

ABSTRACT:
Dark state exchange saturation transfer (DEST) and lifetime line-broadening (ΔR2, the difference in the measured transverse relaxation rates for the observable species in the presence and absence of exchange with a species characterized by very large intrinsic transverse relaxation rates) have proven to be powerful NMR tools for studying exchange phenomena between a NMR visible species and a high-molecular weight, “dark”, NMR invisible state. However, in the exchange regime, where the transverse spin relaxation rates in the bound state are smaller than the strength of the DEST saturation radio frequency field, typically corresponding to systems below ∼6 MDa, the combination of DEST and ΔR2 data, while sufficient to define the apparent association rate unambiguously constant, cannot determine the population of the bound state pB and values independently. We show that the latter exchange and relaxation parameters can be decorrelated by the measurement of the maximal value of the contribution of the fast-relaxing magnetization component to the total NMR signal, , an observable that is directly proportional to pB. When integrated into the analysis of DEST/ΔR2 data, provides an indispensable source of information for quantitative studies of exchange involving high-molecular-weight dark states. We demonstrate the utility of this approach by investigating the binding kinetics of two huntingtin exon-1-derived peptides to small unilamellar lipid vesicles (SUV), ∼ 31 nm in diameter and 4.3 MDa in molecular weight. The interaction of the N-terminal amphiphilic domain of huntingtin exon-1 with membrane surfaces promotes polyglutamine-mediated aggregation and, as such, is thought to play a role in the etiology of Huntington’s disease, an autosomal dominant fatal neurodegenerative condition. The first peptide comprises the 16-residue N-terminal amphiphilic domain (httNT) alone, while the second contains an additional seven residue polyglutamine tract at the C-terminus (httNTQ7). At a peptide-to-lipid molar ratio of 1:4, the population of peptide bound to the SUV surface is substantial, ∼ 7−8%, while exchange between the free and SUV-bound peptide is slow on the relaxation time-scale (kex ∼ 200 s−1). The last two C-terminal residues of httNT and the last 9 of httNTQ7 remain flexible in the SUV-bound form due to transient detachment from the lipid surface that occurs on a time-scale several-fold faster than binding.
Graphical Abstract
INTRODUCTION
Dark state exchange saturation transfer (DEST) and lifetime line broadening (ΔR2) provide a powerful means of probing exchange dynamics and atomic resolution dynamics involving the interaction of a NMR visible species with a very high-molecular weight NMR invisible “dark” state.1,2 (Note that ΔR2 is defined as the difference in transverse relaxation rates measured on the observable species in the presence and absence of exchange, arising from a significantly higher intrinsic transverse relaxation rate in the “dark” state.) Typically, DEST profiles acquired at two radio frequency (RF) saturation fields and ΔR2 values measured at two static spectrometer fields are analyzed together with the aim of quantitatively describing the exchange equilibrium in question; namely, extracting the rate constants of all involved processes, the populations of all states, and the residue-specific transverse spin relaxation rates of the minor, “dark” species.3 Initial applications of the DEST/ΔR2 methodology focused on exchange between monomeric amyloid-β peptides free in solution and bound to the surface of amyloid-β protofibrils.1,2,4 More recent examples have involved the interaction of intrinsically disordered peptides and small protein domains with the ∼800 kDa chaperonin GroEL.5–7 In these latter studies, however, full characterization of the exchanging system required supplementing the DEST and ΔR2 data with exchange-induced chemical shift and/or Carr−Purcell−Meiboom−Gill (CPMG) relaxation dispersion measurements. 8–10 In this article, we illustrate some limitations of the DEST/ΔR2 methodology in applications involving exchanging systems with medium-sized dark states (1−6 MDa), provide a semiquantitative explanation of their origin, and describe the manner and conditions under which these limitations can be overcome.
Here we focus on the characterization of the binding kinetics of two peptides derived from exon-1 of the huntingtin gene (htt) to unilamellar lipid vesicles. Polyglutamine expansion of more than 35 glutamines, immediately C-terminal to the 16-residue N-terminal amphiphilic sequence (httNT), results in a fatal neurodegerative condition known as Huntington’s disease.11 The two peptides are httNT and httNTQ7, the latter containing a seven-residue glutamine repeat. Interaction of the httNT domain with membrane surfaces promotes polyglut-amine-mediated aggregation.12,13 Recently, we showed, using chemical exchange saturation transfer (CEST) NMR and Q-band-pulsed electron paramagnetic resonance (EPR), that both peptides form a well-defined α-helical structure when bound to lipid micelles and, in addition, can dimerize in the bound state.14 As small unilamelar lipid vesicles (SUV) have a molecular weight of ∼4.3 MDa and are ∼4-fold larger in diameter than micelles (∼31 versus ∼8 nm), the same experimental approach cannot be used to study the exchange of httNT and httNTQ7 with their SUV-bound form, and hence, experiments such as DEST and ΔR2 are required. However, in the exchange regime where the transverse spin relaxation rate of the bound state is smaller than the strength of the RF saturation field, DEST and ΔR2 data are only sensitive to the product of and the population pB of the bound state, and hence, and pB cannot be determined independently. We show that this problem can be resolved by supplementing the DEST and ΔR2 data with the measurement of residue-specific maximal (on-resonance) values of the contribution of the fast-relaxing component of magnetization, , to the total NMR signal undergoing slow chemical exchange.15 As provides a good measure of pB, simultaneous analysis of DEST, ΔR2, and allows pB and to be decorrelated from one another. In the absence of other experimental data on the exchanging system under consideration, such as chemical exchange induced shifts and/or CPMG relaxation dispersions,5,7 which may not be readily available for systems in exchange with very high-molecular weight (>1 MDa) dark states, serves as a valuable addition to the array of existing NMR observables that report on the parameters of exchange involving NMR-dark systems.
