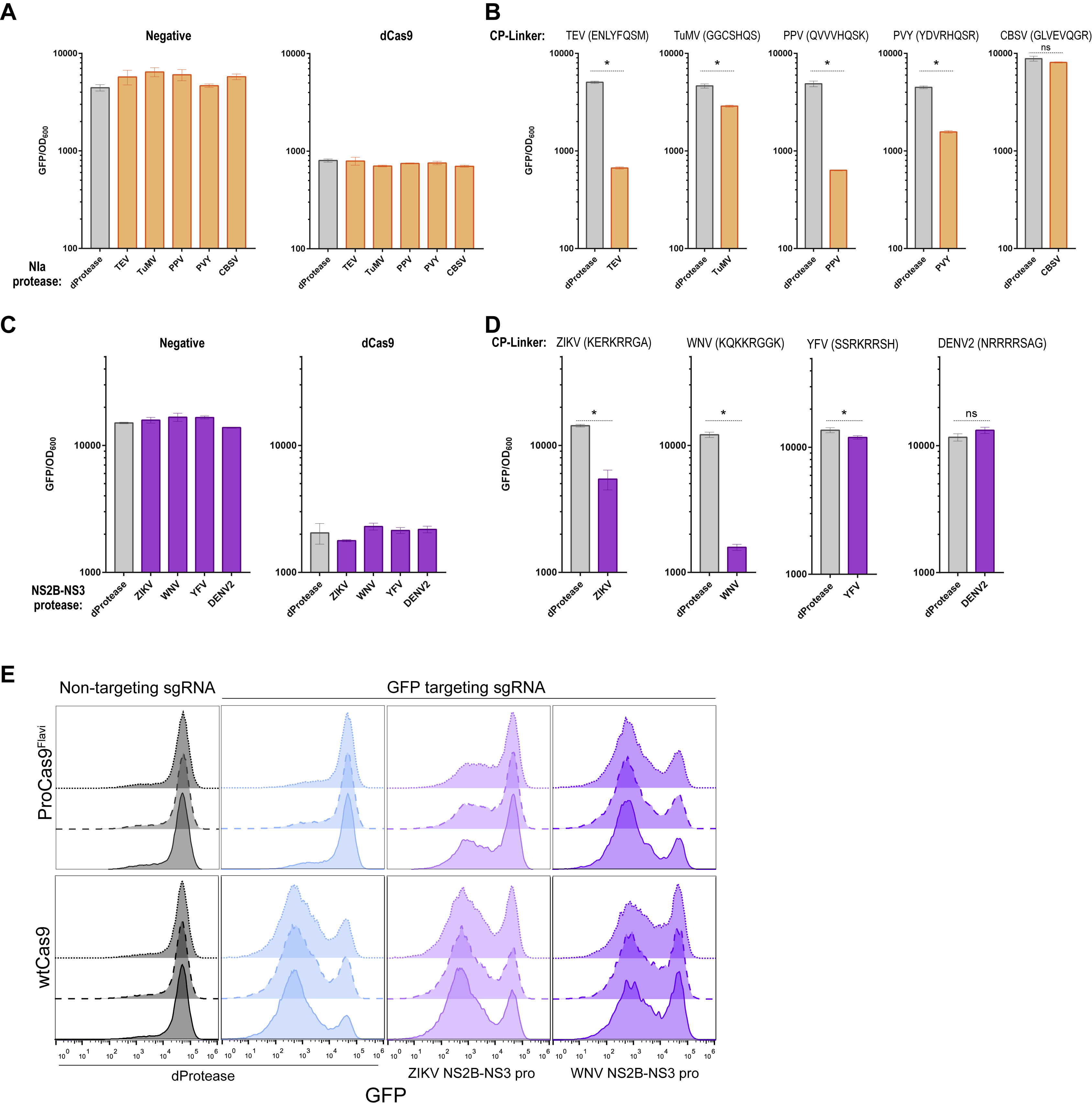
We next sought to determine if this uncaging mechanism could be generalized to other families of proteases. For example, the human rhinovirus 3C is responsible for about 30% of cases of the common cold and contains a well-studied protease (3Cpro), unrelated to that from TEV (Skern, 2013). Thus, we replaced the TEV linker sequence with the 8 AA recognition site for 3Cpro (LEVLFQ/GP) in the six Cas9-CPs and tested for bacterial CRISPRi activity with and without active protease. Protease-dependent activation of Cas9-CPs was observed, with varying amounts of turn-on in activity, thus demonstrating that the mechanism can be extended to other proteases. Cas9-CP199 again had the greatest response (Figure S3E) and was used for all experiments described below.
Next, we sought to apply our protease sensing Cas9-CPs (hereafter ProCas9s) to agriculturally and medically relevant viruses. We examined the Potyvirus proteases from Turnip Mosaic Virus (TuMV), Plum Pox Virus (PPV), Potato Virus Y (PVY), and Cassava Brown Streak Virus (CBSV), plant viruses responsible for significant crop losses each year (Seon Han et al., 2013; Tomlinson et al., 2018). The NIa protease genes from these viruses were cloned for co-expression in conjunction with our ProCas9s. Cognate protease cleavage sites (Methods) were used as the CP linker in Cas9-CP199, yielding the respective ProCas9s that were systematically tested against all co-expressed Nla proteases (Figure 3A, controls in Figures S4A and S4B). CRISPRi experiments revealed a general trend of proteases activating their respective ProCas9 and also that the PPV linker (QVVVHQ/SK) enabled a ProCas9 response to three different Nla proteases with distinct specificity from TEV (Figures 3A and 3B). We term this variant ProCas9Poty for a Cas9 that can recognize and respond to a number of agriculturally important Potyvirus proteases.
Figure 3. Generation of ProCas9s for sensing and responding to Potyvirus and Flavivirus proteases.

(A) Heat map depicting the fold activation of a suite of ProCas9 CP linkers for Potyviral NIa proteases. Data is normalized to a non-active protein expression control (dTEV) in an E. coli based CRISPRi GFP repression assay. Darker coloration indicates greater activity (n = 2).
(B) Endpoint analysis of the E. coli CRISPRi assay utilizing the linker derived from Plum Pox Virus (PPV) comparing the response to distinct NIa proteases and a dead protease (n = 3, error bars are s.d., * = p<0.05, ns = not significant, t-test compared to dProtease).
(C) Heat map depicting the fold activation of a suite of ProCas9 CP linkers for Flavivirus NS2B-NS3 proteases, normalized to a non-active protein expression control (dTEV) in an E. coli based CRISPRi GFP repression assay. Darker coloration indicates greater activity (n = 2).
(D) Endpoint analysis of the E. coli CRISPRi assay utilizing the linker derived from West Nile Virus (WNV) showing the response to distinct NS2B-NS3 proteases and a dead protease (n = 3, error bars are s.d., * = p<0.05, ns = not significant, t-test compared to dProtease).
(E) Schematic of the constructs used for the transient transfection and testing in HEK293T cells.
(F) Mammalian GFP disruption assay (Figures S2A-C). HEK293T-based reporter cells were transfected with the indicated sgRNAs, WT Cas9 or a ProCas9 variant, and the respective proteases Reduction in GFP positive cells indicates genome cleavage by a Cas9 construct (n = 3, error bars are s.d., * = p<0.05, t-test compared to dProtease).
(G) Flow cytometry plots from (F) with overlay of GFP-targeting (pink) vs. non-targeting (black) ProCas9Flavi systems, demonstrating a small degree of background activity.
(H) Truncation of ProCas9 AA linker sequence to prevent leakiness.
(I) Leakiness and orthogonality of the original and shortened ProCas9Flavi constructs. Displayed as percent GFP disrupted via normalization to the non-targeting guide for each construct-protease pairing. In addition to the deactivated protease (dProtease) control, the active Potyvirus NIa proteases were used to assess orthogonality (n = 3, error bars are s.d., * = p<0.05, ns = not significant, t-test).
We repeated this process with a set of proteases from the medically important Flavivirus genus. Briefly, the capsid protein C cleavage sequences from Zika Virus (ZIKV), West Nile Virus (WNV, Kunjin strain), Dengue Virus 2 (DENV2), and Yellow Fever Virus (YFV) (Bera et al., 2007; Kümmerer et al., 2013) were used as the CP linker sequence to generate a set of Flavivirus-specific ProCas9s. In the viral life cycle, these cleavage sequences are cut by the NS2B-NS3 protease from the respective virus to mature the polyprotein (Kümmerer et al., 2013). Screening of these Flavivirus ProCas9 variants against their cognate proteases revealed a variant - hereafter called ProCas9Flavi - that possesses a WNV linker sequence (KQKKR/GGK) and was activated by NS2B-NS3 proteases from both Zika and WNV (Figures 3C, 3D, S4C, and S4D). We did not observe any activation with the CBSV, DENV2, or YFV proteases; this may be due to non-optimal CP linkers, poor expression of the cognate proteases, or a steric hindrance blocking the protease from reaching the CP linker site.
The function of ProCas9s in eukaryotic cells was next validated and optimized using a transient transfection system in our HEK293T-based GFP disruption assay (Figures 3E, S2B, and S2C). In this model, expression of either ProCas9Poty or ProCas9Flavi resulted in GFP disruption only in the presence of the active proteases (Figures 3F and S4E). Nevertheless, we also observed a small amount of leaky activation (~5%) in the absence of protease activity (Figures 3F and 3G). Hence, we tested whether progressively shortening the distance between the original N- and C- termini by 2, 4 or 6 AA would reduce unwanted background activity (Figure 3H). While removing 2 AA from ProCas9Flavi had no apparent effect, removing 6 AA (ProCas9Flavi-S6) significantly reduced activity levels for non-active or non-corresponding active proteases while still enabling a response, albeit weaker, to both ZIKV and WNV (corresponding) proteases (Figure 3I). Thus, linker ‘tightening’ optimization provides an additional safety mechanism, allowing a ProCas9 to exist in cells with little risk of un-triggered genome cleavage activity.
ProCas9 can be stably integrated into mammalian genomes without leaky activity
A prerequisite to the use of activatable genome editors in sensing or molecular recording applications is that they possess low background activity under stable expression conditions. To confirm that ProCas9s function accordingly, we built lentiviral vectors expressing ProCas9 from either a weak EF1a core promoter (EFS) or strong full-length EF1a promoter, along with sgRNAs driven from a U6 promoter, and tested ProCas9Flavi and ProCas9Flavi-S6 activity in HEK-RT1 reporter cells (Figure 4A). When measured six to ten days post-transduction, none of the four tested ProCas9 constructs showed any background activity (Figure 4B), indicating that the systems are not leaky.
Figure 4. ProCas9 stably integrated into mammalian genomes can sense and respond to Flavivirus proteases.

(A) Genomic integration and testing of Flavivirus protease-sensitive ProCas9s. HEK-RT1 genome editing reporter cells are stably transduced with various ProCas9 lentiviral vectors, followed by puromycin selection of ProCas9 cell lines. These cell lines are then either tested for leaky ProCas9 activity in the absence of a stimulus, or stably transduced with a vector expressing the indicated proteases, followed by assessment of genome editing using the GFP reporter.
(B) Leakiness assessment of ProCas9 variants expressed from either the EFS or EF1a promoter. HEK-RT1 reporter cells were stably transduced with the indicated ProCas9 variants or Cas9-wt. Genome editing activity was quantified at the indicated days post-transduction. Error bars indicate the standard deviation of triplicates.
(C) Leakiness assessment at the endogenous PCSK9 locus. HepG2 cells stably transduced with the indicated sgRNAs and ProCas9 variants or Cas9-wt. Cells were selected on puromycin and harvested at day 8 post-transduction for T7E1 analysis.
(D) Mutational patterns and editing efficiency at the PCSK9 locus of samples shown in (C). Indels were quantified using TIDE. For clarity, the fraction of non-edited cells is represented as negative percentages.
(E) ProCas9 leakiness quantification, as in (C), in A549 and HAP1 cells. Cells were selected on puromycin and harvested at day 7 post-transduction for T7E1 analysis.
(F) Quantification of Flavivirus ProCas9 activation in response to various control (dTEV, pCF708) or Flavivirus (ZIKV, pCF709; WNV, pCF710) proteases. ProCas9 reporter cell lines were stably transduced with the indicated protease vectors. At day 3 post-transduction, cells were treated with doxycycline to induce GFP reporter expression. Error bars indicate the standard deviation of triplicates. Significance was assessed by comparing each sample to its respective dTEV control (unpaired, two-tailed t-test, n = 3, * = p<0.05, ns = not significant).
(G) Genome editing activity in Flavivirus ProCas9 reporter cell lines, as in (F), at day 4 or 8 post-transduction.
(H) Protease-sensitive editing at the endogenous PCSK9 locus. T7E1 assay of A549 and HAP1 Flavivirus ProCas9 cell lines (sgNT, sgPCSK9-4) stably transduced with the indicated mTagBFP2-tagged viral proteases. At day 4 post-transduction, mTagBFP2-positive cells were sorted and harvested for T7E1 analysis.
