

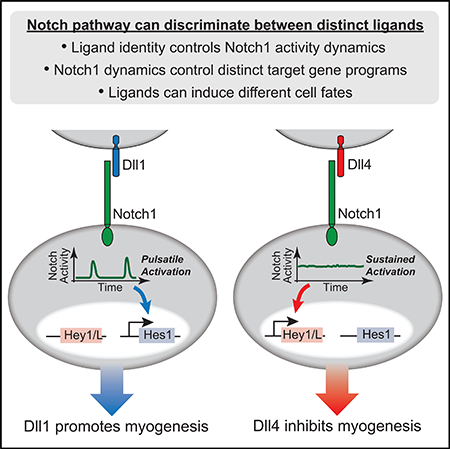
SUMMARY
The Notch signaling pathway comprises multiple ligands that are used in distinct biological contexts. In principle, different ligands could activate distinct target programs in signal-receiving cells, but it is unclear how such ligand discrimination could occur. Here, we show that cells use dynamics to discriminate signaling by the ligands Dll1 and Dll4 through the Notch1 receptor. Quantitative single-cell imaging revealed that Dll1 activates Notch1 in discrete, frequency-modulated pulses that specifically upregulate the Notch target gene Hes1. By contrast, Dll4 activates Notch1 in a sustained, amplitude-modulated manner that predominantly upregulates Hey1 and HeyL. Ectopic expression of Dll1 or Dll4 in chick neural crest produced opposite effects on myogenic differentiation, showing that ligand discrimination can occur in vivo. Finally, analysis of chimeric ligands suggests that ligand-receptor clustering underlies dynamic encoding of ligand identity. The ability of the pathway to utilize ligands as distinct communication channels has implications for diverse Notch-dependent processes.
In Brief
Notch ligands activate distinct targets through the same Notch receptor by triggering pulsatile or sustained activation dynamics.
Graphical Abstract
INTRODUCTION
In metazoans, the Notch signaling pathway enables communication between neighboring cells. It plays critical roles in the development and maintenance of most tissues (Bray, 2016; Guruharsha et al., 2012), and its dysregulation has been implicated in a variety of diseases, making it an important therapeutic target (Andersson and Lendahl, 2014). In mammals, Notch signaling can be activated by four different transmembrane ligands: Dll1, Dll4, Jag1, and Jag2. When these ligands interact with Notch receptors expressed on the surface of neighboring receiver cells, they induce cleavage of the receptor. This releases the Notch intracellular domain (NICD), which translocates to the nucleus and, in complex with CSL/RBPjk, activates Notch target genes (Figure 1A). In principle, different ligands could be used to activate distinct target programs, and thus could constitute distinct “communication channels.”
Figure 1. Dll1 and Dll4 Activate Notch1 with Pulsatile and Sustained Dynamics, Respectively.

(A) Both Dll1 (blue) and Dll4 (red) activate the Notch1 receptor (green) to induce proteolytic release of the Notch intracellular domain (NICD), but are used in different biological contexts (blue and red boxes, bottom). The released NICD translocates to the nucleus and, in complex with CSL/RBPjκ (yellow), activates Notch target genes (white).
(B) Left: Engineered CHO-K1 “sender” cell lines contain stably integrated constructs expressing Dll1 (blue) or Dll4 (red), each with a co-translational (T2A, brown) H2B-mCh readout (purple), from a 4epi-Tetracycline (4epi-Tc) inducible promoter. Right: “Receiver” cells stably express a chimeric receptor combining the Notch1 extracellular domain (Notch1ECD) with a Gal4 transcription factor (orange), which can activate a stably integrated fluorescent H2B-3xCitrine reporter gene (chartreuse).
(C) Left (schematics): A minority of receiver cells (green) are co-cultured with an excess of either Dll1 (blue) or Dll4 (red) sender cells. Right: Filmstrips showing representative sustained (top, Dll4 senders) or pulsatile (bottom, Dll1 senders) response of a single receiver cell (center, automatically segmented nucleus outlined in white). Grey channel shows DIC images of cells, while the rate of increase in Citrine fluorescence, scaled to 25%–75% of its total range, is indicated using green pseudo-coloring. See also Movies S1 and S2.
(D) Left: Representative traces showing total nuclear Citrine fluorescence levels (top) or corresponding derivatives of the total Citrine (dCitrine/dt), i.e., promoter activity (bottom), in individual receiver cells activated by Dll4. Right: Average values of total fluorescence (top) and promoter activity (bottom) in receiver cells activated by Dll4. Solid traces represent medians, lighter shades indicate SEM, and gray shading indicates SD. n, number of traces included in the alignment. See STAR Methods for alignment and normalization procedure.
(E) Left: Corresponding plots (as in D) showing total nuclear Citrine fluorescence levels (top) and promoter activity (bottom) in individual receiver cells in co-culture with Dll1. Right: Average values of total fluorescence (top) and promoter activity (bottom) in receiver cells activated by Dll1. The percentage value (60%) in the plots on right indicates the fraction of receiver traces included in the alignment (STAR Methods, see also Figure S1F).
(F) 95th percentile of (absolute, non-normalized) promoter activity values between 0 and 7.5 hr (after alignment) in the traces included in (D) and (E). This time window is chosen to simultaneously estimate the promoter activity at the peak of Dll1 pulses and at steady-state levels of Dll4 signaling. Solid horizontal lines represent medians, while the boxes delineate 25th–75th percentile values. p value calculated by two-sided Kolmogorov-Smirnov (K-S) test.
See also Figures S1 and S2.
Indeed, ligand-specific effects of Notch signaling have been observed in multiple contexts and occur even with close paralogs like Dll1 and Dll4 (Figure 1A). For example, Dll4 is unable to replace Dll1 function in many tissues, leading to embryonic lethality in mice when knocked into the Dll1 locus (Preuße et al., 2015). Dll1 and Dll4 also have opposing effects on muscle differentiation: Dll1 expressed in the neural crest induces differentiation of muscle progenitors in somites (Rios et al., 2011), while Dll4 expressed in endothelial cells can revert this fate in committed skeletal myoblasts, diverting them to form pericytes instead (Cappellari et al., 2013). Puzzlingly, although Dll1 and Dll4 can behave differently under certain conditions, they appear to function interchangeably in others. For example, when overexpressed, both ligands promote T cell differentiation of primary hematopoietic stem cells in culture, but appear to do so with different efficiencies (Mohtashami et al., 2010).
How could different ligands induce different responses in signal-receiving cells? Due to the proteolytic mechanism by which all ligands activate Notch, information regarding ligand identity must be represented in the levels or dynamics of NICD in signal-receiving cells. In fact, the Dll1 and Dll4 extracellular domains differ by more than 10-fold in their affinity for Notch (Andrawes et al., 2013), which could lead to differences in their signaling strength (NICD levels). However, several aspects of the Notch pathway also suggest a potential sensitivity to dynamics. Cleaved NICD has a short half-life, enabling its concentration to respond rapidly to changes in Notch activation (Fryer et al., 2004; Housden et al., 2013; Ilagan et al., 2011). Similarly, the canonical Notch target genes Hes1 and Hes5 have short mRNA and protein half-lives and their levels oscillate in many contexts (Kobayashi and Kageyama, 2014). While dynamics has been shown to play critical roles in other signaling contexts (Purvis and Lahav, 2013), it has not been systematically investigated in the Notch pathway.
Here, by quantitatively analyzing Notch1 activation in individual cells, we show that Dll1 and Dll4 generate distinct patterns of direct target gene expression by encoding ligand identity in Notch1 activation dynamics. Specifically, Dll1 induces pulses of Notch activation, while Dll4 induces sustained activity. These dynamics are in turn decoded to control relative levels of Hes1 and Hey1/L target gene expression. Notch activity in receiving cells is thus inherently multi-dimensional, possessing both an activation type (pulsatile or sustained) and an activation level. This ability to respond in a ligand-specific fashion enables signal sending cells to use different ligands to activate distinct Notch target programs in receiving cells, effectively expanding the number of communication channels in the Notch pathway.
RESULTS
Dll1 and Dll4 Signal through Notch1 with Different Dynamics
In order to directly compare Notch1 signaling by Dll1 and Dll4 at the single cell level, we constructed “sender” and “receiver” cell lines in CHO-K1 cells (Figure 1B). Sender cells expressed either Dll1 or Dll4 along with a co-translational H2B-mCherry readout, under control of a 4epi-tetracycline (4epi-Tc)-induced promoter (LeBon et al., 2014; Sprinzak et al., 2010). We engineered receiver cells to express chimeric Notch1 receptors whose intracellular domain is replaced by the transcription factor Gal4 (Lecourtois and Schweisguth, 1998; Sprinzak et al., 2010; Struhl and Adachi, 1998), along with an H2B-3xCitrine fluorescent protein reporter that can be activated by Gal4 (Figure 1B). This “diverted” reporter system enables readout of Notch activity without activation of endogenous Notch targets, avoiding potential complications due to downstream feedback interactions.
To compare dynamics of signaling by Dll1 and Dll4, we used time-lapse microscopy of sender-receiver co-cultures. In these experiments, a minority of receiver cells were co-cultured with an excess of either Dll1 or Dll4 sender cells so that each receiver cell was in continuous contact with one or more sender cells (Figure 1C). Increases in the level of stable H2B-3xCitrine fluorescence in receiver cells reflect the activity of Gal4 released from activated receptors. Specifically, the rate of increase in total fluorescence (dCitrine/dt, “promoter activity”) is controlled by the concentration of released Gal4 (Figures S1A and S1B). We therefore estimated the Notch activity from the time derivative of each fluorescent protein trace, computed by calculating the change in total nuclear fluorescence from one time point to the next (30 min apart, see STAR Methods).
Under these experimental conditions, Dll4-expressing sender cells activated receivers in a sustained fashion. After plating, individual receiver cells activated Citrine production and continued to actively produce Citrine for the duration of the experiment (Figures 1C and 1D; Movie S1). The sustained nature of Dll4 signaling was also reflected in the average response of these cells (Figure S1C). To extract stereotyped features of the average response shape, independent of cell-cell variation in signaling amplitude and timing of activation, we normalized each trace by its maximal level and temporally aligned the resulting traces at the point of activation (Figure S1D; STAR Methods). This procedure sharpened the sustained nature of response to Dll4 (Figure 1D).
In contrast, in co-culture with Dll1-expressing senders the same receiver cells activated in discrete, transient pulses (Figures 1C and 1E; Movie S2). In each pulse, the rate of Citrine production increased transiently, and then returned to baseline, displaying a characteristic shape (Figures 1E, S1C, and S1D). Pulses occurred in an unsynchronized fashion, initiating at different times in different receiver cells and could occur throughout the experiment (Figure S1E). Most cells under these conditions displayed a single pulse during the experiment (60% of traces), while two pulses could be detected in other traces (35%) (Figure S1F; Movie S3). Dll1 pulses displayed peak amplitudes comparable to the amplitude observed during the corresponding period of Dll4 signaling (Figure 1F). These results indicate that Dll1 activates Notch1 through stochastic stereotyped pulses.
In order to better understand pulsatile Dll1 signaling dynamics, we sought to estimate the duration of the underlying pulse of Notch activation, accounting for the half-lives of Gal4 protein and H2B-Citrine mRNA, which extend the duration of the observed reporter pulse. We used a mathematical model of reporter activation (STAR Methods) to analyze the decay of Citrine production rate following inhibition of Notch signaling (Figures S1G and S1H) and computed values for the half-lives of Gal4 protein (~4 hr, 95% confidence interval [CI] [3.8 hr, 4 hr]) and H2B-3xCitrine mRNA (~3.4 hr, 95% CI [3.4 hr, 3.5 hr]). Together with the measured duration (~12 hr full-width at half-maximum [FWHM]) and rise-time (~6 hr, “trise”) of the Dll1-induced reporter activity pulses (Figure S1I), this enabled us to estimate an upper bound of ~1 hr on the duration of the underlying signaling events (Figure S1J). Simulations showed that pulses briefer than this would produce indistinguishable reporter dynamics (Figure S1K). As discussed more below, these brief pulses likely represent events in which multiple Notch receptors are activated (cleaved) simultaneously.
