

Abstract
Heart disease remains a leading killer in western society and irreversibly impacts the lives of millions of patients annually. While adult mammals do not possess the ability to regenerate functional cardiac tissue, neonatal mammals are capable of robust cardiomyocyte proliferation and regeneration within a week of birth. Given this change in regenerative function through development, the extracellular matrix (ECM) from adult tissues may not be conducive to promoting cardiac regeneration, although conventional ECM therapies rely exclusively on adult-derived tissues. Therefore the potential of ECM derived from neonatal mouse hearts (nmECM) to prevent adverse ventricular remodeling in adults was investigated using an in vivo model of acute myocardial infarction (MI). Following a single administration of nmECM, we observed a significant improvement in heart function while adult heart-derived ECM (amECM) did not improve these parameters. Treatment with nmECM limits scar expansion in the left ventricle and promotes revascularization of the injured region. Furthermore, nmECM induced expression of the ErbB2 receptor, simulating a neonatal-like environment and promoting neuregulin-1 associated cardiac function. Inhibition of the ErbB2 receptor effectively prevents these actions, suggesting its role in the context of nmECM as a therapy. This study shows the potential of a neonatal-derived biological material in vivo, diverting from the conventional use of adult-derived ECM therapies in research and the clinic.
Keywords: extracellular matrix, decellularization, neonatal, myocardial infarction, cardiac regeneration
Graphical Abstract
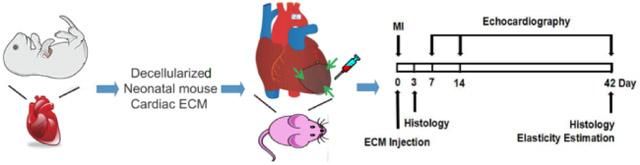
1. Introduction
Coronary heart disease (CHD) remains among the leading causes of death worldwide and accounts for over $100B in direct costs in the United States annually [1]. Myocardial infarction (MI) is one manifestation of this disease characterized by widespread necrosis, chronic inflammation, and the deposition of fibrotic scar tissue. Cardiac function continuously worsens to the point of heart failure and, ultimately, death. Currently available therapies are limited thus far, with a mortality rate of 40% within 5 years post-MI [1]. The success of cell-based therapies in preclinical and clinical trials have been extensively reviewed [2–5], but these approaches are encountering obstacles to their widespread therapeutic translation due to arrhythmias present in large animal models, cell availability for large-scale manufacturing, and low engraftment levels [6]. Additionally, evidence suggests that the implanted cells primarily serve as a source of paracrine signaling rather than as an engrafted proliferative/differentiative source [7]. Therefore, acellular approaches have been proposed as an alternative route to prevent adverse ventricular remodleing post-MI. The extracellular matrix (ECM) is a complex mixture of proteins, glycosaminoglycans (GAGs), and signaling molecules that dictate nearly all aspects of cell behaviors in the body. Biological materials derived from ECM offer similar capabilities to promote pro-repair cellular behavior following injury. Indeed ECM derived from adult porcine tissue is able to improve healing in both small and large animals [8–12]. However, like humans, adult pig hearts rely on scar deposition rather than functional regeneration to respond to myocardial injuries such as MI [13,14]. Other species through development and evolution including: zebrafish, goldfish, newts, salamanders, and neonatal rodents and pigs instead exhibit full cardiac regeneration after injury without the trace of a scar through robust proliferation of cardiomyocytes [14–20]. The exact mechanism of this regenerative process is not yet fully understood, but evidence suggests factors found in the ECM contribute to this response [21–25]. Therefore, it is possible that utilizing cardiac-derived ECM from these regenerative species as a biomaterial could promote functional repair of non-regenerative mammalian hearts. We have previously shown that ECM derived from the zebrafish heart and brain improve cardiac function and optic nerve regeneration, respectively [25,26]. Neonatal rodents exhibit a similar capability to regenerate heart tissue after 10% resection, but the ECM from this immature species has not yet been used as a biomaterial to promote cardiac regeneration in vivo.
Since the discovery of neonatal cardiac regeneration in rodents [20], multiple groups have attempted to identify the proteins responsible for stimulating regeneration in neonatal mice when their adult counterparts exhibit a completely different response [22,23,27–33]. Comparisons in gene and protein expression between adult and neonatal hearts after injury have shown a vast number of differences in their response to injury [29,30,34,35], although the underlying factors causing these protein changes remain unknown. Most of these studies also focus on cellular-level responses to injury and largely neglect the overall contribution of the extracellular matrix. Multiple groups, however, have isolated the ECM from these neonatal mice and used them as a biomaterial for in vitro cardiomyocyte proliferation [23,36]. These studies showed increased cardiomyocyte proliferation in response to neonatal ECM relative to adult mouse ECM [23,36]. Additionally, agrin acting through the Hippo signaling pathway was identified as a key proteoglycan in the ECM necessary to induce cardiomyocyte proliferation in vitro [23]. However, the activity of whole neonatal cardiac ECM on cells in vivo after MI has not yet been explored, and alternative signaling pathways stimulated by neonatal ECM to prevent widespread remodeling in adult models have not yet been identified.
ECM-derived biomaterials have been investigated for decades, and various ECM products have been successfully introduced into clinical practice. The tissues used to develop these products are from adult humans and pigs, and they are relatively thick. Therefore production of these materials largely relies on detergents, acids, and other chemicals to penetrate through the entire tissue [37]. While this results in a successfully decellularized material, these compounds are known disrupters of protein structure and function, which can reduce their efficacy after implantation.
In the present study we have established a protocol to decelluarize neonatal mouse hearts without the use of detergents or chemicals known to degrade proteins with the ECM. We investigated the bioactivity of neonatal mouse cardiac ECM (nmECM) on endothelial cell populations in vitro. The activity of nmECM was further explored in an in vivo model of MI in adult mice, which typically exhibit excessive scarring and decreased cardiac function. Additionally, the epidermal growth factor receptor tyrosine kinase (Erbb) family was identified as critical to the activity of nmECM in vivo as a complement to the recently identified Hippo signaling pathway [23]. Our study reveals the potential of nmECM as a new biomaterial that could be used to inspire new therapies for treating CHD.
2. Materials and Methods
2.1. Animal Use
All animal procedures were reviewed and approved by the Institutional Animal Care and Use Committee at the University of Pittsburgh prior to starting experiments. Mice were kept under standard caging conditions with unrestricted access to food and water. Both male and female neonatal BALB/cJ mice were sacrificed no more than one day after birth to harvest cardiac nmECM. A total of 30 neonatal mice were needed for this study. An additional 90 male BALB/cJ mice (Jackson Laboratory, Bar Harbor, ME, USA) at 9-12 weeks old were used.
2.2. Harvesting Cardiac Tissues for Decellularization
Neonatal mice were sacrificed by decapitation and immersed in 70% ethanol for decontamination before removing hearts with sterile tools. Adult mice were sacrificed by asphyxiation and their hearts removed aseptically. Hearts were washed 3 times in water containing 4% (v/v) antibiotic-antimycotic (A/A) solution (Gibco Laboratories, Gaithersburg, MD, USA) and 100μg/ml gentamycin immediately after harvest. The left ventricle was then dissected aseptically and minced to <1mm3 pieces. Minced heart tissue was washed twice each in water containing 4%, 2%, and 1% A/A solution and 100μg gentamycin (A/A/G solution) and frozen at −80°C until use.
2.3. Decellularization of Mouse Cardiac ECM
Decellularization of mouse cardiac ECM was performed as previously described with slight modification [25]. After thawing, minced tissue was washed once in 1% A/A/G solution and put through 3 freeze-thaw cycles. Each cycle consisted of suspending the tissue in 1% A/A/G solution, submerging in liquid nitrogen for 10 minutes, and then thawing at 37°C. Between each cycle, the supernatant was carefully removed and replaced with fresh 1% A/A/G solution to remove cellular material. Samples were then incubated in red blood cell lysis buffer twice for 20 minutes each to ensure lysis of erythrocytes. The tissue was then incubated in a cocktail containing 250 U/ml deoxyribonuclease I (DNase I, Invitrogen, Carlsbad, CA, USA) and 25 U/ml ribonuclease (RNase, Invitrogen) in 1x DNase buffer (Invitrogen) at 37°C for 1 hour. After removal of the supernatant, this step was repeated to ensure adequate removal of cellular material. The DNase/RNase cocktail was removed, and samples were washed thrice with 0.9% saline to remove any traces of DNase, RNase, and cellular material. After removal of supernatant, ECM samples were lyophilized, then crushed into a fine powder using a liquid nitrogen-cooled mortar (Bel-Art Products, Wayne, NJ, USA). ECM powder was stored at −80°C until use. The entire ECM preparation was performed aseptically.