(I) ProCas9Flavi activation by Flavivirus (Flavi) proteases. *, small subunit of the activated ProCas9Flavi (29 kDa). **, large subunit of the activated ProCas9Flavi (137 kDa).
(J) Immunoblotting for Cas9 in HEK293T co-transfected with plasmids expressing Cas9-wt or ProCas9Flavi and dTEV or WNV proteases. The C-Cas9 (clone 10C11-A12) antibody recognize the large subunit of the activated ProCas9Flavi (**, 137 kDa). The Flag-tag (clone M2) antibody recognizes the small subunit of the activated ProCas9Flavi (*, 29 kDa). ***, likely small-subunit-ProCas9Flavi-T2A-mCherry (55 kDa). Protein ladders indicate reference molecular weight markers.
To further confirm these findings at an endogenous locus, we targeted the non-essential PCSK9 locus in the hepatocellular carcinoma cell line HepG2. Eight days after stable transduction with ProCas9Flavi, ProCas9Flavi-S6 or WT Cas9 PCSK9 editing efficiency was assessed by T7 endonuclease 1 (T7E1) assay (Figure 4C). While WT Cas9 showed high levels of editing, no leakiness was observed with any of the ProCas9 constructs. TIDE analysis (Brinkman et al., 2014) was used to quantify editing outcome (Figure 4D), revealing 71.1% editing with WT Cas9 (11.6% non-edited, 17.3% undetected in the −10 to +10 nucleotide indel range) and confirming the absence of background editing with the ProCas9 constructs. Finally, editing at the PCSK9 locus was also tested in the lung carcinoma cell line A549 and the haploid chronic myeloid leukemia derived line HAP1, two cell lines often used for Flavivirus assays (Figure 4E). Again, the ProCas9 constructs displayed no background activity.
Genomic ProCas9 can be activated by Flavivirus proteases to induce target editing
An activatable switch for molecular sensing must display repeatable induction upon stimulation. In an initial test, HEK-RT1 reporter lines (Figure 4B) containing stably integrated Flavivirus ProCas9s were transiently transfected with vectors expressing dTEV, ZIKV, and WNV proteases, each tagged with mTagBFP2 to enable tracking of activity (Figures 4A and S5A). Two days post-transfection, the GFP reporter was induced by doxycycline treatment for 24 hours, and quantified for editing efficiency by flow cytometry in mTagBFP2-positive cells (Figure S5B). While dTEV protease expression did not lead to genome editing in any reporter cell line, both ZIKV and WNV protease activity led to genome editing, especially with the ProCas9Flavi system. Not surprisingly, the ProCas9Flavi system driven by the stronger EF1a promoter showed the highest genome editing efficiency (Figure 4F, S5B). Together, this suggests that ProCas9 constructs can sense and record Flavivirus protease activity associated with transient expression.
To mimic a viral infection more closely, we next evaluated whether a stably integrated viral vector expressing Flavivirus proteases could also activate ProCas9Flavi enzymes. To generate viral particles, HEK293T packaging cell lines were transfected with dTEV, ZIKV or WNV protease-encoding lentiviral vectors (Figure S5C). Expressing the NS2B-NS3 or NS3 protease is known to be toxic (Ramanathan et al., 2006) and we observed a similar effect with ZIKV and WNV proteases, which led to reduced viral titers and target cell transduction efficiency (Figure S5D). Nevertheless, we were able to stably transduce the HEK-RT1-ProCas9 reporter cell lines with protease constructs and followed the effects of dTEV, ZIKV and WNV protease expression (Figure 4F). While the dTEV protease did not lead to any editing, both the ZIKV and WNV proteases induced genome editing in all four tested ProCas9 lines, with the strongest effect (over 25% editing) again observed with the EF1a-ProCas9Flavi system induced by the WNV protease.
To assess the dynamic range of ProCas9Flavi induction, the above experiments were repeated out to eight days (Figure 4G). Here, stable expression of the WNV protease led to ~35% genome editing when sensed by the EF1a-ProCas9Flavi system. In further tests, we tested an EF1a-ProCas9Flavi construct that did not contain any nuclear localization sequence (NLS, Figure S5E) and observed that WNV protease-mediated induction was reduced compared to NLS containing constructs (Figure S5F). Finally, we qualitatively confirmed these results, based on mTagBFP2-positive cells expressing the protease, using a T7E1 assay (Figure S5G).
As with background activity testing, the activation of ProCas9s by proteases was further validated by targeting the endogenous PCSK9 locus (Figure 4H). Qualitative T7E1 based analysis showed that while no genome editing was observed with a non-targeting guide, the EF1a-ProCas9Flavi system equipped with a guide targeting PCSK9 (sgPCSK9-4) showed clear genome editing in the presence of WNV protease, but not a negative control (dTEV). Together with the absence of leakiness, this clearly demonstrates that ProCas9 can be stably integrated into mammalian genomes to sense, record and respond to endogenous or exogenous protease activity.
Mechanism of ProCas9 activation in mammalian cells
Conceptually, the underlying idea of ProCas9s is that they are present in cells in an inactive, or “vigilant”, state due to the linker sterically inhibiting activity (Figure 4I). The presence of a cognate protease recognizes the peptide linker, relieves inhibition through target cleavage, and leads to an “active” ProCas9 composed of two distinct subunits. To explore this hypothesis, we co-transfected HEK239T cells with vectors expressing either Cas9-wt or ProCas9Flavi and the dTEV or WNV protease (Figure S6A). Immunoblotting with antibodies for the full length Cas9-wt and vigilant ProCas9Flavi - as well as both the small (~29 kDa) and large (~137 kDa) subunit of active ProCas9Flavi - showed that Cas9-wt and ProCas9Flavi are expressed to comparable extents in the absence of a cognate protease (Figures 4J and S6B). In the presence of the WNV protease, however, the vast majority of vigilant ProCas9Flavi was activated and observed as two distinct subunits, confirming the hypothesized mechanism.
Rapid CRISPR-Cas controlled cell depletion
There are many types of outputs a molecular sensor such as ProCas9 could actuate. One unique effect would be to induce cell death upon sensing viral infection, as a form of altruistic defense. Since activated ProCas9 is capable of inducing DNA double-strand breaks, we sought to identify sgRNAs that could induce rapid cell death. As Flaviviruses replicate rapidly upon target cell infection, such sgRNAs would have to kill their host cells in less time. Targeting essential genes such as the single-stranded DNA binding protein RPA1, which is involved in DNA replication, could be one option. Alternatively, targeting highly repetitive sequences within a cell’s genome to induce massive DNA damage and cellular toxicity could be another avenue. Indeed, sgRNAs targeting even only moderately amplified loci have been shown to lead to cell depletion under certain conditions (Wang et al., 2015), independent of whether the sgRNA targets a gene or intergenic region. While these effects have been observed over long assay periods, targeting highly repetitive sequences might provide sufficient DNA damage to trigger rapid cell death.
To compare the two strategies, both HEK293T and HAP1 cells were stably transduced to express WT Cas9 and an sgRNA coupled to an mCherry fluorescence marker (Figure 5A). The effect of guide RNA expression on cell viability was assessed using a competitive proliferation assay in which cells expressing a specific sgRNA were mixed with parental cells expressing only Cas9-wt, and the mCherry-positive population was followed over time. Negative control guides targeting an olfactory receptor gene (sgOR2B6-1, sgOR2B6-2) showed no depletion. As expected, guide RNAs targeting the essential RPA1 gene depleted over the eight-day assay period. To potentially accelerate depletion, we also designed and tested several sgRNAs targeting repetitive sequences in the human genome (~125,000-300,000 target loci each, Methods), which could cause CRISPR-Cas induced death by editing or ‘CIDE’. Indeed, CIDE guide RNAs (sgCIDE-1, sgCIDE-2, sgCIDE-4, sgCIDE-5) led to rapid elimination of the mCherry-positive population (Figure 5A) and show promise as a simple genetic output module for an altruistic defense system based on CRISPR-Cas mediated cell death.
Figure 5. ProCas9 enables genomically encoded programmable response systems.

(A) CRISPR-Cas programmed cell depletion. HEK293T and HAP1 cells expressing Cas9-wt were transduced with mCherry-tagged sgRNAs. After mixing with parental cells, the fraction of mCherry-positive cells was quantified over time. Different sgRNAs targeting a neutral gene (sgOR2B6), an essential gene (sgRPA1), >100,000 genomic loci (sgCIDE) and a non-targeting control (sgNT) were compared. Error bars indicate the standard deviation of triplicates.
(B) Competitive proliferation assay analogous to (A), conducted in HEK293T and HAP1 cells expressing the ProCas9Flavi system. Note, sgCIDE positive cells show little or no depletion because the ProCas9Flavi is in its inactive, vigilant state.
(C) ProCas9Flavi activation by Flavivirus proteases expressed from genomically integrated lentiviral vectors.
(D) Competitive proliferation assay in HEK293T ProCas9Flavi cells expressing the indicated mCherry-tagged sgRNAs, or a non-targeting control (sgNT) used for normalization. Cells were partially transduced with lentiviral vectors expressing a GFP-tagged dTEV or WNV protease, and cell depletion quantified by flow cytometry. Note, the WNV protease leads to protective cell death (altruistic defense) in sgCIDE expressing cells through activation of the ProCas9Flavi system. Error bars indicate the standard deviation of triplicates. Significance was assessed by comparing each sample to its respective dTEV control (unpaired, two-tailed t-test, n = 3, * = p<0.05, ns = not significant).
Genomic ProCas9 can sense Flavivirus proteases and mount an altruistic defense
CIDE as an output constrains the performance of ProCas9. The system must remain off in order to minimize genomic damage, yet be vigilant to respond to a stimulus. To develop this protease-induced altruistic defense platform, we assessed whether stable expression of the best CIDE guide RNAs (sgCIDE-2, sgCIDE-4) in conjunction with a genomically integrated ProCas9Flavi cassette was viable in the absence of a stimulus (Figure 5B). Competitive proliferation assays analogous to the ones run with WT Cas9 showed that in the presence of ProCas9Flavi only minimal amounts of cell depletion were observed.
Finally, we tested induction of this stably integrated altruistic defense system by Flavivirus proteases (Figure S7A). Using the same cell lines (expressing ProCas9Flavi) as above, we observed that stable transduction with vectors expressing either a control (dTEV) or Flavivirus (WNV) protease led to specific cell depletion only when both the WNV protease was present and the system was programmed with one of the two CIDE sgRNAs (Figures 5C, 5D, and S7B). Hence, these results confirmed that our Flavivirus ProCas9 system can be stably integrated into the genome of a host cell to detect predefined protease activity and mount a programmed defense, only in the presence of a specific stimulus of interest.
Discussion
Here we demonstrate that the large, multi-domain, and highly allosteric enzyme Cas9 is amenable to circular permutation via protein engineering, without apparent loss of its functions. By systematically creating and testing the sequence of Cas9 for circular permutation, we identified a comprehensive suite of novel variants that are efficient at genome binding and cleavage, with the added benefit of redistributed new N- and C- termini across Cas9’s topology (Figure 6). Additionally, we show that Cas9 circular permutants can be rewired into molecular recording devices, termed ProCas9s, that can sense proteases – including those from Flaviviruses and Potyviruses – to stably record their activity in the genome or mount a pre-programmed defense. Importantly, the modularity of this system enables simple re-design of the ProCas9 sensing activity by swapping of the CP linker and, as such, could respond to any exogenous or endogenous sequence-specific protease. Thus, the system may be used to sense and report cell-intrinsic pathway activity e.g. for molecular screening and drug discovery, or serve as a means for cell-type-specific Cas activation after general delivery of an editing complex to a target tissue or organ.
Figure 6. Application of Cas9 circular permutants.

Diagram showing various uses of Cas9 circular permutants (Cas9-CPs) as single-molecule sensor-effectors for protease tracing and molecular recording, or as optimized scaffolds for modular CP-fusion proteins with novel and enhanced functionalities.
The ProCas9s ability to serve as a detector of pathogen activity is intriguing as it could enable their use as a fully modular, genomically encoded immune system with both a designable input and programmable output. For example, many plants are known to contain protease-gated transcription factors that activate a protective hypersensitivity response when cleaved by a pathogen protease (Ade et al., 2007; Chisholm et al., 2005) and one of these proteins has even been adapted for the recognition of a non-native protease (Kim et al., 2016). Nevertheless, this system’s output is constrained by the DNA-binding specificity of the transcription factor. In contrast, ProCas9s are a simple effector with a designable input and programmable output that should work in every organism CRISPR proteins have been shown to operate in. As one example, here we show how ProCas9s can be tuned to serve as altruistic defense systems to protect a population of human cells by self-elimination of the few that are expressing a Flavivirus protease - mimicking an infection. Thus, we have demonstrated an initial proof-of-concept for a fully synthetic and customizable resistance gene. Hence, it should be straightforward to transition this self-targeting system into a platform that can induce expression or suppression of genes to mount a systemic immune response, or to activate a synthetic cellular program to track pathogen invasion. Such a strategy for pathogen detection is broadly applicable, as many pathogens express proteases during host infection (Alfano and Collmer, 2004; Gao et al., 1994; Hartmann and Lucius, 2003).