We next asked whether the apparently sustained Dll4 signaling could be explained as a series of Dll1-like pulses, occurring at an elevated rate (Figure S2A). We computationally generated pulse trains composed of pulses with the same shape and amplitude distribution observed for Dll1 pulses (Figure S2B; STAR Methods). We varied both the regularity of the pulses, using dynamic models ranging from periodic to Poisson distributed, as well as the pulse frequency (or mean interval between pulses) within each model, and analyzed the amplitude and temporal (“intra-trace”) variability of the simulated pulse trains (Figures S2C and S2D). Higher pulse frequencies lead to greater pulse overlap, increasing signaling amplitude, while reducing the temporal variability of signaling (Figure S2E). Critically, tuning pulse frequency low enough to match the observed mean Dll4 signaling amplitude generated significantly greater temporal variability than observed experimentally (Figures S2E, inset, and S2F), suggesting that the observed sustained Dll4 signaling cannot be explained as a series of Dll1-like pulses. Furthermore, the difference in experimentally observed Dll1 and Dll4 dynamics was preserved even when the time resolution of the reporter was improved from 6–12 hr (trise–FWHM, Figure S1G) to 2.5–6 hr by destabilizing the Citrine mRNA (Figures S2G–S2I). Taken together, these data and analysis strongly suggest that Dll1 and Dll4 activate Notch1 with distinct dynamics, Dll1 through brief pulses, and Dll4 in a sustained fashion. We note, however, that this does not rule out the possibility that Dll4 signaling originates from a series of smaller pulses (in the extreme limit, individual ligand-receptor activation events can be thought of as small, discrete “pulses”).
Dll1 Levels Modulate Pulse Frequency, while Dll4 Levels Modulate Signaling Amplitude
We next asked how the expression level of each ligand in the sender cell modulated signaling dynamics. To isolate signaling events produced by individual sender cells, we reversed the conditions of the assay, co-culturing an excess of receiver cells with a minority of sender cells (STAR Methods). We analyzed Dll1 senders across a >10-fold range of Dll1 expression levels (Figure S2J). Over this range, most receiver cells activated in pulses (Figure 2A, bottom panels; Movie S4), which maintained the same stereotyped shape and duration (Figure 2B, right panels) and showed a 1.6-fold increase in amplitude (Figure 2C, right panels). At the same time, we observed a stronger increase in the number of activated receiver cells with increasing Dll1 expression, reflecting an increase in pulse frequency (Figure 2D). Together, these results indicate that Dll1 expression levels modulate signaling predominantly through the frequency of stereotyped signaling pulses (Figure 2E, left panel).
Figure 2. Differences in Dll1 and Dll4 Dynamics Are Preserved across a Range of Ligand Expression Levels, and Ligand-Levels Modulate These Dynamics in Different Ways.

(A) Left: Schematic of co-culture assay showing Dll1 (blue) or Dll4 (red) sender cells surrounded by receiver cells (green). Right: Filmstrips showing sustained or pulsatile responses in a single receiver cell (green, automatically segmented nucleus outlined in white) neighboring either Dll4 (top, nuclei pseudo-colored in red) or Dll1 (bottom, nuclei pseudo-colored in blue) sender cells. The gray channel shows DIC images, in which other receiver cells can be seen. Intensity of green in the receiver cell indicates promoter activity scaled to 25%–75% of its range. See also Movies S4 and S5.
(B) Median response profiles in individual receiver cells co-cultured with sender cells expressing low, medium, or high levels of Dll4 (left) or Dll1 (right). See Figures S2J and S2K for ligand expression levels in each group. Solid traces represent medians, light colored regions indicate SEM, gray shading indicates SD. n values indicate number of receiver cell responses included in the alignment. The percentage values in the Dll1 plots indicate the fraction of receiver traces included in the alignment (STAR Methods).
(C) Left: Comparison of maximal promoter activities (95th percentile of promoter activity values in each trace) in activated receiver cells adjacent to sender cells expressing no ligand (black), or low (red), medium (pink), or high (dark red) levels of Dll4 (same designations as used in B). Right: Similar comparison for Dll1. Grey circles represent individual responses, solid horizontal lines represent medians, while the boxes delineate 25th–75th percentile values. p values calculated by two-sided K-S test. Not significant (ns), p > 0.01.
(D) Median values of the number of receiver cells activated by isolated Dll1 sender cells expressing low, medium, or high levels of co-translational H2B-mCherry and their progeny during a 25 hr experiment under excess receiver conditions. Error bars represent SEM.
(E) Schematic: Summary of Dll1 and Dll4 modulation. Dll1 levels primarily control rate or frequency of stereotyped pulses, while Dll4 levels control amplitude of sustained signal.
See also Figures S2J–S2L and Movie S3.
Unlike Dll1, Dll4 showed sustained activation in the excess receiver assay across all levels of Dll4 expression analyzed (Figures 2A, 2B, and S2K; Movie S5). We observed a systematic increase in peak (Figure 2C, left panels) and median (Figure S2L) signaling amplitude with increasing Dll4 expression level over a 10-fold range (Figure S2K). Together, these results indicate that Dll1 and Dll4 produce qualitatively different signaling dynamics across a broad range of expression and signaling levels and modulate those dynamics in distinct ways, with Dll1 mainly controlling the frequency of stereotyped pulses and Dll4 controlling the amplitude of sustained signaling (Figure 2E).
Pulsatile and Continuous Notch Signals Can Elicit Distinct Transcriptional Responses
We next asked whether the different dynamics produced by Dll1 and Dll4 activation could regulate distinct sets of target genes and thereby allow cells to discriminate between the ligands. To directly test the effect of NICD dynamics on target gene expression, we took advantage of the fact that truncated Notch1 receptors lacking most of their extracellular domain (N1ΔECD) are constitutively active, but can be inhibited by DAPT (Fortini et al., 1993; Kopan et al., 1996) (Figure 3A). Cells expressing N1ΔECD can therefore be activated for different durations and to varying levels by controlling DAPT concentration in the media for corresponding time intervals (STAR Methods).
Figure 3. Pulsatile and Sustained Notch Activation Can Regulate Different Sets of Target Genes.

(A) C2C12 cells were engineered to expressed Notch1 receptors lacking the extracellular domain (N1DECD, green). This receptor is inactive in the presence of the γ-secretase inhibitor DAPT (red), but constitutively active when DAPT concentration is reduced in the culture medium.
(B) Comparison of transcript levels in C2C12-N1ΔECD cells at 1 hr or 6 hr after DAPT removal. The blue line represents equal expression at 1 hr and 6 hr, and the gray lines represent 5-fold changes in either direction. Circled genes are putative direct Notch targets. The blue circle highlights target genes that are upregulated >5-fold at 1 hr but not 6 hr, while red circles indicate target genes that are upregulated >5-fold only after 6 hr. See also Figure S3 and Table S1.
(C) qPCR time course measurement of Hes1 (blue), Hey1 (orange), and HeyL (yellow) mRNA levels following complete DAPT removal at t = 0 hr.
(D) Duration dependence of Hes1 (blue) and Hey1 (orange) response to DAPT removal for 5 min, 15 min, or 30 min followed by replenishment (“Pulse”), or no replenishment until the 1 hr or 4 hr measurement (“Sustained”). Error bars represent SEM calculated from duplicate experiments (n = 2).
See also Figure S4.
We stably expressed N1ΔECD in C2C12 cells, where the binding of the NICD-CSL complex to target gene promoters has been previously characterized using chromatin immunoprecipitation sequencing (ChIP-seq) (Castel et al., 2013). Using RNA sequencing (RNA-seq) (STAR Methods), we identified genes that were upregulated at early time points (1 hr or 6 hr) following Notch activation by DAPT removal (Figures S3A and S3B; Table S1). We focused specifically on putative direct Notch targets previously shown to bind the CSL-NICD complex in this cell line (Castel et al., 2013). Other genes that were activated were not considered for further analysis because they are not known Notch targets; several of these genes have been shown to be induced by growth factor signaling, suggesting that they could have induced by media change during DAPT removal (Allan et al., 2001; Gururajan et al., 2008; Kesarwani et al., 2017).
Interestingly, even direct Notch target genes responded to activation of the pathway at different times (Figure 3B). Hes1, but not the other target genes, was rapidly activated, showing strong (~10-fold) upregulation by 1 hr (Figures 3B and S3A; Table S1). Other Notch targets such as Hey1, HeyL, Jag1, and Nrarp responded later, showing little change at 1 hr, but strong upregulation by 6 hr (Figures 3B and S3B; Table S1). In order to follow the early and later phases of response in finer detail, we carried out a real-time qPCR time course measurement of Hes1, Hey1, and HeyL mRNA levels following DAPT removal (Figure 3C). Hes1 expression increased rapidly, within 30 min, and its levels peaked at 1 hr. By contrast, Hey1/L levels did not significantly increase until the end of the Hes1 activation pulse, at 2 hr, after which they continued to rise until reaching a steady state around 4 hr.
These results suggested the possibility that brief (<1 hr) pulses of Notch activation could selectively activate Hes1, with the other targets requiring longer durations of Notch signaling. To test this hypothesis, we used real-time qPCR to analyze the response of Hes1 and Hey1/L to varying durations and amplitudes of Notch activation (STAR Methods). We observed that Hes1 activation was relatively insensitive to the duration of Notch activation and could be induced strongly by brief pulses (5–30 min) and by sustained activation (Figure 3D). On the other hand, Hey1 and HeyL were more sensitive to duration, accumulating continuously as long as Notch activation was maintained (Figures 3D and S3C).
In order to isolate the effects of signaling duration from those of signal amplitude, we compared Hey1/L expression at the same instantaneous NICD concentrations but after different durations of NICD exposure (Figures S3D–S3G). Specifically, we compared a brief pulse of NICD generated by total DAPT removal for 15 min, with a longer (3 hr) duration of NICD activity generated by partial removal of DAPT to 0.3 μM. These two perturbations produce the same final concentration of NICD but differ in the duration of NICD activity (Figures S3D and S3E). If NICD concentration alone controlled Hey1/L expression, then the two conditions should produce similar rates of Hey1/L synthesis (Figure S3F, top). By contrast, a requirement for sustained NICD activity would lead to a greater rate of Hey1/L expression in the prolonged case (Figure S3F, bottom). For each condition, we measured the increase of Hey1/L levels in a 30-min window in order to estimate new Hey1/L expression at the corresponding time-point (Figure S3F). We observed increased Hey1/L expression only at the 3 hr time point, indicating that an extended period of activity is required for efficient activation (Figure S3G). Higher NICD concentrations were not able to overcome the requirement for extended activation, as a 30-min pulse of total DAPT withdrawal, which produced higher NICD concentrations, did not increase Hey1/L expression (Figures S3D and S3G). NICD concentration did, however, affect the maximum induction levels of the Hes/Hey genes under sustained activation (Figure S3H). Finally, we note that the weakness of the Hey1/L response to brief activation pulses was not due to insufficient NICD, as the Notch1ΔECD system produces more NICD from DAPT withdrawal over 30 min than observed in Notch1-expressing receiver cells co-cultured with sender cells expressing maximal levels of Dll4 (Figure S3I). Together, these results indicate that pulsatile and sustained Notch dynamics are decoded into distinct gene expression patterns, with Hes1 responding strongly even to brief pulses and Hey1 and HeyL requiring sustained activation.
