

Abstract
The serine/threonine kinase Akt plays a central role in cell proliferation, survival and metabolism and its hyperactivation is linked to cancer progression. Here we report that Akt undergoes K64 methylation by SETDB1, which is crucial for cell membrane recruitment, phosphorylation and activation of Akt upon growth factor stimulation. Furthermore, we reveal an adaptor function of histone demethylase JMJD2A, which recognizes Akt K64 methylation and recruits E3 ligase TRAF6 and Skp2-SCF to the Akt complex, independently of its demethylase activity, thereby initiating K63-linked ubiquitination, cell membrane recruitment and activation of Akt. Notably, cancer associated Akt mutant (E17K) displays enhanced K64 methylation, leading to its hyper-phosphorylation and activation. SETDB1-mediated Akt K64 methylation is upregulated and correlated with Akt hyperactivation in non-small-cell lung carcinoma (NSCLC), promotes tumor development and predicts poor outcome. Collectively, these findings reveal complicated layers of Akt activation regulation coordinated by SETDB1-mediated Akt K64 methylation to drive tumorigenesis.
Introduction
The Akt kinase serves as a central node for cell proliferation, survival and cell metabolism important for tumorigenesis1, 2. Recent studies reveal K63-linked ubiquitination of Akt as a critical event for cell membrane translocation, T308 phosphorylation and activation of Akt, apart from PI3K-mediated PIP3 production1, 3–5. TRAF6 and Skp2-SCF were identified as two E3 ubiquitin ligases mediating K63-linked ubiquitination and activation of Akt in response to growth factor insulin-like growth factor-1 (IGF-1) and epidermal growth factor (EGF), respectively3, 4. Growth factors trigger the association of E3 ligases with Akt, thereby promoting K63-linked ubiquitination of Akt3, 4. While K63-linked ubiquitination is required for Akt cell membrane recruitment and activation, it does not affect Akt-PIP3 binding3, 4. Thus, Akt-PIP3 binding and K63-linked ubiquitination appear to be two distinct and early events crucial for Akt membrane recruitment and activation. However, it remains unclear how growth factors trigger the interaction of Akt with its E3 ligase to elicit K63-linked ubiquitination.
Lysine methylation of non-histone proteins is involved in numerous molecular events including protein-protein interaction, protein stability, protein subcellular localization, and transcription6–11. While substantial number of the protein lysine methyltransferases (PKMTs) has been identified in human genome, only few non-histone proteins are known methylated by a limited number of PKMTs12, 13. Whether or not Akt methylation occurs and plays an important role in Akt signaling and tumorigenesis remains to be determined. In this study, we identified SETDB1 (also known as ESET or KMT1E) is an Akt interacting protein, which methylates Akt at K64 to elicit Akt ubiquitination, cell membrane recruitment, phosphorylation and activation upon stimulation with growth factors. We demonstrated that SETDB1-mediated K64 methylation of Akt serves as a scaffold to recruit histone demethylase JMJD2A (also known as KDM4A), which then brings Akt’s E3 ligases such as TRAF6 and Skp2-SCF to the Akt complex, thereby promoting Akt K63-linked ubiquitination, cell membrane recruitment and activation as well as tumorigenesis. Our study therefore identifies SETDB1-mediated Akt K64 methylation as an essential step for K63-linked ubiquitination and activation of Akt in response to stimulation with growth factors.
Results
SETDB1 interacts with Akt and is required for Akt activation.
To better understand regulatory modes for Akt phosphorylation and activation, we conducted a systematic mass spectrometry analysis to identify novel Akt interacting proteins by using 293T cells stably expressing HA-Akt1. Interestingly, one candidate Akt1 interacting protein was SETDB1, belonging to the SET-domain proteins and serving as a histone H3 lysine 9-specific methyltransferase (Supplementary Fig. 1a, Supplementary Table. 1)14. We confirmed the interaction between endogenous Akt and SETDB1 by the co-immunoprecipitation assay (Fig. 1a) and demonstrated the direct binding between Akt and SETDB1 by in vitro binding assay (Fig. 1b). However, SETDB1 was not a substrate of Akt, as the in vitro kinase assay showed that recombinant active Akt1 could directly phosphorylate GSK3β, known to be an Akt substrate, but not SETDB1 (Fig. 1c).
Figure. 1. SETDB1 interacts with Akt and is required for Akt activation.

(a) Whole cell extracts (WCE) of HEK293 cells was collected and subjected to co-immunoprecipitation (Co-IP) assays and Immunoblotting (IB). (b) Immunoprecipitated Flag-SETDB1 from HEK293 cell transfected with Flag-SETDB1 were incubated with GST-Akt1 WT or GST purified from engineering bacteria for in vitro binding assay, followed by immunoprecipitation (IP) Flag-SETDB1 and IB analysis. (c) In vitro kinase assay shows Akt phosphorylates GSK3β but not SETDB1, as determined by Phospho-Serine/Threonine (p-S/T) antibody. (d) HEK293 cells were serum-starved for 1 day, treated with 50 ng/ml IGF-1 for various times, and WCE were collected for immunoprecipitation (IP) with SETDB1, followed by IB analysis. (e) Setdb1+/− and Setdb1−/− MEFs were serum-starved, treated with IGF-1 for 15 mins, and WCE were collected for IB analysis. (f) HEK293 cells silenced with control (Shluc) or SETDB1 ShRNA (#1 and #2) were serum-starved, treated with IGF-1 for 15 mins, and WCE were collected for IB analysis. (g) HEK293 cells silenced with control (Shluc) or SETDB1 ShRNA (#1) were serum-starved, treated with EGF for 5 mins, and WCE were collected for IB analysis. (h) HEK293 cells transfected with Vector (Vec) or SETDB1 were serum-starved, treated with IGF-1 for 15 mins, and WCE were collected for IB analysis. (i) HEK293 cells transfected with Vector (Vec), Flag-SETDB1 (WT, 1 μg, 3μg and 5μg) and Flag-SETDB1 H1224K (H1224K) were harvested for IB analysis. IB data shown in a-i represent results from 3 independent experiments. Unprocessed blots are shown in Supplementary Figure 8.
Notably, the interaction of endogenous Akt and SETDB1 was promoted by IGF-1 stimulation (Fig.1d). Since SETDB1 interacts with Akt upon growth factor stimulation, we determined whether SETDB1 regulates Akt activation by loss and gain of function approaches. Setdb1−/− mouse embryonic fibroblasts (MEFs) and SETDB1 knockdown cells displayed impairment of Akt T308 phosphorylation, an essential phosphorylation event for Akt activation, upon IGF-1 and EGF stimulation (Fig. 1e–g, Supplementary Fig. 1b). However, Akt S473 phosphorylation was only slightly affected by SETDB1 knockdown (Fig.1g). Moreover, overexpression of wild-type SETDB1, but not SETDB1 H1224K mutant deficient for methyltransferase activity14, enhanced basal and IGF-1-induced Akt T308 phosphorylation (Fig.1h, 1i, Supplementary Fig. 1c). Collectively, these data suggest that SETDB1 is required for Akt T308 phosphorylation in response to growth factor stimulation.
SETDB1 triggers Akt K64 methylation to mediate Akt phosphorylation
Since SETDB1 is known to induce di-methylation of p5315 and interacts with Akt, we examined whether Akt is a substrate of SETDB1. Notably, both IGF-1 and EGF treatment induced Akt tri-methylation, correlated with Akt phosphorylation and activation (Fig. 2a, 2b, Supplementary Fig. 1d, Supplementary Table. 2). Among three Akt isoforms1, 2, basal and STEDB1-mediated tri-methylation of Akt were much pronounced in Akt1 than Akt2 and Akt3 (Fig. 2c). In vitro methylation assay revealed that SETDB1, but not SETDB1 H1224K mutant, could tri-methylate or di-methylate Akt1 directly (Fig. 2d). Moreover, Setdb1−/− MEFs and SETDB1 knockdown cells displayed impairment of Akt tri-methylation upon IGF-1 and EGF stimulation (Fig. 2e–g), while overexpression of SETDB1 enhanced Akt tri-methylation upon IGF-1 stimulation (Fig.2h). These data along with above findings suggest that SETDB1 is an Akt methyltransferase responsible for Akt tri-methylation in response to growth factor stimulation.
Figure. 2. SETDB1 triggers Akt K64 methylation leading to Akt phosphorylation.