EXPERIMENTAL SECTION
Expression and Purification of httNTQn Peptides.
Huntingtin exon-1 peptides httNTQn (n = 0 or 7, where n is the number of C-terminal polyglutamine repeats) were expressed and 15N-labeled in E. coli as GB1−httNTQn fusion proteins and purified as described previously.14 NMR samples comprised uniformly 15N-labeled peptides at a final concentration of 300 μM (after the addition of liposome solutions). All NMR samples were prepared in 20 mM phosphate buffer (Na2HPO4/NaH2PO4) pH 6.5, 50 mM NaCl, and 10% D2O/ 90% H2O (v/v). Peptide concentrations were determined by UV absorption at a wavelength of 205 nm.16
Preparation and Characterization of SUVs.
The sodium salts of 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho(1′-rac-glycerol) (POPG), 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), and cholesterol were purchased from Avanti Polar Lipids (Alabaster, AL). SUV particles were prepared by dissolving the same amount (35% mol/mol) of POPG and POPC in a round-bottom flask. Cholesterol (30% mol/mol) was added to the lipid mixture to reduce the permeability of vesicles upon httNTQn binding.13 Homogenous SUV preparations were obtained by sonication as described previously.17 The size distribution of the SUV particles (mean diameter ∼31 nm with a polydispersity index PDI = 0.22) was obtained on 1 mM samples (in lipids) at 10 °C from analysis of dynamic light scattering (DLS) data obtained with a Zetasizer Nano ZS (Malvern Instruments, 4 mW He−Ne laser, λ0 = 633 nm, θ = 173°).
NMR Measurements.
NMR experiments were recorded at 10 °C using a Bruker Avance-III spectrometer equipped with a TCI z-axis gradient cryogenic probe operating at a 1H Larmor frequency of 600.82 MHz. All measurements were performed on a 300 μM httNTQn (n = 0 or 7) peptide in the presence of SUVs at a 1:4 peptide:lipid molar ratio. On-resonance 15N-R1ρ measurements were carried out using the pulse sequence described by Yuwen et al.15 A 15N spin-lock field strength of 2.0 kHz was applied for spin-lock durations (Trelax) of 20, 40, 60, 80, and 100 ms. The values of the constant time during which longitudinal relaxation occurs (Trelax,max) and the equilibration delay (τex) were set to 120 and 50 ms, respectively. A reference spectrum (Trelax = 0) was acquired with the 15N spin-lock as well as adiabatic half-passage (AHP) pulses prior to and after each spin-lock period omitted. Two spin-lock elements were used in each relaxation delay (see the pulse-scheme in Figure S5 of Yuwen et al.15), effectively amplifying the value of approximately 2-fold. Each 2D experiment comprised 128* × 512* complex points in the indirect (15N) and direct (1H) dimensions, respectively. Sixty-four scans per increment, with an interscan delay of 2.5 s, were used, resulting in a total acquisition time of ∼11.4 h per 2D experiment. The values of were measured by extrapolation of the exponential decays to time zero.15 The values of Trelax were corrected for the duration of the two AHP pulses (2.4 ms total). 15N-R2 values for the httNTQn peptides in the absence and presence of SUVs were obtained from 15N-R1ρ and R1 measurements recorded at both 600 and 800 MHz as described previously.6,17 ΔR2 values were calculated as the difference between 15N-R2 values obtained in the presence and absence of SUVs.
Two-dimensional 15N-DEST measurements were performed by applying continuous wave (CW) saturation at RF field strengths of 500 and 750 Hz for a duration of 0.7 s at the following offsets from the carrier frequency (in kHz): −22.0, −20.0, −18.0, −16.0, −14.0, −12.0, −10.0, −8.0, −6.0, −5.0, −4.0, −3.0, −2.0, −1.0, −0.5, 0.0, 0.5, 1.0, 2.0, 3.0, 4.0, 5.0, 6.0, 8.0, 10.0, 12.0, 14.0, 16.0, 18.0, 20.0, and 22.0. The carrier was set to 121.5 ppm for 15N and 4.7 ppm for 1H. Reference experiments were performed applying the same RF field strengths but with the offset moved to −1 MHz. A total of 110* × 512* complex data points were acquired in the indirect (15N) and direct (1H) dimensions with respective acquisition times of 71 and 64 ms, resulting in a total measurement time of ∼3 h per 2D experiment.
NMR spectra were processed with the nmrPipe/nmrDraw suite of programs, and uncertainties in the extracted relaxation rates were estimated from Monte Carlo simulations.18
Simultaneous Analysis of 15N-DEST, ΔR2, and Data.