Before use, ECM samples were thawed, resuspended in 0.9% saline (or cell media for in vitro assays), and sonicated for 15 minutes in a bath sonicator. Large particles were pelleted and removed by centrifuging the suspension at 300 × g. The ECM suspension was then used immediately.
2.4. Composition Analysis of ECM Samples
Both adult and neonatal mouse ECM samples were analyzed for gross collagen, elastin, glycosaminoglycan (GAG) content. To determine collagen content, a Sircol collagen assay kit (Biocolor Ltd, Carrickfergus, County Antrim, UK) was used according to the manufacturer’s instructions. A Fastin™ elastin kit (Biocolor Ltd) was used to determine elastin content using the manufacturer’s instructions. GAG content was quantified using the Blycan™ sulfatate GAG assay kit (Biocolor Ltd.). Absorbance values for each of these assays were measured with a SynergyMX plate reader (Biotek, Winooski, VT, USA).
2.5. Measurement of Endothelial Cell Activity in vitro
Human umbilical vein endothelial cells (HUVEC, ATCC, Manassas, VA) were cultured in EGM-2 media (Lonza, Basel, Switzerland) and grown on standard tissue culture polystyrene dishes for all experiments. Cell activity was measured using a modified previously described protocol [25]. Briefly, 1,000 cells were plated in triplicate in a standard 96-well plate overnight to allow cell attachment. Cells were washed with PBS and the media replaced with treatment-specific media. As a negative control to stimulate nutrient deprivation, EBM-2 media (Lonza) supplemented with 2% fetal bovine serum (FBS) was used. Cells treated with ECM received 2% FBS media containing 25μg (250μg/mL) amECM or nmECM. EBM-2 media supplemented with 10% FBS was used as a positive control. Cultures were incubated 4 days in 2.5% O2 conditions to stimulate hypoxia, and cell activity was then measured with the CellTiter 96® AQueous Cell proliferation assay (Promega, Madison, WI, USA). Cell activity was normalized to the positive control containing 10% FBS. ECM concentrations were determined from a previously optimized protocol [25].
2.6. Measurement of HUVEC Migration in vitro
A Boyden chamber assay was used to determine the effect of nmECM on HUVEC migration as described previously [25]. EBM-2 supplemented with 2% FBS was used in all treatment groups for this assay. HUVECs were plated in the top chamber of a MilliCell® 24-well cell culture insert at 10,000 cells/cm2. The bottom well was loaded with media alone, media supplemented with 125μg amECM, or media supplemented with 125μg nmECM (250μg/mL, n=4 for each treatment group). After 6 hours, membranes were removed from culture media and non-migrated cells removed from the top of the membrane with a cotton-tipped swab. The membranes were submerged in methanol for 10 minutes to fix migrated cells and then stained with Quant-IT PicoGreen dsDNA reagent (Thermo Fisher Scientific, Waltham, MA, USA). Three fluorescent images of each well were captures with a Nikon Eclipse Ti microscope paired with NIS Elements software (Nikon, Tokyo, Japan) and the number of migrated cells in each image counted manually and averaged for each well. The number of migrated cells is normalized to cells incubated without ECM. ECM concentrations were determined from a previously optimized protocol [25].
2.7. Mouse model of MI
MI was induced in mice as previously described [38,39]. Briefly, anesthesia was induced by inhalation of 4% isoflurane and maintained under 2% isoflurane in oxygen after intubation. A left thoracotomy was performed to visualize the heart, and a permanent MI was induced by ligation of the left anterior descending coronary artery with a 7-0 suture. Successful suture placement was confirmed by blanching of the downstream tissue, indicating impaired perfusion. Mice were treated with either 30 μl saline (negative control), 500 μg amECM suspended in 30 μl saline, or 500 μg nmECM suspended in 30 μl saline by direct injection at three points in the ischemic heart tissue (one injection in the infarct zone, two in the border zone) immediately after injury. After closing the chest cavity and suturing the muscle and skin layers, the mouse recovered.
To inhibit ErbB2 activity, a subset of mice were injected with AG825 (Santa Cruz Biotechnology, Dallas, TX, USA). Immediately after administering treatment, AG825 dissolved in DMSO was injected intraperitoneally at 5 mg/kg as previously reported [40,41]. The use of DMSO in this model has previously shown no appreciable effect on cardiac function relative to untreated mice [25]. ECM doses were determined according to a previously published protocol using zebrafish ECM prepared in a similar manner [25].
2.8. Live Animal Echocardiography Imaging
B-mode and M-mode echocardiography were used to assess cardiac function over time in mice before injury and 5 days, 2 weeks, and 6 weeks post-MI as described previously [39,42]. Briefly, anesthetized mice were placed on a heated stage equipped for echocardiography. A high-resolution ultrasound scanner (Vevo 2100; VisualSonics, Torotonto, Ontaria, Canada) equipped with a 30MHz transducer (MS400; VisualSonics) was used to make all imaging measurements. End systolic and end diastolic diameter (ESD and EDD, respectively) were measured from short-axis M-mode scans; a series of ten consecutive heart cycles were analyzed and averaged for each animal. End systolic and end diastolic areas (EDA and ESA, respectively) were taken as the maximum and minimum LV area in short-axis B-mode imaging scans, respectively. Fractional shortening (FS), fractional area change (FAC), and ejection fraction (EF) were measured as previously described [43,44].
In vivo strain measurements were quantified using short axis B-mode echocardiography images in the VevoStrain software package (VisualSonics). Three animals were analyzed in each group as previously described [45,46]. Briefly, radial and circumferential strain within the ventricle wall was measured using speckle tracking at 5 points around the ventricle: one in the injured region and four in the uninjured region. The strain value at the injured region was normalized to the average of the four measurements taken in the uninjured region to determine ventricle wall stiffness relative to healthy cardiac tissue in the same heart.
2.9. Histology and Immunofluorescent Staining
Mice were sacrificed 3 days and 6 weeks post-MI for histological analysis. After inducing deep anesthesia, an intracardiac injection of 1M potassium chloride in saline was administered to arrest the heart in diastole. Hearts were harvested, embedded in O.C.T. compound (Sakura Finetek USA Inc., Torrance, CA, USA) and flash-frozen in liquid nitrogen-cooled 2-methylbutane before cryosectioning at an 8 μm thickness. Serial sections from the apex to level of ligation were placed on same slide. Sections were fixed with a mixture of ice-cold methanol and acetone for 5 minutes or in 4% paraformaldehyde in phosphate-buffered saline (PBS) for 8 minutes at room temperature immediately before staining. Masson’s trichrome stain was performed according to a standard protocol before mounting and imaging. Fibrotic areas stained in blue were measured to calculate the percent of the ventricle circumference covered by scar tissue.
To quantify revascularization of the tissue, endothelial cells were stained with a goal polyclonal CD31 antibody (1:100, AF3628; R&D Systems, Minneapolis, MN, USA) followed by a goat anti-rabbit AlexaFluor594 antibody (1:400, A-11058; Life Technologies). Smooth muscle cells surrounding the endothelial cells were detected with a mouse anti-mammalian alpha-smooth muscle actin (αSMA) (diluted at 1:100; A5228; Sigma-Aldrich, St. Louis, MO, USA) followed by goat anti-mouse Alexa488 secondary antibody (1:400, A-11001; Life Technologies). Angiogenesis was quantified by the density of CD31+ endothelial cells and αSMA+ vessles in the infarct region and the border zones. It was additionally quantified by the density of CD31+ blood vessels and blood vessels expressing αSMA.
The presence of macrophages as a sign of long-term inflammation in the cardiac tissue was detected with a rat anti-mouse CD68 pan macrophage antibody (1:200, ab53444; Abcam, Cambridge, MA, USA) followed by a goat anti-rat AlexaFluor488 antibody (1:400; A-11006; Life Technologies). Macrophage density in the injured region was counted as the number of CD68+ cells present within a field of view and averaged across samples.