Dll1 and Dll4 Induce Different Gene Responses
Based on the different responses of Hes1 and Hey1/L to Notch dynamics, we hypothesized that Dll1 signaling could activate Hes1 without significantly inducing the Hey genes, while Dll4 could more strongly upregulate Hey1/L, even at similar Hes1 induction levels. To test this hypothesis, we used a C2C12 cell line constitutively expressing wild-type Notch1, with its endogenous Notch2 knocked down by small interfering RNA (siRNA) (STAR Methods). We first verified that the dynamic differences between Dll1 and Dll4 activation of Notch1 are preserved in this cell line, even at similar mean levels of Notch activity (Figures S4A–S4C). We then co-cultured this cell line with CHO-K1 cells expressing Dll1, Dll4, or no ligand, and measured Hes1, Hey1, and HeyL mRNA levels by real-time qPCR (Figures S4D and S4E). We found that for the same, reproducible, 1.6-fold upregulation in mean Hes1 levels, Dll4 induced ~3- to 5-fold more Hey1/L than Dll1 did (Figure S4E). This result is consistent with the different signaling dynamics of Dll1 and Dll4 inducing different Hes/Hey expression regimes. By contrast, signaling levels (amplitudes) do influence the levels of both Hes and Hey1/L expression, but do so proportionately (Figure S3H), and therefore cannot explain the disproportionate induction of these gene sets by Dll1 and Dll4.
Further, we used a complementary imaging approach to analyze the effects of single (or few) sender cells on neighboring receivers, by using plating conditions that allowed the two cell types to contact each other predominantly along a linear interface (Figure S4F; STAR Methods). Gene expression was analyzed by hybridization chain reaction-fluorescence in situ hybridization (HCR-FISH), which provides an amplified single-cell readout of specific mRNA levels (Choi et al., 2010, 2016). In these experiments, we similarly observed that Dll4 senders, but not Dll1 senders, strongly upregulated Hey1/L in neighboring receiver cells (Figures S4G and S4H). Changes in Hes1 mRNA levels were more difficult to observe at the single cell level using this technique, due to the basal expression of Hes1 (Table S1) and the stochastic, unsynchronized nature of Dll1 pulses. Nevertheless, these results further support the conclusion that Dll1 and Dll4 activate different Hes/Hey gene expression regimes, with Dll4 producing a higher expression of Hey1/L compared to Dll1 at similar Hes1 levels.
Dll1 and Dll4 Direct Opposite Fates In Vivo
We next sought to test the ability of Notch receiving cells to distinguish between Dll1 and Dll4 in the in vivo context of embryonic myogenesis in chick somites. In the developing embryo, it has been shown that Dll1 expressed in migrating neural crest cells signals to Notch1 expressed in the dorsomedial lip (DML) of the neighboring somite. This interaction promotes differentiation of Pax7+ progenitor cells in the DML by upregulating the muscle regulatory factors Myf5 and MyoD1, likely via Hes1 (Rios et al., 2011) (Figure 4A). Critically, in this system, transient activation of the Notch pathway enables normal muscle differentiation, while sustained activation inhibits this process (Rios et al., 2011).
Figure 4. Dll1 Expression in the Chick Neural Crest Promotes Myogenesis but Dll4 Inhibits It.

(A) Developing chick embryo (dorsal view schematic). Dll1 (blue cells in 3) is expressed in a fraction of neural crest cells (gray, see 2, 3). These cells activate Notch1-expressing Pax7+ progenitor cells in the dorsomedial lip (DML, magenta) of the somite. When activated, these progenitor cells (green, 3) upregulate Hes1 and the muscle regulatory gene MyoD1.
(B–D) Representative images showing effects of Dll1 or Dll4 electroporation into the neural crest, on Hes1, Hey1, and MyoD1 expression in the DML. White arrows indicate the somites on the electroporated side. The dotted lines indicate the DMLs of somites or the central line of the neural tube.
(B) Top: Dll1-T2A-EGFP (i, blue), electroporated into the left side of the neural tube, is expressed in the neural tube and neural crest, resulting in upregulation of Hes1 (ii, red) and MyoD1 (iii, green) in the somites on the electroporated (left) side compared to the right side, which serves as negative control. Bottom: When Dll4-T2A-EGFP (iv, blue) is electroporated, Hey1 (v, red) is upregulated on the electroporated side, and MyoD1 (vi, green) expression is decreased.
(C) Dll1-T2A-EGFP (blue, left) electroporation does not affect expression of Hey1 (red, right) in adjacent somites.
(D) Dll4-T2A-EGFP (blue, left) electroporation increases expression of Hes1 (red, right) in adjacent somites.
Our results thus far suggest that transient and sustained Notch activation are intrinsic properties of the Dll1 and Dll4 ligands, respectively. Therefore, we predicted that the pulsatile dynamics of Dll1 would promote myogenic fate, while the sustained dynamics produced by Dll4 would inhibit myogenesis in the same cells. To test this possibility, we electroporated either Dll1 or Dll4 into the neural crest unilaterally in stage HH 12–13 chick embryos, using the other side as a negative control (Elena de Bellard and Bronner-Fraser, 2005; Rios et al., 2011). 20 hr later, we measured expression levels of Notch targets (Hes1, Hey1, or HeyL) and MyoD1 in the adjacent somites using whole-mount HCR-FISH (Figure S5A; STAR Methods). Consistent with previously published results (Rios et al., 2011), ectopic Dll1 expression in the neural crest systematically upregulated Hes1 in the somite (Figures 4B, i and ii, and quantification in S5C) and frequently increased MyoD1 in adjacent somites (Figures 4B, iii, and S5C; Table 1) or maintained its levels (Figure S5C; Table 1). As expected, ectopic Dll1 expression did not significantly alter Hey1 levels (Figures 4C and S5C). On the other hand, ectopic Dll4 expression consistently increased Hey1 (Figures 4B, iv and v, and S5C) and HeyL (Figure S5B), in addition to Hes1 (Figures 4D and S5C). Importantly, Dll4 also strongly decreased MyoD1 in the majority of neighboring somites (Figures 4B, vi, and S5C; Table 1). Thus, Dll1 and Dll4 induced opposite effects on cell fate in the same Notch1-expressing somite cell population that received the signal. While a role for differences in signaling levels between the two ligands in this context cannot be directly excluded, it is striking that these responses, observed in an in vivo context, matched the differences in dynamics and target specificity observed in cell culture systems.
Table 1.
Quantification of Changes in MyoD1 Expression in Embryos Electroporated with Dll1 or Dll4
| MyoD1 Levels in Somites on Electroporated Side Relative to Control Side | ||||
|---|---|---|---|---|
| Ligand | No. Showing Increase (% of total) | No. Showing No Change (% of total) | No. Showing Decrease (% of total) | Total |
| Dll1 | 21 (34.4) | 30 (49.1)a | 10 (16.3) | 61 |
| Dll4 | 9 (14.8) | 12 (19.6) | 40 (65.6)a | 61 |
For each treatment, 61 pairs of somites across 11 Dll1-expressing or 10 Dll4-expressing embryos were scored blindly for differences in HCR-FISH signal between the electroporated side and the control side (see STAR Methods). Entries show the number (and percentage) of somite pairs that show an increase, decrease, or no change in MyoD1 expression on the electroporated side.
Indicates most frequent category for each ligand.
Ligand Intracellular Domains Influence Dynamics through Differences in Transendocytosis
To gain insight into how Dll1 and Dll4 control Notch activation dynamics, we asked whether the dynamic mode was determined by the ligand intracellular domain (ICD) or extracellular domain (ECD). We constructed two chimeric Delta ligands, Dll1ECD-Dll4ICD and Dll4ECD-Dll1ICD, by exchanging the ICDs of Dll1 and Dll4 (STAR Methods) and stably expressed them in sender cell lines (as in Figure 1B), obtaining cell surface levels similar to those of their wild-type counterparts (Figure S5D; STAR Methods).
We first compared Dll4ECD-Dll1ICD with Dll4 using the excess receiver co-culture assay. Unlike Dll4, the Dll4ECD-Dll1ICD ligand generated pulsatile activation, showing that the Dll1 ICD can strongly alter the activation dynamics of the Dll4 ligand (Figure 5A). The amplitude of these pulses was ~3-fold greater than signaling amplitude generated by Dll4 at the highest expression levels analyzed here, suggesting that pulsatile Dll4ECD-Dll1ICD dynamics could not be explained by a reduction in Dll4 signaling strength. In parallel, we compared Dll1 and Dll1ECD-Dll4ICD using the excess sender co-culture assay. With this chimeric ligand, most signaling occurred in a sustained fashion, but at an amplitude slightly lower than the peak amplitude of Dll1 signaling (Figure 5B). This result indicates that the Dll4 ICD can convert the dynamics of Dll1 to a more sustained behavior, even at comparable mean signaling strengths. Furthermore, consistent with the idea that dynamics strongly impact target gene expression, the Dll1ECD-Dll4ICD chimeric ligand, like Dll4, produced more Hey1/L expression than Dll1 at a similar level of Hes1 activation (Figure S5E, bottom panel inset). Additionally, it was not possible to match Dll1-induced Hes/Hey gene expression levels by varying the expression level of the chimeric ligand (thus varying signal amplitude), suggesting that this ligand produces a qualitatively distinct Hes/Hey gene expression response compared to Dll1 (Figure S5E). Together, these results indicate that the ligand ICD plays an important role in determining dynamic signaling mode of the ligand (pulsatile or sustained) and downstream gene expression.
Figure 5. Ligand Intracellular Domains Control Dynamic Signaling Mode and Influence Transendocytosis Patterns.

(A and B) Dll4ECD-Dll1ICD and Dll1ECD-Dll4ICD were constructed by exchanging the intracellular domain (ICD) of Dll4 with that of Dll1.
(A) Median response profiles in activated receiver cells co-cultured with Dll4 sender cells (red, top left) or Dll4ECD-Dll1ICD sender cells (magenta, right) under excess receiver conditions (as in Figure 2). Solid traces represent medians, lighter colored regions represent SEM, and gray shading represents SD. n, number of cell traces included in the alignment. See STAR Methods for alignment and normalization procedures. Bottom left: 95th percentile of (absolute, non-normalized) promoter activity values between 0 and 7.5 hr (after alignment) in individual traces included in the averaging. Solid horizontal lines represent medians, while the boxes delineate 25th–75th percentile values. p value calculated by two-sided K-S test.
(B) Corresponding response profiles (right, top left) and amplitudes (bottom left) in activated receiver cells co-cultured with Dll1 sender cells (blue) or Dll1ECD-Dll4ICD sender cells (purple) under excess sender conditions.
(C) Representative images of “excess sender” co-cultures of receiver cells (R) expressing full-length Notch1 and sender cells (S) expressing either Dll4ECD-Dll1ICD (left) or Dll4 (Dll4ECD-Dll4ICD, right), immunostained for Notch1ECD. Examples of dispersed, low intensity staining or higher-intensity puncta are indicated by the white circles.
(D) Left: Median values of number of puncta detected (see STAR Methods) in Dll1ICD (blue) or Dll4ICD (red) sender cells neighboring receiver cells. Right: Median values of the (background subtracted) mean pixel intensity of dispersed signal (see STAR Methods) within Dll1ICD (blue) or Dll4ICD (red) sender cells that neighbor receiver cells. Error bars represent SEM. p value calculated using the two-sided K-S test.
(E) Schematic: Proposed differences in the abilities of ligands containing the Dll1 (blue) and Dll4 (red) ICDs to initiate transendocytosis in different clustering states.
See also Figure S6.