All experimental data (15N- DEST, ΔR2, and ) were fit simultaneously by minimizing the following error function:
| (1) |
where the first term accounts for the ΔR2 data, the second for the DEST normalized intensities κ, and the third for . The indices i, k, and j refer to the residue number, DEST offset, and saturation RF field strength (500 and 750 Hz), respectively; α1−3 represent empirically determined factors used to appropriately weight the different data types and have values of 3, 0.5, and 1000, respectively; and σ denotes the uncertainty of each data type. The high value of the weighting factor used for the term that accounts for in eq 1 is justified by the apparent absence of contradictions between the information content of and the other two types of data, i.e., the information content of the DEST/ΔR2 data may be considered to be “separated” from that of . The set of global variable parameters for the interconversion comprised The set of global variable parameters for the equilibrium comprised The uncertainties in the values of the optimized parameters, corresponding to confidence intervals of ±1 standard deviation, were determined from the variance−covariance matrix of the nonlinear fits. Minimization of the error function F was performed using an in-house Matlab program (Math-Works Inc., MA).
RESULTS AND DISCUSSION
Problem of Correlated Parameters in DEST/ΔR2 Analysis of Exchange Data.
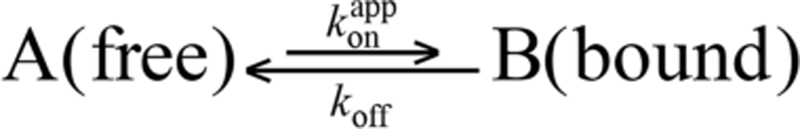
The first applications of the DEST/ΔR2 methodology focused on systems where the transverse relaxation rate of the “dark” state, , was very large, typically reaching tens of thousands per second, so that ≫ 2πω1, where ω1 is the strength of the applied RF field in the DEST experiments, with saturation RF field strengths typically not exceeding ∼750 Hz (∼4700 rad/s).1,2,4 The very high transverse relaxation rates in the dark state ensured that accurate kinetic parameters of exchange, as well as the values themselves, could be extracted from the combined analysis of DEST profiles acquired at two RF field strengths and ΔR2 profiles acquired at two spectrometer fields. When the DEST/ΔR2 methodology is applied to exchanging systems with ≪ 2πω1, such as complexes of the 800 kDa chaperonin GroEL with amyloid-β peptide5 and Fyn-SH3 domains,6,7 where the typical average 15N- value is ∼900 s−1 (at 10 °C and 900 MHz spectrometer field) and the lowest ω1 field strength is typically ∼500 Hz (∼3140 rad/s), simultaneous extraction of the kinetic parameters of exchange (namely, the dissociation rate constant, koff, and consequently the population of the dark, bound state, pB) together with the values in the bound state is no longer possible without additional information. The latter can come, for example, from CPMG relaxation dispersion and exchange-induced chemical shift measurements.5–7 In terms of data modeling, the problem boils down to the high degree of correlation between the fitted parameters, pB ∼ 1/koff and , so that only a quantity proportional to the product pB can be derived from analysis of the DEST/ΔR2 data.
Exactly the same underlying problem is apparent in the current studies of the binding of huntingtin exon-1 peptides, httNT and httNTQ7, to SUV lipid vesicles. The SUVs used here are 31 nm in diameter and have a molecular weight of ∼4.3 MDa, and exchange arising from the binding is slow on both the chemical shift and relaxation time scales. For this particle size range and exchange regime, CPMG dispersion and exchange-induced shift data, even in the presence of large differences in chemical shifts between free and bound states of the peptides, are of little utility as their magnitudes are predicted to be unmeasurably small.
Examples of 15N-DEST profiles for Lys5 and Ser12 of httNT in the presence of SUVs at two RF field strengths are shown in Figure 1A, and 15N-ΔR2 profiles for both httNT and httNTQ7 are displayed in Figure 1B. With the exception of the C-terminal residues (15−16 for httNT and 15−23 for httNTQ7 are displayed in Figure 1B. With the exception of the C-terminal residues (15−16 for httNT and 15−23 for httNTQ7 that require a separate treatment), the 15N-DEST and ΔR2 data for residues 3−14 can be well fit simultaneously to a two-state exchange model between free (A) and bound (B) states (Scheme 1) by propagation of a set of Bloch-McConnell differential equations19 to yield apparent association rates, , of 14.1 ± 0.03 and 17.1 ± 0.05 s−1 for httNT and httNTQ7, respectively. However, the dissociation rate constant (koff), and hence pB, as well as the residue specific values are associated with very large uncertainties. This is due to the fact that the 15N-DEST/ΔR2 data can be fit equally well (with practically the same value of the resulting χ2) by a continuum of {pB;} combinations, as shown in Figure 2, where a series of fits were performed with pB fixed at different values (x-axis), and the resulting average values, , are plotted on the y-axis.
Figure 1.