Others have recently adapted constitutively expressed CRISPR systems to target pathogenic viruses directly (Baltes et al., 2015; Chaparro-Garcia et al., 2015; Kennedy et al., 2014). However, these systems utilize a fully active nuclease gated only by sequence recognition. The sustained expression of Cas9 both increases the risks of off-target effects and promotes evolution of the targeted viruses. Indeed, recent reports (Chaparro-Garcia et al., 2015; Mehta et al., 2018) highlight this phenomenon, which may represent an unintended consequence of utilizing CRISPR systems in a pathogen-directed fashion. In contrast, the ProCas9 system allows programming a response to a viral infection akin to innate immunity, where a self-directed response can be activated to minimize the opportunity for evasive viral hypermutation and resistance.
Additionally, the Cas9-CPs serve as a diverse set of protein scaffolds for advanced CRISPR-Cas fusion proteins. The natural N- and C-termini are fixed for all proteins. Our work paves the way for making a new class of CP-based CRISPR tools with optimized N- and C-termini for fusions. In Cas9, for example, the native termini are ~50 Å apart, requiring long linkers for fusions that seek access to the DNA (Gaudelli et al., 2017; Guilinger et al., 2014; Komor et al., 2016; Tsai et al., 2014a). The dearth of options when attempting to build new Cas9 fusions may explain the relative lack of activity or undesired side effects of many compound constructs. Indeed, dCas9 activators need numerous domains (up to 24) (Chavez et al., 2015; Tanenbaum et al., 2014) or combinations of guide RNAs for high activity (Hilton et al., 2015). dCas9-FokI fusions are not as efficient at indel induction as Cas9 itself (Guilinger et al., 2014; Tsai et al., 2014a), and the base editing cytidine deaminase fusions, which result in strong C to T editing within a 12 bp target window, may also cause deamination up to 15 bp outside of the Cas9 target sequence (Komor et al., 2016). Circular permutation of Cas9 yields a new class of scaffolds with N- and C- termini within 5 Å of the bound target or non-target strand, which may remedy current steric limitations.
Taken together, a more holistic approach to Cas9 tool building – one that includes engineering of both the fusion scaffold and fusion domain – enables a more proficient generation of modular and customizable CRISPR-Cas9 effectors. Our work lays the foundation for this process by providing both a blueprint for the circular permutation of Cas9, as well as by providing a resource of functionally active Cas9-CPs for advanced fusion proteins. Additionally, we present the concept of ProCas9 variants that can be enzymatically activated by sequence-specific proteases to serve as molecular recorders or tissue-specific effectors.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, David F. Savage (savage@berkeley.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Bacterial strains and media
For in-vivo E. coli screening, fluorescence measurements, and cell proliferation assays, we used MG1655 with a chromosomally integrated and constitutively expressed GFP and RFP (Oakes et al., 2014; Qi et al., 2013). EZ-rich defined growth medium (EZ-RDM, Teknoka) was used for all liquid culture assays and plates were made using 2xYT. Plasmids used were based on a 2 plasmid system as reported previously (Oakes et al., 2014, 2016; Qi et al., 2013) containing Cas9 and variants on a selectable CmR marker and plasmids with sgRNAs and proteases with AmpR markers, representative sequences of which can be found in Table S1. The antibiotics were used to verify transformation and to maintain plasmid stocks. No blinding or randomization was done for any of the experiments reported.
Mammalian cell culture
All mammalian cell cultures were maintained in a 37°C incubator, at 5% CO2. HEK293T (293FT; Thermo Fisher Scientific, #R70007) human kidney cells and derivatives thereof were grown in Dulbecco’s Modified Eagle Medium (DMEM; Corning Cellgro, #10-013-CV) supplemented with 10% fetal bovine serum (FBS; Seradigm, #1500-500), and 100 Units/ml penicillin and 100 μg/ml streptomycin (100-Pen-Strep; Gibco #15140-122). HepG2 human liver cells (ATCC, #HB-8065) and derivatives thereof were cultured in Eagle's Minimum Essential Medium (EMEM; ATCC, #30-2003) supplemented with 10% FBS and 100-Pen-Strep. A549 human lung cells (ATCC, #CCL-185) and derivatives thereof were grown in Ham’s F-12K Nutrient Mixture, Kaighn’s Modification (F-12K; Corning Cellgro, #10-025-CV) supplemented with 10% FBS and 100-Pen-Strep. HAP1 cells (kind gift from Jan Carette, Stanford) and derivatives thereof were grown in Iscove’s Modified Dulbecco’s Medium (IMDM; Gibco #12440-053 or HyClone #SH30228.01) supplemented with 10% FBS and 100-Pen-Strep. HAP1 cells had been derived from the near-haploid chronic myeloid leukemia cell line KBM7 (Carette et al., 2011). Karyotyping analysis demonstrated that most cells (27 of 39) were fully haploid, while a smaller population (9 of 39) was haploid for all chromosomes except chromosome 8, like the parental KBM7 cells. Less than 10% (3 of 39) were diploid for all chromosomes except for chromosome 8, which was tetraploid.
A549 cells were authenticated using short tandem repeat DNA profiling (STR profiling; UC Berkeley Cell Culture/DNA Sequencing facility). STR profiling was carried out by PCR amplification of nine STR loci plus amelogenin (GenePrint 10 System; Promega #B9510), fragment analysis (3730XL DNA Analyzer; Applied Biosystems), comprehensive data analysis (GeneMapper software; Applied Biosystems), and final verification using supplier databases including American Type Culture Collection (ATCC) and Deutsche Sammlung von Mikroorganismen und Zellkulturen (DSMZ).
HEK293T, HEK-RT1, HEK-RT6, HepG2, A549, and HAP1 cells were tested for absence of mycoplasma contamination (UC Berkeley Cell Culture facility) by fluorescence microscopy of methanol fixed and Hoechst 33258 (Polysciences #09460) stained samples.
METHOD DETAILS
Transposon library construction
To begin, a dCas9 flanked by BsaI restriction enzyme sites was inserted into a pUC19 based plasmid. A modified transposon with R1 and R2 sites based on (Jones et al., 2016) (Table S1), containing a chloramphenicol antibiotic resistance marker, p15A origin of replication, TetR and TetR/A promoter, was built using custom oligos and standard molecular biology techniques. It was then cleaved from a plasmid using HindIII and gel purified. This linear transposon product was used in overnight in vitro reactions (0.5 molar ratio transposon to 100 ng dCas9-Puc19 plasmid) using 1 μl of MuA Transposase (F-750, Thermo Fisher) in 10 replicates. The transposed DNA was purified and recovered, Plasmids were electroporated into custom made electrocompetent MG1655 E. coli (Oakes et al., 2014) using a BTX Harvard apparatus ECM 630 High Throughput Electroporation System and titered on carbenicillin (Carb) and chloramphenicol (CM) to ensure >100x coverage of the library size (13,614). These cells were then outgrown for 12 hours and selected for via Carb and CM markers to ensure growth of transposed members. After isolating transposed plasmids via miniprep (Qiagen), the original Puc19 backbone was removed via BsaI cleavage and dCas9 proteins transposed with a new plasmid backbone were selected via a 0.7% TAE agarose gel. The linear fragments were then ligated overnight with annealed and phosphorylated oligos coding for GGS linkers of 5, 10, 15 and 20 AA using a BsaI Golden Gate reaction. Completed libraries were purified, electroporated into the Mg1655 RFP and GFP screening strain containing a RFP-repressing sgRNA and titered on Carb and CM to ensure >5x coverage of the library size (8,216).
Screening for Cas9 circular permutants (Cas9-CPs)
Screens were performed in a similar manner to previous reports (Oakes et al., 2014, 2016), briefly biological duplicates of Cas9-CP libraries with a RFP guide RNA were transformed (at >5x library size) into MG1655 with genetically integrated and constitutively expressed GFP and RFP. Cells were grown overnight in EZ-RDM + Carb, CM and 200 nM Anhydrotetracycline (aTc) inducer. E. coli were then sorted based on gates for RFP but not GFP repression, collected, and resorted immediately to further enrich for functional Cas9-CPs (Figure S1C). Double sorted libraries were then grown out and DNA was collected for sequencing. This DNA was also re-transformed onto plates and individual clones were picked for further analysis.
Deep sequencing library preparation
This method was modified from previous Tnseq protocols (Coradetti et al., 2018). Briefly, the transposed plasmids were sheared to ~300bp using a S220 Focused-ultrasonicator (Covaris) and purified in between each of the following steps using Agencourt AMPure XP beads (Beckman Coulter). Following shearing, fragments were end-repaired and A-tailed according to NEB manufacturers protocols and then universal adapters were ligated on in a 50 ul quick ligase reaction at RT. Finally fragments from each library were amplified in a 20-cycle reaction with Indexed Illumina primers that annealed upstream of the new CP start codon and in the universal adapter (Table S1). PCRs were cleaned again and analyzed for primer dimers via an Agilent Bioanalyzer DNA 1000 chip. Sequencing was performed at the QB3 Vincent J. Coates Genomics Sequencing Laboratory on a HiSeq2500 in a 100 bp run.
Deep sequencing analysis
Demultiplexed reads from the HiSeq2500 were assessed using FastQC to check basic quality metrics. Reads for each sample were then trimmed using a custom python script. The trimmed sequences were mapped to the dCas9 nucleic acid sequence using BWA via a custom python wrapper script to determine the amino acid position in dCas9 corresponding to the starting amino acid position in the dCas9-CP permutant. The resulting alignment files were then processed using a custom python script to calculate the abundance of each dCas9-CP permutant in a given library sample. Fold-changes for each dCas9-CP permutant between pre- and post-library sorts along with significance values for each enrichment were calculated (Table S2) using the DESeq package in R (Anders and Huber, 2010). Due to ambiguity in transposon sequence, insertion site calls in Table S2 are one greater (sites: n+1) than the variants named in Table 1. As per the DESeq guidelines, count data from technical sequencing replicates were summed to create one unique replicate before running through the DESeq pipeline. All relevant sequencing data and Cas9-CP analysis scripts are available at https://github.com/SavageLab/Cas9-CP.
E. coli CRISPRi GFP repression assay
Assays were performed similar to previous descriptions (Oakes et al., 2016). To measure the ability of a circular permutant to bind to and repress DNA expression, cells were co-transformed with a Cas9 permutant plasmid with aTc inducible promoter and a single guide RNA plasmid for RFP or GFP that, in the case of the ProCas9 assays, also contained the active or inactive proteases on an IPTG-inducible promoter. Endpoint Assay: Cells were picked in biological triplicate into 96 well plates containing 500 uL EZ MOPS plus Carb and CM. Plates were grown in 37°C shakers for twelve hours. Next, cells were diluted 1:1000 in 500 uL EZ MOPS plus Carb, CM, IPTG and aTc. 200 nM aTc was used to induce Cas9-CPs or ProCas9s and 50 uM IPTG levels was used to induce the proteases in a 2mL deep well blocks and shaken at 750 rpm at 37°C. After an eigh t-twelve hr induction and growth period, 20 uL of cells were added to 80 uL of water and put into a 96-well microplate reader (Tecan M1000) at 37°C and read immediately. Each well was measured for optical density at 600 nm and GFP or RFP fluorescence. GFP expression was normalized by dividing it with OD600. In the case of the time course assays (Figures S3B and S3C), 150 uL of the 1:1000 dilution was used and placed into a black walled clear bottom plate (3631-Corning) and directly into the Tecan M1000 for a 130x 600 sec kinetic cycle of reading. For E. coli single cell analysis (Figure S3C), cells from the endpoint time course were run on a Sony SH800 to capture 100,000 events per sample.
E. coli genomic cleavage assay
Assays were performed as previously described (Oakes et al., 2016) E. coli containing sgRNA plasmids targeting a genomically integrated GFP were made electrocompetent and transformed with 10 ng of the the various Cas9-CP plasmids or controls using electroporation. After recovery in 1 mL SOC media for 1 hr, cells were plated in technical triplicate of tenfold serial dilutions onto 2xYT agar plates with antibiotics selection for both plasmids and aTc induction at 200 nM. Plates were grown at 37°C overnight and CFU/mL was determined. A reduction in CFUs indicated genomic cleavage and cell death.
E. coli western blotting
After CRISPRi repression assays for TEV linker Pro-Cas9s, 40 uL of cell culture was pelleted and resuspended in SDS loading buffer for further analysis. SDS samples were loaded into 4-20% acrylamide gels (BioRad) for electrophoresis. After transfer to membranes (Trans-Blot Turbo- BioRad), blots were washed 3x with 1xTBS + 0.01% Tween 20, blocked with 5% milk for 1.5 hr and then a 1:1000 of HRP-conjugated DYKDDDDK Tag (Anti-Flag) antibody (Cell Signaling Technology, #2044) was incubated for twenty-four hours at 4°C. Antibod ies were washed away with 3x TBST and detected using Pierce ECL Western Blotting Substrate (Thermo Fisher).