How could ligand ICDs, functioning within sending cells, determine the dynamics of Notch activity in receiving cells? Based on previous work showing that the ligand ICD mediates receptor transendocytosis (Chitnis, 2006; Weinmaster and Fischer, 2011), we reasoned that the differences in dynamics between Dll1ICD and Dll4ICD ligands might reflect distinct modes of transendocytosis. We therefore compared transendocytosis in Dll1ICD and Dll4ICD sending cells, by immunostaining the Notch1ECD in sender-receiver co-cultures followed by confocal imaging (Figure S5F; STAR Methods).
We first compared Dll4 and Dll4ECD-Dll1ICD. At the interface between receivers and senders expressing either ligand, we observed regions of intense Notch1ECD staining, which colocalized with ligand staining (Figure S6A). This is consistent with previous observations of Notch ligand-receptor “clustering” at points of intercellular contacts (Bardot et al., 2005; Meloty-Kapella et al., 2012; Nichols et al., 2007). Within the sender cells, we observed two distinct types of staining for transendocytosed receptors: (1) dispersed, low-intensity staining that lacked apparent structure, and (2) discrete, high-intensity puncta that typically spanned >10 pixels (in three dimensions), possessed >100-fold higher cumulative intensities (Figures 5C and S6D), and co-localized with the endocytosis marker Rab5 (Figure S6B).
The generally pulsatile Dll1 ICD was strongly associated with the punctate endocytosis patterns in a signaling context. Dll4ECD-Dll1ICD senders adjacent to receivers showed a significant increase in the levels of punctate, but not dispersed, staining, relative to sender cells not adjacent to receivers (Figure S6C). Importantly, when compared at expression levels that produced similar Notch activity (Figure S6E), Dll4ECD-Dll1ICD sender cells exhibited more puncta per cell compared to Dll4 senders (Figure 5D, left). Wild-type Dll1 ligand also exhibited puncta (Figures S6F and S6G). Furthermore, the relative number of puncta per sender cell between Dll4ECD-Dll1ICD and Dll1 (Figure S6G, right) was similar to the ratio of their pulse rates (Figure S6H), while dispersed staining levels were similar. These results show that pulsatile signaling correlates with the appearance of punctate transendocytosis patterns. By contrast, Dll4 sender cells showed elevated levels of dispersed staining relative to sender cells not adjacent to receivers (Figure S6C) and also relative to Dll4ECD-Dll1ICD sender cells at the same mean signaling activity (Figure 5D, right), suggesting that dispersed staining reflects sustained signaling.
Together, these data suggest a model for how different ligands could generate different Notch activity dynamics in signal receiving cells through differences in transendocytosis patterns. In this model, the Dll1 ICD preferentially activates in the context of a ligand-receptor cluster (Figure 5E, top panel). A typical signaling event would involve the simultaneous activation of multiple receptors by interacting ligands within a single cluster, thereby releasing multiple NICDs at the same time to generate a pulse of signaling in the receiving cell (Figure S6I). In the sending cell, these events would produce transendocytic vesicles containing many receptor ECDs (punctate staining). By contrast, while the Dll4 ICD can also form clusters (Figures 5C and S6A), it would not require clustering for activation. It could thus predominantly activate in the context of smaller complexes, or individual ligand-receptor pairs (Figure 5E, bottom panel). This would enable Dll4 ICD to generate sustained Notch signal in the receiver cell (Figure S6I), consisting of a relatively steady “trickle” of receptor transendocytosis events, each generating a transendocytic vesicle containing a smaller number of receptor ECDs, leading to more dispersed staining in the sending cell.
DISCUSSION
The use of multiple channels is a fundamental aspect of engineered communication systems and could similarly provide powerful capabilities for intercellular communication. We find here that Dll1 and Dll4 can function as distinct communication channels in the Notch pathway by activating Notch1 with distinct dynamics (Figures 1 and 2) that can then be decoded into different patterns of Hes and Hey target gene expression (Figure 3) and cell fate (Figure 4).
While ligands differ in their mean amplitude of signaling, several lines of evidence show that downstream programs are particularly sensitive to dynamics. First, direct manipulation of signaling dynamics through the Notch1ΔECD system (Figure 3) demonstrates that even at pulse amplitudes larger than those occurring during intercellular signaling in co-cultures, the duration of NICD pulses strongly affect target gene activation patterns, with Hey1/L activation occurring only after a delay. This time-dependence cannot be explained by a slow ramp-up in NICD levels (Figures S3D–S3G). The role of dynamics is further supported by analysis of gene expression induced by Dll1 and Dll1ECD-Dll4ICD, which share the same extracellular domain, and therefore the same affinity for Notch1, but differ in their intracellular domains and signaling dynamics (Figures 5B and S5E). Overall, while amplitude undoubtedly plays an important role, these results are consistent with dynamic encoding and strongly argue against an exclusively amplitude-based scheme for ligand discrimination.
Dynamic encoding can be explained by a simple model based on previous observations that Notch ligands and receptors spontaneously assemble into ligand-receptor clusters at cell-cell interfaces. In the model, a Dll1-mediated pulse occurs when receptors in the cluster activate in a coordinated manner, releasing a burst of NICD (Figures 5E and S6I). The key requirement of the model is that the Dll1 ICD does not efficiently initiate transendocytosis until clusters reach a critical size, ensuring that most signaling occurs in pulses. By contrast, Dll4 may cluster, but would not require clustering for activation, and therefore be able to generate sustained signaling through activation of individual ligand-receptor complexes or smaller clusters (Figures 5E and S6I). Future studies should provide a more complete understanding of the molecular and biophysical basis of encoding by directly testing the sensitivity of transendocytosis to ligand-receptor clustering and elucidating the mechanism and dynamics of the clustering process (Seo et al., 2017).
Decoding of Notch dynamics is evident in the distinct responses of Hes and Hey Notch target genes to different durations of Notch activation (Figure 3). Known features of the Hes/Hey system, including the short half-life and negative autoregulation of Hes1 (Hirata et al., 2002), and negative cross-regulation between Hes1 and Hey1/L could play roles in decoding (Fischer and Gessler, 2007; Heisig et al., 2012; Kobayashi and Kageyama, 2014). The homologous Drosophila Hairy/E(spl) Notch target genes also show differential responses to different durations of Notch activation (Housden et al., 2013; Krejcí et al., 2009), suggesting that dynamic ligand discrimination could have existed ancestrally. A more complete and quantitative understanding of Hes/Hey interactions, including dimerization and cross-regulation, could provide insight into the decoding of Notch dynamics.
The ability of the Notch pathway to either promote or inhibit somite myogenesis, depending on the activating ligand (Figure 4), challenges the view that Notch activity promotes a single fate in any given context and shows that a seemingly minor change in ligand usage (i.e., from Dll1 to Dll4) can have dramatic consequences. Such contrasting roles for Notch ligands have also been reported in other contexts (Gama-Norton et al., 2015). The distinct effects of different ligands on cellular responses could have implications for therapeutic interventions targeting Notch signaling and for directed differentiation applications that require control of Notch-dependent cell fate decisions (Andersson and Lendahl, 2014; Behar et al., 2013; Dahlberg et al., 2011; Mohtashami et al., 2010). We note that, despite their intrinsic differences, there are cases where Dll1 can partially compensate for Dll4 (Mohtashami et al., 2010). This may be because at high expression levels, Dll1 pulses from multiple sender cells effectively ‘merge’ and thereby become indistinguishable from sustained activation produced by Dll4.
The use of dynamics to transmit multiple signals through the same pathway occurs in other systems (Purvis and Lahav, 2013) including p53 (Batchelor et al., 2011; Purvis et al., 2012), NFAT (Noren et al., 2016; Yissachar et al., 2013), nuclear factor κB (NF-κB) (Cheong et al., 2008; Covert et al., 2005), growth factor signaling (Marshall, 1995; Santos et al., 2007), and yeast stress response (Hansen and O’Shea, 2016; Hao and O’Shea, 2011), suggesting it is a broadly useful strategy. Dynamic encoding could be particularly beneficial when the amplitude of signaling is difficult to control precisely, due to variability in expression or cell contact. Signaling pathways such as transforming growth factor β (TGF- β), bone morphogenetic protein (BMP), and Wnt, like Notch, also utilize multiple ligands capable of interacting with multiple receptors (Antebi et al., 2017). This raises the question of whether these different ligands can be discriminated by signal-receiving cells and, if so, whether this discrimination involves dynamics. Finally, pulsatile and sustained signaling could also provide different patterning capabilities in highly dynamic Notch-dependent patterning processes such as neurogenesis (Imayoshi and Kageyama, 2014), lateral inhibition (Barad et al., 2010; Cohen et al., 2010), and somitogenesis (Oates et al., 2012; Pourquié, 2011). Ultimately, the discovery that the Notch pathway can transmit more and different types of information than previously suspected should help to explain how it enables such an extraordinary range of outcomes, in development and physiology.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
“Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Michael Elowitz (melowitz@caltech.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Gene constructs
All constructs used in this paper were assembled using standard restriction enzyme-based cloning and/or Gibson cloning (Gibson et al., 2009). pcDNA3-hNECD-Gal4 (Figures 1, 2, and 5) has been described previously (Sprinzak et al., 2010). The H2B-3xCitrine fluorescent reporter (Figures 1, 2, and 5) was constructed by cloning 3 repeats of mCitrine in frame with H2B, downstream of a UAS promoter. The mRNA destabilized version of this reporter was constructed by fusing the 3’UTR of mouse Hes1 downstream of the STOP codon. Ligand constructs were cloned into pcDNA5 or piggyBac plasmids (System Biosciences Inc.) by fusing the complete rat Dll1 (kind gift from G.Weinmaster) or human Dll4 cDNA in frame with T2A-H2B-mCherry, downstream of a previously described inducible pCMV-TO promoter (Sprinzak et al., 2010). We note that hDll1 shows the same pulsatile behavior described here for rDll1. Chimeric ligands (Figure 5) were constructed by exchanging the intracellular domains of rDll1 (aa 561 – 714) and hDll4 (aa 551 – 685). The hN1ΔECD gene (Figure 3) was cloned from hN1 (kind gift from J. Aster) by removing residues 22–1716 and fused in frame with myc-T2A-H2B-mCherry, downstream of the CMV-TO promoter in a piggyBac construct. Constructs used for in ovo electroporation (Figure 4) were made by cloning rDll1 or hDll4 cDNA (minus stop) upstream of, and in frame with, T2A-EGFP in a pCI-CAGG plasmid.
Tissue culture and Cell lines
CHO-K1 (Hamster cells, RRID:CVCL_0214, ATCC Catalog No. CCL-61) or CHO- TREx (RRID:CVCL_D586, Invitrogen) cells and their derivatives were grown on tissue-culture grade plastic plates (Thermo Scientific) in Alpha MEM Earle’s Salts (Life Technologies), supplemented with 10% Tet System Approved FBS (ClonTech), 100 U/ml penicillin, 100 ug/ml streptomycin, 0.292 mg/ml L-glutamine (GIBCO).
C2C12 cells (Mouse cells, RRID:CVCL_0188, ATCC Catalog No. CRL-1772) were grown in DMEM (Life Technologies), supplemented with 20% Tet System Approved FBS (ClonTech), 100 U/ml penicillin, 100 ug/ml streptomycin, 0.584 mg/ml L-glutamine (GIBCO). C2C12 media was used for CHO-K1 + C2C12 co-culture assays (Figure S4). All cells were grown at 37° C in 5% CO2 in a humidified atmosphere. Cells were passaged every 2–3 days, depending on confluency, using 0.05% or 0.25% Trypsin-EDTA (Life Technologies).