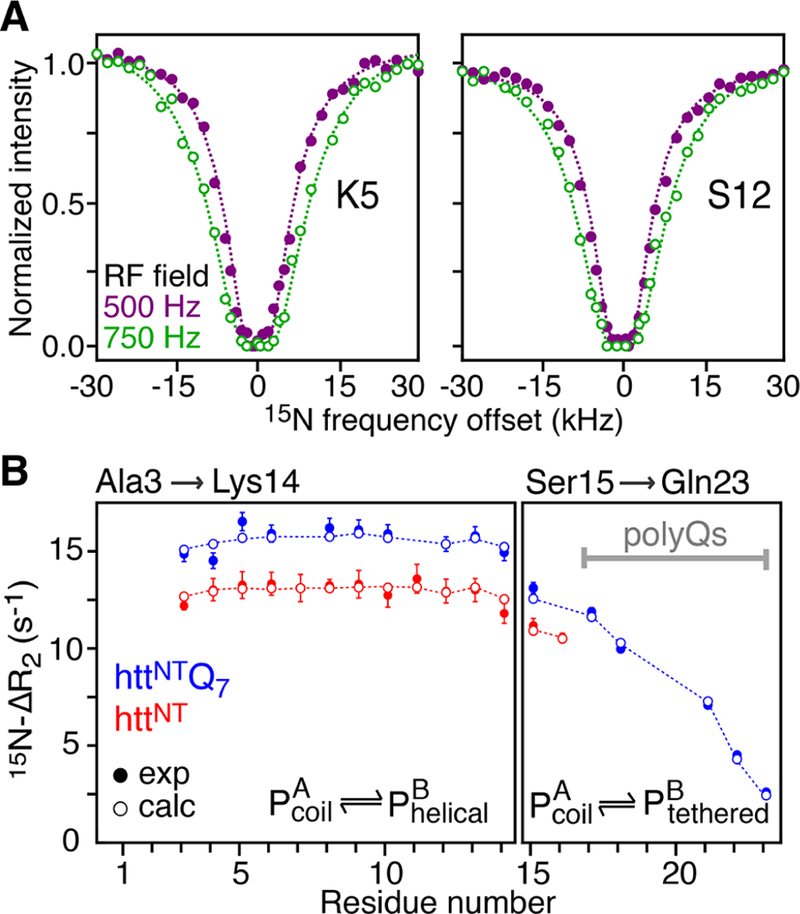
Binding of httNT and httNTQ7 to SUVs characterized by relaxation-based NMR experiments. (A) Examples of 15N-DEST profiles acquired on Lys5 and Ser12 of httNT in the presence of SUVs at RF field strengths of 500 and 750 Hz shown as filled-in purple and open green circles, respectively. (B) 15N-ΔR2 profiles for httNT and httNTQ7 in the presence of SUVs. Experimental data are shown as red and blue filled-in circles for httNT and httNTQ7, respectively. Dashed lines in (A) and open circles in (B) represent best-fits of the 15N-DEST and 15N-ΔR2 data to a two-state exchange model. Residues that do not fit well to this model and required a separate treatment (see text) are shown in the right panel. The data were recorded at 600 MHz and 10 °C on a 300 μM peptide sample in the presence of SUVs at a 1:4 molar ratio of peptide to lipid (on a lipid molecule basis).
Figure 2.
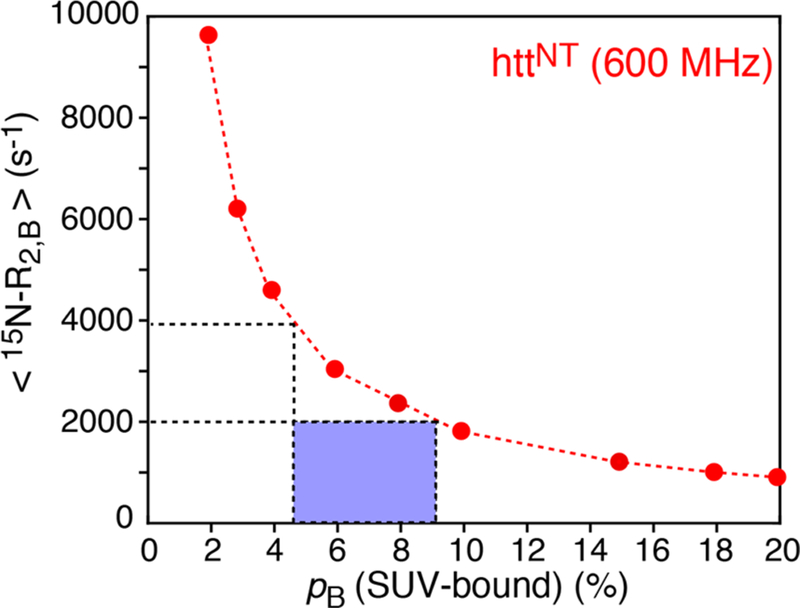
Average values obtained from a fit of the 15N-DEST/ΔR2 data of httNT in the presence of SUVs (600 MHz; 10 °C) to a two-state exchange model (y-axis), plotted versus the population of SUV-bound httNT (pB) which was fixed in the fit (x-axis). For a particle of 31 nm in diameter, one can estimate a range of 4.5−9 μs for the rotational correlation time, corresponding to ⟨15N-⟩ values of 2000−4000 s−1. The bound population pB for this range of ⟨15N‑⟩ values spans ∼9−4.5% (enclosed in the blue box).
Thus, without prior knowledge of , the value of pB (and therefore koff) cannot be determined unambiguously. Of note, the same indeterminacy was described in our earlier studies on the interaction of the Δ57VPL mutant of the Fyn-SH3 domain with GroEL.7 The problem is akin to that arising with the interpretation of ΔR2 data in the limit of fast exchange on the transverse relaxation time scale, where ΔR2 = pB , and only the product of the two parameters can be extracted. Numerical simulations using synthetic data sets comprising well-digitized 15N-DEST profiles at three saturation field strengths and ΔR2 at a single spectrometer field, generated using the parameters of exchange of httNT binding to SUVs with 1% random noise superimposed on the data, show that accurate values of pB and can be reliably extracted from the DEST/ΔR2 fits when is at least a factor of approximately four times higher than the maximal RF field strength used in the DEST experiments Β1,max = 2πω1,max (i.e., larger than ∼20 000 s−1 for a maximal RF saturation field strength of 750 Hz). However, we would predict, on the basis of these simulations, that as long as just exceeds B1,max ( > 2πω1,max) for high-quality 15N-DEST profiles acquired with at least two RF field strengths, the errors in the values of pB, koff, and will remain relatively small and likely within the uncertainties of the fit.