Proliferating cardiomyocytes were detected by first preparing samples with the mouse-on-mouse antibody staining kit per manufacturer’s instructions (Vector Laboratories, Burlingame, CA, USA). A mouse cTnT primary antibody (1:200, ab10214; Abcam) followed by goat anti-mouse Alexa488 secondary antibody (1:400, A-11001; Life Technologies) were used to mark cardiomyocytes. A rabbit polyclonal Ki67 antibody (1:200, ab15580; Abcam) along with a donkey anti-rabbit AlexaFluor488 antibody were used to detect proliferating cells. The number of CTnT+/Ki67+ cells were counted in both the infarct and border zone of mice with and without ErbB2 inhibition.
ErbB2-expressing cardiomyocytes were detected using the same CTnT marker described above. ErbB2 was detected using a chicken polyclonal ErbB2 antibody (1:100, ab14027; Abcam) followed by a goat anti-chicken AlexaFluor594 antibody (1:400, A-11042; Life Technologies). The density of ErbB2-expressing cardiomyocytes was quantified by counting the number of CTnT+/ErbB2+ cells within the infarct and border zones.
Nuclei in all sections were marked with 4’,6-diamidino-2-phenylindole (DAPI) in addition to the antibodies described above. The infarct zone was defined as the visually damaged region of the ventricle; the border zone was defined as the field of view immediately adjacent to the visibly damaged region of the ventricle. Five images from the border and infarct zones were analyzed for each sample. All imaging was performed with a Nikon Eclipse Ti inverted microscope equipped with NIS Elements software (Nikon).
2.10. Western Blot
Proteins were extracted from neonatal and adult mouse ventricles using RIPA lysis buffer (ProteinSimple, San Jose, CA, USA). 50 μg protein was separated on a polyacrylamide gel, transferred to a PVDF membrane, and stained with a rabbit polyclonal NRG1 antibody (1:300, ab27303; Abcam). A horseradish peroxidase-conjugated secondary antibody (1:1000, SC-2004; Santa Cruz Biotechnology) and reaction with horseradish peroxidase (HRP) allowed visualization of the NRG1 band. The membrane was imaged with a ChemiDic XRS+ imaging system (Bio-Rad Laboratories, Hercules, CA, USA), and ImageJ was used to measure band density. A total of four neonatal and adult hearts were each analyzed for NRG1 levels.
2.11. Statistical Analysis
Data is presented as mean ± standard deviation (SD). A two-tailed t-test was used to detect significant differences when only two groups were compared with a threshold p ≤ 0.05. One-way analysis of variance (ANOVA) was used to detect differences in histological and immunofluorescent measurements followed by Tukey’s post hoc analysis when two or more treatment groups were compared. Differences in echocardiography-based measurements were detected by two-way mixed ANOVA (comparisons between timepoints and treatment groups) followed by pairwise comparisons with a Bonferroni correction. All analyses were performed with SigmaStat 3.5 (Systat Software, San Jose, CA, USA), GraphPad Prism 5.0 (La Jolla, CA, USA), and SPSS v21 (IBM, Armonk, NY, USA).
3. Results
3.1. Neonatal and adult-derived tissues are distinct biomaterials
Neonatal and adult mouse hearts were decelluarized using a physical rather than detergent-based method. The final yield of lyophilized ECM was approximately 6-8% the wet weight of the starting tissue. More importantly, the DNA content within the material was 33ng/mg lyophilized ECM, well below the threshold for decellularized materials [37]. Basic characterization of the ECM powders revealed that nmECM contains 50.7% the concentration of collagen (amECM: 408.8 ± 25.81 μg/mg, nmECM: 207.3 ± 23.54 μg/mg, n = 4, p < 0.01), 182.3% the concentration of elastin (amECM: 45.7 ± 9.51 μg/mg, nmECM: 83.33 ± 5.03 μg/mg, n = 4, p < 0.01), and 154.0% the concentration of glycosaminoglycans (GAGs) relative to amECM (amECM: 2.35 ± 0.58 μg/mg, nmECM: 3.62 ± 0.26 μg/mg, n = 4, p < 0.05) (all measurements normalized to dry ECM weight). Although this analysis only includes 3 of the most abundant ECM components, these results suggest drastic differences in ECM composition between neonatal and adult-derived heart ECM.
3.2. Neonatal ECM improves cardiac function in vivo
An adult mouse MI model was used to determine the effect of nmECM on cardiac function. Either saline, amECM, or nmECM was injected directly into the injured ventricle immediately after MI induction. Echocardiography measurements were taken at 5 days, 2 weeks, and 6 weeks post-MI. Short-axis echocardiography revealed that while saline-treated mice exhibited enlargement of the left ventricle at diastole (end diastolic area, EDA) (Figure 1A,B), this adverse effect was drastically reduced in mice treated with single administration of nmECM at the 6 week timepoint (p < 0.05 at 2 weeks relative to saline, p < 0.01 at 6 weeks relative to saline and amECM). A similar trend was seen in end systolic area (ESA) measurements (p < 0.01 at 6 weeks relative to saline and amECM) (Figure 1C). Fractional area change (FAC), fractional shortening (FS), and ejection fraction (EF), measurements of cardiac contractile function, showed continuous decline in saline-treated mice. Mice treated with amECM did not show significant improvement in any of these measurements up to 6 weeks post-MI. However, nmECM-treated mice showed a significant increase in all these parameters relative to saline and amECM at both 2 and 6 weeks post-MI (FAC: p < 0.05 relative to saline at 2 weeks, p < 0.01 relative to saline and amECM at 6 weeks; FS and EF: p < 0.05 relative to saline and amECM at 2 weeks, p < 0.01 relative to saline and amECM at 6 weeks) (Figure 1D-F). These results indicate the ability of nmECM to better maintain the native dimension and function of the heart following injury relative to adult-derived ECM or saline.
Figure 1. In vivo echocardiographic analysis of cardiac function.

(A) Short-axis B-mode echocardiography images of the left ventricle at diastole showing expansion of the left ventricle 6 weeks post-injury. Quantification of end diastolic (B) and systolic (C) area show reduced ventricle size in nmECM-treated mice. Cardiac function, shown as fractional area change (D), fractional shortening (E), and ejection fraction (F), is not preserved in saline or amECM-treated mice but significantly improved in nmECM-treated mice. (n = 8 per group; *p < 0.05, **p < 0.01 relative to saline; #p < 0.05, ## p < 0.01 relative to amECM).
3.3. Neonatal ECM reduces MI-induced fibrosis
MI in adult mammals results in the deposition of a collagen-rich scar at the site of infarction. Over time, this scar expands through adverse ventricular remodeling, ultimately resulting in a ventricle wall that is much stiffer and thinner than healthy myocardium. To examine the effect of nmECM on this response, Masson’s trichrome staining was used on samples taken 6 weeks post-MI (Figure 2A). Quantifying the fraction area of the ventricle occupied by fibrotic tissue in blue revealed that nearly 60% of the ventricle area consisted of scar tissue in saline-treated mice, and this was not significantly different in amECM-treated mice. However, treatment with nmECM drastically reduced the fractional area occupied by a collagen-rich scar (p < 0.01 relative to saline, p < 0.05 relative to amECM) (Figure 2B). Additionally, histology showed a visibly thinner ventricle wall in saline-treated mice relative to either ECM treatment (Figure 2A). The chronic presence of phagocytic cells at 6 weeks post-MI can at least partially explain this effect. CD68+ cells were seen across all treatment groups following injury (Figure 2C), but the cell density was reduced in nmECM-treated mice relative to both saline and amECM treatments (p < 0.01 relative to saline, p < 0.05 relative to amECM) (Figure 2D).
Figure 2. Fibrosis and inflammation in response to MI.

nmECM prevents widespread scar expansion and inflammation in the infarcted heart. (A) Representative images of Masson’s trichrome staining reveals scar expansion through the ventricle wall 6 weeks post-injury (scale bar = 1mm). (B) Quantification of fibrotic area reveals a significant reduction in scar tissue in nmECM-treated mice. (C) CD68+ macrophages were stained (scale bar = 50μm) and quantified (D) in the infarct zone 6-weeks post-MI, revealing a significant reduction in the number of macrophages in nmECM-treated mice. (n = 4 per treatment group; **p < 0.01 relative to saline; #p < 0.05 relative to amECM)
Presence of a collagen scar within the ventricle wall increases stiffness and reduces the ability of the heart to contract. To examine the mechanical effect of this collagen scar in vivo, circumferential and radial strain measurements were taken via echocardiography analysis. Strain measurements were taken at the infarct zone of each mouse 6 weeks post-MI and normalized to the strain values measured at 4 non-infarcted sections of the ventricle wall (Figure 3A). At 6 weeks post-MI, both radial and circumferential strain values at the infarct zone in all 3 groups were lower than the uninjured tissue, likely due to the presence of the scar in the infarct zone (Figure 3B-D). Among treatment groups, saline-treated mice exhibited lowest strain values in both the radial and circumferential directions. amECM significantly increased these values relative to saline (p < 0.05 in radial and circumferential strain), and nmECM further increased strain values relative to both saline and amECM treatment (p < 0.01 relative to saline, p < 0.05 relative to amECM in radial and circumferential strain) (Figure 3B,C). These increased strain values show a partial retention of ventricle elasticity due to ECM treatments, although this is more pronounced under nmECM. Together this data shows a reduction in scar expansion and ventricle stiffening due to nmECM treatment.