Cell line engineering
All cell lines used in this paper contained stable integrations of transgenes, and were typically clonal populations. To create each stable cell line, the following steps were followed: 1) Cells were first transfected with 800–1000 ng of plasmid DNA using Lipofectamine 2000 or Lipofectamine LTX. 2) 24 h later, cells were transferred to selection media containing 600 ug/ml Geneticin, 500 ug/ml Hygromycin, 400 ug/ml Zeocin, or 10 ug/ml Blasticidin as appropriate. 3) After selection for 1–2 weeks, the resulting polyclonal populations stably expressing the transgene were allowed to recover for ~1 week. 4) Single clones were isolated through the technique of limiting dilution. 5) Single clonal populations were screened for desired behavior, usually high expression (for constitutive genes) or low background expression of the transgene and large dynamic range (for inducible genes and reporter genes). Cell lines incorporating multiple transgenes were constructed by sequential rounds of this process. For piggybac constructs, the initial transfection comprised of the target plasmid along with the construct expressing the piggybac transposase, typically in a 1:1 or 2:1 molar ratio.
Chicken embryos
Fertile chicken (Gallus gallus) eggs, purchased from commercial sources, were incubated at in a humidified 37 C incubator, and staged by the criteria of Hamburger and Hamilton (HH) (Hamburger and Hamilton, 1992). Embryos were electroporated at stage 12–13, replaced in the incubator, and dissected 20h later.
METHOD DETAILS
Co-culture assays and time-lapse microscopy
Used in Figures 1, 2, 5, S1, S2, and S5
Surface treatment
In preparation for plating of cells, glass-bottom multi-well plates (MatTek, No. 1.5 glass, 10 mm radius) were coated with 5 ug/ml Hamster Fibronectin (Oxford Biomedical Research) diluted in 1x Phosphate-Buffered Saline (PBS) for 1h at room temperature.
Cell culture
After trypsinization, sender cells (pre-induced for > 48h with 4-epiTc, Sigma) or CHO-K1 cells were mixed in suspension with similarly trypsinized receiver cells at a ratio of 100:1 or 1:100, for excess sender or excess receiver assays, respectively. A total of 8×104 cells (60% confluence) were plated for each experiment, with continued 4-epiTc induction when appropriate. Imaging commenced 2–4h post-plating.
Time-lapse microscopy
Movies were acquired at 20X (0.75 NA) on an Olympus IX81 inverted epi-fluoresence microscope equipped with hardware autofocus (ZDC2) and an environmental chamber maintaining cells at 37C, 5% CO2. Automated acquisition software (METAMORPH, Molecular Devices) was used to acquire images every 30 min in multiple colors (YFP, RFP, CFP) or differential interference contrast (DIC), from multiple stage positions.
Plate-bound Dll1 assay
Used in Figures S1D and S1E
Recombinant human Dll1ext-Fc fusion proteins (kind gift from I. Bernstein) were diluted to 1 ug/ml in PBS, and the solution was used to coat the tissue-culture surface. After 1h incubation at room temperature, the solution was removed, and cells were plated for the experiment.
Image segmentation, tracking, and single-cell fluorescence calculation
Used in Figures 1, 2, 5, S1, S2, and S5
Custom MATLAB code (2013a, MathWorks) was used to segment cell nuclei in images based on constitutive CFP/RFP fluorescence or background YFP fluorescence. The segmentation procedure uses edge detection, adaptive thresholds, and the Watershed algorithm to detect nuclear edges. Nuclear segments were then matched in pairs of images corresponding to consecutive time frames, and thus tracked through the duration of the movie. Single-cell tracks were subsequently curated manually. In particular, there were periods where any given cell could not be automatically segmented (typically due to high density) but could be visually followed. In such cases, the tracks corresponding to the cell prior to and after such time frames were manually linked if fewer than ~5 frames were missing.
Fluorescence data was extracted from nuclear segments by calculating the integrated fluorescence within the segment and subtracting a background fluorescence level estimated from the local neighborhood of the segment. This fluorescence was linearly interpolated across time frames where nuclei could not be segmented automatically. Division events were detected automatically, and fluorescence traces were corrected for cell division by adding back fluorescence lost to sister cells. The resulting ‘continuized’ traces were smoothed and the difference in fluorescence between consecutive time frames was calculated. A smoothed version of this difference was used as the rate of change or promoter activity of the fluorescence.
Analysis of single-cell traces
Used in Figures 1, 2, 5, S1, S2, and S5
Alignment
For each receiver cell trace, including those of cells in control conditions (showing background fluorescence levels) an average rate of fluorescence increase (‘average slope’) was calculated by dividing the change in total fluorescence of the reporter by the duration of the trace. Traces showing activation were automatically selected for further analysis based on their average slopes surpassing a threshold value, chosen to be higher than average slopes observed in receiver cells under control conditions. Activating traces were aligned at the point of activation, defined as the time point when their promoter activity crosses an absolute threshold level, chosen based on typical promoter activities corresponding to background activity. Note that activations occurring during the first 15h of the movie were typically not considered, to eliminate transient effects produced by cell transfer to imaging conditions. The same thresholds were always used when direct comparisons were made between ligands or conditions, and we verified (by varying threshold levels) that qualitative results did not depend strongly on the choice of threshold.
For C2C12 dynamics (Figure S4) promoter activity could not be reliably used to align traces due to the low levels of reporter activity and resulting noise in the promoter activity data. These traces were instead aligned based on when the total fluorescence levels increased a threshold level.
Double-pulse alignment
In order to align traces showing two pulses in response to Dll1 (Figure S1D) at the second pulse, the following procedure was used: the first activation was determined using the usual procedure (see above). Traces were then normalized by the peak activity (‘Peak1’, 95th percentile) in the 0–7.5h window during which the first pulse is expected to reach maximum levels. Starting at 7.5h, i.e., after the peak of the first pulse, traces were re-aligned at the point when the subsequent promoter activity values cross Peak1, and re-normalized to the 90th percentile of values in the period from 7.5h (relative to the first activation point) to the end of the trace.
Normalization
When applied, the object of normalizing the response trace by its amplitude is to demonstrate its stereotyped features, such are relative rise time and duration. Un-normalized averaging would distort the shape of the response because higher-amplitude signals are also prolonged, since the timescales of the reporter are fixed by the half-lives of its components (Gal4 protein, H2B-3xCitrine mRNA) and do not scale with amplitude. Traces were typically normalized to the 90th percentile value during the analysis time window, except in Figure S2H, where traces were normalized to the 90th percentile value occurring within 15h after activation.
Amplitudes
While normalized traces were used to make comparisons of the stereotyped shapes of responses (see above), absolute values of promoter activity, calculated from non-normalized promoter activity, are reported in all amplitude comparisons. Except in Figure 2C, this amplitude represents the 95th percentile of (absolute, non-normalized) promoter activity values between 0 and 7.5h (after alignment) in the traces. This time window is chosen to simultaneously estimate the promoter activity at the peak of pulses and at steady-state levels of sustained signaling. In Figure 2C, the amplitude represents the 95th percentile of promoter activity values during the 25h after activation (the period over which activities are averaged).
Trace filtering
In Figure 1D, traces were included in the Dll1 alignment if the median promoter activity between 20–25h fell below 50% of the peak activity (95th percentile) in the 0–7.5h period (after alignment). This criterion was designed to automatically detect single pulses in the data. In Figure 2B traces were only included in the Dll1 alignment if the normalized value at 20h fell below 0.7. This filter eliminates traces consisting of multiple pulses, especially in the high Dll1 cases. A similar filter applied to Dll4 traces reveals a small fraction of cells activated transiently, but displaying qualitatively different behavior, such as a systematic increase in duration and amplitude with increasing Dll4 levels in senders. For C2C12 experiments in Figures 3G and 3H, activating cells were identified based on an increase in total fluorescence levels above a threshold.
Estimating Gal4 and mRNA half-lives, Related to Figure S1H
For this model, we assume that the free Gal4 protein produced due to cleavage of N1ECD-Gal4 degrades with first-order kinetics with rate γGal4 after inhibition of the pathway using DAPT, at time 0h.
Reporter mRNA m is produced through non-cooperative binding of Gal4 to the promoter, with dissociation constant K and maximum rate βm. m is degraded with rate constant γm.
The parameters γGal4; K and γm were calculated by fitting the Citrine mRNA m to the experimentally measured decay in Citrine fluorescence rate using the lsqnonlin function in MATLAB. The fit was constrained using bounds for γGal4 and γm of log(2)/5h – log(2)/3h, based on Sprinzak et al. (2010) and Bintu et al. (2016). Bootstrapped 95% confidence intervals were computed from 100 iterations of fitting 30 points, chosen randomly with replacement, out of a total 50 measured time points.
Mathematical model for estimating duration of Notch activation, Related to Figure S1J
For this model, we assume that Gal4 is produced at a rate βGal4 for a duration τact, and degrades with first-order kinetics with rate γGal4.
Reporter mRNA m is produced through non-cooperative binding of Gal4 to the promoter, with dissociation constant K and maximum rate βm. m is degraded with rate constant γm.
For the results of Figure S1, βGal4 = 1, βm = 1, and K = 6.6 (also fitted in Figure S1E), and estimated mean values from Figure S1E were used for the Gal4 and mRNA degradation rates.
Simulations of Dll1 pulse trains and analysis, Related to Figures S2A–S2F
This model constructs pulse-trains composed of Dll1-like pulses occurring at various frequencies and regularities based on each of three underlying pulse models, and analyzes the features of the resulting simulated signaling traces.
Pulse train construction (Figure S2B)
For each simulation we construct 200 pulse trains. Each pulse train is constructed from a series of pulses with the average Dll1 promoter activity pulse shape (Figure S1I), scaled by an amplitude randomly sampled from the empirically measured distribution of Dll1 pulse amplitudes (from the Figure 1D dataset). The first pulse occurs at 0h, representing activation at time 0 in the aligned Dll4 traces. Subsequently, new pulses are introduced after successive time intervals τ chosen based on one of the underlying pulse models (see below), and the composite signal is constructed until it extends at least 10h beyond the 25h time period averaged in Figure 1D.
Feature analysis (Figure S2D)
For each trace, two features are analyzed:
Amplitude: The amplitude of each constructed trace is its median value over 25h.
Intra-trace variability: After calculation of the amplitude, each trace is normalized to its 90th percentile value. For each point t in this trace, the local temporal variability is estimated by the standard deviation of values in a 10h window starting at t. The overall intra-trace variability calculated for each trace is the median of the local variability value at each point, calculated by moving a 10h time window through the trace.
For each simulation (200 constructed traces), the medians of the calculated amplitudes and intra-trace variability are tabulated, and the SEM calculated.
Pulse models (Figure S2C)
Three models are considered for the underlying pulsing process:
Periodic model: In this model, the interval τ between adjacent pulses is fixed at a value Tperiod, that can range from 1h to 8h. Since the Dll1 pulse decay becomes apparent after 7.5 h (Figure 1D), intervals greater than 8h will result in pulse trains in which the individual pulses can be clearly discerned in each trace, and the average behavior will show oscillations. Since neither individual Dll4 traces, nor the average shape display overt oscillatory features, values for Tperiod greater than 8h are not considered in the simulation.
Poisson model: In this model, the interval between successive pulses i and i+1, τi, represents the inverse of a pulse rate, ri, drawn from a Poisson distribution with parameter, λ, ranging from 1/h-1/15h.
Mixed model: In these models, the interval t between adjacent pulses is drawn from a normal distribution with mean Tperiod (range 1h - 15h) and standard deviation σ (2.5h or 5h). This model therefore combines the regular pulsing inherent to the periodic model with the trace-to-trace variability of the Poisson model (thus preventing ‘constructive interference’ of pulse peaks, which would lead to apparent oscillations in the average signal shape).
For every parameter value (Tperiod, λ, or σ, as appropriate) in each of the models, 36 simulations were run and the average of the median amplitudes and median intra-trace variabilities (see above) were calculated. These values are plotted in Figure S2E.