Given the molecular size of the SUVs, is predicted to lie within a range of 2000−4000 s−1, with values of 4.5−9% for pB (as shown by the dashed lines and blue box in Figure 2). The corresponding range for koff is ∼350−180 s−1, indicating that exchange is slow on the relaxation time-scale, which is in agreement with the observed absence of the dependence of ΔR2 on the static spectrometer field.
Quantitative Analysis and Resolution of the Uncertainties.
To gain deeper quantitative insight into the nature of the uncertainty described above, we draw an analogy with the recent work of Kay and co-workers,15 who probed a slowly exchanging system involving two unequally populated states of a protein via off-resonance R1ρ experiments. In the limit where ω1 ≫ and in the absence of chemical shift differences between the interconverting species (Δω = 0), the 6 × 6 Bloch-McConnell exchange matrix describing the evolution of magnetization in a two-state system (Scheme 1) that exchanges slowly (ω1 ≫ koff) can be approximated by15
| (2) |
where MA and MB are the transverse magnetizations of states A (free) and B (bound), respectively, and θ is the angle between the spin-locked magnetization and the z-axis of the laboratory frame. The longitudinal relaxation rates of states A and B ( and ) as well as the transverse relaxation rate of state A, , are assumed to be zero in this approximation. The solutions of this set of equations are
| (3) |
with small (“slow”) and large (“fast”) eigenvalues, λs and λf, given by
| (4) |
where the exchange rate and the population of state B, Expressions for the coefficients A−D in eq 3 in the absence of chemical shift differences between states are given by
| (5) |
From eqs 2–4, given that −λs represents the effective relaxation rate measured as a function of the offset angle θ, it follows that the contribution to the transverse relaxation rate arising from exchange, Rex, in the limit of skewed populations (pB ≪ pA) can be expressed as15
| (6) |
Equation 2 and its solutions in eqs 3–5 are a good approximation to the full Bloch-McConnell equations in the ω1 ≫ ,kex for analysis of off-resonance R1ρ experiments where for each value of the offset angle θ, the magnetization is aligned along the effective RF field (i.e., “locked”).15 In contrast, in exchange saturation transfer experiments, such as DEST, the magnetization is saturated in the course of the relaxation period by application of a continuous RF field resulting in the angle between the magnetization and the z-axis of the laboratory frame becoming time-dependent. Therefore, we use the approximations above solely for illustrative purposes. Nevertheless, using the expressions for relaxation rates in the rotating frame, R1ρ = R1cos2 θ + R2,eff sin2 θ, where R2,eff = + Rex, and Rex is calculated using eq 6, we can quantitatively reproduce the DEST profiles generated using the full set of Bloch-McConnell equations as long as the values are sufficiently large (> ∼500 s−1), indicating that the relationships above in general and eq 6 in particular serve as good approximations for reproducing DEST profiles under conditions that are relevant for the present study, i.e., when ω1 > > kex and > ∼ 500 s−1. Equation 6 is especially useful in this regard as it shows that whereas the value of in the denominator of the second term is largely defined by the ΔR2 data, the product pB in the denominator of the first term is the only quantity that can be determined with certainty from DEST profiles. (Note that ΔR2 in our notation is the measured difference between the transverse relaxation rates measured on the observable species in the presence and the absence of exchange, as opposed to the difference − in Yuwen et al.15) Equation 6 thus serves as a clear illustration of the uncertainties in the values of pB and derived from the DEST data as described above.
To decorrelate pB and , we make use of another observable quantity introduced by Kay and co-workers, Cfast,15 that relates to the fractional contribution of the fast decaying component to the total signal intensity, namely, (B + D) in eq 3 if the sum of all components (A + B + C + D) is normalized to unity. As can be seen from eq 3, the decay of “spin-locked” magnetization for each state in the R1ρ experiments is inherently biphasic. Since the magnetization is carefully equilibrated before and after each relaxation period (“spin-lock” element) of the R1ρ measurements such that after each relaxation period and ,15 where T is the duration of relaxation delay, the Cfast component can be reliably estimated by extrapolation of the R1ρ decay curve to zero time, provided that normalization of signal intensities is performed relative to those measured in the absence of relaxation period (i.e., at T = 0, corresponding to A + B + C + D in eq 3). Using the expressions in eqs 4 and 5, it is straightforward to show that in the limit Δω = 0, Cfast has the form
| (7) |
Note that eq 7 is different from the corresponding eq 11 in Yuwen et al.15 by a factor of “2” in the second term of the denominator, as no assumptions about λf were made in its derivation (the full expression for λs,f in eq 4 was used). Equation 7 shows that Cfast is directly proportional to pB and, importantly, pB is not “mixed” with anywhere in this relationship.