Figure 3. In vivo analysis of myocardial strain.

(A) Representative B-mode echocardiography images show the five regions of interest selected at diastole and systole for analysis of myocardial strain and consists of the infarct area and four uninjured areas to serve as internal controls of relative strain. Quantification of radial (B) and circumferential (C) strain show increased wall stiffness thickness in response to amECM, although this effect is further exacerbated in nmECM-treated mice. (D) Representative graphs of radial and circumferential strain are shown within the infarct area (navy) relative to the uninjured areas (yellow, green, red, light blue) in response to each treatment (n = 3 per treatment group; *p < 0.05, **p < 0.01 relative to saline; #p < 0.05 relative to amECM).
3.4. Neonatal ECM promotes angiogenesis and endothelial cell activity
Human umbilical vein endothelial cells (HUVEC) were grown in vitro to determine the effect of nmECM on their migration and metabolism. In nutrient-deprived cell media, HUVEC migration increased over 5-fold relative to saline when treated with nmECM (p < 0.01 relative to saline, p < 0.05 relative to amECM) while amECM did not stimulate cell migration (Figure 4A). Cells were also grown in both hypoxic (2.5% O2) and nutrient-deprived media to measure metabolic activity in stressed conditions. In cells treated with saline, this resulted in a significant reduction in cell activity relative to cells grown in nutrient-rich media. amECM treatment did not drastically improve metabolic activity in these cells, but nmECM treatment increased cell activity relative to both saline and amECM treatments (p < 0.01 relative to saline, p < 0.05 relative to amECM) (Figure 4B). These data suggest the capacity of nmECM to stimulate migration and metabolic activity of vascular endothelial cells that could contribute to angiogenesis.
Figure 4. In vitro and in vivo stimulation of angiogenesis.

(A) In vitro HUVEC migration assays show significantly higher migration in response to nmECM than saline or amECM. (B) In oxygen-deprived (2.5% O2) and nutrient-deprived culture conditions, nmECM significantly preserves cell activity relative to amECM or 2% FBS control. (C) Representative images of CD31+ endothelial cells and surrounding αSMA+ smooth muscle cells reveal vasculature in uninjured hearts and in response to each treatment after MI (scale bar = 50μm). Quantification of CD31+ (D) and αSMA+ (E) cells reveal increased density of vascular cells in response to both amECM and nmECM, although nmECM produces significantly more than amECM-treated mice. The density of CD31+ blood vessels (F) and blood vessels surrounded by αSMA-expressing cells (G) showed a similar trend. (n = 4 per treatment group in all panels; *p < 0.05, **p < 0.01 relative to saline; #p < 0.05 relative to amECM).
In vivo, CD31+ endothelial cells (EC) and αSMA+ smooth muscle cells were identified 6 weeks post-MI with immunofluorescent staining (Figure 4C). The density of CD31+ EC and blood vessel structures both increased as a result of amECM treatment relative to saline (p < 0.05), but treatment with nmECM surpassed both saline and amECM (p < 0.01 relative to saline, p < 0.05 relative to amECM) (Figure 4D,F). Additionally, the density of αSMA+ cells and small blood vessels beyond capillaries, indicated by ECs surrounded by smooth muscle cells, similarly increased as a result of ECM treatment (amECM p < 0.05 relative to saline; nmECM p < 0.01 relative to saline, p < 0.05 relative to amECM) (Figure 4E,G). Together this data shows the ability of nmECM to stimulate angiogenesis and vasculogenesis in nutrient-deprived conditions such as the post-MI heart.
3.5. Erbb signaling is vital to nmECM-induced cardiac performance
Erbb signaling has previously been explored in the context of neonatal cardiac regeneration and has been suggested as a vital pathway in this process [27][47]. While high in embryonic and neonatal mouse hearts, ErbB2 expression in cardiomyocytes decreases with age as the mouse loses the ability to regenerate cardiac tissue [27]. We therefore investigated the ability of nmECM to re-induce ErbB2 expression in cardiomyocytes and its role in the effects described above. Immunofluorescent co-staining of cTnT+ cardiomyocytes and ErbB2 revealed as reported previously that uninjured adult mouse cardiomyocytes do not express ErbB2 in detectable levels (Figure 5A) [27]. Even after injury, saline treated mice exhibit very low levels of ErbB2 expression, and amECM does not increase the density of ErbB2-expressing cardiomyocytes. However, treatment with nmECM drastically increases the expression of ErbB2 in cardiomyocytes relative to saline and amECM-treated mice (p < 0.001 relative to saline and amECM) (Figure 5B,C). This suggests an ability of neonatal-derived ECM to induce environmental cues resembling the neonatal state of cardiac development.
Figure 5. ErbB2/Neuregulin-1 signaling within the neonatal heart ECM.

(A) Immunostaining reveals no colocalization of ErbB2+ and CTnT+ cells in uninjured adult hearts. (B,C) Representative images of hearts following injury reveal few detectable CTnT+/ErbB2+ cells in saline or amECM treatment groups relative to those treated with nmECM (scale bar = 50μm; n = 4 per treatment group; ***p < 0.001 relative to saline, ###p < 0.001 relative to amECM). (D) Western blotting and (E) densitometry analysis shows increased levels of NRG1 within nmECM relative to amECM. (n = 4 per treatment group; ***p < 0.001 relative to amECM).
The Erbb receptors have multiple extracellular ligands that could be responsible for nmECM-stimulated cardiac repair. One such ligand is neuregulin-1 (NRG1), a mitogen known to induce cardiomyocyte proliferation. NRG1 expression in intact mouse hearts was quantified, which revealed a 5-fold increase in protein expression in neonatal mouse hearts relative to adult (p < 0.001) (Figure 5D,E). Although not exhaustive, this result further validates the possibility that nmECM is acting through Erbb-based signaling to improve cardiac health in adult mice post-MI.
To investigate the effect of ErbB2 upregulation in cardiomyocytes seen after nmECM treatment, an ErbB2 inhibitor was administered directly after establishment of MI. In contrast with the results seen in wild-type mice, inhibition of ErbB2 eliminated any differences seen between treatment groups in echocardiography-based measurements such as ESA, EDA, FAC, and EF (Figure 6A-D). On a cellular level, nmECM treatment did not increase angiogenesis in the infarct zone when ErbB2 was inhibited 6 weeks post-MI (Figure 6E,F) or stimulate cardiomyocyte proliferation 3 days post-MI (Figure 6G,H). This reveals widespread effects both on cell activity and cardiac function as a result of Erbb pathway disruption in mice treated with nmECM. This indicates that Erbb signaling is at least partially responsible for nmECM-stimulated improvements in cardiac function.
Figure 6. Effect of ErbB2 inhibition on cardiac function with ECM treatment.

Ventricle dilation at diastole (A) and systole (B) are no longer inhibited in ErbB2-inhibited mice treated with nmECM. (C) FAC and (D) EF are similar in all treatment groups when an ErbB2 inhibitor is administered at the time of MI. (E,F) Immunostaining of CD31+ and αSMA+ cells 6 weeks post-MI show similar numbers of blood vessels in all treatment groups after ErbB2 inhibition. (G,H) Immunostaining of CTnT+/Ki67+ in mice following ErbB2 inhibition shows similar numbers of proliferating cardiomyocytes 3 days post-MI regardless of treatment group (scale bar = 50μm; n = 4 per treatment group; p > 0.05 for all measurements).