NIa protease cleavage sites
NIa protease cleavage sites – i.e. the CP linkers – were identified from previous reports (TuMV, 7 AA; (Kim et al., 2016), by using the sequence between the P3 and 6KI genes annotated in NCBI (PPV, PVY, CBSV), or from previously identified Potyvirus protease consensus sequences (Seon Han et al., 2013).
Lentiviral vectors
A lentiviral vector referred to as pCF204, expressing a U6 driven sgRNA and an EFS driven Cas9-P2A-Puro cassette, was based on the lenti-CRISPR-V2 plasmid (Sanjana et al., 2014), by replacing the sgRNA with an enhanced Streptococcus pyogenes Cas9 sgRNA scaffold (Chen et al., 2013). The pCF704 and pCF711 lentiviral vectors, expressing a U6-sgRNA and an EFS driven ProCas9 variant, were derived from pCF204 by swapping wild-type Cas9 for the respective ProCas9 variant. The pCF712 and pCF713 vectors were derived from pCF704 and pCF711, respectively, be replacing the EF1a-short promoter (EFS) with the full-length EF1a promoter. The lentiviral vector pCF732 was derived from pCF712 by removal of the ProCas9’s nuclear localization sequences (NLSs). Vectors not containing a guide RNA, including pCF226 (Cas9-wt) and pCF730 (ProCas9Flavi), were derived from pCF204 and pCF712, respectively, through KpnI/NheI-based removal of the U6-sgRNA cassette and blunt ligation. The guide RNA-only vector pCF221, encoding a U6-sgRNA cassette and an EF1a driven mCherry marker, is loosely based on the pCF204 backbone and guide RNA cassette. Lentiviral vectors expressing viral proteases, including pCF708 expressing an EF1a driven mTagBFP2-tagged dTEV protease, pCF709 expressing an EF1a driven mTagBFP2-tagged ZIKV NS2B-NS3 protease, and pCF710 expressing an EF1a driven mTagBFP2-tagged WNV protease, are all based on the pCF226 backbone. The GFP-tagged protease vectors pCF736 and pCF738 are derived from pCF708 and pCF710, respectively, by swapping mTagBFP2 with GFP. All vectors were generated using custom oligonucleotides (IDT), gBlocks (IDT), standard cloning methods, and Gibson assembly techniques and reagents (NEB). Vector sequences are provided (Table S1).
Design of sgRNAs
Standard sgRNA sequences were either designed manually, using CRISPR Design (crispr.mit.edu), or using GuideScan (Perez et al., 2017). When editing endogenous genes, sgRNAs were often designed to target evolutionarily conserved regions in the 5’ proximal third of the gene of interest. The following sequences were used: sgGFP1 (CCTCGAACTTCACCTCGGCG), sgGFP2 (CAACTACAAGACCCGCGCCG), sgGFP9 (CCGGCAAGCTGCCCGTGCCC), sgOR2B6-1 (CATTATTCTAGTGTCACGCC), sgOR2B6-2 (GGGTATGAAGTTTGGTGTCC), sgPCSK9-4 (CCGGTGGTCACTCTGTATGC), sgPuro5 (TGTCGAGCCCGACGCGCGTG), sgPuro6 (GCTCGGTGACCCGCTCGATG), sgRPA1-1 (ACAAAAGTCAGATCCGTACC), sgRPA1-2 (TACCTGGAGCAACTCCCGAG). All sgRNAs were designed with a G preceding the 20 nucleotide guide for better expression from U6 promoters.
To enable rapid CRISPR-Cas controlled cell depletion, through a strategy that we termed Cas-induced death by editing or ‘CIDE’, we designed sgRNAs (sgCIDEs) directed again highly repetitive sequences in the human genome. In brief, using GuideScan (Perez et al., 2017) we identified the most frequently occurring Streptococcus pyogenes Cas9 sgRNA target sites (5’-NGG-3’ PAM) in the hg38 assembly (Genome Reference Consortium Human Build 38) of the human genome. From this list we eliminated sequences containing extended homomeric stretches (>4 A/T/C/G), and empirically validated two sequences with slightly over 125,000 target loci (sgCIDE-4, CGCCTGTAATCCCAGCACTT; sgCIDE-5, CCTCGGCCTCCCAAAGTGCT) and two sequences with approximately 300,000 target loci (sgCIDE-1, TGTAATCCCAGCACTTTGGG; sgCIDE-2, TCCCAAAGTGCTGGGATTAC). All four sgCIDEs led to rapid cell depletion when expressed in presence of active Cas9.
Lentiviral transduction
Lentiviral particles were produced in HEK293T cells using polyethylenimine (PEI; Polysciences #23966) based transfection of plasmids. HEK293T cells were split to reach a confluency of 70-90% at time of transfection. Lentiviral vectors were co-transfected with the lentiviral packaging plasmid psPAX2 (Addgene #12260) and the VSV-G envelope plasmid pMD2.G (Addgene #12259). Transfection reactions were assembled in reduced serum media (Opti-MEM; Gibco #31985-070). For lentiviral particle production on 10 cm plates, 8 μg lentiviral vector, 4 μg psPAX2 and 2 μg pMD2.G were mixed in 2 ml Opti-MEM, followed by addition of 42 μg PEI. After 20-30 min incubation at room temperature, the transfection reactions were dispersed over the HEK293T cells. Media was changed 12 hr post-transfection, and virus harvested at 36-48 hr post-transfection. Viral supernatants were filtered using 0.45 μm cellulose acetate or polyethersulfone (PES) membrane filters, diluted in cell culture media if appropriate, and added to target cells. Polybrene (5 μg/ml; Sigma-Aldrich) was supplemented to enhance transduction efficiency, if necessary.
Transduced target cell populations (HEK293T, A549, HAP1, HepG2 and derivatives thereof) were usually selected 24-48 hr post-transduction using puromycin (InvivoGen #ant-pr-1; HEK293T, A549 and HepG2: 1.0 μg/ml, HAP1: 0.5 μg/ml) or hygromycin B (Thermo Fisher Scientific #10687010; 200-400 μg/ml).
Rapid mammalian genome editing reporter assay
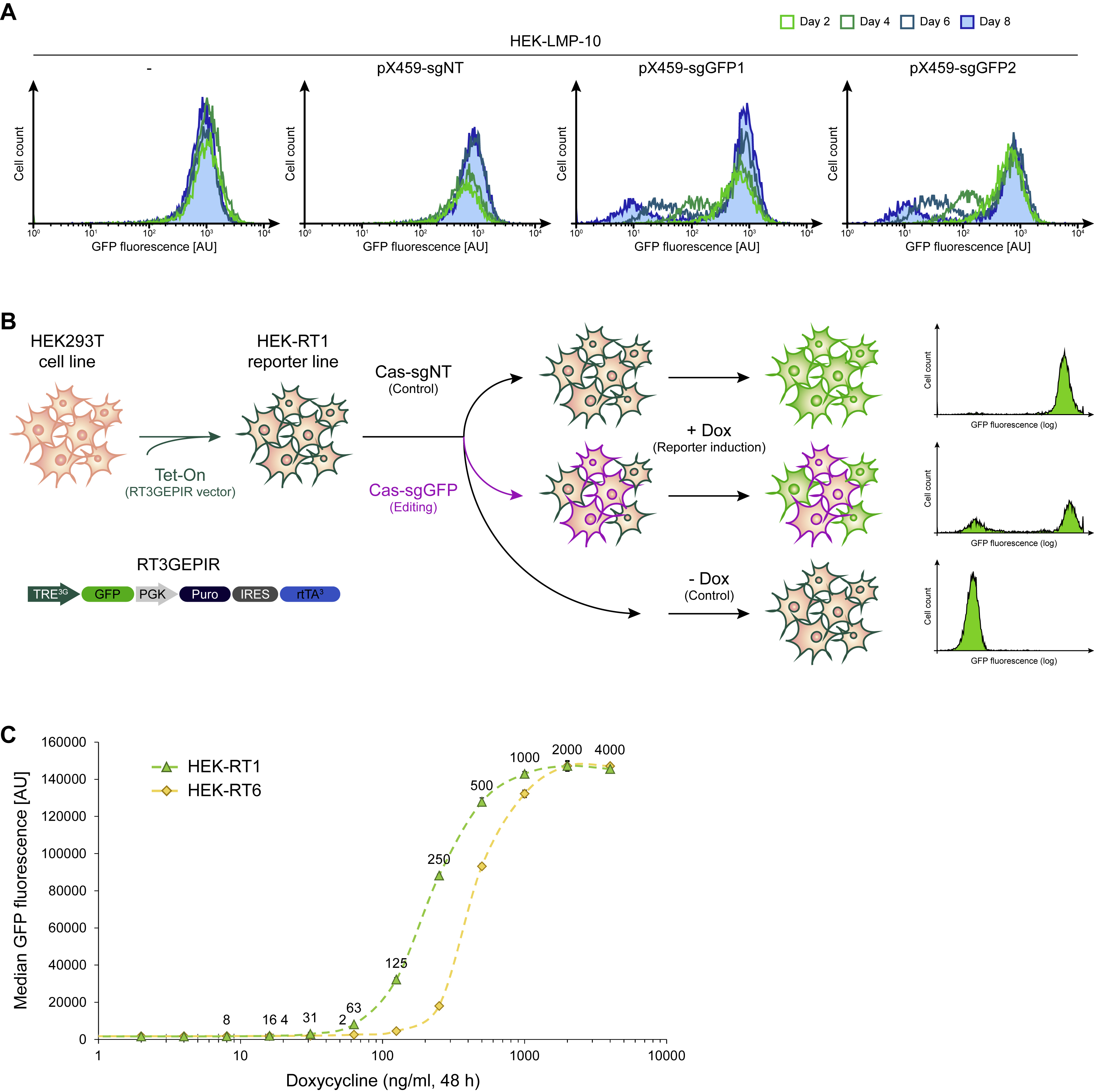
To establish a rapid and quantitative way to reliably assess genome editing efficiency from various CRISPR-Cas constructs in mammalian cells, we decided to build a fluorescence-based reporter assay. Assays leveraging editing-based disruption of a constitutively expressed fluorescence marker have been built before. However, such assays show a long detection lag time as the genetic disruption of a locus coding for the fluorescent marker will not immediately lead to a reduction in the fluorescence signal, due to the remaining presence of intact transcripts and protein half-life. To quantify this effect, we stably transduced HEK293T cells with a retroviral vector (LMP-Pten.1524) constitutively expressing GFP (Fellmann et al., 2013), and established monoclonal derivatives. The best performing cell line was termed HEK-LMP-10. When editing this reporter line with a vector (pX459, Addgene #48139) expressing wild-type Streptococcus pyogenes Cas9 and guide RNAs targeting the reporter (sgGFP1, sgGFP2), or a non-targeting control (sgNT), the editing detection lag – defined as the time between introduction of an editing reagent and complete loss of fluorescence signal in edited cells – was up to eight days (Figure S2A). Hence, this type of assay is inconvenient for rapid quantification of editing efficiency. Conversely, assays relying on frameshift mutations to activate a fluorescence reporter often require specific guide RNA sequences and only get activated with the faction of edits that lead to the required frameshift, thus introducing a quantification bias.
To overcome this limitation, we decided to build an inducible genome editing reporter cell line comprising a fluorescence marker that is not expressed in the default state but can be induced following a defined time of potential genome editing (Figure S2B). In this scenario, unedited cells will rapidly turn positive, while non-edited cells remain fluorophore negative. Specifically, inducible monoclonal HEK293T-based genome editing reporter cells, referred to as “HEK-RT1”, were established in a two-step procedure. In the first step, puromycin resistant monoclonal HEK-RT3-4 reporter cells were generated (Park et al., 2018). In brief, HEK293T human embryonic kidney cells were transduced at low-copy with the amphotropic pseudotyped RT3GEPIR-Ren.713 retroviral vector (Fellmann et al., 2013), comprising an all-in-one Tet-On system enabling doxycycline-controlled GFP expression. After puromycin (2.0 μg/ml) selection of transduced HEK239Ts, 36 clones were isolated and individually assessed for i) growth characteristics, ii) homogeneous morphology, iii) sharp fluorescence peaks of doxycycline (1 μg/ml) inducible GFP expression, iv) relatively low fluorescence intensity to favor clones with single-copy reporter integration, and v) high transfectability. HEK-RT3-4 cells are derived from the clone that performed best in these tests.
Since HEK-RT3-4 are puromycin resistant, in the second step, monoclonal HEK-RT1 and analogous sister reporter cell lines were derived by transient transfection of HEK-RT3-4 cells with a pair of vectors encoding Cas9 and guide RNAs targeting puromycin (sgPuro5, sgPuro6), followed by identification of monoclonal derivatives that are puromycin sensitive. In total, eight clones were isolated and individually assessed for i) growth characteristics, ii) homogeneous morphology, iii) doxycycline (1 μg/ml) inducible and reversible GFP fluorescence, and v) puromycin and hygromycin B sensitivity. The monoclonal HEK-RT1 and HEK-RT6 cell lines performed best in these tests and were further evaluated in a doxycycline titration experiment (Figure S2C), showing that both reporter lines enable doxycycline concentration-dependent induction of the fluorescence marker in as little as 24-48 hours. The HEK-RT1 cell line was chosen as rapid mammalian genome editing reporter system for all further assays.