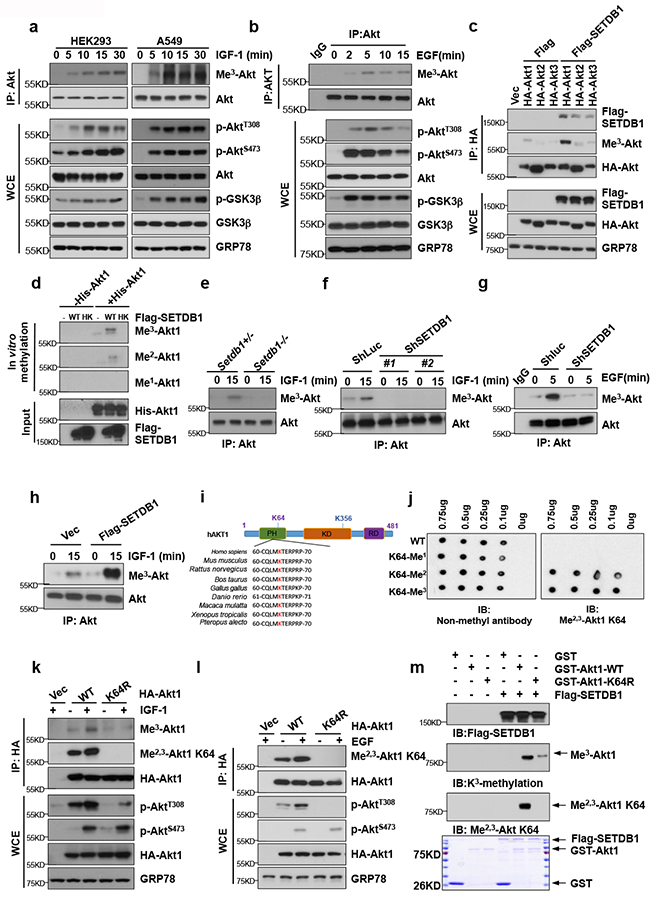
(a-b) HEK293 cells or A549 cells were serum-starved, treated with IGF-1 (a) or EGF (b) for various times, and WCE were collected for IP with Akt antibody, followed by IB analysis. (c) HEK293 cells transfected with indicated plasmids and WCE were collected for IP with HA antibody, followed by IB analysis. (d) Immunoprecipitated Flag-SETDB1 or Flag-SETDB1 H1224K from HEK293 cells transfected with Flag-SETDB1 or Flag-SETDB1 H1224K were incubated with S-adenosyl-L-methionine along with Akt protein for in vitro methylation of Akt. The methylated proteins were separated by SDS-PAGE, and Akt methylation was analyzed by IB analysis. (e) Setdb1+/− and Setdb1−/− MEFs were serum-starved, treated with IGF-1 for 15 mins, and WCE were collected for IP with Akt antibody, followed by IB analysis. (f-g) HEK293 cells silenced with control (Shluc) or SETDB1 ShRNA (#1 and #2) were serum-starved, treated with IGF-1 for 15 mins (f) or EGF for 5 mins (g), and WCE were collected for IP with Akt antibody, followed by IB analysis. (h) HEK293 cells transfected with Vector (Vec) or SETDB1 were serum-starved, treated with IGF-1 for 15 mins, WCE were collected for IP with Akt antibody, followed by IB analysis. (i) The Akt K64 site amino acid in different species. (j) Dot blot validates Akt K64 Di-/Tri-methylation antibody. PEP00022 Affinity Purified Antibody was Me2,3-Akt K64 antibody, while PEP00021 Affinity Purified Antibody was Non-methyl antibody, which recognizes the peptide N-CQLMKTERPRP. WT Akt peptide, K64-Me1 peptide, K64-Me2 peptide, and K64-Me3 peptide were synthesized by LifeTein Lab. (k-l) HEK293 cells transfected with indicated plasmids were serum-starved, treated with IGF-1 for 15 mins (k) or EGF for 5 mins (l), and WCE were collected for IP with HA antibody, followed by IB analysis. (m) Immunoprecipitated Flag-SETDB1 from HEK293 cell transfected with Flag-SETDB1 were incubated with S-adenosyl-L-methionine along with GST-Akt1 WT or GST-Akt1 K64R purified from engineering bacteria for in vitro methylation of Akt. The methylated proteins were separated by SDS-PAGE, and Akt methylation was analyzed by IB analysis. All IB data in this figure represent results from 3 independent experiments. Unprocessed blots are shown in Supplementary Figure 8.
To identify Akt1 methylation sites, we found multiple lysine residues on Akt including K64, K112, K154, K183, and K400 could be methylated by mass spectrometry analysis (Supplementary Fig. 1e). However, by comparing control cells with SETDB1 knockdown cells, only Akt K64 methylation was SETDB1 dependent (Supplementary Fig. 1f). Akt K64 located in pleckstrin homology (PH) domain, which is essential for Akt-PIP3 lipid binding and protein-protein interaction, is highly conserved from Xenopus to human (Fig. 2i). We therefore focused on Akt K64 methylation and examined its role in Akt activation. We generated a K64-specific methylation antibody of Akt that recognized K64 di-methylation and tri-methylation of Akt (anti-Me2,3-Akt K64) (Fig. 2j–l). Using both lysine tri-methylation antibody and anti-Me2,3-Akt K64 antibody, we found that Akt K64 methylation was induced by both IGF-1 and EGF, and mutation of K64 to arginine (R) impaired Akt K64 methylation (Fig. 2k, 2l). Consistent with the results that SETDB1 deficient cells exhibited defect in Akt T308 phosphorylation and activation, loss of Akt K64 methylation also impaired Akt T308 phosphorylation (Fig. 2k, 2l). In vitro methylation assay revealed that SETDB1 could tri-methylate or di-methylate Akt1 at K64 (Fig. 2m). Collectively, SEDTB1-mediated K64 methylation of Akt is induced by growth factor stimulation and plays a crucial role in Akt T308 phosphorylation.
Akt K64 methylation is critical for Akt activation and cell membrane localization
To corroborate the role of Akt K64 methylation in Akt phosphorylation, we employed DLD Akt1/2−/− cells and found that Akt1 K64 methylation, Akt1 T308 phosphorylation and Akt1 substrate Foxo3a phosphorylation were induced in DLD-1 Akt1/2−/− cells restored with Akt1, but not with Akt1 K64R (Fig. 3a). E17K is the most common Akt mutation identified in numerous human cancers16, 17, correlated with constitutive Akt T308 phosphorylation and activation and greater oncogenic potential3, 18. Interestingly, we found that Akt1 E17K mutant displayed not only hyper-phosphorylation of Akt at T308, but also higher Akt K64 methylation compared to wild-type Akt, likely due to the enhancement of the interaction between Akt and SETDB1 (Fig. 3b). Conversely, loss of Akt K64 methylation impaired heightened Akt tri-methylation and T308 phosphorylation in Akt1 E17K mutant (Fig. 3b). Consistently, Akt methylation was reduced in Akt1 E17K/K64R mutant (Fig. 3c). Furthermore, we obtained the same result in NIH3T3 cells with stable expression of Akt1 WT, Akt1 E17K or Akt1 E17K/K64R mutant (Fig. 3d). Hence, K64 hyper-methylation of Akt contributes to hyper-phosphorylation of Akt1 E17K mutant.
Figure. 3. Akt K64 methylation is critical for Akt activation and membrane localization.

(a) The WCE from DLD-1 Akt1/2−/− cells restored with Akt1 (WT) or Akt1 K64R (K64R) serum-starved and treated with IGF-1 for 15 mins and 30 mins were collected and subjected to IB analysis, the Akt T308 band intensity was analyzed by Image J and the intensity was adjusted by beta-actin, the average intensity data was got from three replicate experiments and the relative ratio was shown. (b) HEK293 cells were transfected with Vector (Vec), HA-Akt1 (WT) or HA-Akt1 E17K (E17K), and WCE were collected for IP with HA antibody, followed by IB analysis. (c) HEK293 cells transfected with Vector (Vec), HA-Akt1 (WT), HA-Akt1 E17K (E17K) or HA-Akt1 E17K/K64R (E17K/K64R) were serum-starved, treated with IGF-1 for 10 mins, and WCE were collected for IP with HA antibody, followed by IB analysis. (d) IB of lysed NIH3T3 cells transfected with Vector (Vec), HA-Akt1 (WT), HA-Akt1 E17K (E17K) and HA-Akt1 E17K/K64R (E17K/K64R). (e) The membrane fractions from DLD-1 Akt1/2−/− cells restored with Akt1 (WT) or Akt1 K64R (K64R) serum-starved and treated with IGF-1 for 15 mins and 30 mins were collected and subjected to IB analysis. (f) The membrane and cytosolic fractions from HEK293 cells silenced with control (Shluc) or SETDB1 ShRNA (#1) serum-starved and treated with IGF-1 for 15 mins and 30 mins were collected and subjected to IB analysis. All IB data in this figure represent results from 3 independent experiments. Unprocessed blots are shown in Supplementary Figure 8.