4. Discussion
Unlike non-mammalian models of cardiac regeneration such as the zebrafish and salamander, mammals only exhibit this potential in fetal and neonatal stages of life [14,16–20]. Within days of birth, the response to cardiac injury shifts from regeneration to a fibrotic response. This provides an interesting model to determine changes in gene and protein expression that lead to these diverging phenomena, although relatively few studies focus on changes in the ECM that are involved with these changes. We proposed that the ECM at each stage of development provides biological cues to promote regeneration or fibrosis and hypothesized that ECM derived from neonatal mice can prevent widespread ventricular remodeling following injury in an adult model of MI. Previous studies, particularly those investigating mesenchymal stem cells, have consistently shown that young ECM is able stimulate expansion of adult cells and contribute to osteogenesis more effectively than adult ECM [48–50]. Placental ECM has also been proposed to assist in a number of medical applications [51,52]. Together these studies suggest a distinct advantage of using young and fetal ECM over standard adult sources. Although we are not the first to utilize neonatal-derived ECM for cardiac disease [23,36], this is the first report of utilizing whole ECM in vivo as a biomaterial to maintain cardiac structure and function. This study elucidates the functional-level responses to cardiac injury in adult mice when treated with neonatal or adult mouse ECM and identifies an suggests a signaling pathway that partially accounts for these differences.
Tissue decellularization must strike a balance between removing cellular components and preserving bioactivity. Adverse outcomes have been observed as a consequence of tissues that haven’t been sufficiently decellularized [53,54]. Multiple reviews have thoroughly discussed the effect of different decellularization methods on final biomaterials[37,55]. The present study utilized a detergent-free decellularization method in order to preserve bioactivity of the matrix, and we saw sufficient removal of DNA within our tissue according to previously published standards [37]. In order to ensure other cellular components are not stimulating an adverse immune response, future studies will thoroughly investigate the host response to this material after implantation.
Treatment of acute MI in adult mice revealed that nmECM prevents widespread ventricular remodeling and its associated decline in ventricular up to 6 weeks in vivo. This occurs through prevention of scar expansion and increased angiogenesis into the MI region. Functionally, treatment with nmECM preserved ventricle elasticity and cardiac contractile function measured as EF, FAC, and FS. In contrast, amECM did not significantly improve these parameters on a cellular or functional level relative to treatment with saline. The performance of amECM is surprisingly minimal considering the large body of work that has shown improved cardiac performance using adult mammal-derived ECM [8–10,12,56–58]. These differences could arise from the dose, method of decellularization, or timing of intervention/analysis, but further studies into these discrepancies could provide further insight into the mechanisms underlying successful ECM-based therapies. Additionally, we have not yet optimized the ECM dose in this MI model; the use of slightly different models such as the MI-reperfusion model [59] or different ECM doses could potentially affect the results reported here. Singelyn et al. [59] uses a slightly lower dose of adult porcine ECM (450μg) but utilizes a MI-reperfusion model, delays treatment for 2 weeks after infarct, and decellularizes tissue using different methods from this study. This introduces many confounding variables between these studies and makes direct comparisons difficult. Further extrapolating this results to a porcine study [8] is even more difficult. A future study comparing the efficacy of different decellularization methods in vivo could allow for more direct comparisons; additionally it would provide the field with crucial data to consider when designing new ECM therapies. This study does not attempt to discredit these previous studies using adult-derived ECM sources but rather to provide evidence that alternative ECM sources can be studied in order to improve the response of adult mammals to MI.
Gross content of structural proteins and GAGs were significantly different between adult and neonatal ECM; nmECM contains a higher concentration of GAGs, which are known to protect soluble signaling factors and facilitate interactions with their receptors [60–62]. Previous studies have shown that ECMs containing GAGs are able to sequester growth factors from their surroundings [63,64]. It is therefore possible that GAGs present in nmECM are able to increase the bioactivity of signaling factors present in the MI region to facilitate a more effective response. We additionally detected higher concentrations of elastin in nmECM versus amECM. Previous studies suggest that high levels of elastin preserves myocardial structure and function after MI [65], which is consistent with our findings after nmECM treatment.
The effect of nmECM on chronic presence of macrophages was quantified by the number of CD68+ cells present within the infarct 6 weeks post-MI. Macrophages play a crucial role in both neonatal and adult heart regeneration [66,67]. However, chronic presence of macrophages has also been linked to increased ventricular remodeling and interstitial fibrosis [67]. This study reports only the presence of macrophages at 6 weeks post-MI, well after resolution of the acute inflammatory phase. Therefore a reduced number of macrophages was interpreted as a positive outcome, especially since the use of neonatal ECM prevented widespread fibrosis. A future study looking at the time-course of inflammation is warranted to understand how neonatal ECM affects the immune response to MI in adult mammals.
Cardiomyocyte proliferation is a controversial area within the field of cardiac regeneration, especially in adult animals after MI. Although previous studies have shown that nmECM likely stimulates cardiomyocyte proliferation in vitro [23,36], our future studies will investigate this effect in vivo to determine if nmECM is able to stimulate a similar effect in the complex post-MI environment and if it is sufficient to stimulate functional cardiac repair.
A recent study identified agrin as a vital mediator of cardiac regeneration in neonatal mice and showed that administration of agrin in adult mice following MI promotes robust cardiomyocyte proliferation and preservation of cardiac function while preventing fibrosis [23]. Here we sought to investigate an additional signaling pathway known to be vital in neonatal cardiac regeneration. Many protein-level differences are seen between adult and neonatal tissues that are known to contribute to regeneration [15], and several could be playing a role in the differences seen in the present study. Signaling through ErbB2 at the neonatal stage is vital to cardiac regeneration, and the reduction of ErbB2 expression during development corresponds with the switch from cardiac regeneration to fibrosis in mice following the neonatal stage [27,47,68]. A previous study has shown that reestablishment of ErbB2 in adult cardiomyocytes after injury promotes cardiac regeneration in vivo [27]. Therefore, we determined the role of nmECM and amECM on ErbB2 expression following MI. Immunostaining clearly revealed that nmECM stimulates expression of ErbB2 in adult mice, which is not seen in uninjured mice or injured mice treated with saline or amECM. To further validate that reexpression of ErbB2 is at least partially responsible for the effects seen in mice treated with nmECM, adult mice were treated with an ErbB2 inhibitor in addition to nmECM or amECM. These mice treated with nmECM no longer exhibited any improvements in cardiac function or angiogenesis, indicating the Erbb signaling pathway is crucial to preventing adverse remodeling in response to nmECM after MI in adult mammals.
The ErbB2 co-receptor interacts with several ligands when complexed with Erbb1/3/4, including NRG1. Previous reports have shown that overexpression of NRG1 enhances cardiomyocyte dedifferentiation and proliferation [21], and that ErbB2 expression is necessary for NRG1-mediated cardiomyocyte proliferation. After identifying the increase in ErbB2 expression in nmECM-treated mice, the potential role of the NRG1 ligand was further investigated. Western blot revealed that neonatal ECM contained significantly higher concentrations of NRG1 than adult ECM; this result paired with the increased expression of ErbB2 shows evidence that Erbb and NRG1 signaling are at least partially responsible for the improved cardiac function seen in nmECM-treated animals.
Although the increase in ErbB2 expression is interesting, especially when paired with the increase in NRG1 seen in nmECM, these findings do not fully elucidate the mechanism of action behind nmECM. While ErbB2 upregulation does increase the ability of NRG1 to induce cardiomyocyte proliferation [28], NRG1 alone is not thought to increase expression of ErbB2 [27], indicating that other unknown mechanisms are likely responsible for this change. Additionally, while NRG1 is the Erbb ligand most typically associated with the heart, additional ligands such as epidermal growth factor, transforming growth factor alpha, and neuregulins 2-4 have not yet been investigated in the context of neonatal ECM and could also be contributing. Further studies to identify the roles of each of these ligands and each Erbb dimer will reveal the mechanistic pathway associated with improved cardiac health following treatment with nmECM. Additionally, further studies are needed to elucidate the underlying mechanism that results in ErbB2 reexpression in cardiomyocytes after nmECM treatment, which is a necessary component of Erbb-based signaling in the heart.
While ErbB2 is the focus of this study, it is entirely possible that other signaling pathways may also be at play. Indeed agrin within the neonatal ECM is already known to signal through a different mechanism [23]. Indeed Bassat et al. [23] has already reported mass spectrometry data comparing ECM from different neonatal stages of development and listed multiple other proteins that may be playing a role. Future studies using a more holistic approach to compare ECM components between neonatal and adult mice can similarly elucidate additional signaling pathways, proteins, and growth factors crucial for stimulating cardiac repair with neonatal ECM.