Genome editing analysis using the mammalian HEK-RT1 reporter assay
When employing the HEK-RT1 genome editing reporter assay to quantify WT Cas9 (Cas9-wt) and ProCas9 variant activity following stable genomic integration, HEK-RT1 reporter cells were transduced with the indicated Cas-wt/ProCas9 and sgRNA lentiviral vectors (Table S1) and selected on puromycin. A guide RNA targeting the GFP fluorescence reporter (sgGFP9) was compared to a non-targeting control (sgNT). We used the non-targeting control in all assays for normalization, in case not all non-edited cells turned GFP positive upon doxycycline treatment, though usual reporter induction rates were above 95%. GFP expression in HEK-RT1 reporter cells was induced for 24-48 hr using doxycycline (1 μg/ml; Sigma-Aldrich), at the indicated days post-editing. Percentages of GFP-positive cells were quantified by flow cytometry (Attune NxT, Thermo Fisher Scientific), routinely acquiring 10,000-30,000 events per sample. When quantifying ProCas9 activation by mTagBFP2-tagged proteases, GFP fluorescence was quantified in mTagBFP2-positive cells. In all cases, editing efficiency was reported as the difference in percentage of GFP-positive cells between samples expressing a non-targeting guide (sgNT) and samples expressing the sgGFP9 guide targeting the GFP reporter.For ProCas9 GFP disruption assays following transfection of the tested components (Figure 3F), transfection-based plasmids were designed and cloned using standard molecular biology techniques to express either ProCas9-T2A-mCherry and a single guide RNA, or the protease of interest-P2A-mTagBFP2 (Table S1). Transient assays were performed as follows: in triplicate the reporter cell line HEK-RT1 was seeded at 20-30 thousand cells per well into 96-well plates and transfected using 0.5 uL of Lipofectamine 2000 (Thermo Fisher Scientific), 12.5 ng of the WT Cas9 or ProCas9 plasmid and 14 ng of the Protease plasmid (2x molar ratio), following the manufacturer's protocol. 24 hours later the media was changed and doxycycline was added to induce GFP expression. 48 hours following induction the cells were gated for mCherry (WT Cas9, ProCas9) expression and analyzed using flow cytometry for GFP depletion. At least 10,000 events were collected for each sample.
Mammalian flow cytometry and fluorescence microscopy
Flow cytometry (Attune Nxt Flow Cytometer, Thermo Fisher Scientific) was used to quantify the expression levels of fluorophores (mTagBFP2, GFP/EGFP, mCherry) as well as the percentage of transfected or transduced cells. For the HEK-RT1 genome editing reporter cell line, flow cytometry was used to quantify the percentage of GFP-negative (edited) cells, 24-48 hr after doxycycline (1 μg/mL) treatment to induce GFP expression. Phase contrast and fluorescence microscopy was carried out following standard procedures (EVOS FL Cell Imaging System, Thermo Fisher Scientific), routinely at least 48 hr post-transfection or post-transduction of target cells with fluorophore expressing constructs.
Mammalian immunoblotting
HEK293T (293FT; Thermo Fisher Scientific) were co-transfected with the indicated plasmids expressing Cas9-wt or ProCas9-Flavi and plasmids expressing dTEV or WNV protease. HEK293T cells were split to reach a confluency of 70-90% at time of transfection. For transfections in 6-well plates, 1 μg Cas9-sgRNA vector and 0.75 μg protease vector (if applicable) were mixed in 0.4 ml Opti-MEM, followed by addition of 5.25 μg polyethylenimine (PEI; Polysciences #23966). After 20-30 min incubation at room temperature, the transfection reactions were dispersed over the HEK293T cells. Media was changed 12 hr post-transfection. At 36 hr post-transfection, HEK293T were washed in ice-cold PBS and scraped from the plates. Cell pellets were lysed in Laemmli buffer (62.5 mM Tris-HCl pH 6.8, 10% glycerol, 2% SDS, 5% 2-mercaptoethanol). Equal amounts of protein were separated on 4-20% Mini-PROTEAN TGX gels (Bio-Rad, #456-1095) and transferred to 0.2 μm PVDF membranes (Bio-Rad, #162-0177). Blots were blocked in 5% milk in TBST 0.1% (TBS + 0.01% Tween 20) for 1 hr; all antibodies were incubated in 5% milk in TBST 0.1% at 4°C overnight; blots were washed in TBST 0.1%. The abundance of β-actin (ACTB) was monitored to ensure equal loading (Figure S6B). Immunoblotting was performed using the antibodies: mouse monoclonal Anti-Flag-M2 (Sigma-Aldrich, #1804, clone M2, 1:500; https://www.sigmaaldrich.com/content/dam/sigma-aldrich/docs/Sigma/Bulletin/f1804bul.pdf), mouse monoclonal C-Cas9 Anti-SpyCas9 (Sigma-Aldrich, #SAB4200751, clone 10C11-A12, 1:500; https://www.sigmaaldrich.com/content/dam/sigma-aldrich/docs/Sigma/Datasheet/10/sab4200751dat.pdf), mouse monoclonal N-Cas9 Anti-SpyCas9 (Novus Biologicals, #NBP2-36440, clone 7A9-3A3, 1:500; https://www.novusbio.com/PDFs2/NBP2-36440.pdf), HRP-conjugated mouse monoclonal Anti-Beta-Actin (Santa Cruz Biotechnology, #sc-47778 HRP, clone C4, 1:250; https://datasheets.scbt.com/sc-47778.pdf), and HRP-conjugated sheep Anti-Mouse (GE Healthcare Amersham ECL, #NXA931; 1:5000; https://es.vwr.com/assetsvc/asset/es_ES/id/9458958/contents). Blots were exposed using Amersham ECL Western Blotting Detection Reagent (GE Healthcare Amersham ECL, #RPN2209) and imaged using a ChemiDoc MP imaging system (Bio-Rad). Protein ladders were used as molecular weight reference (Bio-Rad, #161-0374).
Mammalian competitive proliferation assay
For assessment of CRISPR-Cas programmed cell depletion using guide RNAs targeting an essential gene (RPA1) or sgCIDEs targeting hundreds of thousands of loci within the genome, cells were stably transduced with a lentiviral vector expressing Cas9-wt (pCF226) or ProCas9Flavi (pCF730), and selected on puromycin. Subsequently, these cell lines were further stably transduced with vectors expressing various mCherry-tagged sgRNAs and analyzed as follows: 1) After mixing sgRNA expressing populations with parental cells, the fraction of mCherry-positive cells was quantified over time. Different sgRNAs targeting a neutral gene (sgOR2B6), an essential gene (sgRPA1), >100,000 genomic loci (sgCIDE) and a non-targeting control (sgNT) were compared. 2) Alternatively, the cell lines were partially transduced with lentiviral vectors expressing a GFP-tagged dTEV (pCF736) or WNV (pCF738) protease, and cell depletion quantified by flow cytometry. We quantified depletion of protease-expressing (GFP+) cells among the sgRNA-positive (mCherry+) population.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis
Specific statistical tests used are indicated in all cases. Propagation of uncertainty was taken into consideration when reporting data and their uncertainty (standard deviation) as functions of measurement variables. Unless otherwise noted, error bars indicate the standard deviation of triplicates, and significance was assessed by comparing samples to their respective controls using unpaired, two-tailed t-tests (alpha = 0.05). Genome editing quantification using TIDE was carried out as recommended (Brinkman et al., 2014). In brief, indels ranging from −10 to +10 nucleotides were quantified. Parental cells were used as reference for normalization. When reporting TIDE editing efficiencies, only indels with p-values < 0.01 in at least one replicate were considered true.
DATA AND SOFTWARE AVAILABILITY
To identify functional Cas9 circular permutants (Cas9-CPs), fold-changes for each dCas9-CP between pre- and post-library sorts along with significance values for each enrichment were calculated (Table S2). Cas9-CP analysis scripts are available at https://github.com/SavageLab/Cas9-CP. All relevant sequencing data have been deposited in the National Institutes of Health (NIH) Sequencing Read Archive (SRA) at https://www.ncbi.nlm.nih.gov/bioproject/PRJNA505363 under ID code 505363, Accession code PRJNA505363.
Supplementary Material
Supplemental Table S1. Related to Figure 1-5. Prokaryotic and eukaryotic vectors.
Name, key features, length and sequence of prokaryotic and eukaryotic vectors used.
Supplemental Table S2. Related to Figure 1. Cas9 circular permutation screen.
Deep sequencing analysis of Cas9 circular permutation library screen.
Supplemental Figure S1. Related to Figure 1. Cas9 circular permutation.
(A) Detailed schematic of the transposition method used to build the Cas9-CP libraries, REs = Restriction Enzyme sites.
(B) Schematic and uncropped gel of the PCR system used to validate the creation of CP libraries.
(C) Schematic and flow cytometry from the screen and enrichment of active Cas9-CPs in all four Cas9-CP libraries.
(D) Endpoint values for 13 new Cas9-CPs in a 12 hr E. coli CRISPRi DNA binding and RFP repression system compared with WT dCas9 and a protein expression vector control (n = 3).
(E) Alternate view of the model of new Cas9-CP termini (in red) based on PDB ID:5F9R. The HNH domain has been removed to clearly demonstrate the new termini flanking either side of the non-targeting (nt) DNA strand. Inset highlights distances between various new Cas9-CP termini and R-loop.
(F) Deep sequencing analysis and log2-fold change for new termini in the 20 AA library as mapped onto the primary sequence of Cas9. Red bars indicate clusters of CPs in specific domains.
(G) Overlay of enrichment values for Domain Insertion (DI, Oakes et al., 2016) and CP, demonstrating clustering of events.
{kind=link}
Supplemental Figure S2. Related to Figure 1. Mammalian genome editing reporter cell lines.
(A) Flow cytometry time course of GFP fluorescence decay after editing. Monoclonal HEK-LMP-10 reporter cells stably expressing GFP were transfected with a vector (pX459) expressing wild-type Cas9 and the indicated sgRNAs targeting the reporter, or a negative control (sgNT). Note, full fluorescence decay after transfection of editing reagents took up to eight days.
(B) Schematic showing the concept of a rapid mammalian genome editing reporter assay. Monoclonal reporter cell lines were established by stably integrating and all-in-one Tet-On cassette enabling doxycycline-inducible GFP expression, followed by selection and characterization of single clones. To assess editing efficiency of novel variants, reporter cells are transduced with Cas constructs of interest and guide RNAs targeting GFP, or a non-targeting control. At 24+ hours post-transduction, the GFP fluorescence reporter is induced by doxycycline treatment for 24-48 hr and genome editing quantified by flow cytometry.
(C) Activation curves (doxycycline titration) of two monoclonal genome editing reporter cell lines. HEK-RT1 and HEK-RT6 reporter cell lines were treated with the indicated doxycycline concentrations for 48 hours. The median GFP fluorescence intensity was quantified by flow cytometry and normalized to parental HEK293T cells. Both cell lines show full reporter induction at 1000-2000 ng/ml doxycycline and similar EC50 values (HEK-RT1: 214.5 +/− 2.3 ng/ml; HEK-RT6: 433.0 +/− 9.5 ng/ml).
{kind=link}
Supplemental Figure S3. Related to Figure 2. CP linker length and activation.
(A) Schematic of the PCR system and uncropped gel of the PCRs for each library, in biological replicate, pre and post sorting.
(B) Fold changes of the TEV based activation of CP-TEV linker clones from Figure 2C.
(C) Time course values from the CRISPRi assay in Figure 2C, demonstrating constancy of activity for clones with TEV (blue) vs dTEV (grey).
(D) Single cell analysis of Cas9-CP-TEV linkers.
(E) Endpoint analysis of an E. coli CRISPRi based GFP expression assay with all six Cas9-CPs containing a 8 AA 3C linker (LEVLFQ/GP) in the presence of a functional 3C protease (3C pro, green) or a deactivated TEV protease with a catalytic triad mutant C151A (dProtease, gray).
{kind=link}
Supplemental Figure S4. Related to Figure 3. ProCas9 specificity assessment.
(A) Endpoint analysis of an E. coli CRISPRi based GFP expression assay with negative and positive controls in the presence of all NIa proteases to determine if any protease changes the GFP expression levels.
(B) Endpoint analysis of an E. coli CRISPRi based GFP expression assay for each Cas9-CP-Potyviral linker against its respective protease. Significance was assessed by comparing each sample to its respective dProtease control (unpaired, two-tailed t-test, n = 3, * = p<0.05, ns = not significant).
(C) Endpoint analysis of an E. coli CRISPRi based GFP expression assay with negative and positive controls in the presence of all Flavirus NS2B-NS3 proteases to determine if any protease changes the GFP expression levels.