We then sought to understand how SEDTB1-mediated Akt K64 methylation regulates Akt T308 phosphorylation and activation in response to growth factor stimulation. We detected Akt K64 methylation in the cytosol (Fig. 3f) and confirmed the K64 methylated Akt was recruited to the cell membrane upon growth factor treatment by using biochemical fractionations/western blot analysis and immunofluorescence assay (Fig. 3e, Supplementary Fig.2a). SETDB1 was detected mainly in the cytosolic fraction, but only weak signal of SETDB1 could be detected in the cell membrane fractionation (Fig. 3f), and SETDB1 deficiency impaired IGF-mediated Akt cell membrane recruitment and T308 phosphorylation (Fig. 3f, Supplementary Fig.2b). Collectively, these results indicate that SETDB1-mediated Akt methylation at K64, occurred likely in the cytosol, is crucial for Akt activation and cell membrane localization upon growth factor stimulation.
SETDB1-mediated Akt K64 methylation promotes tumorigenesis
To determine oncological consequences of SETDB1-mediated Akt K64 methylation, we employed NSCLC A549 cell model. Knockdown of SETDB1 reduced IGF-1-induced Akt K64 methylation and T308 phosphorylation, and T308 phosphorylation was rescued by restoration of constitutively active of Akt (Myr-Akt) (Fig. 4a, 4b). Accordingly, cell proliferation and in vitro colony formation were reduced in SETDB1 knockdown cells, and restoration of Akt activity by Myr-Akt rescued these defects (Fig. 4c, 4d). Moreover, glucose uptake and lactate production were reduced in SETDB1 knockdown cells compared with control cells (Fig. 4e, 4f). In xenograft tumor models, SETDB1 knockdown cells exhibited much lower tumorigenic potential than control cells (Fig. 4g, Supplementary Fig. 3a, 3b), correlated with reduced Akt K64 methylation and T308 phosphorylation in xenograft tumor tissues (Fig. 4h). Moreover, restoration of Akt activation in SETDB1 knockdown cells by Myr-Akt partially rescued the defect in tumorigenic potential (Fig. 4i, Supplementary Fig. 3b). Thus, SETDB1 appears to promote tumorigenesis by inducing Akt K64 methylation and activation.
Figure. 4. SETDB1-mediated Akt K64 methylation promotes tumorigenesis, correlates with Akt activation and predicts poor survival of cancer patients.

(a) A549 cells silenced with control (Shluc) or SETDB1 ShRNA (#1 and #2) were serum-starved, treated with IGF-1 for 15 mins, and WCE were collected for IP with Akt antibody, followed by IB analysis. (b) A549 cells were silenced with control (Shluc) or SETDB1 ShRNA (#1, #2) along with or without Myr-Akt transfection, and WCE was collected for IB analysis. (c) Colony formation assay in A549 cells with Shluc, SETDB1 knockdown or SETDB1 knockdown plus Myr-Akt overexpression. (d) Cell number was counted in A549 cells with control (Shluc), SETDB1 ShRNA (#1) or SETDB1 ShRNA (#1) plus Myr-Akt overexpression. (e) Glucose uptake was measured in A549 cells silenced with control (Shluc) or SETDB1 ShRNA (#1). Cells treated with glucose-free medium for 4 hours were added with the fluorescent analog NBDG for 60 mins, and glucose uptake was quantified by FACS analysis (**** p<0.0001). (f) Lactate production was measured in A549 cells silenced with control (Shluc) or SETDB1 ShRNA (#1, #2). In d-e, data shown represent mean ± s.d (n = 3 independent experiments), in f data shown represent mean ± s.d (n = 4 independent experiments). (g) Tumor volume of xenograft tumors derived from A549 cells silenced with control (Shluc) or SETDB1 ShRNA (#1, #2), data shown represent mean ± s.d, n = 5 mice for each group, **** p<0.0001). (h) IB of lysed xenograft tumors derived from A549 cells silenced with control (Shluc) or SETDB1 ShRNA (#1, #2). (i) Tumor volume of xenograft tumors derived from A549 cells silenced with control (Shluc), SETDB1 ShRNA (#1) and SETDB1 ShRNA (#1) plus Myr-Akt overexpression, data shown represent mean ± s.d, n = 7 mice for each group, **** p<0.0001). (j) NSCLC tumors and paired normal lung tissues were extracted and subjected to IB analysis. (k) Histological and quantification analysis of Akt K64 di-methylation/tri-methylation and Akt pT308 expressions in NSCLC patients with low or high expression of SETDB1. Red box is the enlarge view and scale bar represents 100 μm. 1 section was analyzed for each patient sample and 156 patient samples were collected for this assay. (l) Kaplan-Meier plot analysis of 111 cases NSCLC patients with low or high expression of Akt K64 methylation. IB data in a, b, h and j represent results from 3 independent experiments. Statistical significance in d-g, i, and l was assessed using Student’s two-tailed t-test. Statistics source data in d-g, i, k, and l are shown in in Supplementary Table.4. Unprocessed blots are shown in Supplementary Figure 8.
To determine the role of Akt K64 methylation in oncogenic Akt-mediated tumorigenic potential, we performed anchorage-independent soft agar assay in NIH3T3 cells with stable overexpression of Akt, cancer-associated mutant E17K or E17K/K64R double mutant. While overexpression of Akt was insufficient to drive NIH3T3 cell transformation, overexpression of Akt E17K mutant could drive cell transformation. Remarkably, loss of Akt K64 methylation impaired transformation potential of E17K mutant (Supplementary Fig. 3c). Collectively, these results suggest that SETDB1-mediated Akt K64 methylation contributes to oncogenic activity of Akt E17K cancer-associated mutant.
SETDB1-mediated Akt K64 methylation correlates with Akt activation and predicts poor survival of NSCLC patients
By examining cBioPortal (http://www.cbioportal.org) and Oncomine databases, we found that SETDB1 was not only up-regulated in diverse human cancers, but also overexpressed in different types of lung cancers (Supplementary Fig. 4a, 4b)19, 20. KM-PLOT database21 revealed that higher expression of SETDB1 was associated with poor survival outcome of NSCLC patients (Supplementary Fig. 4c). Moreover, higher expression of SETDB1 was also associated with poor survival outcome in other cancer types using Human Protein Atlas database22 (Supplementary Fig. 4d). We detected SETDB1 expression and Akt K64 methylation and T308 phosphorylation in fresh NSCLC specimens and found that SETDB1 was overexpressed in most of paired lung tumor tissues compared with normal lung tissues (Fig. 4j). Of note, Akt K64 methylation was upregulated in lung tumor tissues compared with adjacent normal counterparts and positively correlated with SETDB1 expression, except for the P1 sample in which N and T displayed Akt1 E17K mutation (Fig. 4j). Immunohistochemistry (IHC) staining of 156 NSCLC specimens revealed that SETDB1 expression and Akt K64 methylation were upregulated in NSCLC patients (Fig. 4k). Notably, the expression level of SETDB1 and Akt K64 methylation also correlated with Akt pT308 level (Fig. 4k). SETDB1 staining was detected in the cytoplasm and nucleus, whereas Akt K64 methylation and T308 phosphorylation were detected in the cell membrane (Fig. 4k). Importantly, Kaplan-Meier survival analysis showed that Akt K64 methylation was associated with poor survival outcome in 111 NSCLC patients (Fig. 4l). Collectively, these data underscore the importance of SETDB-1-mediated Akt K64 methylation in Akt T308 phosphorylation and survival outcome in NSCLC patients.
Akt K64 methylation is essential for Akt-E3 ligase interaction and Akt K63-linked ubiquitination
We sought to explore the mechanism by which Akt K64 methylation regulates Akt cell membrane recruitment. Consistent with the finding that Akt K64 methylation is critical for Akt cell membrane recruitment, co-immunoprecipitation assay revealed that endogenous PDK1 interacted with Akt1, but not with Akt1 K64R (Fig. 5a). However, there was no difference in the in vitro binding of recombinant PDK1 with Akt1 WT and Akt1 K64R isolated from cells transfected with Flag-Akt1 WT or Flag-Akt1 K64R. Furthermore, in vitro PIP3-binding assay revealed that Flag-Akt1 WT or Flag-Akt1 K64R isolated from cells bind to PIP3 with the same efficiency (Fig. 5b, 5c). These data indicate that the defect in cell membrane recruitment of Akt K64R likely contributes to its reduction in PDK1 binding.
Figure. 5. Akt K64 methylation is essential for Akt K63-linked ubiquitination and Akt-E3 ligase interaction.