5. Conclusion
This study demonstrates that ECM derived from the neonatal mouse heart prevents widespread ventricular remodeling in adult mice following myocardial infarction. MI-associated ventricle wall stiffening, fibrosis, and worsening EF all decreased after a single treatment with nmECM. Adult-derived ECM, on the other hand, did not significantly improve any of these parameters in vivo relative to saline in the time course of this study. Moreover, we identified the Erbb signaling pathway as a vital component of nmECM-induced improvements and NRG1 as one ligand playing a role in this response. Overall, this study demonstrates the utility of neonatal ECM in vivo to heal cardiac tissues after injury and shows the potential of neonatal-derived materials as a biomaterial in adult mammals.
Statement of Significance.
The of use extracellular matrix biomaterials to aid tissue repair has been previously reported in many forms of injury. The majority of ECM studies to date utilized ECM derived from adult tissues that are not able to fully regenerate functional tissue. In contrast, this study tests the ability of ECM derived from a regenerative organ, the neonatal heart, to stimulate functional cardiac repair after MI. This study is the first to test its potential in vivo. Our results indicate that extracellular factors present in the neonatal environment can be used to alter the healing response in adults, and we have identified the role of ErbB2 in neonatal ECM-based cardiac repair.
Acknowledgements
This study was supported by the National Institutes of Health (grant numbers 1S10RR027383-01, 5T32HL007208, 5T32EB003392-13) and an internal fund from University of Pittsburgh.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Works Cited
- [1].Benjamin EJ, Salim Virani CS, Chair Clifton Callaway C-VW, Chamberlain AM, Alexander Chang MR, Susan Cheng M, Stephanie Chiuve ME, Mary Cushman S, Francesca Delling FN, Rajat Deo M, Sarah de Ferranti MD, Jane Ferguson FF, Fornage M, Cathleen Gillespie F, Carmen Isasi MR, Monik Jiménez FC, Lori Chaffin Jordan S, Judd SE, Daniel Lackland M, Judith Lichtman FH, Lynda Lisabeth F, Simin Liu F, Chris Longenecker FT, Lutsey PL, Jason Mackey FS, Matchar DB, Matsushita K, Michael Mussolino FE, Khurram Nasir F, Martin FO, Palaniappan LP, Ambarish Pandey F, Pandey DK, Mathew Reeves FJ, Ritchey MD, Carlos Rodriguez MJ, Gregory Roth FA, Wayne Rosamond FD, Uchechukwu Sampson FK, Gary Satou FM, Svati Shah FH, Nicole Spartano FL, Tirschwell DL, Tsao CW, Jenifer Voeks MH, Willey JZ, John Wilkins MT, Jason Wu FH, Heather Alger FM, Wong SS, Paul Muntner F, Heart Disease and Stroke Statistics-2018 Update A Report From the American Heart Association, Circulation. 137 (2018) 67–492. doi: 10.1161/CIR.0000000000000558. [DOI] [PubMed] [Google Scholar]
- [2].Higuchi A, Ku N-J, Tseng Y-C, Pan C-H, Li H-F, Kumar SS, Ling Q-D, Chang Y, Alarfaj AA, Munusamy MA, Benelli G, Murugan K, Stem cell therapies for myocardial infarction in clinical trials: bioengineering and biomaterial aspects, Lab. Investig. 97 (2017) 1167–1179. doi: 10.1038/labinvest.2017.100. [DOI] [PubMed] [Google Scholar]
- [3].Krishna KA, Krishna KS, Berrocal R, Rao KS, Sambasiva Rao KRS, Myocardial infarction and stem cells., J. Pharm. Bioallied Sci 3 (2011) 182–8. doi: 10.4103/0975-7406.80761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Madigan M, Atoui R, Therapeutic Use of Stem Cells for Myocardial Infarction, Bioeng. (Basel, Switzerland: ). 5 (2018). doi: 10.3390/bioengineering5020028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Satessa Jima GD, Stem Cell Therapy for Myocardial Infarction: Challenges and Prospects, J. Stem Cell Res. Ther. 05 (2015) 1–5. doi: 10.4172/2157-7633.1000270. [DOI] [Google Scholar]
- [6].Nguyen PK, Neofytou E, Rhee J-W, Wu JC, Potential Strategies to Address the Major Clinical Barriers Facing Stem Cell Regenerative Therapy for Cardiovascular Disease: A Review., JAMA Cardiol. 1 (2016) 953–962. doi: 10.1001/jamacardio.2016.2750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Bellamy V, Vanneaux V, Bel A, Nemetalla H, Emmanuelle Boitard S, Farouz Y, Joanne P, Perier M-C, Robidel E, Mandet C, Hagège A, Bruneval P, Larghero J, Agbulut O, Menasché P, Long-term functional benefits of human embryonic stem cell-derived cardiac progenitors embedded into a fibrin scaffold, J. Hear. Lung Transplant. 34 (2015) 1198–1207. doi: 10.1016/J.HEALUN.2014.10.008. [DOI] [PubMed] [Google Scholar]
- [8].Seif-Naraghi SB, Singelyn JM, Salvatore MA, Osborn KG, Wang JJ, Sampat U, Kwan OL, Strachan GM, Wong J, Schup-Magoffin PJ, Braden RL, Bartels K, DeQuach JA, Preul M, Kinsey AM, DeMaria AN, Dib N, Christman KL, Safety and efficacy of an injectable extracellular matrix hydrogel for treating myocardial infarction, Sci Transl Med. 5 (2013) 173ra25. doi: 10.1126/scitranslmed.3005503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Singelyn JM, DeQuach JA, Seif-Naraghi SB, Littlefield RB, Schup-Magoffin PJ, Christman KL, Naturally derived myocardial matrix as an injectable scaffold for cardiac tissue engineering, Biomaterials. 30 (2009) 5409–5416. doi: 10.1016/j.biomaterials.2009.06.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Wainwright JM, Hashizume R, Fujimoto KL, Remlinger NT, Pesyna C, Wagner WR, Tobita K, Gilbert TW, Badylak SF, Right Ventricular Outflow Tract Repair with a Cardiac Biologic Scaffold, Cells Tissues Organs. 195 (2012) 159–170. doi: 10.1159/000331400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].D’Amore A, Yoshizumi T, Luketich SK, Wolf MT, Gu X, Cammarata M, Hoff R, Badylak SF, Wagner WR, Bi-layered polyurethane - Extracellular matrix cardiac patch improves ischemic ventricular wall remodeling in a rat model, Biomaterials. 107 (2016) 1–14. doi: 10.1016/j.biomaterials.2016.07.039. [DOI] [PubMed] [Google Scholar]
- [12].Robinson KA, Li J, Mathison M, Redkar A, Cui J, Chronos NA, Matheny RG, Badylak SF, Extracellular matrix scaffold for cardiac repair, Circulation. 112 (2005) I135–43. doi: 10.1161/CIRCULATIONAHA.104.525436. [DOI] [PubMed] [Google Scholar]
- [13].Dib N, Diethrich EB, Campbell A, Gahremanpour A, McGarry M, Opie SR, A percutaneous swine model of myocardial infarction, J. Pharmacol. Toxicol. Methods. 53 (2006) 256–263. doi: 10.1016/J.VASCN.2005.10.005. [DOI] [PubMed] [Google Scholar]
- [14].Ye L, D’Agostino G, Loo SJ, Wang CX, Su LP, Tan SH, Tee GZ, Pua CJ, Pena EM, Cheng RB, Chen WC, Abdurrachim D, Lalic J, Tan RS, Lee TH, Zhang J, Cook SA, Early Regenerative Capacity in the Porcine Heart, Circulation. (2018) CIRCULATIONAHA.117.031542. doi: 10.1161/CIRCULATIONAHA.117.031542. [DOI] [PubMed] [Google Scholar]
- [15].Uygur A, Lee RT, Mechanisms of Cardiac Regeneration., Dev. Cell. 36 (2016) 362–74. doi: 10.1016/j.devcel.2016.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Poss KD, Wilson LG, Keating MT, Heart regeneration in zebrafish., Science. 298 (2002) 2188–90. doi: 10.1126/science.1077857. [DOI] [PubMed] [Google Scholar]
- [17].Grivas J, Haag M, Johnson A, Manalo T, Roell J, Das TL, Brown E, Burns AR, Lafontant PJ, Cardiac repair and regenerative potential in the goldfish (Carassius auratus) heart., Comp. Biochem. Physiol. C. Toxicol. Pharmacol. 163 (2014) 14–23. doi: 10.1016/j.cbpc.2014.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Becker RO, Chapin S, Sherry R, Regeneration of the ventricular myocardium in amphibians., Nature. 248 (1974) 145–7. http://www.ncbi.nlm.nih.gov/pubmed/4818918 (accessed October 31, 2018). [DOI] [PubMed] [Google Scholar]