(D) Endpoint analysis of an E. coli CRISPRi based GFP expression assay for each Cas9-CP-Flaviviral linker against its respective protease. Significance was assessed by comparing each sample to its respective dProtease control (unpaired, two-tailed t-test, n = 3, * = p<0.05, ns = not significant).
(E) Raw Flow cytometry plots from Figure 3F demonstrating the always on nature of WT Cas9 and the activation of ProCas9Flavi in the presence of Flavivirus proteases.
{kind=link}
Supplemental Figure S5. Related to Figure 4. ProCas9 activation by Flavivirus proteases.
(A) Fluorescence analysis of the indicated HEK-RT1 based cell lines stably expressing a ProCas9 variant and an sgRNA targeting the reporter (sgGFP9) or a non-targeting control (sgNT). All cell lines were either non-transfected or transfected with vectors expressing the dTEV (pCF708), ZIKV (pCF709) or WNV (pCF710) protease. The percentage mTagBFP2+ cells was measured three days post-transfection along with the median fluorescence intensity (MFI) of the mTagBFP2+ cells. AU, arbitrary units. Error bars indicate the standard deviation of triplicates.
(B) Activation of Flavivirus ProCas9 by transfection of various proteases. ProCas9 cell lines were transiently transfected to express the indicated mTagBFP2-tagged viral proteases. At day 2 post-transfection, cells were treated with doxycycline for 24 hr to induce GFP reporter expression. GFP fluorescence was quantified in mTagBFP2-positive cells, for samples expressing either a non-targeting guide (sgNT) or sgGFP9 targeting the reporter. Editing efficiency is reported as the normalized difference between the two in each case. Error bars indicate the standard deviation of triplicates. Significance was assessed by comparing each sample to its respective dTEV control (unpaired, two-tailed t-test, n = 3, * = p<0.05, ns = not significant).
(C) Fluorescence imaging of mTagBFP2 in HEK293T cells 36 hr after transfection of the indicated lentiviral plasmids expressing viral proteases. Lentiviral helper plasmids were co-transfected in each case. Scale bar: 400 μm.
(D) Fluorescence analysis of the indicated HEK-RT1-ProCas9 reporter cell lines expressing an sgRNA targeting the reporter (sgGFP9) or a non-targeting control (sgNT). All cell lines were either non-transduced or stably transduced with vectors expressing the dTEV (pCF708), ZIKV (pCF709) or WNV (pCF710) protease. The percentage mTagBFP2+ cells was quantified four days post-transduction along with the median fluorescence intensity (MFI) of the mTagBFP2+ cells. AU, arbitrary units. Error bars indicate the standard deviation of triplicates.
(E) Schematic vector maps.
(F) Activity comparison of Flavivirus ProCas9 constructs with and without nuclear localization sequences (NLSs). Genome editing efficiency was assessed in the indicated HEK-RT1-ProCas9 reporter cell lines at day 4 post-transduction of the indicated proteases, followed by 24 hr of GFP reporter induction. Error bars indicate the standard deviation of triplicates. Significance was assessed by comparing each sample to its respective dTEV control (unpaired, two-tailed t-test, n = 3, * = p<0.05, ns = not significant).
(G) T7E1 assay of samples shown in (F). Note that while the flow cytometry-based editing quantification was based on cells expressing the respective proteases (mTagBFP2+), the T7E1 assay is based on the total population of cells.
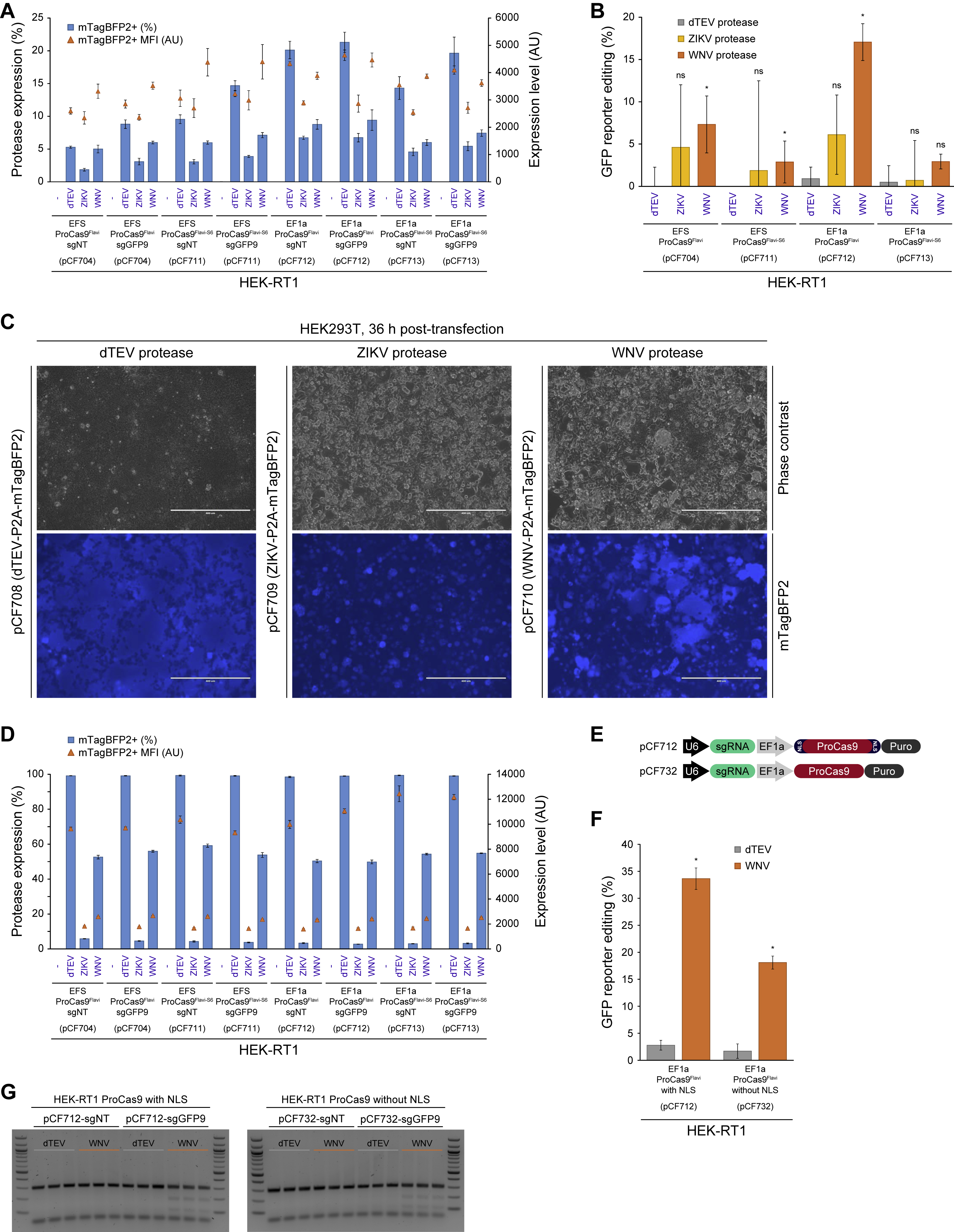{kind=link}
Supplemental Figure S6. Related to Figure 4. Mechanism of ProCas9 activation.
(A) Phase contrast and fluorescence imaging in HEK293T cells 36 hr after co-transfection of the indicated plasmids expressing Cas9-wt (pCF204-sgGFP9) or ProCas9Flavi (pBLO43.3-sgGFP9) and plasmids expressing the dTEV (pCF783) or WNV (pCF785) proteases. Scale bars: 400 μm.
(B) Immunoblotting for Cas9 in HEK293T co-transfected with the indicated plasmids expressing Cas9- wt or ProCas9Flavi (including sgGFP9) and plasmids expressing the dTEV or WNV proteases. The N-Cas9 (clone 7A9-3A3) antibody recognize the large subunit of the activated ProCas9Flavi (**, 137 kDa). Beta-actin (ACTB, 42 kDa) was used as loading control. Protein ladders indicate reference molecular weight markers.
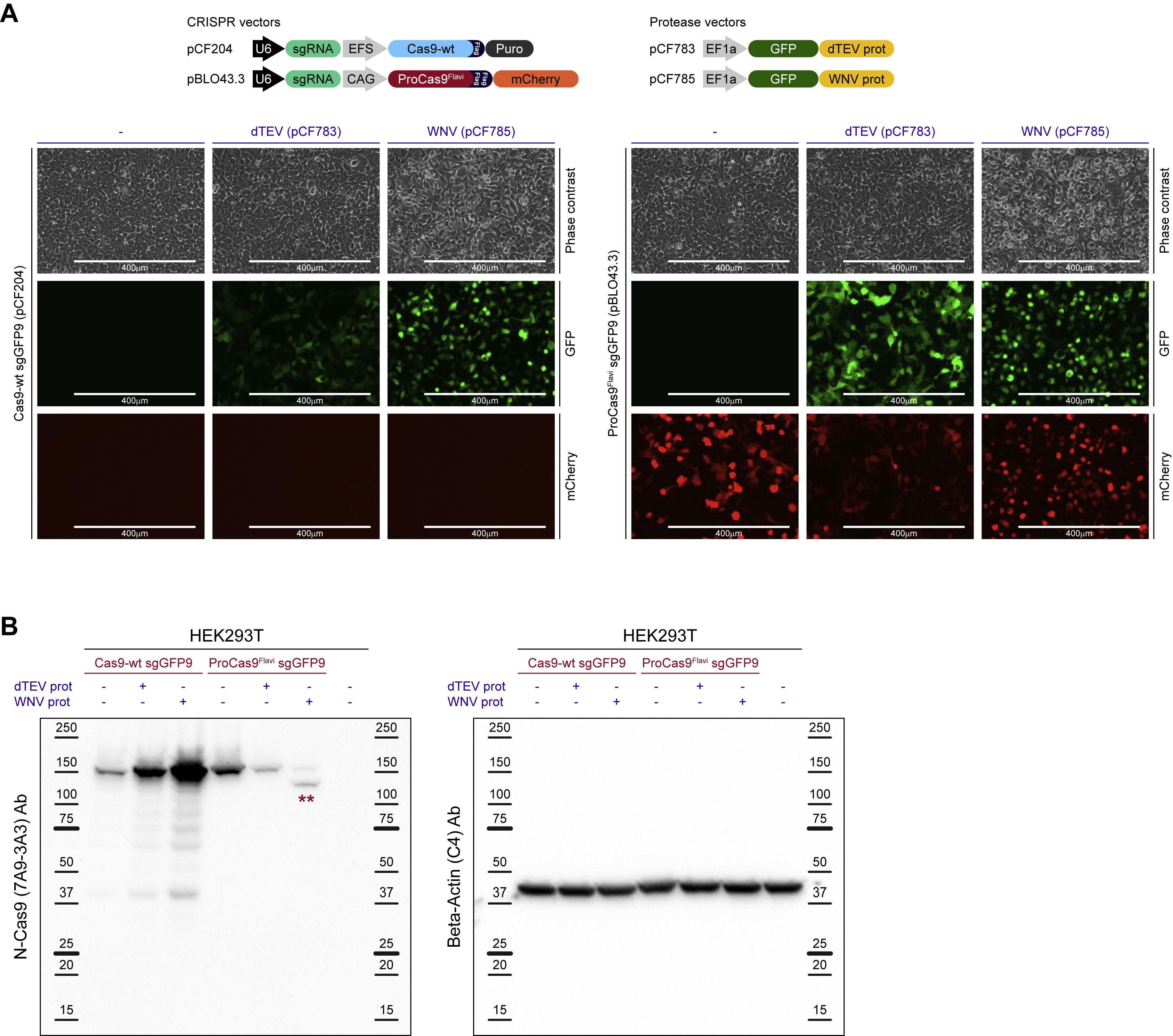{kind=link}
Supplemental Figure S7. Related to Figure 5. ProCas9-based altruistic defense systems.
(A) Transfection of protease expression vectors in virus packaging cell lines. GFP fluorescence imaging in HEK293T cells 42 hr after transfection of the indicated lentiviral plasmids expressing viral proteases. Lentiviral helper plasmids were co-transfected in each case. Scale bar: 400 μm.
(B) Competitive proliferation assay in HAP1 ProCas9Flavi (pCF730) cell lines expressing the indicated mCherry-tagged controls (sgOR2B6-1, sgOR2B6-2) or guide RNAs targeting highly repetitive sequences (sgCIDE-2, sgCIDE-4), or a non-targeting control (sgNT) used for normalization. The cell lines were partially transduced with lentiviral vectors expressing a GFP-tagged dTEV (pCF736) or WNV (pCF738) protease, and cell depletion quantified by flow cytometry. Shown is the normalized (sgRNA/sgNT) depletion of protease-expressing (GFP+) cells among the sgRNA-positive (mCherry+) population. Error bars indicate the standard deviation of triplicates. Significance was assessed by comparing each sample to its respective dTEV control (unpaired, two-tailed t-test, n = 3, * = p<0.05, ns = not significant).