(a) HEK293 cells were transfected with indicated plasmids and WCE were collected for IP with PDK1 antibody, followed by IB analysis. (b-c) Immunoprecipitated Flag-Akt1 WT or Flag-Akt1 K64R from HEK293 cell transfected with Flag-Akt1 WT or Flag-Akt1 K64R were incubated with PDK1 (b) or PIP3 (c) Beads in vitro, followed by IB analysis. (d-e) IB of lysed HEK293 cells transfected with indicated plasmids along with His-Ub. Ni-nitrilotriacetic acid (NTA), nicked bead pulldown. (f) HEK293 cells were silenced with control (Shluc) or SETDB1 ShRNA (#1) along with HA-Akt and His-Ubiquitin (His-Ub), and WCE were collected for nicked bead pulldown and IB analysis. (g) HEK293 cells silenced with control (Shluc) or SETDB1 ShRNA (#1, #2) were transfected with His-Ubiquitin(His-Ub), serum-starved, treated with IGF-1 for 30 mins, and WCE were collected for IP with Akt antibody, followed by IB analysis. (h) Setdb1+/− and Setdb1−/− MEFs cells were serum-starved, treated with IGF-1 for 30 mins, and WCE were collected for IP with Akt antibody, followed by IB analysis. (i) HEK293 cells were serum-starved, treated with IGF-1 for indicated time points, and WCE were collected for IP with Akt or SETDB1 antibody, followed by IB analysis. The Akt ubiquitination band intensity was adjusted by IP Akt, Akt band intensity was adjusted by IP SETDB1, Akt K64 methylation and Akt T308 phosphorylation band intensity was adjusted by WCE Akt, the average intensity data were obtained from three replicate experiments and the relative ratio was shown. (j) HEK293 cells were serum-starved for 12 hours and treated with IGF-1 at different time points before harvesting to separate cell fractionations for IB analysis. (k) HEK293 cells transfected with indicated plasmids were serum-starved, treated with IGF-1 for 30 mins, and WCE were collected for IP with HA antibody, followed by IB analysis. (l) HEK293 cells transfected with indicated plasmids were serum-starved, treated with EGF for 5mins and 10 mins, and WCE were collected for IP with HA antibody, followed by IB analysis. IB data shown in a-l represent results from 3 independent experiments. Unprocessed blots are shown in Supplementary Figure 8.
We then determined the crosstalk between Akt tri-methylation and Akt K63-linked ubiquitination. Of note, K63-linked ubiquitination of Akt was markedly reduced in Akt K64R mutant (Fig. 5d). Akt ubiquitination was significantly increased in Akt1 E17K mutant, but decreased in Akt1 E17K/K64R mutant (Fig. 5e). Consistently, K63-linked ubiquitination was markedly reduced in SETDB1 knockdown cells and Setdb1−/− MEFs (Fig. 5f–h), while Akt K8R mutant, an Akt K63-linked ubiquitination defective mutant, did not affect Akt K64 methylation (Supplementary Fig. 5a), suggesting that SETDB1-mediated Akt tri-methylation is an upstream event of K63-linked ubiquitination of Akt. To further validate this notion, the kinetics and chronological sequence of endogenous SETDB1/Akt binding, K64 methylation, ubiquitination, membrane translocation and phosphorylation of Akt1 were examined (Fig. 5i, 5j). Specifically, we detected SETDB1/Akt interaction and Akt K64 methylation 5 mins after IGF-1 treatment, followed by Akt K63-linked ubiquitination and T308 phosphorylation (Fig. 5i), indicating that Akt K64 methylation is the earlier event induced by growth factor. Moreover, Akt K64 methylation was correlated with T308 phosphorylation and cell membrane localization of Akt (Fig. 5j). Hence, SETDB1-mediated Akt K64 methylation is indispensable for K63-linked ubiquitination of Akt.
To elucidate the mechanism by which Akt K64 methylation regulates K63-linked ubiquitination and phosphorylation of Akt by growth factor stimulation, we determined whether Akt K64 methylation orchestrates the interaction of Akt with its E3 ligases TRAF6 and Skp2-SCF. While Akt readily interacted with TRAF6 and Skp2, Akt1 K64R mutant showed marked reduction in interacting with TRAF6 and Skp2 under basal conditions and IGF-1 or EGF treatment (Fig. 5k,5l, Supplementary Fig. 5b, 5c). These data underscore the crucial role of Akt K64 methylation in E3 ligases recruitment to Akt, thereby triggering K63-linked ubiquitination of Akt.
JMJD2A recognizes K64 methylated Akt and facilities the Akt-E3 ligase interaction.
Since TRAF6 or Skp2 does not contain any domain that can recognize methylated proteins, we reasoned that TRAF6 or Skp2 may require an adaptor to bridge the methylated form of Akt and TRAF6/Skp2. We hypothesized that methylation of Akt at K64 may serve as a scaffold to recruit an adaptor of TRAF6/Skp2 that recognizes methylated Akt and in turn facilitates the recruitment of TRAF6 or Skp2 to Akt for eliciting K63-linked ubiquitination of Akt. To test this hypothesis, we conducted the unbiased mass spectrometry analysis to identify factors that bridge K64 methylated Akt and TRAF6/Skp2. We identified more than 300 proteins that interacted with Akt1 WT, but not with Akt1 K64R mutant. Of note, Skp2 and TRAF6 were detected among them (Fig. 6a, Supplementary Table. 2), further supporting the notion that Akt K64 methylation acts to facilitate the recruitment of E3 ligases Skp2 and/or TRAF6 to Akt for eliciting Akt ubiquitination.
Figure. 6. JMJD2A recognizes K64 methylated Akt and facilities the Akt-E3 ligase interaction.

(a) HEK293 cells were transfected with HA-Akt1 WT or HA-Akt1 K64R for 48 hours, WCE were collected for IP with HA antibody, followed by mass spectrometry analysis for detecting binding proteins. Some proteins that only bind to Akt1 WT are listed, such as JMJD2A, Skp2 and TRAF6. (b) HEK293 cells transfected with Vector (Vec) or HA-Akt, and WCE were collected for Co-IP with HA antibody, followed by IB analysis with indicated proteins. (c) HEK293 cells transfected with Vector (Vec) or Flag-JMJD2A, and WCE were collected for Co-IP with Flag antibody, followed by IB analysis indicated proteins. (d) HEK293 cells were silenced with control (Shluc) or JMJD2A ShRNA (#1), and WCE were collected for Co-IP with Akt antibody, followed by IB analysis indicated proteins. (e) HEK293 cells silenced with control (Shluc) or JMJD2A ShRNA (#1) were serum-starved, treated with EGF for 10 mins, WCE were collected for IP with Akt antibody, followed by IB analysis. (f) HEK293 cells transfected with Vector (Vec), HA-Akt1 or HA-Akt1 K64R were serum-starved for 1 day, treated with 50 ng/ml IGF-1 for various times, and WCE were collected for IP with HA antibody, followed by IB analysis indicated proteins. (g) HEK293 cells were transfected with Vector (Vec), HA-Akt1 (WT) or HA-Akt1 K64R (K64R), WCE were collected for IP with JMJD2A antibody, followed by IB analysis. (h) HEK293 cells silenced with control (Shluc) or SETDB1 ShRNA (#1) were serum-starved, treated with IGF-1 for 30 mins, and WCE were collected for IP with JMJD2A antibody, followed by IB analysis. (i) Immunoprecipitated HA-Akt1 WT and HA-Akt1 K64R from HEK293 cell transfected with HA-Akt1 WT or HA-Akt1 K64R were incubated with recombinant JMJD2A purified from engineering bacteria for in vitro binding assay, followed by IP with HA and IB analysis. (j) HEK293 cells silenced with control (Shluc) or SETDB1 ShRNA (#1) were serum-starved, treated with EGF for 10 mins, and WCE were collected for IP with Akt antibody, followed by IB analysis. IB data shown in b-j represent results from 3 independent experiments. Unprocessed blots are shown in Supplementary Figure 8.
Interestingly, we found that JMJD2A, a KDM family protein that recognizes and demethylates methylated proteins, was a potential interacting protein of wild-type Akt1, but not of Akt1 K64R (Supplementary Fig.1a, Fig. 6a, Supplementary Table. 1, 2). We confirmed the endogenous interaction of JMJD2A with Akt and TRAF6/Skp2 by co-immunoprecipitation assay (Fig. 6b, 6c). Notably, we observed marked reduction of Akt interaction with TRAF6 or Skp2 in JMJD2A knockdown cells (Fig.6d, 6e), suggesting that JMJD2A mediates the interaction of Akt with its E3 ligases.