- [19].Oberpriller JO, Oberpriller JC, Response of the adult newt ventricle to injury, J. Exp. Zool. 187 (1974) 249–259. doi: 10.1002/jez.1401870208. [DOI] [PubMed] [Google Scholar]
- [20].Porrello ER, Mahmoud AI, Simpson E, Hill JA, Richardson JA, Olson EN, Sadek HA, Transient Regenerative Potential of the Neonatal Mouse Heart, Science (80-. ). 331 (2011) 1078–1080. doi: 10.1126/science.1200708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Polizzotti BD, Ganapathy B, Walsh S, Choudhury S, Ammanamanchi N, Bennett DG, dos Remedios CG, Haubner BJ, Penninger JM, Kühn B, Neuregulin stimulation of cardiomyocyte regeneration in mice and human myocardium reveals a therapeutic window., Sci. Transl. Med 7 (2015) 281ra45. doi: 10.1126/scitranslmed.aaa5171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Chen Z, Xie J, Hao H, Lin H, Wang L, Zhang Y, Chen L, Cao S, Huang X, Liao W, Bin J, Liao Y, Ablation of periostin inhibits post-infarction myocardial regeneration in neonatal mice mediated by the phosphatidylinositol 3 kinase/glycogen synthase kinase 3β/cyclin D1 signalling pathway, Cardiovasc. Res. 113 (2017) 620–632. doi: 10.1093/cvr/cvx001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Bassat E, Mutlak YE, Genzelinakh A, Shadrin IY, Baruch Umansky K, Yifa O, Kain D, Rajchman D, Leach J, Riabov Bassat D, Udi Y, Sarig R, Sagi I, Martin JF, Bursac N, Cohen S, Tzahor E, The extracellular matrix protein agrin promotes heart regeneration in mice, Nature. 547 (2017) 179–184. doi: 10.1038/nature22978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Missinato MA, Tobita K, Romano N, Carroll JA, Tsang M, Extracellular component hyaluronic acid and its receptor Hmmr are required for epicardial EMT during heart regeneration, Cardiovasc. Res. 107 (2015) 487–498. doi: 10.1093/cvr/cvv190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Chen CW, Wang Z, Missinato MA, Park DW, Long DW, Liu HJ, Zeng X, Yates NA, Kim K, Wang Y, Decellularized Zebrafish Cardiac Extracellular Matrix Induces Mammalian Heart Regneration, Submitted. (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Kim S-M, Long DW, Tsang MWK, Wang Y, Zebrafish extracellular matrix improves neuronal viability and network formation in a 3-dimensional culture, Biomaterials. 170 (2018) 137–146. doi: 10.1016/j.biomaterials.2018.04.009. [DOI] [PubMed] [Google Scholar]
- [27].D’Uva G, Aharonov A, Lauriola M, Kain D, Yahalom-Ronen Y, Carvalho S, Weisinger K, Bassat E, Rajchman D, Yifa O, Lysenko M, Konfino T, Hegesh J, Brenner O, Neeman M, Yarden Y, Leor J, Sarig R, Harvey RP, Tzahor E, ERBB2 triggers mammalian heart regeneration by promoting cardiomyocyte dedifferentiation and proliferation, Nat. Cell Biol. 17 (2015) 627–638. doi: 10.1038/ncb3149. [DOI] [PubMed] [Google Scholar]
- [28].Wadugu B, Kühn B, The role of neuregulin/ErbB2/ErbB4 signaling in the heart with special focus on effects on cardiomyocyte proliferation, Am. J. Physiol. Circ. Physiol. 302 (2012) H2139–H2147. doi: 10.1152/ajpheart.00063.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Liu M, Zhu J-G, Yu Z-B, Song G-X, Shen Y-H, Liu Y-Q, Zhu C, Qian L-M, QIAN L-M, Identification of differentially expressed genes involved in transient regeneration of the neonatal C57BL/6J mouse heart by digital gene expression profiling., Mol. Med. Rep. 9 (2014) 2111–6. doi: 10.3892/mmr.2014.2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Quaife-Ryan GA, Sim CB, Ziemann M, Kaspi A, Rafehi H, Ramialison M, El-Osta A, Hudson JE, Porrello ER, Multicellular Transcriptional Analysis of Mammalian Heart Regeneration., Circulation. 136 (2017) 1123–1139. doi: 10.1161/CIRCULATIONAHA.117.028252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Heallen T, Morikawa Y, Leach J, Tao G, Willerson JT, Johnson RL, Martin JF, Hippo signaling impedes adult heart regeneration., Development. 140 (2013) 4683–90. doi: 10.1242/dev.102798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Ahmed A, Wang T, Delgado-Olguin P, Ezh2 is not required for cardiac regeneration in neonatal mice, PLoS One. 13 (2018) e0192238. doi: 10.1371/journal.pone.0192238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Porrello ER, Mahmoud AI, Simpson E, Johnson BA, Grinsfelder D, Canseco D, Mammen PP, Rothermel BA, Olson EN, Sadek HA, Regulation of neonatal and adult mammalian heart regeneration by the miR-15 family, Proc. Natl. Acad. Sci. 110 (2013) 187–192. doi: 10.1073/pnas.1208863110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Gomes RSM, Skroblin P, Munster AB, Tomlins H, Langley SR, Zampetaki A, Yin X, Wardle FC, Mayr M, "Young at heart": Regenerative potential linked to immature cardiac phenotypes., J. Mol. Cell. Cardiol. 92 (2016) 105–8. doi: 10.1016/j.yjmcc.2016.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Lalowski MM, Björk S, Finckenberg P, Soliymani R, Tarkia M, Calza G, Blokhina D, Tulokas S, Kankainen M, Lakkisto P, Baumann M, Kankuri E, Mervaala E, Characterizing the Key Metabolic Pathways of the Neonatal Mouse Heart Using a Quantitative Combinatorial Omics Approach., Front. Physiol. 9 (2018) 365. doi: 10.3389/fphys.2018.00365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Williams C, Quinn KP, Georgakoudi I, Black LD 3rd, Young developmental age cardiac extracellular matrix promotes the expansion of neonatal cardiomyocytes in vitro, Acta Biomater. 10 (2014) 194–204. doi: 10.1016/j.actbio.2013.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Crapo PM, Gilbert TW, Badylak SF, An overview of tissue and whole organ decellularization processes, Biomaterials. 32 (2011) 3233–3243. doi: 10.1016/j.biomaterials.2011.01.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Chen CW, Okada M, Proto JD, Gao XQ, Sekiya N, Beckman SA, Corselli M, Crisan M, Saparov A, Tobita K, Peault B, Huard J, Human Pericytes for Ischemic Heart Repair, Stem Cells. 31 (2013) 305–316. doi: 10.1002/stem.1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Chen WC, Lee BG, Park DW, Kim K, Chu H, Kim K, Huard J, Wang Y, Controlled dual delivery of fibroblast growth factor-2 and Interleukin-10 by heparin-based coacervate synergistically enhances ischemic heart repair, Biomaterials. 72 (2015) 138–151. doi: 10.1016/j.biomaterials.2015.08.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Miyagawa S, Katsu Y, Watanabe H, Iguchi T, Estrogen-independent activation of erbBs signaling and estrogen receptor α in the mouse vagina exposed neonatally to diethylstilbestrol, Oncogene. 23 (2004) 340–349. doi: 10.1038/sj.onc.1207207. [DOI] [PubMed] [Google Scholar]
- [41].Akhtar S, Yousif MHM, Chandrasekhar B, Benter IF, Activation of EGFR/ERBB2 via Pathways Involving ERK1/2, P38 MAPK, AKT and FOXO Enhances Recovery of Diabetic Hearts from Ischemia-Reperfusion Injury, PLoS One. 7 (2012) e39066. doi: 10.1371/journal.pone.0039066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Chu H, Chen CW, Huard J, Wang Y, The effect of a heparin-based coacervate of fibroblast growth factor-2 on scarring in the infarcted myocardium, Biomaterials. 34 (2013) 1747–1756. doi: 10.1016/j.biomaterials.2012.11.019. [DOI] [PubMed] [Google Scholar]