{kind=link}
Highlights.
Cas9 can be circularly permuted (Cas9-CP) for optimized fusion protein construction
Cas9 circular permutation enables the engineering of protease-activated ProCas9s
ProCas9s can sense and respond to protease activity, such as during viral infection
Cas9-CPs provide a toolkit of safer and more efficient genome modifying enzymes
ACKNOWLEDGMENTS
B.L.O. and K.L.T. are supported by the Innovative Genomic Institute Entrepreneurial fellowship program. C.F. is supported by a US National Institutes of Health K99/R00 Pathway to Independence Award (K99GM118909, R00GM118909) from NIGMS. R.Y. is a Simons Foundation Fellow of the LSRF. J.A.D. is an Investigator of the Howard Hughes Medical Institute (HHMI), and this study was supported in part by HHMI. D.F.S. is supported by a US National Institutes of Health New Innovator Award (1DP2EB018658-01) from the NIBIB. This work used the Vincent J. Coates Genomics Sequencing Laboratory at UC Berkeley, supported by NIH S10 Instrumentation Grants S10RR029668 and S10RR027303. We thank Mary West and the CIRM/QB3 Shared Stem Cell Facility / High-Throughput Screening Facility for technical support as well as Timothy Brown (Thermo Fisher Scientific) for flow cytometry support. We thank Alexendar Reinaldo Perez for help with using the GuideScan software. We thank Jan Carette for providing HAP1 cells.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
SUPPLEMENTAL INFORMATION
Supplemental Information includes seven figures and two tables and can be found with this article online.
DECLARATION OF INTERESTS
UC Regents have filed a patent related to this work on which B.L.O., J.A.D. and D.F.S. are inventors. B.L.O. is a co-founder and employee of Scribe Therapeutics. C.F. is a co-founder of Mirimus, Inc. K.L.T. is an employee of Scribe Therapeutics. J.A.D. is a co-founder of Caribou Biosciences, Editas Medicine, Intellia Therapeutics, Scribe Therapeutics, and Mammoth Biosciences. J.A.D. is a scientific advisory board member of Caribous Biosciences, Intellia Therapeutics, eFFECTOR Therapeutics, Scribe Therapeutics, Synthego, Metagenomi, Mammoth Biosciences and Inari. J.A.D is a member of the board of directors at Driver and Johnson & Johnson. D.F.S. is a co-founder of Scribe Therapeutics and a scientific advisory board member of Scribe Therapeutics and Mammoth Biosciences. All other authors declare no competing interests.
REFERENCES
- Ade J, DeYoung BJ, Golstein C, and Innes RW (2007). Indirect activation of a plant nucleotide binding site-leucine-rich repeat protein by a bacterial protease. Proc. Natl. Acad. Sci. U. S. A 104, 2531–2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfano JR, and Collmer A (2004). Type III secretion system effector proteins: double agents in bacterial disease and plant defense. Annu. Rev. Phytopathol 42, 385–414. [DOI] [PubMed] [Google Scholar]
- Anders S, and Huber W (2010). Differential expression analysis for sequence count data. Genome Biol. 11, R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders C, Niewoehner O, Duerst A, and Jinek M (2014). Structural basis of PAM-dependent target DNA recognition by the Cas9 endonuclease. Nature 513, 569–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baltes NJ, Hummel AW, Konecna E, Cegan R, Bruns AN, Bisaro DM, and Voytas DF (2015). Conferring resistance to geminiviruses with the CRISPR-Cas prokaryotic immune system. Nature Plants 1, 15145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beernink PT, Yang YR, Graf R, King DS, Shah SS, and Schachman HK (2001). Random circular permutation leading to chain disruption within and near alpha helices in the catalytic chains of aspartate transcarbamoylase: effects on assembly, stability, and function. Protein Sci. 10, 528–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bera AK, Kuhn RJ, and Smith JL (2007). Functional Characterization of cis and trans Activity of the Flavivirus NS2B-NS3 Protease. J. Biol. Chem 282, 12883–12892. [DOI] [PubMed] [Google Scholar]
- Brinkman EK, Chen T, Amendola M, and van Steensel B (2014). Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 42, e168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler JS, Mitrea DM, Mitrousis G, Cingolani G, and Loh SN (2009). Structural and Thermodynamic Analysis of a Conformationally Strained Circular Permutant of Barnase. Biochemistry 48, 3497–3507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carette JE, Raaben M, Wong AC, Herbert AS, Obernosterer G, Mulherkar N, Kuehne AI, Kranzusch PJ, Griffin AM, Ruthel G, et al. (2011). Ebola virus entry requires the cholesterol transporter Niemann-Pick C1. Nature 477, 340–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaparro-Garcia A, Kamoun S, and Nekrasov V (2015). Boosting plant immunity with CRISPR/Cas. Genome Biol. 16, 254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavez A, Scheiman J, Vora S, Pruitt BW, Tuttle M, P R Iyer E, Lin S, Kiani S, Guzman CD, Wiegand DJ, et al. (2015). Highly efficient Cas9-mediated transcriptional programming. Nat. Methods 12, 326–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B, Gilbert LA, Cimini BA, Schnitzbauer J, Zhang W, Li G-W, Park J, Blackburn EH, Weissman JS, Qi LS, et al. (2013). Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system. Cell 155, 1479–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chisholm ST, Dahlbeck D, Krishnamurthy N, Day B, Sjolander K, and Staskawicz BJ (2005). Molecular characterization of proteolytic cleavage sites of the Pseudomonas syringae effector AvrRpt2. Proc. Natl. Acad. Sci. U. S. A 102, 2087–2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, et al. (2013). Multiplex Genome Engineering Using CRISPR/Cas System. Science 339, 819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coradetti ST, Pinel D, Geiselman GM, Ito M, Mondo SJ, Reilly MC, Cheng Y-F, Bauer S, Grigoriev IV, Gladden JM, et al. (2018). Functional genomics of lipid metabolism in the oleaginous yeast Rhodosporidium toruloides. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis KM, Pattanayak V, Thompson DB, Zuris J. a., and Liu DR (2015). Small molecule–triggered Cas9 protein with improved genome-editing specificity. Nat. Chem. Biol 11, 316–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fellmann C, Hoffmann T, Sridhar V, Hopfgartner B, Muhar M, Roth M, Lai DY, Barbosa IAM, Kwon JS, Guan Y, et al. (2013). An optimized microRNA backbone for effective single-copy RNAi. Cell Rep. 5, 1704–1713. [DOI] [PubMed] [Google Scholar]
- Fellmann C, Gowen BG, Lin P-C, Doudna JA, and Corn JE (2016). Cornerstones of CRISPR–Cas in drug discovery and therapy. Nat. Rev. Drug Discov 16, 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao M, Matusick-Kumar L, Hurlburt W, DiTusa SF, Newcomb WW, Brown JC, McCann PJ, Deckman I, and Colonno RJ (1994). The protease of herpes simplex virus type 1 is essential for functional capsid formation and viral growth. J. Virol 68, 3702–3712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaudelli NM, Komor AC, Rees HA, Packer MS, Badran AH, Bryson DI, and Liu DR (2017). Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature 551, 464–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert LA, Horlbeck MA, Adamson B, Villalta JE, Chen Y, Whitehead EH, Guimaraes C, Panning B, Ploegh HL, Bassik MC, et al. (2014). Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation. Cell 159, 647–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilinger JP, Thompson DB, and Liu DR (2014). Fusion of catalytically inactive Cas9 to FokI nuclease improves the specificity of genome modification. Nat. Biotechnol 32, 577–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann S, and Lucius R (2003). Modulation of host immune responses by nematode cystatins. Int. J. Parasitol 33, 1291–1302. [DOI] [PubMed] [Google Scholar]
- Hemphill J, Borchardt EK, Brown K, Asokan A, and Deiters A (2015). Optical Control of CRISPR/Cas9 Gene Editing. J. Am. Chem. Soc 137, 5642–5645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilton IB, D’Ippolito AM, Vockley CM, Thakore PI, Crawford GE, Reddy TE, and Gersbach CA (2015). Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat. Biotechnol 33, 510–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, and Charpentier E (2012). A Programmable Dual-RNA–Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science 337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, East A, Cheng A, Lin S, Ma E, and Doudna J (2013). RNA-programmed genome editing in human cells. Elife 2, e00471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson RJ, Lin SR, and Raines RT (2006). A ribonuclease zymogen activated by the NS3 protease of the hepatitis C virus. FEBS J. 273, 5457–5465. [DOI] [PubMed] [Google Scholar]
- Jones AM, Mehta MM, Thomas EE, Atkinson JT, Segall-Shapiro TH, Liu S, and Silberg JJ (2016). The Structure of a Thermophilic Kinase Shapes Fitness upon Random Circular Permutation. ACS Synth. Biol 5, 415–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy EM, Kornepati AVR, Goldstein M, Bogerd HP, Poling BC, Whisnant AW, Kastan MB, and Cullen BR (2014). Inactivation of the human papillomavirus E6 or E7 gene in cervical carcinoma cells by using a bacterial CRISPR/Cas RNA-guided endonuclease. J. Virol 88, 11965–11972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K, Park SW, Kim JH, Lee SH, Kim D, Koo T, Kim K-E, Kim JH, and Kim J-S (2017). Genome surgery using Cas9 ribonucleoproteins for the treatment of age-related macular degeneration. Genome Res. 27, 419–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SH, Qi D, Ashfield T, Helm M, and Innes RW (2016). Using decoys to expand the recognition specificity of a plant disease resistance protein. Science 351, 684–687. [DOI] [PubMed] [Google Scholar]
- Komor AC, Kim YB, Packer MS, Zuris JA, and Liu DR (2016). Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature advance on. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kümmerer BM, Amberg SM, and Rice CM (2013). Chapter 687 - Flavivirin In Handbook of Proteolytic Enzymes, Rawlings ND, and Salvesen G, eds. (Academic Press; ), pp. 3112–3120. [Google Scholar]
- Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, and Church GM (2013). RNA-Guided Human Genome Engineering via Cas9 Prashant. Science 339, 823–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta D, Stürchler A, Hirsch-Hoffmann M, Gruissem W, and Vanderschuren H (2018). CRISPR-Cas9 interference in cassava linked to the evolution of editing-resistant geminiviruses. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta MM, Liu S, and Silberg JJ (2012). A transposase strategy for creating libraries of circularly permuted proteins. Nucleic Acids Res. 40, e71–e71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakes BL, Nadler DC, and Savage DF (2014). Protein Engineering of Cas9 for Enhanced Function. Methods Enzymol. 546C, 491–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakes BL, Nadler DC, Flamholz A, Fellmann C, Staahl BT, Doudna JA, and Savage DF (2016). Profiling of engineering hotspots identifies an allosteric CRISPR-Cas9 switch. Nat. Biotechnol 34, 646–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park HM, Liu H, Wu J, Chong A, Mackley V, Fellmann C, Rao A, Jiang F, Chu H, Murthy N, et al. (2018). Extension of the crRNA enhances Cpf1 gene editing in vitro and in vivo. Nat. Commun 9, 3313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattanayak V, Lin S, Guilinger JP, Ma E, Doudna JA, and Liu DR (2013). High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nat. Biotechnol 31, 839–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez AR, Pritykin Y, Vidigal JA, Chhangawala S, Zamparo L, Leslie CS, and Ventura A (2017). GuideScan software for improved single and paired CRISPR guide RNA design. Nat. Biotechnol 35, 347–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plainkum P, Fuchs SM, Wiyakrutta S, and Raines RT (2003). Creation of a zymogen. Nat. Struct. Biol 10, 115–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi LS, Larson MH, Gilbert L. a., Doudna J. a., Weissman JS, Arkin AP, and Lim W. a. (2013). Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 152, 1173–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian Z, and Lutz S (2005). Improving the catalytic activity of Candida antarctica lipase B by circular permutation. J. Am. Chem. Soc 127, 13466–13467. [DOI] [PubMed] [Google Scholar]
- Ramanathan MP, Chambers JA, Pankhong P, Chattergoon M, Attatippaholkun W, Dang K, Shah N, and Weiner DB (2006). Host cell killing by the West Nile Virus NS2B-NS3 proteolytic complex: NS3 alone is sufficient to recruit caspase-8-based apoptotic pathway. Virology 345, 56–72. [DOI] [PubMed] [Google Scholar]
- Richter F, Fonfara I, Gelfert R, Nack J, Charpentier E, and Möglich A (2017). Switchable Cas9. Curr. Opin. Biotechnol 48, 119–126. [DOI] [PubMed] [Google Scholar]
- Roybal KT, Rupp LJ, Morsut L, Walker WJ, McNally KA, Park JS, and Lim WA (2016). Precision Tumor Recognition by T Cells With Combinatorial Antigen-Sensing Circuits. Cell 164, 770–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjana NE, Shalem O, and Zhang F (2014). Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seon Han J, Kim D-H, and Yong Choi K (2013). Chapter 544 - Potyvirus NIa Protease In Handbook of Proteolytic Enzymes, Rawlings ND, and Salvesen G, eds. (Academic Press; ), pp. 2427–2432. [Google Scholar]
- Skern T (2013). Chapter 537 - Picornain 3C In Handbook of Proteolytic Enzymes, Rawlings ND, and Salvesen G, eds. (Academic Press; ), pp. 2396–2402. [Google Scholar]
- Staahl BT, Benekareddy M, Coulon-Bainier C, Banfal AA, Floor SN, Sabo JK, Urnes C, Munares GA, Ghosh A, and Doudna JA (2017). Efficient genome editing in the mouse brain by local delivery of engineered Cas9 ribonucleoprotein complexes. Nat. Biotechnol 35, 431–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanenbaum ME, Gilbert LA, Qi LS, Weissman JS, and Vale RD (2014). A Protein-Tagging System for Signal Amplification in Gene Expression and Fluorescence Imaging. Cell 159, 635–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomlinson KR, Bailey AM, Alicai T, Seal S, and Foster GD (2018). Cassava brown streak disease: historical timeline, current knowledge and future prospects. Mol. Plant Pathol 19, 1282–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai SQ, Wyvekens N, Khayter C, Foden JA, Thapar V, Reyon D, Goodwin MJ, Aryee MJ, and Joung JK (2014a). Dimeric CRISPR RNA-guided FokI nucleases for highly specific genome editing. Nat. Biotechnol 32, 569–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai SQ, Zheng Z, Nguyen NT, Liebers M, Topkar VV, Thapar V, Wyvekens N, Khayter C, Iafrate a. J., Le LP, et al. (2014b). GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat. Biotechnol 33, 187–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Birsoy K, Hughes NW, Krupczak KM, Post Y, Wei JJ, Lander ES, and Sabatini DM (2015). Identification and characterization of essential genes in the human genome. Science 350, 1096–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitehead TA, Bergeron LM, and Clark DS (2009). Tying up the loose ends: circular permutation decreases the proteolytic susceptibility of recombinant proteins. Protein Eng. Des. Sel 22, 607–613. [DOI] [PubMed] [Google Scholar]
- Yu Y, and Lutz S (2011). Circular permutation: a different way to engineer enzyme structure and function. Trends Biotechnol. 29, 18–25. [DOI] [PubMed] [Google Scholar]
- Zuris JA, Thompson DB, Shu Y, Guilinger JP, Bessen JL, Hu JH, Maeder ML, Joung JK, Chen Z-Y, and Liu DR (2015). Cationic lipid-mediated delivery of proteins enables efficient protein-based genome editing in vitro and in vivo. Nat. Biotechnol 33, 73–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table S1. Related to Figure 1-5. Prokaryotic and eukaryotic vectors.