Bootstrapped analysis of variability in measured Dll4 signaling trace (Figure S2F)
Finally, for direct comparison to simulation data, the Dll4 dataset of traces (200 traces in total) was subsampled 30 times (50 traces per sample) to generate a bootstrapped distribution of measured median intra-trace variability, and a corresponding median value was calculated. This bootstrapped median is compared to simulation data in Figure S2F.
Sender cell categorization in excess receiver assays
Used in Figures 2 and S2
Dll1- and Dll4-T2A-H2B-mCherry sender cells were induced with different 4epi-Tc concentrations, to access their full dynamic range of ligand expression. Following co-culture with receiver cells and timelapse analysis, individual sender cell nuclei were automatically segmented, and mCherry levels were calculated. At the same time, each receiver cell response was automatically associated with the closest sender cell. All data, across 4epi-Tc induction levels, were then pooled, and sender cells re-categorized into ‘low’, ‘medium’, or ‘high’ expression along with their associated receiver cell responses. This process of pooling and recategorization was necessary because of the broad, overlapping distributions in mCherry expression produced by 4epi-Tc treatment.
Detection of surface ligand
Used in Figure S5D
Recombinant mouse Notch1ext-Fc chimeric protein (R&D Systems) was used for surface-detection of ligands at a concentration of 10 ug/ml, based on a previously described protocol (LeBon et al., 2014). Sender cells were first cultured and induced with 4epiTc for 48h, then transferred from media to blocking solution (2% FBS in Phosphate Buffered Saline, PBS) for 30 min at room temperature (RT). Cells were then incubated with recombinant mouse Notch1ext-Fc protein in binding solution (blocking solution containing 100 ug/ml CaCl2, R&D Systems) for 45 min at RT. Following this, cells were washed 3x with binding solution, then incubated with anti-mouse secondary antibody conjugated to AlexaFluor-488 (1:1000 dilution, Life Technologies) for 30 min. Cells were then trypsinized and analyzed using flow cytometry.
C2C12 N1ΔECD activation assays
The procedure for activating the Notch pathway in C2C12-hN1ΔECD cells was as follows: Cells were cultured in 10 μM DAPT (Sigma-Aldrich) until the experiment. In order to wash out DAPT, cells were washed quickly twice and a third time for 5 min with media at room temperature. Finally, cells were incubated in medium containing the appropriate activating DAPT concentration (0, 0.3, or 0.5 μM) at 37 C for the required activation duration (5 min, 15 min, 30 min, or until RNA extraction, i.e., sustained). In order to generate a pulse of activation, medium was then replaced with fresh 10 μM DAPT medium.
RNaseq
RNA was prepared using the RNeasy kit (QIAGEN) and submitted to the Caltech sequencing core facility, where cDNA libraries for RNaseq were prepared according to standard Illumina protocols. 100 base single-end read (100SR) sequencing was performed on a HiSeq2500 machine at the same facility. Reads were assembled, aligned, and mapped to the mouse genome (mm9 assembly) on a local instance of the Galaxy server, using Tophat. Cufflinks was used to calculate FPKM values.
In the analysis, we focused first on genes that showed > 5 fold-changes in their FKPM values (highlighted in Table S1). We further narrowed our subsequent analyses to the transcription factors Hes1, Hey1, and HeyL, because their promoters were shown to directly bind NICD by ChIP-Seq, they show early and strong (> 10-fold) responses to NICD, and they are key factors mediating Notch responsive behaviors in many contexts. These are also the only Hes and Hey family genes that activate in response to Notch in C2C12 cells (Castel et al., 2013). The RNaseq experiment did show upregulation of other genes, but we did not focus on them either because they were not transcription factors (such as Jag1 or Nrarp), or were not direct NICD targets based on the ChIP-Seq data.
RT-qPCR
RNA was prepared using the RNeasy kit (QIAGEN). cDNA was prepared from 500ng RNA using the iScript cDNA synthesis kit (Bio-Rad). 0.5 μL cDNA was used per 10 μL RT-qPCR reaction mix containing 1X iqSYBR Green Supermix (Bio-Rad) and 450 nM total forward and reverse primers. Reactions were performed on a BioRad CFX Real-Time PCR Detection System using a 2-step amplification protocol, with the following thermocycling parameters: 95 C, 3 min followed by 40 cycles of 95 C, 10 s (melting) and 55 C, 30 s (annealing + extension). All reactions were performed in duplicate.
Western blot analysis of NICD
Used in Figure S3
For this analysis, 0.5×106 – 1×106 cells were trypsinized after treatment, spun down in excess PBS, and lysed using Lithium Dodecyl Sulfate (LDS) buffer also containing reducing agents (DTT + 2-Mercaptoethanol) and Protease Inhibitors (Roche). Standard procedure was used for LDS-PAGE gel electrophoresis and transfer to nitrocellulose (iBlot, Thermo Fisher Scientific). Cleaved NICD (1:1000, Cell Signaling Technology, Catalog # D3B8) and GAPDH (1:5000, Abcam, Catalog #6C5) were detected using monoclonal antibodies. The blots were subsequently stained using HRP-conjugated secondary antibodies and detected using the Enhanced Chem-iLuminescence system (Pierce).
CHO-C2C12 co-culture assay
Used in Figure S4.
In preparation for the co-culture, C2C12-hN1 cells (4–6×104 cells in 12 well multi-well plate wells) were transfected with 60 pmol siRNA directed against mouse Notch2 (5’-UGAACUUGCAGGAUGGGUGAAGGUC-3’), using Lipofectamine RNAiMAX (Life Technologies). 24h later, 3×104 CHO-K1 based Dll1- and Dll4- sender cells (pre-induced for > 48h) were plated within the two chambers of ibidi culture inserts (Ibidi USA) on hamster fibronectin-treated (5 μg/ml in PBS, incubated for 3–5h at RT) surfaces of 24-well glass bottom plate wells. Once cells had attached to the surface (< 6h), inserts were removed and previously prepared C2C12-hN1 cells were plated, in 5 μM DAPT media, at high density so as to cover the gaps on the surface. After 12h, DAPT was washed out and cells were allowed to signal for 6h, after which the cultures were fixed in 4% formaldehyde at room temperature for 10 mins.
in ovo Electroporation
Batches of eggs were selected at random for electroporation with either Dll1 or Dll4, and the final data represents experiments conducted on at least two separate batches. The neural tubes of HH stage 12–13 embryos were injected with plasmid DNA (5 mg/ml) and electroporated by applying a series of current pulses (25V, 5x, 30 ms pulses separated by 100 ms) at the level of the pre-somitic mesoderm. 20h post-electroporation, embryos were screened for GFP fluorescence. Healthy embryos showing strong fluorescence in the neural crest were dissected (to remove extra-embryonic tissue) in Ringer’s solution and transferred to freshly prepared 4% paraformaldehyde, on ice. Embryos were fixed overnight at 4 C.
Hybridization Chain Reaction Fluorescence In Situ Hybridization
The hybridization chain reaction fluorescence in situ hybridization (HCR-FISH) protocol was based on a previously described protocol (Choi et al., 2016). Briefly, in situ HCR-FISH detection involves the following steps: 1. Dehydration and rehydration of embryos in MeOH, 2. Overnight hybridization with probes at 45 C, 3. Removal of unbound excess probes through washes at 45 C, 4. Overnight amplification at room temperature, and 5. Removal of excess amplifier. Each gene of interest was detected using 6 probes. At most three genes were detected simultaneously, typically EGFP, MyoD1, and Hes1, Hey1, or HeyL. After HCR processing, portions of the embryos anterior to the forelimbs were removed. Embryos were then mounted on glass-bottom multiwell plates in 1% agarose, with the dorsal surface in contact with the glass.
Confocal laser-scanning microscopy of embryos
Samples were imaged on a Zeiss LSM700 or using a 20x (0.8 NA) dry objective. For embryos, Z stacks were acquired using Zen software (ZEISS) and 3D-reconstructed in Imaris 8.0 (Bitplane). Optical slices in Imaris were used to remove obscuring auto-fluorescence from residual extra-embryonic tissue in the reconstructed images, without affecting signal in the areas of interest. For cell-culture Z stacks, the sum was projected in 2D using ImageJ.
Quantitation of effect on MyoD1 and Notch targets
Blind scoring of embryos for changes in MyoD1 (Used in Table 1)
3D images of transverse optical sections of the interlimb region of the trunk (containing 3–5 pairs of somites per image), were sorted randomly, and then scored blindly for differences in somite MyoD1 levels between the electroporated and control sides of the embryo. The scoring procedure was as follows: any features that might reveal the specific experimental perturbation (Dll1 or Dll4 ectopic expression), such as image filenames, differences in pseudo-color attributes, or information from secondary channels, were removed before the files were re-ordered using a pseudorandom sequence. Subsequently, images were scored blindly, comparing MyoD1 signal in somites on the electroporated side with signal in the corresponding somites on the control side, as long as the two somites were level with each other. This requirement minimizes imaging artifacts. Finally, sample images were re-matched with the perturbation type and scores were tallied. The number of embryos scored per condition (11 Dll1 expressing embryos, 10 Dll4 expressing embryos, 61 somites for each perturbation) is standard for this type of quantification (Rios et al., 2011).
Quantification of fold-changes in MyoD1, Hes1, and Hey1 gene-expression (Used in Figure S5C)
The DML regions of the somites on the electroporated and control sides were manually identified in Z-projections of 3D-reconstructed confocal images (see above), and the maximal HCR-FISH staining intensities (90th percentile values within identically-sized areas on both sides) were calculated. The reported fold-changes represent the ratio of these values for electroporated versus control DMLs.
Immunofluorescence detection of transendocytosed Notch in co-cultures
Used in Figures 5 and S5
Sender cells and receiver cells were co-cultured on glass-bottom dishes, in the excess sender configuration, as described above. After 24h of co-culture, cells were fixed in 4% formaldehyde (diluted in PBS). All subsequent steps were carried out in blocking solution (2% Bovine Serum Albumin diluted in PBS). Following 1h of incubation at room temperature, samples were incubated overnight at 4 C with 1:250 mouse anti-hNotch1 (Biolegend Catalog No. 352014, RRID AB_10899408). Samples were then washed and incubated in an anti-mouse secondary antibody conjugated to Alexa Fluor 488 (Life Technologies). After room temperature washes, samples were permeabilized in 0.3% Triton X-100 (Sigma-Aldrich) for 1h. Samples were then again incubated in 1:250 anti-hNotch1 overnight at 4C, following which they were incubated in Alexa Fluor 647 conjugated anti-mouse antibody (Life Technologies).
Confocal imaging and quantification of transendocytosed Notch
Used in Figures 5 and S5
Immunostained cultures (see above) were imaged as Z stacks (0.8 μm intervals) on an LSM800 inverted confocal microscope using a 100x (1.3 NA, oil-immersion) objective. Sender cells abutting receiver cells (or distant from them, for background estimation) were manually segmented in ImageJ software, and stacks composed of 5 slices each were exported to MATLAB. In MATLAB, pixels within the stacks were categorized as being either intracellular, or belonging to the cell surface, based on the intensity of pre-permeabilization stain. Only cells that showed mean dispersed staining intensities higher than the median of the background staining levels were included in further analysis. This selected cells that were likely to be active senders (especially in the Dll1 case); we verified that none of the cells eliminated at this step displayed puncta. Next, in order to identify puncta, the bwconncomp function in the Image Processing Toolbox was used to assess 3-D connectivities of intracellular pixels possessing intensities above a fixed threshold and to group them into puncta of sizes > 6 pixels. Several threshold/puncta size combinations were tested; one pair of values that returned puncta numbers most consistent with visual estimation was chosen. Qualitative conclusions remained the same for a range of threshold/size values. Pixels with intensities below the threshold, or failing to be included in such puncta were deemed part of the ‘dispersed’ staining.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistics
The non-parametric two-sided KS-test was typically used to compare the distributions of receiver activation amplitudes in response to different sender cell lines. All pairwise comparisons between samples fulfilled the criterion n1*n2/(n1 + n2) ≥ 4, where n1 and n2 represent the number of data points in two samples. Under this condition the KS-statistic is greater than the twice the inverse of the Kolmogorov statistic, and the calculated P-value is accurate. The non-parametric nature of the KS-test obviates the need to make assumptions regarding the shape of the distributions being compared. Furthermore, since the KS-test compares the distributions directly, and not the mean values, it is sensitive to differences in variance. Where the distribution itself is not shown, variance in the distribution is displayed as standard deviations or s.e.m. The number of samples (‘n’) used for calculating statistics is indicated in the Figures or accompanying legends.