The variation of Cfast as a function of offset frequency (angle θ) is predicated on the value of kex/ (eq 7). For small values of kex/, as is the case in the present study (typically, kex/ < 0.1 for the httNT and httNTQ7 peptides in the presence of SUVs), the profiles of Cfast versus offset frequency show little variability. Figure 3A shows the profiles of Cfast as a function of 15N-frequency offset simulated using a variant of eq 7, where the assumption that Δω = 0 is dropped (see eq 12 in Yuwen et al.15) for Lys5 of httNT and httNTQ7. The simulations were performed using two spin-lock periods effectively amplifying the value of Cfast approximately 2-fold, and using the chemical shifts of the bound state of Lys5 available from our previous study of httNTQn peptides binding to lipid micelles.14 The curves obtained via propagation of the full Bloch-McConnell matrix are practically indistinguishable from those shown in Figure 3A. The maxima of the Cfast profiles are only very slightly shifted from the position of the carrier (assumed to be at 0 Hz, which coincides with the chemical shift of the major free state A) toward the chemical shifts of the minor bound state (Δω/2π = −212 Hz for Lys5). In this context, it is important to note that although httNTQn peptides are known to form helices on the surfaces of lipid-based particles14 resulting in large (and negative) 15N chemical shift changes in the bound state, the difference in chemical shifts between the free and bound states is largely of no consequence in the present study, as all the experimental techniques employed here are either insensitive to chemical shift changes, such as DEST and Cfast, or the effect of these changes is suppressed by strong spin-lock RF fields, as in the case of ΔR2 measurements.
Figure 3.
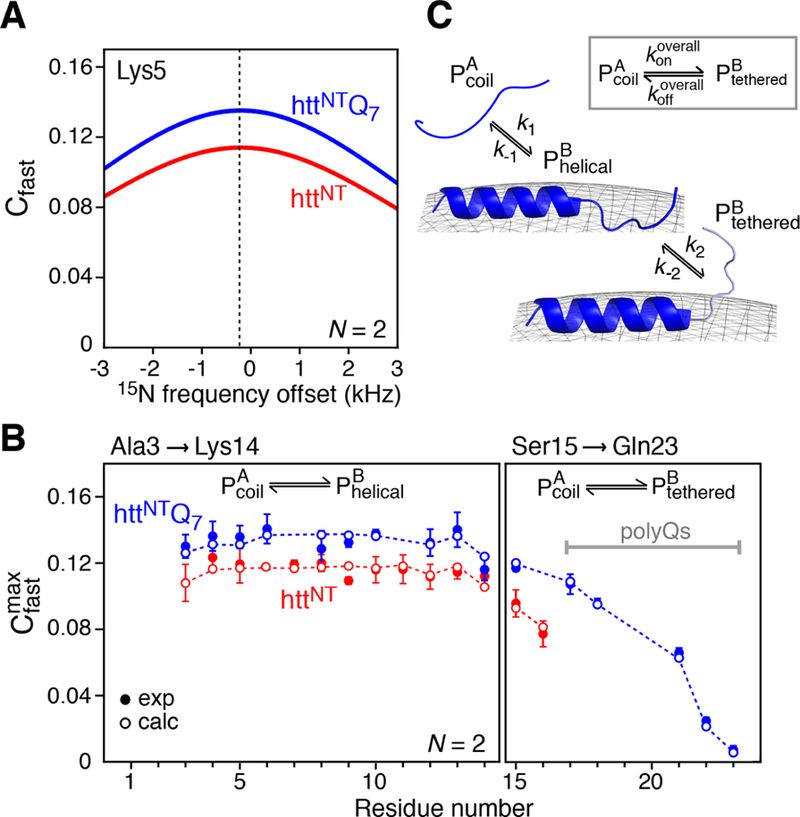
Simulated and experimental Cfast profiles for the binding of httNT and httNTQ7 to SUVs. (A) Simulated profiles of Cfast as a function of 15N frequency offset for Lys5 of httNT (red) and httNTQ7 (blue) in the presence of SUV vesicles at a molar ratio of 1:4 peptide to lipid. The curves were calculated using a variant of eq 7 that accounts for Δω ≠ 015) with the following values for the exchange parameters (where the first value refers to httNT and the second to httNTQ7): kex = 204.7/208.3 s−1, pB = 0.07/0.08, = 1.5/1.5 s− 1, = 2.4/3.6 s− 1, = 2300/2300 s−1, ω1 field strength = 2000 Hz, number of spin locks N = 2, and Δω/(2π) = −211.8 Hz (−3.47 ppm at 600 MHz). (B) Experimental 15N- profiles measured for httNT (red filled-in circles) and httNTQ7 (blue filled circles) in the presence of SUVs. The open circles are the best-fit profiles obtained by including into the target function together with the DEST and ΔR2 data (see Experimental Section). (C) Kinetic scheme used for modeling the binding of httNT and httNTQ7 to the surface of SUVs. The data for residues 3−14 are fit to a two-state exchange between the free peptide and membrane-bound helical peptide For residues 15−16 of httNT and 15−23 of httNTQ7, there is an additional process involving the interconversion between and , where the residues in the state are not attached to the membrane but tethered via the membrane-bound helical residues. The overall process for the conversion of to (via ) is shown in the inset.
As practically no information can be gleaned from the dependence of Cfast on the frequency offset for the exchange parameters of httNT and httNTQ7 peptides binding to SUVs, we opted for the measurement of a single Cfast value per residue at or close to the on-resonance frequency. As follows from eq 7, Cfast reaches its maximal value, , on resonance (sin θ = 1). For skewed populations (pB ≪ pA), the expression for can be simplified to
| (8) |
Equation 8 shows that as long as provides a good measure of the population of the bound state pB, even if only a single on-resonance measurement of Cfast is available. Only when the ratio kex/ becomes significantly larger than unity does decrease to unmeasurably low values.