- [43].Pollick C, Hale SL, Kloner RA, Echocardiographic and cardiac Doppler assessment of mice., J. Am. Soc. Echocardiogr. 8 (n.d.) 602–10. http://www.ncbi.nlm.nih.gov/pubmed/9417202 (accessed October 31, 2018). [DOI] [PubMed] [Google Scholar]
- [44].Wandt B, Bojö L, Tolagen K, Wranne B, Echocardiographic assessment of ejection fraction in left ventricular hypertrophy., Heart. 82 (1999) 192–8. http://www.ncbi.nlm.nih.gov/pubmed/10409535 (accessed October 31, 2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].O’Donnell M, Skovoroda AR, Shapo BM, Emelianov SY, Internal displacement and strain imaging using ultrasonic speckle tracking, IEEE Trans. Ultrason. Ferroelectr. Freq. Control. 41 (1994) 314–325. doi: 10.1109/58.285465. [DOI] [Google Scholar]
- [46].Lubinski MA, Emelianov SY, O’Donnell M, Speckle tracking methods for ultrasonic elasticity imaging using short-time correlation, IEEE Trans. Ultrason. Ferroelectr. Freq. Control. 46 (1999) 82–96. doi: 10.1109/58.741427. [DOI] [PubMed] [Google Scholar]
- [47].D’Uva G, Tzahor E, The key roles of ERBB2 in cardiac regeneration., Cell Cycle. 14 (2015) 2383–4. doi: 10.1080/15384101.2015.1063292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Shakouri-Motlagh A, O’Connor AJ, Kalionis B, Heath DE, Improved ex vivo expansion of mesenchymal stem cells on solubilized acellular fetal membranes, J. Biomed. Mater. Res. Part A. 107 (2019) 232–242. doi: 10.1002/jbm.a.36557. [DOI] [PubMed] [Google Scholar]
- [49].Ng CP, Mohamed Sharif AR, Heath DE, Chow JW, Zhang CB, Chan-Park MB, Hammond PT, Chan JK, Griffith LG, Enhanced ex vivo expansion of adult mesenchymal stem cells by fetal mesenchymal stem cell ECM, Biomaterials. 35 (2014) 4046–4057. doi: 10.1016/J.BIOMATERIALS.2014.01.081. [DOI] [PubMed] [Google Scholar]
- [50].Sun Y, Li W, Lu Z, Chen R, Ling J, Ran Q, Jilka RL, Chen X-D, Rescuing replication and osteogenesis of aged mesenchymal stem cells by exposure to a young extracellular matrix, FASEB J. 25 (2011) 1474–1485. doi: 10.1096/fj.10-161497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Choi JS, Kim JD, Yoon HS, Cho YW, Full-Thickness Skin Wound Healing Using Human Placenta-Derived Extracellular Matrix Containing Bioactive Molecules, Tissue Eng. Part A. 19 (2013) 329–339. doi: 10.1089/ten.tea.2011.0738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Schneider KH, Aigner P, Holnthoner W, Monforte X, Nürnberger S, Rünzler D, Redl H, Teuschl AH, Decellularized human placenta chorion matrix as a favorable source of small-diameter vascular grafts, Acta Biomater. 29 (2016) 125–134. doi: 10.1016/J.ACTBIO.2015.09.038. [DOI] [PubMed] [Google Scholar]
- [53].Morris AH, Chang J, Kyriakides TR, Inadequate Processing of Decellularized Dermal Matrix Reduces Cell Viability In Vitro and Increases Apoptosis and Acute Inflammation In Vivo., Biores. Open Access. 5 (2016) 177–87. doi: 10.1089/biores.2016.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Zheng MH, Chen J, Kirilak Y, Willers C, Xu J, Wood D, Porcine small intestine submucosa (SIS) is not an acellular collagenous matrix and contains porcine DNA: Possible implications in human implantation, J. Biomed. Mater. Res. Part B Appl. Biomater. 73B (2005) 61–67. doi: 10.1002/jbm.b.30170. [DOI] [PubMed] [Google Scholar]
- [55].Keane TJ, Swinehart IT, Badylak SF, Methods of tissue decellularization used for preparation of biologic scaffolds and in vivo relevance, Methods. 84 (2015) 25–34. doi: 10.1016/j.ymeth.2015.03.005. [DOI] [PubMed] [Google Scholar]
- [56].Seif-Naraghi SB, Salvatore MA, Schup-Magoffin PJ, Hu DP, Christman KL, Design and characterization of an injectable pericardial matrix gel: a potentially autologous scaffold for cardiac tissue engineering, Tissue Eng Part A. 16 (2010) 2017–2027. doi: 10.1089/ten.TEA.2009.0768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Mewhort HEM, Svystonyuk DA, Turnbull JD, Teng G, Belke DD, Guzzardi DG, Park DS, Kang S, Hollenberg MD, Fedak PWM, Bioactive Extracellular Matrix Scaffold Promotes Adaptive Cardiac Remodeling and Repair, JACC Basic to Transl. Sci. 2 (2017) 450–464. doi: 10.1016/j.jacbts.2017.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Okada M, Payne TR, Oshima H, Momoi N, Tobita K, Huard J, Differential efficacy of gels derived from small intestinal submucosa as an injectable biomaterial for myocardial infarct repair, Biomaterials. 31 (2010) 7678–7683. doi: 10.1016/j.biomaterials.2010.06.056. [DOI] [PubMed] [Google Scholar]
- [59].Singelyn JM, Sundaramurthy P, Johnson TD, Schup-Magoffin PJ, Hu DP, Faulk DM, Wang J, Mayle KM, Bartels K, Salvatore M, Kinsey AM, Demaria AN, Dib N, Christman KL, Catheter-deliverable hydrogel derived from decellularized ventricular extracellular matrix increases endogenous cardiomyocytes and preserves cardiac function post-myocardial infarction., J. Am. Coll. Cardiol. 59 (2012) 751–63. doi: 10.1016/j.jacc.2011.10.888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Pellegrini L, Role of heparan sulfate in fibroblast growth factor signalling: a structural view, Curr Opin Struct Biol. 11 (2001) 629–634. http://www.ncbi.nlm.nih.gov/pubmed/11785766. [DOI] [PubMed] [Google Scholar]
- [61].Peysselon F, Ricard-Blum S, Heparin-protein interactions: from affinity and kinetics to biological roles. Application to an interaction network regulating angiogenesis, Matrix Biol. 35 (2014) 73–81. doi: 10.1016/j.matbio.2013.11.001. [DOI] [PubMed] [Google Scholar]
- [62].Capila I, Linhardt RJ, Heparin-protein interactions, Angew Chem Int Ed Engl. 41 (2002) 391–412. http://www.ncbi.nlm.nih.gov/pubmed/12491369. [DOI] [PubMed] [Google Scholar]
- [63].Sonnenberg SB, Rane AA, Liu CJ, Rao N, Agmon G, Suarez S, Wang R, Munoz A, Bajaj V, Zhang S, Braden R, Schup-Magoffin PJ, Kwan OL, DeMaria AN, Cochran JR, Christman KL, Delivery of an engineered HGF fragment in an extracellular matrix-derived hydrogel prevents negative LV remodeling post-myocardial infarction, Biomaterials. 45 (2015) 56–63. doi: 10.1016/j.biomaterials.2014.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Seif-Naraghi SB, Horn D, Schup-Magoffin PJ, Christman KL, Injectable extracellular matrix derived hydrogel provides a platform for enhanced retention and delivery of a heparin-binding growth factor, Acta Biomater. 8 (2012) 3695–3703. doi: 10.1016/j.actbio.2012.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Mizuno T, Yau TM, Weisel RD, Kiani CG, Li R-K, Elastin stabilizes an infarct and preserves ventricular function., Circulation. 112 (2005) I81–8. doi: 10.1161/01.CIRCULATIONAHA.105.523795. [DOI] [PubMed] [Google Scholar]
- [66].Aurora AB, Porrello ER, Tan W, Mahmoud AI, Hill JA, Bassel-Duby R, Sadek HA, Olson EN, Macrophages are required for neonatal heart regeneration., J. Clin. Invest. 124 (2014) 1382–92. doi: 10.1172/JCI72181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Liu J, Wang H, Li J, Inflammation and Inflammatory Cells in Myocardial Infarction and Reperfusion Injury: A Double-Edged Sword., Clin. Med. Insights. Cardiol. 10 (2016) 79–84. doi: 10.4137/CMC.S33164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Force T, Krause DS, Van Etten RA, Molecular mechanisms of cardiotoxicity of tyrosine kinase inhibition, Nat. Rev. Cancer. 7 (2007) 332–344. doi: 10.1038/nrc2106. [DOI] [PubMed] [Google Scholar]