We then determined whether SETDB1-mediated Akt K64 methylation regulates the interaction between Akt and JMJD2A. JMJD2A interacted with Akt, but not with Akt K64 mutant, both in IGF-1 treatment and serum culture conditions (Fig. 6f, 6g). While the interaction of JMJD2A with TRAF6 remained unchanged upon IGF-1 treatment, its interaction with Akt was markedly impaired upon SETDB1 knockdown (Fig. 6h). In vitro binding assay showed that recombinant JMJD2A could interact with Akt, but not Akt K64R mutant, isolated from 293 cells transfected with HA-Akt or HA-Akt K64R (Fig. 6i). Moreover, the interaction of Akt with Skp2 was reduced in SETDB1 knockdown cells upon EGF treatment (Fig. 6j). Thus, SETDB1-mediated Akt K64 methylation is critical for the interaction of Akt with JMJD2A, thereby recruiting TRAF6 or Skp2 to trigger K63-linked ubiquitination of Akt upon growth factor stimulation.
JMJD2A non-demethylase function contributes to Akt ubiquitination and activation.
JMJD2A emerges as a novel player in cancer development, largely dependent on its demethylase activity23. We then determined whether JMJD2A acts through its demethylase activity to orchestrate Akt activity. JMJD2A and JMJD2A H188A mutant deficient in demethylase activity increased Akt T308 phosphorylation to a similar degree (Fig. 7a), indicating that JMJD2A promotes Akt activity independently of its demethylase activity. Upon IGF-1 treatment, overexpression of JMJD2A led to enhanced Akt activation, while knockdown of JMJD2A reduced Akt phosphorylation and its substrate phosphorylation (Fig. 7b). However, IGF-1-mediated Akt K64 methylation remained unchanged in JMJD2A knockdown cells (Fig. 7c), suggesting that JMJD2A regulates Akt activity downstream of Akt K64 methylation.
Figure. 7. JMJD2A non-demethylase function contributes to Akt ubiquitination and activation.

(a) HEK293 cells were transfected with Vector, pBabe-JMJD2A or pBabe-JMJD2A H188A, and WCE were collected, followed by IB analysis. (b) HEK293 cells transfected with Vector (Vec) or Flag-JMJD2A were serum-starved, treated with IGF-1 for various times, and WCE were collected, followed by IB analysis. (c) HEK293 cells silenced with control (Shluc) or JMJD2A ShRNA (#1, #2) were serum-starved, treated with IGF-1 for 15 mins, and WCE were collected, followed by IB analysis. (d) HEK293 cells silenced with control (Shluc) or JMJD2A ShRNA (#1, #2) were serum-starved, treated with IGF-1 for 15 mins or 30 mins, and cell membrane fractionations were collected for IB analysis. (e) Primary JMJD2Alox/lox MEFs cells were infected with retrovirus control (MSCV) or retrovirus Cre (Cre), and WCE were collected for IP with Akt antibody, followed by IB analysis. (f) IB of lysed HEK293 cells silenced with control (Shluc) or JMJD2A ShRNA (#1) transfected with HA-Akt1 along with His-Ub, serum-starved and treated with IGF-1 for 30 mins. (g) Glucose uptake was measured in A549 cells silenced with control (Shluc) or JMJD2A ShRNA (#1). Cells treated with glucose-free medium for 4 hours were added with the fluorescent analog NBDG for 60 mins, and glucose uptake was quantified by FACS analysis, data shown represent mean ± s.d. (n=3 independent experiments, **** p<0.0001, Student’s two-tailed t-test). (h) Colony formation assay in A549 cells with Shluc, SETDB1 knockdown or JMJD2A knockdown plus Myr-Akt overexpression. (i) Cell number was counted in A549 cells with control (Shluc), JMJD2A ShRNA (#1) or JMJD2A ShRNA (#1) plus Myr-Akt overexpression. Data shown represent mean ± s.d. (n=3 independent experiments, Student’s two-tailed t-test). (j) The working model of SETDB1-mediatd Akt K64 methylation in growth factor-mediated Akt ubiquitination, cell membrane recruitment and activation. IB data in a-f represent results from 3 independent experiments. Statistics source data for g and i are shown in Supplementary Table.4. Unprocessed blots are shown in Supplementary Figure 8.
Since JMJD2A is critical for Akt ubiquitination and activation by facilitating E3 ligase-Akt interaction upon growth factor stimulation, we expect that JMJD2A should regulate the recruitment of K64 methylated Akt and Akt to the cell membrane. Indeed, JMJD2A knockdown impaired the recruitment of K64 methylated Akt and Akt to the cell membrane upon growth factor treatment (Fig. 7d). In JMJD2Alox/lox primary MEFs, knockout of JMJD2A by Cre recombinase expression also decreased Akt ubiquitination and T308 phosphorylation (Fig. 7e, Supplementary Fig. 6a). In JMJD2A knockdown cells, IGF-1-indcued Akt ubiquitination and T308 phosphorylation were impaired (Fig. 7f), and JMJD2A restoration in JMJD2A knockdown cells rescued Akt phosphorylation (Supplementary Fig. 6b).
Finally, we determined oncogenic role of JMJD2A and its association with Akt activation. In A549 cells with JMJD2A knockdown, Akt phosphorylation was reduced (Supplementary Fig. 6c). Accordingly, the glucose uptake decreased in JMJD2A knockdown A549 cells compared with control cells (Fig. 7g). While JMJD2A knockdown reduced colony formation and cell proliferation, the restoration of Akt activity by Myr-Akt rescued these defects (Fig. 7h,7i). In xenograft tumor models, we found that JMJD2A knockdown cells exhibited much lower tumorigenic potential than control cells (Supplementary Fig. 6d, 6e). Remarkably, restoration of Akt activity in JMJD2A knockdown cells by Myr-Akt partially rescued the defect (Supplementary Fig. 6d, 6e). We assessed the expression of JMJD2A in patient Tissue Microarrays (TMAs) and showed that the expression of JMJD2A was positively correlated with Akt1 T308 phosphorylation (Supplemental Table.3). Taken together, these results indicate that JMJD2A acts as an adaptor to promote Akt activation and oncogenic activity.
Discussion
Our current study provides novel insight into the regulation of K63-linked ubiquitination of Akt by revealing that Akt K64 methylation is an essential event for Akt and its E3 ligase interaction. We show that SETDB1-dependent K64 methylation of Akt upon growth factor stimulation is recognized by JMJD2A, resulting in subsequent recruitment of E3 ligases to elicit K63-linked ubiquitination and activation of Akt. It should be noted that JMJD2A interacts with Skp2 and TRAF6 E3 ligases independently of growth factor stimulation and Akt K64 methylation. Therefore, SETDB1-mediated K64 methylation of Akt represents a sensing signal to drive the interaction of Akt with its E3 ligase in response to growth factor stimulation. Although PI3K connects growth factor receptors to Akt activation24–26, we found that PI3K is not required for SETDB1-mediated Akt K64 methylation (Supplementary Fig. 7a). Moreover, Akt R25C mutant, deficient for PIP3 binding and cell membrane localization, displayed the same level of Akt K64 methylation as WT Akt (Supplementary Fig. 7b). Thus, PI3K activity, Akt-PIP3 binding and Akt cell membrane localization appear to be dispensable for SETDB1-mediated Akt K64 methylation.
The methylation has emerged as a prevalent posttranslational modification that regulates a variety of non-histone proteins7, 9, 11, 15, 27, 28, while little is known about SETDB1 non-histone substrates. In this study, we identify Akt as a novel non-histone substrate of SETDB1 and demonstrate that SETDB1-mediated Akt K64 methylated is a key event for Akt ubiquitination and activation in response to growth factor stimulation. This study expands our understanding of the substrates and function of SETDB1, which is amplified and overexpressed in many cancer types15, 29, 30. Consistently, we show that SETDB1 is overexpressed in diverse cancers including NSCLC and its overexpression predicts poor survival outcome. Notably, we found that SETDB1-mediated Akt K64 methylation is upregulated in NSCLC and correlated with Akt activation. Moreover, increased levels of Akt K64 methylation confer poor prognosis in NSCLC patients. The transcriptional regulation by SETDB1 via inducing histone H3 K9 methylation is thought to be a primary action for SETDB1-mediated biological functions14, 31, 32. However, we demonstrate that activation of Akt through SETDB1-mediated Akt K64 methylation also contributes to SETDB1-mediated oncogenic activity. Thus, SETDB1 appears to act through direct transcriptional and non-transcriptional regulation to impact its oncogenic processes. Our study therefore reveals that oncogenic activity of SETDB1 is partly due to activation of Akt through SETDB1-mediated Akt K64 methylation.