DATA AND SOFTWARE AVAILABILITY
C2C12 hN1ΔECD transcriptomic data
Used in Figures 3 and S3
The accession number for the raw sequencing reads and processed FKPM data reported in this paper is Gene Expression Omnibus (GEO): GSE72847.
Code availability
Image segmentation and cell tracking code used can be accessed at https://github.com/nnandago/cell2017-segtrack. Datasets and processing code is available upon request.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-hNotch1 (extracellular domain) | BioLegend | Cat# 352014; RRID:AB_10899408 |
| Rabbit anti-hN1ICD (V1744) monoclonal [D3B8] | Cell Signaling Technology | Cat# 4147; RRID:AB_2153348 |
| Mouse anti-mGAPDH [6C5] | Abcam | Cat# ab8245; RRID:AB_2107448 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Human DII1ext-IgG | Sprinzak et al., 2010 | N/A |
| Mouse N1ext-mFc | R&D systems | Cat# 5267-TK |
| Hamster Fibronectin | Oxford Biomedical Research | Cat# CT30 |
| DAPT | Sigma Aldrich | Cat# D5942 |
| 4-epi tetracycline Hydrochloride | Sigma Aldrich | Cat# 37918 |
| Critical Commercial Assays | ||
| RNeasy mini kit for RNA extraction | QIAGEN | Cat# 74106 |
| iScript cDNA synthesis kit | Bio-Rad | Cat# 1708890 |
| iQ SYBR Green Supermix | Bio-Rad | Cat# 1708880 |
| DNA HCR kit | Molecular Instruments | N/A |
| Deposited Data | ||
| Raw and analyzed RNaseq data | This paper | GSE72847 |
| Experimental Models: Cell Lines | ||
| CHO-K1 | ATCC | Cat# CCL-61; RRID:CVCL_0214 |
| CHO TREx | Invitrogen | RRID:CVCL_D586 |
| C2C12 | ATCC | Cat# CRL-1772; RRID:CVCL_0188 |
| CHO-K1-TREx + pcDNA3-CMV-hN1ECD-Gal4esn + pEV-UAS-H2B-3xCitrine | This paper | N/A |
| CHO-K1-TREx + pcDNA3-CMV-hN1ECD-Gal4esn + pEV-UAS-H2B-3xCitrine + pGK-H2B-mCherry | This paper | N/A |
| CHO-K1-TREx + pcDNA5-CMV-TO-rDll1-T2A-H2B-mCherry | This paper | N/A |
| CHO-K1-TREx + pcDNA5-CMV-TO-rDll1-T2A-H2BmCherry + pLenti-CAG-H2B-Cerulean | This paper | N/A |
| CHO-K1-TREx + pcDNA5-CMV-TO-hDll4-T2A-H2B-mCherry | This paper | N/A |
| CHO-K1-TREx + pb-CMV-TO-hDll4-T2A-H2BmCherry + pb-CMV-H2B-Cerulean | This paper | N/A |
| CHO-K1-TREx + pb-CMV-TO-Gal4esn-T2A-H2BmCh + UAS-H2B-Citrine | This paper | N/A |
| CHO-K1-TREx + pEF-hN1ECD-Gal4esn + UAS-H2B- 3xCitrine-3′Hes1UTR | This paper | N/A |
| CHO-K1-TREx + pcDNA5-CMV-TO-rDll1ECD- Dll4ICD-T2A-H2B-mCherry | This paper | N/A |
| CHO-K1-TREx + pcDNA5-CMV-TO-hDll4ECD- Dll1ICD-T2A-H2B-mCherry | This paper | N/A |
| CHO-K1-TREx + pcDNA5-CMV-TO-rDll1-FLAG | This paper | N/A |
| CHO-K1-TREx + pcDNA5-CMV-TO-hDll4-FLAG | This paper | N/A |
| CHO-K1-TREx + pb-CMV-hN1-T2A-H2B-mCherry | This paper | N/A |
| CHO-K1-TREx + pcDNA5-CMV-TO- hDll4ECD-Dll1ICD -FLAG | This paper | N/A |
| C2C12 + pb/CMV7-hN1-myc-T2A-H2B-mCherry | This paper | N/A |
| C2C12 + pb-CMV-TO-hN1ΔECD-T2A-H2B-mCherry | This paper | N/A |
| C2C12 + pb/hNECD-Gal4esn-ANK-T2A-H2B-Cer + pEV/UAS-dm-H2B-3xCit | This paper | N/A |
| C2C12 + pb/TO-hDll1-T2A-H2B-mCh-P2A-Hyg | This paper | N/A |
| C2C12 + pb/TO-hDll1-T2A-H2B-mCh-P2A-Hyg | This paper | N/A |
| Experimental Models: Organisms | ||
| Stage 12–13 chicken embryos | This paper | N/A |
| Recombinant DNA | ||
| pcDNA3/CMV-hN1ECD-Gal4esn | Sprinzak et al., 2010 | N/A |
| pEV/UAS-H2B-3xCitrine | LeBon et al., 2014 | N/A |
| pcDNA5/CMV-TO-rDll1-T2A-H2B-mCherry | This paper | N/A |
| pcDNA5/CMV-TO-hDll4-T2A-H2B- mCherry | This paper | N/A |
| pb/CMV-TO-Gal4esn-T2A-H2B- mCherry | This paper | N/A |
| pEV/UAS-H2B-3xCitrine-3′ Hes1 UTR | This paper | N/A |
| pcDNA5/CMV-TO-Dll1ECD-Dll4ICD | This paper | N/A |
| pcDNA5/CMV-TO-Dll4ECD-Dll1ICD | This paper | N/A |
| pcDNA5/CMV-TO-Dll1-FLAG | This paper | N/A |
| pcDNA5/CMV-TO-Dll4ECD-Dll1ICD-FLAG | This paper | N/A |
| pcDNA5/CMV-TO-Dll4-FLAG | This paper | N/A |
| pb/CMV-TO-hN1DECD-T2A-H2B- mCherry | This paper | N/A |
| pb/CMV7-hN1-myc-T2A-H2B- mCherry | This paper | N/A |
| pCI/CAGG-rDll1-T2A-EGFP | This paper | N/A |
| pCI/CAGG-hDll4-T2A-EGFP | This paper | N/A |
| pb/CMV-TO-hDll1-T2A-H2B- mCherry | This paper | N/A |
| pb/CMV-TO-hDll4-T2A-H2B- mCherry | This paper | N/A |
| Oligonucleotides | ||
| siRNA targeting mouse Notch2 5′-UGAACUU GCAGGAUGGGUGAAGGUC-3′ | Invitrogen | N/A |
| mouse Hes1 primer set 1 (Figure 3C) - Forward, 5′-CAACACGACACCGGACAAAC-3′ | IDT DNA | N/A |
| mouse Hes1 primer set 1 (Figure 3C) - Reverse, AAGAATAAATGAAAGTCTAAGCCAA-3′ | IDT DNA | N/A |
| Mouse Hes1 primer set 2 (Figure 3D, S4, 5) – Forward, 5′-AAGAATAAATGAAAGTCTAAGCCAA-3′ | IDT DNA | N/A |
| Mouse Hes1 primer set 2 (Figure 3D, S4, 5) – Reverse, 5′-TTCTTGCCCTTCGCCTCTTC-3′ | IDT DNA | N/A |
| mouse Hey1 primers – Forward, 5′-GCCGAAGTTG CCCGTTATCT-3′ | IDT DNA | N/A |
| mouse Hey1 primers – Reverse, 5′-CGCTGGGATG CGTAGTTGTT-3′ | IDT DNA | N/A |
| mouse HeyL primers – Forward, 5′-GAGCTGAC TTCCCACAACCA-3′ | IDT DNA | N/A |
| mouse HeyL primers – Reverse, 5′-GAGAGG TGCCTTTGCGTAGA-3′ | IDT DNA | N/A |
| mouse SdhA primers - Forward, 5′-AGTGGGCT GTCTTCCTTAAC-3′ | IDT DNA | N/A |
| mouse SdhA primers - Reverse, 5′-GGATTGCTTCT GTTTGCTTGG-3′ | IDT DNA | N/A |
| Software and Algorithms | ||
| Segmentation, tracking, and fluorescence analysis software | This paper | https://github.com/nnandago/cell2017-segtrack |
Highlights.
Dll1 and Dll4 can activate distinct targets through the same Notch receptor
Ligand identity is encoded in pulsatile or sustained Notch activation dynamics
Dynamic encoding involves ligand-receptor clustering
Dll1 and Dll4 induce opposite cell fates during embryonic myogenesis
ACKNOWLEDGMENTS
We thank Mark Budde, Joe Markson, Pulin Li, Yihan Lin, James Linton, Emily Capra, Jordi Garcia-Ojalvo, and Xiaojing Gao for critical feedback on the manuscript, and Young-Wook Jun, Roy Kishony, Irv Bernstein, Stephen Blacklow, and Elizabeth Jensen for helpful discussions. Harry Choi and Colby Calvert, Caltech Flow Cytometry Facility, Caltech Biological Imaging Facility, and the Millard and Muriel Jacobs Genetics and Genomics Laboratory at Caltech provided essential technical assistance. This work was supported by the Defense Advanced Research Projects Agency (HR0011-16-0138), by the NIH (R01 HD075335), and the NSF (EFRI 1137269). N.N. was a Howard Hughes Medical Institute International Student Research fellow.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information includes six figures, one table, and five movies and can be found with this article online at https://doi.org/10.1016/j.cell.2018.01.002.