Name, key features, length and sequence of prokaryotic and eukaryotic vectors used.
Supplemental Table S2. Related to Figure 1. Cas9 circular permutation screen.
Deep sequencing analysis of Cas9 circular permutation library screen.
Supplemental Figure S1. Related to Figure 1. Cas9 circular permutation.
(A) Detailed schematic of the transposition method used to build the Cas9-CP libraries, REs = Restriction Enzyme sites.
(B) Schematic and uncropped gel of the PCR system used to validate the creation of CP libraries.
(C) Schematic and flow cytometry from the screen and enrichment of active Cas9-CPs in all four Cas9-CP libraries.
(D) Endpoint values for 13 new Cas9-CPs in a 12 hr E. coli CRISPRi DNA binding and RFP repression system compared with WT dCas9 and a protein expression vector control (n = 3).
(E) Alternate view of the model of new Cas9-CP termini (in red) based on PDB ID:5F9R. The HNH domain has been removed to clearly demonstrate the new termini flanking either side of the non-targeting (nt) DNA strand. Inset highlights distances between various new Cas9-CP termini and R-loop.
(F) Deep sequencing analysis and log2-fold change for new termini in the 20 AA library as mapped onto the primary sequence of Cas9. Red bars indicate clusters of CPs in specific domains.
(G) Overlay of enrichment values for Domain Insertion (DI, Oakes et al., 2016) and CP, demonstrating clustering of events.
Supplemental Figure S2. Related to Figure 1. Mammalian genome editing reporter cell lines.
(A) Flow cytometry time course of GFP fluorescence decay after editing. Monoclonal HEK-LMP-10 reporter cells stably expressing GFP were transfected with a vector (pX459) expressing wild-type Cas9 and the indicated sgRNAs targeting the reporter, or a negative control (sgNT). Note, full fluorescence decay after transfection of editing reagents took up to eight days.
(B) Schematic showing the concept of a rapid mammalian genome editing reporter assay. Monoclonal reporter cell lines were established by stably integrating and all-in-one Tet-On cassette enabling doxycycline-inducible GFP expression, followed by selection and characterization of single clones. To assess editing efficiency of novel variants, reporter cells are transduced with Cas constructs of interest and guide RNAs targeting GFP, or a non-targeting control. At 24+ hours post-transduction, the GFP fluorescence reporter is induced by doxycycline treatment for 24-48 hr and genome editing quantified by flow cytometry.
(C) Activation curves (doxycycline titration) of two monoclonal genome editing reporter cell lines. HEK-RT1 and HEK-RT6 reporter cell lines were treated with the indicated doxycycline concentrations for 48 hours. The median GFP fluorescence intensity was quantified by flow cytometry and normalized to parental HEK293T cells. Both cell lines show full reporter induction at 1000-2000 ng/ml doxycycline and similar EC50 values (HEK-RT1: 214.5 +/− 2.3 ng/ml; HEK-RT6: 433.0 +/− 9.5 ng/ml).
Supplemental Figure S3. Related to Figure 2. CP linker length and activation.
(A) Schematic of the PCR system and uncropped gel of the PCRs for each library, in biological replicate, pre and post sorting.
(B) Fold changes of the TEV based activation of CP-TEV linker clones from Figure 2C.
(C) Time course values from the CRISPRi assay in Figure 2C, demonstrating constancy of activity for clones with TEV (blue) vs dTEV (grey).
(D) Single cell analysis of Cas9-CP-TEV linkers.
(E) Endpoint analysis of an E. coli CRISPRi based GFP expression assay with all six Cas9-CPs containing a 8 AA 3C linker (LEVLFQ/GP) in the presence of a functional 3C protease (3C pro, green) or a deactivated TEV protease with a catalytic triad mutant C151A (dProtease, gray).
Supplemental Figure S4. Related to Figure 3. ProCas9 specificity assessment.
(A) Endpoint analysis of an E. coli CRISPRi based GFP expression assay with negative and positive controls in the presence of all NIa proteases to determine if any protease changes the GFP expression levels.
(B) Endpoint analysis of an E. coli CRISPRi based GFP expression assay for each Cas9-CP-Potyviral linker against its respective protease. Significance was assessed by comparing each sample to its respective dProtease control (unpaired, two-tailed t-test, n = 3, * = p<0.05, ns = not significant).
(C) Endpoint analysis of an E. coli CRISPRi based GFP expression assay with negative and positive controls in the presence of all Flavirus NS2B-NS3 proteases to determine if any protease changes the GFP expression levels.
(D) Endpoint analysis of an E. coli CRISPRi based GFP expression assay for each Cas9-CP-Flaviviral linker against its respective protease. Significance was assessed by comparing each sample to its respective dProtease control (unpaired, two-tailed t-test, n = 3, * = p<0.05, ns = not significant).
(E) Raw Flow cytometry plots from Figure 3F demonstrating the always on nature of WT Cas9 and the activation of ProCas9Flavi in the presence of Flavivirus proteases.
Supplemental Figure S5. Related to Figure 4. ProCas9 activation by Flavivirus proteases.
(A) Fluorescence analysis of the indicated HEK-RT1 based cell lines stably expressing a ProCas9 variant and an sgRNA targeting the reporter (sgGFP9) or a non-targeting control (sgNT). All cell lines were either non-transfected or transfected with vectors expressing the dTEV (pCF708), ZIKV (pCF709) or WNV (pCF710) protease. The percentage mTagBFP2+ cells was measured three days post-transfection along with the median fluorescence intensity (MFI) of the mTagBFP2+ cells. AU, arbitrary units. Error bars indicate the standard deviation of triplicates.
(B) Activation of Flavivirus ProCas9 by transfection of various proteases. ProCas9 cell lines were transiently transfected to express the indicated mTagBFP2-tagged viral proteases. At day 2 post-transfection, cells were treated with doxycycline for 24 hr to induce GFP reporter expression. GFP fluorescence was quantified in mTagBFP2-positive cells, for samples expressing either a non-targeting guide (sgNT) or sgGFP9 targeting the reporter. Editing efficiency is reported as the normalized difference between the two in each case. Error bars indicate the standard deviation of triplicates. Significance was assessed by comparing each sample to its respective dTEV control (unpaired, two-tailed t-test, n = 3, * = p<0.05, ns = not significant).
(C) Fluorescence imaging of mTagBFP2 in HEK293T cells 36 hr after transfection of the indicated lentiviral plasmids expressing viral proteases. Lentiviral helper plasmids were co-transfected in each case. Scale bar: 400 μm.
(D) Fluorescence analysis of the indicated HEK-RT1-ProCas9 reporter cell lines expressing an sgRNA targeting the reporter (sgGFP9) or a non-targeting control (sgNT). All cell lines were either non-transduced or stably transduced with vectors expressing the dTEV (pCF708), ZIKV (pCF709) or WNV (pCF710) protease. The percentage mTagBFP2+ cells was quantified four days post-transduction along with the median fluorescence intensity (MFI) of the mTagBFP2+ cells. AU, arbitrary units. Error bars indicate the standard deviation of triplicates.
(E) Schematic vector maps.
(F) Activity comparison of Flavivirus ProCas9 constructs with and without nuclear localization sequences (NLSs). Genome editing efficiency was assessed in the indicated HEK-RT1-ProCas9 reporter cell lines at day 4 post-transduction of the indicated proteases, followed by 24 hr of GFP reporter induction. Error bars indicate the standard deviation of triplicates. Significance was assessed by comparing each sample to its respective dTEV control (unpaired, two-tailed t-test, n = 3, * = p<0.05, ns = not significant).
(G) T7E1 assay of samples shown in (F). Note that while the flow cytometry-based editing quantification was based on cells expressing the respective proteases (mTagBFP2+), the T7E1 assay is based on the total population of cells.
Supplemental Figure S6. Related to Figure 4. Mechanism of ProCas9 activation.
(A) Phase contrast and fluorescence imaging in HEK293T cells 36 hr after co-transfection of the indicated plasmids expressing Cas9-wt (pCF204-sgGFP9) or ProCas9Flavi (pBLO43.3-sgGFP9) and plasmids expressing the dTEV (pCF783) or WNV (pCF785) proteases. Scale bars: 400 μm.
(B) Immunoblotting for Cas9 in HEK293T co-transfected with the indicated plasmids expressing Cas9- wt or ProCas9Flavi (including sgGFP9) and plasmids expressing the dTEV or WNV proteases. The N-Cas9 (clone 7A9-3A3) antibody recognize the large subunit of the activated ProCas9Flavi (**, 137 kDa). Beta-actin (ACTB, 42 kDa) was used as loading control. Protein ladders indicate reference molecular weight markers.
Supplemental Figure S7. Related to Figure 5. ProCas9-based altruistic defense systems.
(A) Transfection of protease expression vectors in virus packaging cell lines. GFP fluorescence imaging in HEK293T cells 42 hr after transfection of the indicated lentiviral plasmids expressing viral proteases. Lentiviral helper plasmids were co-transfected in each case. Scale bar: 400 μm.
(B) Competitive proliferation assay in HAP1 ProCas9Flavi (pCF730) cell lines expressing the indicated mCherry-tagged controls (sgOR2B6-1, sgOR2B6-2) or guide RNAs targeting highly repetitive sequences (sgCIDE-2, sgCIDE-4), or a non-targeting control (sgNT) used for normalization. The cell lines were partially transduced with lentiviral vectors expressing a GFP-tagged dTEV (pCF736) or WNV (pCF738) protease, and cell depletion quantified by flow cytometry. Shown is the normalized (sgRNA/sgNT) depletion of protease-expressing (GFP+) cells among the sgRNA-positive (mCherry+) population. Error bars indicate the standard deviation of triplicates. Significance was assessed by comparing each sample to its respective dTEV control (unpaired, two-tailed t-test, n = 3, * = p<0.05, ns = not significant).
Data Availability Statement
To identify functional Cas9 circular permutants (Cas9-CPs), fold-changes for each dCas9-CP between pre- and post-library sorts along with significance values for each enrichment were calculated (Table S2). Cas9-CP analysis scripts are available at https://github.com/SavageLab/Cas9-CP. All relevant sequencing data have been deposited in the National Institutes of Health (NIH) Sequencing Read Archive (SRA) at https://www.ncbi.nlm.nih.gov/bioproject/PRJNA505363 under ID code 505363, Accession code PRJNA505363.