In a recent independent work, Guo et al. identified K140/K142 as the SETDB1-mediated Akt methylation sites, which is required for Akt T308 and S473 phosphorylation and activation33. Their mass spectrometry analysis also identified K64 as the Akt methylation site, consistent with our findings. Although our result in Fig. 2k revealed K64 as the major site for SETDB1-mediated Akt methylation, SETDB1-dependent methylation could also occur at other sites on Akt since there is still weak tri-methylation signal detected in Akt K64R mutant by using the tri-methylation antibody. To provide a comprehensive understanding of Akt methylation and activation, we also examined the Akt K140/K142 methylation and confirmed that K140 and K142 were Akt methylation sites (Supplementary Fig. 7c), and Akt K140/K142 methylation was required for Akt T308 and S473 phosphorylation and activation (Supplementary Fig. 7d). Interestingly, it seems that Akt K64 methylation and Akt K140/142 methylation occur independently and do not affect each other (Supplementary Fig. 7d). Both groups reveal that methylation of Akt is critical for Akt activation, although it involves different lysine residues (K64 versus K140/K142) likely through distinct molecular mechanisms. Our study shows that Akt K64 methylation is crucial for ubiquitination and T308 phosphorylation of Akt but not Akt-PIP3 binding and Akt S473 phosphorylation, while the study by Guo et al. indicates that Akt K140/K142 methylation is required for both Akt T308 and S473 phosphorylation and Akt-PIP3 binding33.
JMJD2A is upregulated in human cancers and display oncogenic activity in mouse tumor models through promoting YAP1 expression and repressing CHD523, 34, 35. While most studies reveal that JMJD2A acts though its demethylase activity to orchestrate gene expression and oncogenic activity35, 36, our study defines a novel adaptor function of JMJD2A in Akt signaling activation independent of demethylase activity. JMJD2A bridges the interaction of Akt with Akt E3 ligases to facilitate Akt ubiquitination and activation. In addition, we demonstrate that JMJD2A displays oncogenic activity partly through promoting Akt activation. Notably, an earlier study revealed that JMJD2A regulates DNA damage signaling independently of its enzymatic activity37. Our study together with this report suggest that JMJD2A may regulate various biological processes through its novel role as an adaptor independently of its enzymatic activity.
In summary, we identify Akt K64 methylation by SETDB1 upon growth factor stimulation as a crucial initiation event for subsequent K63-linked ubiquitination of Akt by recruiting JMJD2A and E3 ligases to the Akt complex, leading to cell membrane recruitment, T308 phosphorylation and activation of Akt (Fig. 7j). Importantly, our study reveals that SETDB1-mediated Akt K64 methylation not only serves as a key signal to drive Akt hyperactivation and promote cancer progression, but also represents a poor prognosis marker for NSCLC patients. Therefore, targeting SETDB1-mediated Akt K64 methylation may be a promising strategy for the treatment of cancers such as NSCLC.
Methods
Cell culture and reagents.
Setdb1+/− and Setdb1−/− mouse embryonic fibroblasts (MEFs) were gifts from Pro.Yoichi Shinkai. Jmjd2aflox/flox MEFs were prepared from Jmjd2aflox/flox mice as previously described38,39. In short, female pregnant mice were sacrificed, and embryos were isolated at 13.5 days after mating. Embryos were digested and the resulting cells were cultured in complete DMEM. All procedures were conducted under the approved protocol of Institutional Animal Care and Use Committee (IACUC). Jmjd2aflox/flox mice were provided by Dr. Zhi-Ping Liu (The University of Texas Southwestern). NIH3T3, A549, HEK293 and 293T cells were got from ATCC and cultured in DMEM containing 10% FBS. DLD-1Akt1/2−/− cell line was gifted by Dr. Bert Vogelstein. All cell lines were routinely checked for mycoplasma contamination and confirmed negative. All cell lines were subjected to routine fingerprinting analyses to confirm identity. pcDNA6-HA-Akt1 K64R, pcDNA6-HA-Akt1 E17K, and pcDNA6-HA-Akt1 E17K/K64R constructs were generated using a site-directed mutagenesis kit (Agilent Technologies, #200518) according to the manufacturer’s standard procedures with pcDNA6-HA-Akt1 as the template. Flag-SETDB1 and Flag-SETDB1 H1224K were obtained from Pro. Frank J. Rauscher III, (His)6-ubiqutin and HA-Akt1 were gifts from Drs. D. Bohmann and M.C. Hung, respectively. Mammalian expression vector pEV3S-Flag-JMJD2A was a gift from Dr. Ralf Janknecht from Oklahoma University. pBabe-JMJD2A was sub-cloned from Flag-JMJD2A using blunt end cloning method. Enzyme dead mutants (H188A) of JMJD2A were generated using a site-directed mutagenesis kit (Agilent Technologies, #200518) according to the manufacturer’s standard procedures with pBabe-JMJD2A as the template. Recombinant Human IGF-I Protein(#291-G1) and Recombinant Human EGF Protein (#236-EG) were obtained from R&D Systems.
Immunoprecipitation (IP), Immunofluorescence(IF) and immunoblotting (IB).
IP, IF and IB were performed essentially as previously described38,39. Each experiment was successfully carried out two to three times. Antiboides were validated by using positive and negative control tissues and cells. For protein-protein interactions, cells were lysed by RIPA lysis buffer [150 mM NaCl, 0.5% NP-40, 5 mM EDTA, protease inhibitor cocktail (Roche)]. The following antibodies were used for IP and IB: anti-Akt antibody (IP: 1:200; IB: 1:1000, Cell Signaling, Cat # 9275), anti-phospho (S473)-Akt antibody (IB: 1:2000, Cell Signaling, Cat # 4060), anti-phospho (T308)-Akt (IB:1:1000, Cell Signaling, Cat # 9275), anti-JMJD2A antibody (IP: 1:100; IB:1:1000, Cell Signaling, Cat # 5328), anti-SETDB1 antibody (IP: 1:100; IB:1:1000, Cell Signaling, Cat # 2196) anti-K63 ubiquitin antibody (IB: 1:1000, Cell Signaling, Cat #12930), anti-N-cadherin (IB: 1:1000, Cell Signaling, Cat #14215), anti-phospho (S9)-GSK3β antibody (IB: 1:1000, Cell Signaling, Cat # 9322), Anti-GSK3β antibody (IB: 1:3000, Cell Signaling, Cat # 5676), anti-tri-methyl Lysine (IB: 1:1000, Cell Signaling, Cat # 14680), anti-Di-methyl Lysine (IB: 1:1000, Cell Signaling, Cat # 14117), anti-mono-methyl Lysine (IB: 1:1000, Cell Signaling, Cat # 14679), anti-Akt K64 Di-/tri-methylation rabbit polyclonal antibody generated by Proteintech company using the peptide N-CQLM-K(Me3)-TERPRP, anti-β-actin antibody (IB:1:1000, Sigma, Cat # A5441), anti-HA antibody (IB/IF:1:1000, Sigma, Cat # H3663), anti-Flag antibody (M2, IP: 1:200; IB: 1:3000, Sigma, Cat # F3165). The western blot band intensity analysis was done by Image J (NIH, Image J).
In vivo methylation assays, In vitro binding assay and Ubiquitination assay.
In vivo methylation assays were performed as described elsewhere40. Each experiment was successfully carried out two to three times. In brief, 293T cells were transfected with the indicated plasmids for 48 h and lysed by RIPA buffer (150 mM NaCl, 0.5% NP-40, 5 mM EDTA, protease inhibitor cocktail (Roche)), followed by IP purification and IB analysis. In vitro methylation assays using purified Flag, Flag-SETDB1 or Flag-SETDB1 H1224K incubated with S-adenosylmethionine (Abcam) and Akt1 protein (Abcam) at 30 °C for 1–1.5 h. Then Stop the reaction by adding 6 μL of 6X SDS protein sample loading buffer (180 mM Tris-HCl, pH 6.8, 30% glycerol, 10% SDS, 0.6 M ditriothreitol (DTT), 0.012% bromophenol blue) and heat at 95 °C for 5 min. Proteins were eluted in SDS-sample buffer, subjected to SDS-PAGE, transferred to PVDF membrane, and immunoblotted with antibodies. In vitro binding assay and ubiquitination assay were performed as previously described38,39,41. Each experiment was successfully carried out two to three times.
Viral infection and Transfection.