Impact of Incorporation of Data in the Analysis of DEST/ ΔR2 Data.
The measurements of for httNT and httNTQ7 peptides in the presence of SUVs were conducted as described in the Experimental section and closely followed the procedures of Yuwen et al.15 To amplify the value of approximately 2-fold, two spin-lock periods were used in the total relaxation delay, each preceded and followed by the magnetization equilibration delay τeq ≥ 2/kex. The experimental 15N- profiles are shown in Figure 3B. Inclusion of an additional term that accounts for the data into the DEST/ΔR2 target function (see the Experimental section for details of the calculations) enables unambiguous determination of pB, and hence koff, as well as the set of residue-specific 15N- values, without compromising the quality of the fit for either the DEST (Figure 1A) or ΔR2 (Figure 1B) data.
The 15N-DEST, ΔR2, and data for Ala3−Lys14 were fitted simultaneously to a two-state exchange model (defined by the rate constants k1 and k−1 in Figure 3C) to yield the following exchange parameters: = 14.1 ± 0.03 and 17.1 ± 0.04 s−1, and k−1 = koff = 190.6 ± 1.3 and 191.2 ± 1.3 s−1 for httNT and httNTQ7, respectively (corresponding to pB values of 6.9 ± 0.1 and 8.2 ± 0.1%).
The C-terminal region for both peptides, comprising residues 15−16 of httNT and 15−23 (encompassing the polyglutamine tract) of httNTQ7 required a separate treatment: the same overall two- state exchange model (shown in the inset of Figure 3C) was used to fit the data for these residues, keeping in mind that the overall exchange process is more complex for these sites and involves initial binding to the surface of SUVs followed by transient, reversible detachment from the lipid surface depicted as the state in Figure 3C. The fits for these residues to the model shown in the inset to Figure 3C yielded = 9.2 ± 0.2 and 13.2 ± 0.1 s−1, and = 144 ± 3 and 157 ± 2 s−1 for httNT and httNTQ7, respectively. The rate constants k2 and k−2 describing the exchange process between membrane-bound (direct-contact) and tethered states of the C-terminal residues can be calculated from the relationships
| (9) |
yielding k2 = 358 ± 13 and 635 ± 14 s−1, and k−2 = 413 ± 22 and 680 ± 24 s−1 for httNT and httNTQ7, respectively. From the values of these rate constants, it follows that reversible detachment of the C-terminal residues from the SUV surface occurs ∼4-to-6-fold faster than the binding itself (first stage of the global process). Further, exchange between the direct-contact and tethered states of the C-terminal residues occurs ∼1.7-fold faster in httNTQ7 than in httNT, consistent with the presence of the seven-residue polyglutamine C-terminal tail, while the slight preference for the direct-contact state is apparent for both peptides.
The 15N- values for SUV-bound httNT and httNTQ7, derived from the combined analysis of 15N-DEST, ΔR2, and data, are shown in Figure 4 as a function of residue number. The average values of 2550 and 2340 s−1 for the membrane-bound states of httNT and httNTQ7 lie within the range predicted for a particle of 31 nm in diameter (Figure 2). The corresponding values for the tethered state are on average 3-to-4-fold lower for both complexes (right panel of Figure 4), as expected since the C-terminal residues are semi-detached from the lipid surface, do not form a helical structure, and are therefore at least partially disordered.
Figure 4.
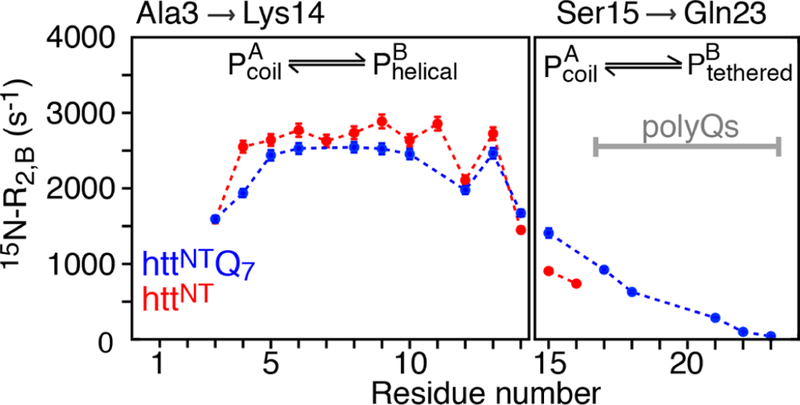
15N- profiles for httNT (red circles) and httNTQ7 (blue circles) bound to SUVs, obtained from combined analysis of 15N-DEST,15N-ΔR2, and 15N- data.