REFERENCES
- Allan AL, Albanese C, Pestell RG, and LaMarre J (2001). Activating transcription factor 3 induces DNA synthesis and expression of cyclin D1 in hepatocytes. J. Biol. Chem 276, 27272–27280. [DOI] [PubMed] [Google Scholar]
- Andersson ER, and Lendahl U (2014). Therapeutic modulation of Notch signalling–are we there yet? Nat. Rev. Drug Discov 13, 357–378. [DOI] [PubMed] [Google Scholar]
- Andrawes MB, Xu X, Liu H, Ficarro SB, Marto JA, Aster JC, and Blacklow SC (2013). Intrinsic selectivity of Notch 1 for Delta-like 4 over Delta-like 1. J. Biol. Chem 288, 25477–25489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antebi YE, Linton JM, Klumpe H, Bintu B, Gong M, Su C, McCardell R, and Elowitz MB (2017). Combinatorial signal perception in the BMP pathway. Cell 170, 1184–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barad O, Rosin D, Hornstein E, and Barkai N (2010). Error minimization in lateral inhibition circuits. Sci. Signal 3, ra51. [DOI] [PubMed] [Google Scholar]
- Bardot B, Mok L-P, Thayer T, Ahimou F, and Wesley C (2005). The Notch amino terminus regulates protein levels and Delta-induced clustering of Drosophila Notch receptors. Exp. Cell Res 304, 202–223. [DOI] [PubMed] [Google Scholar]
- Batchelor E, Loewer A, Mock C, and Lahav G (2011). Stimulus-dependent dynamics of p53 in single cells. Mol. Syst. Biol 7, 488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behar M, Barken D, Werner SL, and Hoffmann A (2013). The dynamics of signaling as a pharmacological target. Cell 155, 448–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bintu L, Yong J, Antebi YE, McCue K, Kazuki Y, Uno N, Oshimura M, and Elowitz MB (2016). Dynamics of epigenetic regulation at the single-cell level. Science 351, 720–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray SJ (2016). Notch signalling in context. Nat. Rev. Mol. Cell Biol 17, 722–735. [DOI] [PubMed] [Google Scholar]
- Cappellari O, Benedetti S, Innocenzi A, Tedesco FS, Moreno-Fortuny A, Ugarte G, Lampugnani MG, Messina G, and Cossu G (2013). Dll4 and PDGF-BB convert committed skeletal myoblasts to pericytes without erasing their myogenic memory. Dev. Cell 24, 586–599. [DOI] [PubMed] [Google Scholar]
- Castel D, Mourikis P, Bartels SJJ, Brinkman AB, Tajbakhsh S, and Stunnenberg HG (2013). Dynamic binding of RBPJ is determined by Notch signaling status. Genes Dev. 27, 1059–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheong R, Hoffmann A, and Levchenko A (2008). Understanding NF-kappaB signaling via mathematical modeling. Mol. Syst. Biol 4, 192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chitnis A (2006). Why is delta endocytosis required for effective activation of notch? Dev. Dyn 235, 886–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi HMT, Chang JY, Trinh A, Padilla JE, Fraser SE, and Pierce NA (2010). Programmable in situ amplification for multiplexed imaging of mRNA expression. Nat. Biotechnol 28, 1208–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi HMT, Calvert CR, Husain N, Huss D, Barsi JC, Deverman BE, Hunter RC, Kato M, Lee SM, Abelin ACT, et al. (2016). Mapping a multiplexed zoo of mRNA expression. Development 143, 3632–3637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen M, Georgiou M, Stevenson NL, Miodownik M, and Baum B (2010). Dynamic filopodia transmit intermittent Delta-Notch signaling to drive pattern refinement during lateral inhibition. Dev. Cell 19, 78–89. [DOI] [PubMed] [Google Scholar]
- Covert MW, Leung TH, Gaston JE, and Baltimore D (2005). Achieving stability of lipopolysaccharide-induced NF-kappaB activation. Science 309, 1854–1857. [DOI] [PubMed] [Google Scholar]
- Dahlberg A, Delaney C, and Bernstein ID (2011). Ex vivo expansion of human hematopoietic stem and progenitor cells. Blood 117, 6083–6090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elena de Bellard M, and Bronner-Fraser M (2005). Neural crest migration methods in the chicken embryo. Methods Mol. Biol 294, 247–267. [DOI] [PubMed] [Google Scholar]
- Fischer A, and Gessler M (2007). Delta-Notch–and then? Protein interactions and proposed modes of repression by Hes and Hey bHLH factors. Nucleic Acids Res. 35, 4583–4596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortini ME, Rebay I, Caron LA, and Artavanis-Tsakonas S (1993). An activated Notch receptor blocks cell-fate commitment in the developing Drosophila eye. Nature 365, 555–557. [DOI] [PubMed] [Google Scholar]
- Fryer CJ, White JB, and Jones KA (2004). Mastermind recruits CycC:CDK8 to phosphorylate the Notch ICD and coordinate activation with turnover. Mol. Cell 16, 509–520. [DOI] [PubMed] [Google Scholar]
- Gama-Norton L, Ferrando E, Ruiz-Herguido C, Liu Z, Guiu J, Islam AB, Lee SU, Yan M, Guidos CJ, López-Bigas N, et al. (2015). Notch signal strength controls cell fate in the haemogenic endothelium. Nat. Commun 6, 8510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson DG, Young L, Chuang R-Y, Venter JC, Hutchison CA 3rd, and Smith HO (2009). Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6, 343–345. [DOI] [PubMed] [Google Scholar]
- Guruharsha KG, Kankel MW, and Artavanis-Tsakonas S (2012). The Notch signalling system: recent insights into the complexity of a conserved pathway. Nat. Rev. Genet 13, 654–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gururajan M, Simmons A, Dasu T, Spear BT, Calulot C, Robertson DA, Wiest DL, Monroe JG, and Bondada S (2008). Early growth response genes regulate B cell development, proliferation, and immune response. J. Immunol 181, 4590–4602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamburger V, and Hamilton HL (1992). A series of normal stages in the development of the chick embryo. 1951. Dev. Dyn 195, 231–272. [DOI] [PubMed] [Google Scholar]
- Hansen AS, and O’Shea EK (2016). Encoding four gene expression programs in the activation dynamics of a single transcription factor. Curr. Biol 26, R269–R271. [DOI] [PubMed] [Google Scholar]
- Hao N, and O’Shea EK (2011). Signal-dependent dynamics of transcription factor translocation controls gene expression. Nat. Struct. Mol. Biol 19, 31–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heisig J, Weber D, Englberger E, Winkler A, Kneitz S, Sung W-K, Wolf E, Eilers M, Wei C-L, and Gessler M (2012). Target gene analysis by microarrays and chromatin immunoprecipitation identifies HEY proteins as highly redundant bHLH repressors. PLoS Genet. 8, e1002728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirata H, Yoshiura S, Ohtsuka T, Bessho Y, Harada T, Yoshikawa K, and Kageyama R (2002). Oscillatory expression of the bHLH factor Hes1 regulated by a negative feedback loop. Science 298, 840–843. [DOI] [PubMed] [Google Scholar]
- Housden BE, Fu AQ, Krejci A, Bernard F, Fischer B, Tavaré S, Russell S, and Bray SJ (2013). Transcriptional dynamics elicited by a short pulse of notch activation involves feed-forward regulation by E(spl)/Hes genes. PLoS Genet. 9, e1003162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilagan MXG, Lim S, Fulbright M, Piwnica-Worms D, and Kopan R (2011). Real-time imaging of notch activation with a luciferase complementation-based reporter. Sci. Signal 4, rs7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imayoshi I, and Kageyama R (2014). bHLH factors in self-renewal, multipotency, and fate choice of neural progenitor cells. Neuron 82, 9–23. [DOI] [PubMed] [Google Scholar]
- Kesarwani M, Kincaid Z, Gomaa A, Huber E, Rohrabaugh S, Siddiqui Z, Bouso MF, Latif T, Xu M, Komurov K, et al. (2017). Targeting c-FOS and DUSP1 abrogates intrinsic resistance to tyrosine-kinase inhibitor therapy in BCR-ABL-induced leukemia. Nat. Med 23, 472–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T, and Kageyama R (2014). Expression dynamics and functions of Hes factors in development and diseases. Curr. Top. Dev. Biol 110, 263–283. [DOI] [PubMed] [Google Scholar]
- Kopan R, Schroeter EH, Weintraub H, and Nye JS (1996). Signal transduction by activated mNotch: importance of proteolytic processing and its regulation by the extracellular domain. Proc. Natl. Acad. Sci. USA 93, 1683–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krejcí A, Bernard F, Housden BE, Collins S, and Bray SJ (2009). Direct response to Notch activation: signaling crosstalk and incoherent logic. Sci. Signal 2, ra1. [DOI] [PubMed] [Google Scholar]
- LeBon L, Lee TV, Sprinzak D, Jafar-Nejad H, and Elowitz MB (2014). Fringe proteins modulate Notch-ligand cis and trans interactions to specify signaling states. eLife 3, e02950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecourtois M, and Schweisguth F (1998). Indirect evidence for Delta-dependent intracellular processing of notch in Drosophila embryos. Curr. Biol 8, 771–774. [DOI] [PubMed] [Google Scholar]
- Marshall CJ (1995). Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal-regulated kinase activation. Cell 80, 179–185. [DOI] [PubMed] [Google Scholar]
- Meloty-Kapella L, Shergill B, Kuon J, Botvinick E, and Weinmaster G (2012). Notch ligand endocytosis generates mechanical pulling force dependent on dynamin, epsins, and actin. Dev. Cell 22, 1299–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohtashami M, Shah DK, Nakase H, Kianizad K, Petrie HT, and Zúñiga-Pflücker JC (2010). Direct comparison of Dll1- and Dll4-mediated Notch activation levels shows differential lymphomyeloid lineage commitment outcomes. J. Immunol 185, 867–876. [DOI] [PubMed] [Google Scholar]
- Nichols JT, Miyamoto A, Olsen SL, D’Souza B, Yao C, and Weinmaster G (2007). DSL ligand endocytosis physically dissociates Notch1 heterodimers before activating proteolysis can occur. J. Cell Biol 176, 445–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noren DP, Chou WH, Lee SH, Qutub AA, Warmflash A, Wagner DS, Popel AS, and Levchenko A (2016). Endothelial cells decode VEGF-mediated Ca2+ signaling patterns to produce distinct functional responses. Sci. Signal 9, ra20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oates AC, Morelli LG, and Ares S (2012). Patterning embryos with oscillations: structure, function and dynamics of the vertebrate segmentation clock. Development 139, 625–639. [DOI] [PubMed] [Google Scholar]
- Pourquié O (2011). Vertebrate segmentation: from cyclic gene networks to scoliosis. Cell 145, 650–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preuße K, Tveriakhina L, Schuster-Gossler K, Gaspar C, Rosa AI, Henrique D, Gossler A, and Stauber M (2015). Context-dependent functional divergence of the Notch ligands DLL1 and DLL4 in vivo. PLoS Genet. 11, e1005328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purvis JE, and Lahav G (2013). Encoding and decoding cellular information through signaling dynamics. Cell 152, 945–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purvis JE, Karhohs KW, Mock C, Batchelor E, Loewer A, and Lahav G (2012). p53 dynamics control cell fate. Science 336, 1440–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rios AC, Serralbo O, Salgado D, and Marcelle C (2011). Neural crest regulates myogenesis through the transient activation of NOTCH. Nature 473, 532–535. [DOI] [PubMed] [Google Scholar]
- Santos SDM, Verveer PJ, and Bastiaens PIH (2007). Growth factorinduced MAPK network topology shapes Erk response determining PC-12 cell fate. Nat. Cell Biol 9, 324–330. [DOI] [PubMed] [Google Scholar]
- Seo D, Southard KM, Kim J-W, Lee HJ, Farlow J, Lee J-U, Litt DB, Haas T, Alivisatos AP, Cheon J, et al. (2017). A mechanogenetic toolkit for interrogating cell signaling in space and time. Cell 169, 1357. [DOI] [PubMed] [Google Scholar]
- Sprinzak D, Lakhanpal A, Lebon L, Santat LA, Fontes ME, Anderson GA, Garcia-Ojalvo J, and Elowitz MB (2010). Cis-interactions between Notch and Delta generate mutually exclusive signalling states. Nature 465, 86–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Struhl G, and Adachi A (1998). Nuclear access and action of notch in vivo. Cell 93, 649–660. [DOI] [PubMed] [Google Scholar]
- Weinmaster G, and Fischer JA (2011). Notch ligand ubiquitylation: what is it good for? Dev. Cell 21, 134–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yissachar N, Sharar Fischler T, Cohen AA, Reich-Zeliger S, Russ D, Shifrut E, Porat Z, and Friedman N (2013). Dynamic response diversity of NFAT isoforms in individual living cells. Mol. Cell 49, 322–330. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
C2C12 hN1ΔECD transcriptomic data
Used in Figures 3 and S3
The accession number for the raw sequencing reads and processed FKPM data reported in this paper is Gene Expression Omnibus (GEO): GSE72847.
Code availability
Image segmentation and cell tracking code used can be accessed at https://github.com/nnandago/cell2017-segtrack. Datasets and processing code is available upon request.