For lentiviral short hairpin RNA (shRNA) infection, 293T cells were prepared 50%−60% confluency and co-transfected with either luciferase (shLuc) or target gene shRNA with packaging plasmid (pHelper) and envelop plasmid (pEnv) by using the calcium phosphate transfection method. Medium was changed 6 hours later. After 48 hours, the virus particles were harvested and used to infect parental cells for another 48 hours. The stably infected cells were then selected by 2 μg/ml puromycin for 5–7 days. ShSETDB1 #1:5’-CCGGCGTGACTTCATAGAGGAGTATCTCGAGATACTCCTCTATGAAGTCACGTTTTTG-3’; ShSETDB1 #2:5’-CCGGGCTCAGATGATAACTTCTGTACTCGAGTACAGAAGTTATCATCTGAGCTTTTTG-3’; ShJMJD2A #1: 5’- CCGGCCGAAACTTCAGTAGATACATCTCGAGATGTATCTACTGAAGTTTCGGTTTTT-3’; ShJMJD2A #2: 5’- CCGGGCCTTGGATCTTTCTGTGAATCTCGAGATTCACAGAAAGATCCAAGGCTTTTT-3’;
For retroviral infection, MSCV and MSCV-Cre were transfected with VSV-G and Gag-Pol retroviral packaged plasmids into the HEK293T cells. Medium was replaced 6 hours later. After 48 hours, the virus particles were collected to infect MEFs for another 48 hours. The cells were selected by 1 μg/ml puromycin for 4–5 days. For transient transfection, plasmids were transfected by using either the calcium phosphate method or Lipofectamine 2000 (Invitrogen) reagent following the manufacturer’s instructions. For calcium phosphate transfection, indicated plasmids were mixed with sterile H2O, 2M CaCl2, 2X HBSS (drop by drop). Mixed solutions were incubated for 30 minutes at room temperature, and added to the cells in DMEM high glucose medium. After 6–8 hours, medium was replaced with fresh regular medium, and transfected cells were harvested 48 hours after transfection.
PIP3 phospholipid binding.
Cells with different transfection or growth factor treatments were lysed by RIPA buffer, and the cell lysates were incubated with control beads or PIP3 beads (Enchelon) overnight. The beads were washed four times with RIPA buffer and subjected to Immunoblotting analysis. Each experiment was successfully carried out two to three times.
Cytosolic and Membrane Fractions.
Cytosolic and membrane fractions were prepared using the Mem-PER™ Plus Membrane Protein Extraction Kit (Thermo Scientific) according to the manufacturers’ standard procedures.
Mass spectrometry analyses.
For mass spectrometry analysis, immunoprecipitation (IP) was performed with the whole cell lysates in different treated cells. The IP proteins were resolved by SDS-PAGE, followed by Coomassie staining. The band containing target protein or whole lane was reduced with 10 mM DTT for 30 minutes, alkylated with 55 mM iodoacetamide for 45 minutes, and in-gel-digested with trypsin enzymes. The resulting peptides were analyzed by Thermo Fisher Orbitrap Elite with Waters NanoAcuity UPLC. MS/MS data were searched against the Uniprot Human protein database (version 20151209 containing 21,024 entries) using Mascot 2.5.1 (Matrix Science) and data analysis was performed using the Scaffold 4.4.8 software (Proteome Software). Peptides and modified peptides were accepted if they passed a 1% FDR threshold. Specially, for Supplementary Fig. 1e we pulled down endogenous Akt by IP assay and subjected to mass spectrometry analysis. The mass spectrum shows a tryptic digest of Akt. For the Supplementary Fig. 1f, we pulled down endogenous Akt by IP assay in shLuc or shSETDB1 HEK293 cells and subjected to the Time-of-flight mass spectrometry (TOFMS) analysis. The investigators were blinded to allocation during experiments and outcome assessment. After the mass spectrometry analysis, we pick up the data with the 3-fold-change cutoff as the potential binding proteins or modifications, then we subject to Co- Immunoprecipitation to further confirm protein-protein binding or modifications.
Patient materials.
156 NSCLC patients without systemic drug therapy before surgery were surgically resected and histologically diagnosed at the third affiliated hospital of SUN Yat-sen University, China during the period 1999–2011. Follow up information is available for 111 patients until 30th December 2011. Informed consent was obtained from patients before surgery for paraffin-embedded (FFPE) lung cancer and fresh lung cancer specimens to be used for this research and approved by the Institutional Review Board of the Institute of Biophysics, SUN Yat-sen University. All samples were reevaluated by experienced pathologist according to the World Health Organization (WHO) grading system and the General Rules for Clinical Lung Cancer. The study is compliant with all relevant ethical regulations involving human participants.
Animal study.
We complied the ethical regulations of the ethical committee of Institute of Biophysics, Huazhong University of Science and Technology. The Institutional Animal Care and Use Committee (IACUC) of Huazhong University of Science and Technology has approved our animal studies. All animal experiments were carried out in accordance with the Animal Study Guidelines of Huazhong University of Science and Technology. Female 5 weeks old BALB/c nude mice were used for xenogrant studies. For in vivo tumorigenesis assays, 5×106 of A549 treated as indicated cells were subcutaneously injected into athymic nude mice (5–8 mice for each group). Tumor size was measured weekly with a caliper, and tumor volume was determined with the standard formula: L ×W2 ×0.52, where L is the longest diameter and W the shortest diameter. For all xenograft studies, mice were euthanized at the ethical endpoint when they failed to meet the predetermined IACUC quality-of-life guidelines. This study is compliant with all relevant ethical regulations regarding animal research.
Immunohistochemical (IHC) staining and Evaluation.
Formalin-fixed, paraffin-embedded tissue sections of 4-μm thick obtained from 156 primary NSCLC tumors tissues were immune-stained using the Gene Tech EnVision™ Detection Kit (GK500710, Gene tech, Shanghai, China). After deparaffinization and dehydration through graded alcohols and xylene, endogenous peroxidase activity and non-specific antigen were blocked with 3% hydrogen peroxide. The sections were heated for 5 min twice at 100 °C with Tris EDTA (pH 9.0) in a microwave oven for antigen retrieval. All sections were incubated with anti-Akt K64 me3 rabbit polyclonal antibody (1:200), anti-SETDB1 rabbit polyclonal antibody (1:100, Cell Signaling), anti-Akt T308 rabbit polyclonal antibody (1:200, Cell Signaling), anti-JMJD2A rabbit polyclonal antibody (1:100, Cell Signaling), at 37℃ for 1h. After washing, the sections were incubated with the Chem-Mate ™ Envision ™/HRP, Rabbit (ENV) reagent at 37℃ for 30min. Rinsed gently with PBS, the sections were visualized by the Chem-Mate TM DAB+ Chromogen, counterstained with hematoxylin, mounted in neutral gum, and analyzed using a bright field microscope.
The immunohistochemically stained tissue sections were reviewed and scored by two independent experienced pathologists blinded to the clinicopathlolgical variables. For each section, 5 high power fields were randomly selected and the staining was evaluated by the extentsity (the percentage of positive tumor cells or normal epithelial cells in relation to the whole tissue area: negative, 0; ≤10%,1; 10–50%,2; ≥51%, 3) and the intensity (absent 0; weak 1; moderate 2; strong 3). The combined score (extensity ×intensity) of 0~4 and 5~9 were defined as low and high expression levels of Akt K64 methylation, Akt T308, JMJD2A and SETDB1, respectively.
Statistics and reproducibility.
For all co-IP and immunostaining were repeated 3 times with similar results obtained. All statistical analyses were processed using SPSS 16.0 statistical software. Spearman’s correlation coefficient was calculated to evaluate the correlation between Akt K64 me3 and SETDB1 expression levels. Survival analysis was performed using Kaplan-Meier method and log-rank test. Overall survival was measured from the date of surgery to the date of death or the date on which the patient was last known to be alive and the data were censored if patients were alive or lost to follow-up. The investigators were blinded to allocation for IHC analyses. P values were generated using a two-sided t-test to calculate statistical significance with P<0.05 representing a statistically significant difference. No statistical method was used to predetermine sample size. No mice that completed in vivo studies were excluded from analyses.
Data availability
Mass spectrometry data have been deposited in ProteomeXchange with the primary accession code PXD011966.
The human lung adenocarcinoma data were derived from the TCGA Research Network: http://cancergenome.nih.gov/. The data-set derived from this resource that supports the findings of this study is available in http://www.cbioportal.org. The human cancer SETDB1 protein expression data were derived from the Human Protein Atlas Network and the supports the findings of this study is available in https://www.proteinatlas.org/ENSG00000143379-SETDB1/pathology.
Source data for Fig. 4, 7 and Supplementary Fig. 3, 6, have been provided in Supplementary Table 4. All other data supporting the findings of this study are available from the corresponding author on reasonable request.
Supplementary Material
Acknowledgments
We are grateful to the members in Lin’s lab for the critical inputs and suggestions. We thank Drs. Zhi-Ping Liu, Dr. Yoichi Shinkai, Dr. Frank J. Rauscher III, Dr. Bert Vogelstein and Dr. Ralf Janknecht for providing mice, cell lines or plasmids. We thank Dr. Eric Spooner for performing Mass Spectrometry assay. We acknowledge the support of Cellular Imaging & flow cytometry Shared of Resource, the Wake Forest Baptist Comprehensive Cancer Center, supported by the National Cancer Institute’s Cancer Center Support Grant (P30CA012197). This work is supported by Start-ups from Wake Forest School of Medicine, NIH grants (R01CA182424 and R01CA193813) to H.K.L and NIH grants (R01CA207098 and R01CA207109) to M.G.L.
Footnotes
Competing interest Statement
The authors declare no competing interests.
References
- 1.Cheung M & Testa JR Diverse mechanisms of AKT pathway activation in human malignancy. Curr Cancer Drug Targets 13, 234–244 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Manning BD & Cantley LC AKT/PKB signaling: navigating downstream. Cell 129, 1261–1274 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yang WL et al. The E3 ligase TRAF6 regulates Akt ubiquitination and activation. Science 325, 1134–1138 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chan CH et al. The Skp2-SCF E3 ligase regulates Akt ubiquitination, glycolysis, herceptin sensitivity, and tumorigenesis. Cell 149, 1098–1111 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang WL, Wu CY, Wu J & Lin HK Regulation of Akt signaling activation by ubiquitination. Cell Cycle 9, 487–497 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hamamoto R, Saloura V & Nakamura Y Critical roles of non-histone protein lysine methylation in human tumorigenesis. Nat Rev Cancer 15, 110–124 (2015). [DOI] [PubMed] [Google Scholar]
- 7.Campaner S et al. The methyltransferase Set7/9 (Setd7) is dispensable for the p53-mediated DNA damage response in vivo. Mol Cell 43, 681–688 (2011). [DOI] [PubMed] [Google Scholar]
- 8.Kunizaki M et al. The lysine 831 of vascular endothelial growth factor receptor 1 is a novel target of methylation by SMYD3. Cancer Res 67, 10759–10765 (2007). [DOI] [PubMed] [Google Scholar]
- 9.Mazur PK et al. SMYD3 links lysine methylation of MAP3K2 to Ras-driven cancer. Nature 510, 283–287 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dasgupta M, Dermawan JK, Willard B & Stark GR STAT3-driven transcription depends upon the dimethylation of K49 by EZH2. Proc Natl Acad Sci U S A 112, 3985–3990 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim E et al. Phosphorylation of EZH2 activates STAT3 signaling via STAT3 methylation and promotes tumorigenicity of glioblastoma stem-like cells. Cancer Cell 23, 839–852 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang X & Bruice TC Enzymatic mechanism and product specificity of SET-domain protein lysine methyltransferases. Proc Natl Acad Sci U S A 105, 5728–5732 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jacob Y et al. Regulation of heterochromatic DNA replication by histone H3 lysine 27 methyltransferases. Nature 466, 987–991 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schultz DC, Ayyanathan K, Negorev D, Maul GG & Rauscher FJ 3rd SETDB1: a novel KAP-1-associated histone H3, lysine 9-specific methyltransferase that contributes to HP1-mediated silencing of euchromatic genes by KRAB zinc-finger proteins. Genes Dev 16, 919–932 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fei Q et al. Histone methyltransferase SETDB1 regulates liver cancer cell growth through methylation of p53. Nat Commun 6, 8651 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim MS, Jeong EG, Yoo NJ & Lee SH Mutational analysis of oncogenic AKT E17K mutation in common solid cancers and acute leukaemias. Br J Cancer 98, 1533–1535 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bleeker FE et al. AKT1(E17K) in human solid tumours. Oncogene 27, 5648–5650 (2008). [DOI] [PubMed] [Google Scholar]
- 18.Carpten JD et al. A transforming mutation in the pleckstrin homology domain of AKT1 in cancer. Nature 448, 439–444 (2007). [DOI] [PubMed] [Google Scholar]
- 19.Gao J et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 6, pl1 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cerami E et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2, 401–404 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gyorffy B, Surowiak P, Budczies J & Lanczky A Online survival analysis software to assess the prognostic value of biomarkers using transcriptomic data in non-small-cell lung cancer. PLoS One 8, e82241 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Uhlen M et al. A pathology atlas of the human cancer transcriptome. Science 357 (2017). [DOI] [PubMed] [Google Scholar]
- 23.Berry WL & Janknecht R KDM4/JMJD2 histone demethylases: epigenetic regulators in cancer cells. Cancer Res 73, 2936–2942 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cantley LC The phosphoinositide 3-kinase pathway. Science 296, 1655–1657 (2002). [DOI] [PubMed] [Google Scholar]
- 25.LC C The phosphoinositide 3-kinase pathway. Science 296, 1655–1657 (2001). [DOI] [PubMed] [Google Scholar]
- 26.Datta SR, B. A, Greenberg ME, Cellular survival: a play in three Akts. Genes & development 13 (1999). [DOI] [PubMed] [Google Scholar]
- 27.Biggar KK & Li SS Non-histone protein methylation as a regulator of cellular signalling and function. Nat Rev Mol Cell Biol 16, 5–17 (2015). [DOI] [PubMed] [Google Scholar]
- 28.Ea CK & Baltimore D Regulation of NF-kappaB activity through lysine monomethylation of p65. Proc Natl Acad Sci U S A 106, 18972–18977 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ceol CJ et al. The histone methyltransferase SETDB1 is recurrently amplified in melanoma and accelerates its onset. Nature 471, 513–517 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rodriguez-Paredes M et al. Gene amplification of the histone methyltransferase SETDB1 contributes to human lung tumorigenesis. Oncogene 33, 2807–2813 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sarraf SA & Stancheva I Methyl-CpG binding protein MBD1 couples histone H3 methylation at lysine 9 by SETDB1 to DNA replication and chromatin assembly. Mol Cell 15, 595–605 (2004). [DOI] [PubMed] [Google Scholar]
- 32.Fritsch L et al. A subset of the histone H3 lysine 9 methyltransferases Suv39h1, G9a, GLP, and SETDB1 participate in a multimeric complex. Mol Cell 37, 46–56 (2010). [DOI] [PubMed] [Google Scholar]
- 33.Jianping Guo XD, Laurent Besnoit, Zheng Nana, Gan Wenjian, Zhang Jian, Guo Ailan, Yuan Min, Liu Pengda, Asara John M., Toker Alex, Shi Yang, Pandolfi Pier Paolo, Wei Wenyi Akt methylation by SETDB1 promotes Akt kinase activity and oncogenic functions. Nature Cell Biology (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim TD et al. Histone demethylase JMJD2A drives prostate tumorigenesis through transcription factor ETV1. J Clin Invest 126, 706–720 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mallette FA & Richard S JMJD2A promotes cellular transformation by blocking cellular senescence through transcriptional repression of the tumor suppressor CHD5. Cell Rep 2, 1233–1243 (2012). [DOI] [PubMed] [Google Scholar]
- 36.Berry WL, Shin S, Lightfoot SA & Janknecht R Oncogenic features of the JMJD2A histone demethylase in breast cancer. Int J Oncol 41, 1701–1706 (2012). [DOI] [PubMed] [Google Scholar]
- 37.Mallette FA et al. RNF8- and RNF168-dependent degradation of KDM4A/JMJD2A triggers 53BP1 recruitment to DNA damage sites. EMBO J 31, 1865–1878 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang WL et al. The E3 ligase TRAF6 regulates Akt ubiquitination and activation. Science 325, 1134–1138 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chan CH et al. The Skp2-SCF E3 ligase regulates Akt ubiquitination, glycolysis, herceptin sensitivity, and tumorigenesis. Cell 149, 1098–1111 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schultz DC, Ayyanathan K, Negorev D, Maul GG & Rauscher FJ 3rd SETDB1: a novel KAP-1-associated histone H3, lysine 9-specific methyltransferase that contributes to HP1-mediated silencing of euchromatic genes by KRAB zinc-finger proteins. Genes Dev 16, 919–932 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cederquist CT et al. Systemic insulin sensitivity is regulated by GPS2 inhibition of AKT ubiquitination and activation in adipose tissue. Mol Metab 6, 125–137 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Mass spectrometry data have been deposited in ProteomeXchange with the primary accession code PXD011966.
The human lung adenocarcinoma data were derived from the TCGA Research Network: http://cancergenome.nih.gov/. The data-set derived from this resource that supports the findings of this study is available in http://www.cbioportal.org. The human cancer SETDB1 protein expression data were derived from the Human Protein Atlas Network and the supports the findings of this study is available in https://www.proteinatlas.org/ENSG00000143379-SETDB1/pathology.
Source data for Fig. 4, 7 and Supplementary Fig. 3, 6, have been provided in Supplementary Table 4. All other data supporting the findings of this study are available from the corresponding author on reasonable request.