CONCLUSIONS
In summary, we have characterized the binding kinetics of two peptides derived from exon-1 of the huntingtin gene, httNT and httNTQ7, to SUV particles 31 nm in diameter and 4.3 MDa in molecular weight, using DEST and lifetime line broadening (ΔR2) supplemented with the measurement of the maximal (on-resonance) value of the contribution of the fast-relaxing component of magnetization, . We show that the measurement of DEST and ΔR2 is not sufficient to unambiguously determine both the transverse relaxation rates in the bound state, , and the population of the bound state, pB, simultaneously in the exchange regime where is smaller than the strength of the DEST saturation RF field (see Figure 2). The measurement of , an observable that is directly proportional to pB, allows one to decorrelate and pB from one another. thus serves as a valuable addition to the array of available experimental NMR observables for studying exchanging systems involving binding to high-molecular-weight, dark states in the 2−6 MDa range, where exchange-induced chemical shifts and/or CPMG relaxation dispersions are not measurable. Under the experimental conditions employed (300 μM peptide at a 1:4 molar ratio of peptide to lipid), exchange between the free huntingtin peptides and the SUV-bound states is slow on the relaxation time-scale (τex ∼ 5 ms, mean < T2 > ∼ 400 μs for the bound state) with bound populations of 7−8%. While the peptide core (residues 3−14) is stably bound to the SUV surface, the C-terminal region of both peptides (residues 15−16 of httNT and residues 15−23 of httNTQ7 encompassing the polyglutamine region) remain flexible in the bound form due to reversible detachment from the surface of SUV particles that occurs on a time-scale ∼4-to-6-fold faster than the binding to the SUV.
Scheme 1
ACKNOWLEDGMENTS
This article is dedicated to William A. Eaton on the occasion of his 80th birthday. We thank Lewis Kay and Tairan Yuwen (University of Toronto) for supplying the pulse code for R1ρ/Cfast measurements and stimulating discussions. This work was supported by funds from the Intramural Program of the NIH, NIDDK, and the Intramural AIDS Targeted Antiviral Program of the Office of the Director of the NIH (to G.M.C.).
Footnotes
Notes
The authors declare no competing financial interest
REFERENCES
- (1).Fawzi NL; Ying J; Torchia DA; Clore GM Kinetics of Amyloid β Monomer-to-Oligomer Exchange by NMR Relaxation. J. Am. Chem. Soc 2010, 132, 9948–9951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Fawzi NL; Ying J; Ghirlando R; Torchia DA; Clore GM Atomic-Resolution Dynamics on the Surface of Amyloid-β Protofibrils Probed by Solution NMR. Nature 2011, 480, 268–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Fawzi NL; Ying J; Torchia DA; Clore GM Probing Exchange Kinetics and Atomic Resolution Dynamics in High Molecular Weight Complexes Using Dark-State Exchange Saturation Transfer NMR Spectroscopy. Nat. Protoc 2012, 7, 1523–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Fawzi NL; Libich DS; Ying J; Tugarinov V; Clore GM Characterizing Methyl-Bearing Side Chain Contacts and Dynamics Mediating Amyloid β Protofibril Interactions Using 13C-Methyl DEST and Lifetime Line Broadening. Angew. Chem., Int. Ed 2014, 53, 10345–10349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Libich DS; Fawzi NL; Ying J; Clore GM Probing the Transient Dark State of Substrate Binding to GroEL by Relaxation-Based Solution NMR. Proc. Natl. Acad. Sci. U. S. A 2013, 110, 11361–11366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Libich DS; Tugarinov V; Clore GM Intrinsic Unfoldase/ Foldase Activity of the Chaperonin GroEL Directly Demonstrated Using Multinuclear Relaxation-Based NMR. Proc. Natl. Acad. Sci. U. S. A 2015, 112, 8817–8823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Libich DS; Tugarinov V; Ghirlando R; Clore GM Confinement and Stabilization of Fyn SH3 Folding Intermediate Mimetics within the Cavity of the Chaperonin GroEL Demonstrated by Relaxation-Based NMR. Biochemistry 2017, 56, 903–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Skrynnikov NR; Dahlquist FW; Kay LE Reconstructing NMR Spectra of “Invisible″ Excited Protein States Using HSQC and HMQC Experiments. J. Am. Chem. Soc 2002, 124, 12352–60. [DOI] [PubMed] [Google Scholar]
- (9).Palmer AG 3rd NMR Characterization of the Dynamics of Biomacromolecules. Chem. Rev 2004, 104, 3623–3640. [DOI] [PubMed] [Google Scholar]
- (10).Anthis NJ; Clore GM Visualizing Transient Dark States by NMR Spectroscopy. Q. Rev. Biophys 2015, 48, 35–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Andresen JM; et al. The Relationship Between CAG Repeat Length and Age of Onset Differs for Huntington’s Disease Patients with Juvenile Onset or Adult Onset. Ann. Hum. Genet 2007, 71, 295–301. [DOI] [PubMed] [Google Scholar]
- (12).Chaibva M; Burke KA; Legleiter J Curvature Enhances Binding and Aggregation of Huntingtin at Lipid Membranes. Biochemistry 2014, 53, 2355–2365. [DOI] [PubMed] [Google Scholar]
- (13).Gao X; Campbell W. A. t.; Chaibva M; Jain P; Leslie AE; Frey SL; Legleiter J Cholesterol Modifies Huntingtin Binding to, Disruption of and Aggregation on Lipid Membranes. Biochemistry 2016, 55, 92–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Ceccon A; Schmidt T; Tugarinov V; Kotler SA; Schwieters CD; Clore GM Interaction of Huntingtin Exon-1 Peptides with Lipid-Based Micellar Nanoparticles Probed by Solution NMR and Q-Band Pulsed EPR. J. Am. Chem. Soc 2018, 140, 6199–6202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Yuwen T; Brady JP; Kay LE Probing Conformational Exchange in Weakly Interacting, Slowly Exchanging Protein Systems via Off-Resonance R1ρ Experiments: Application to Studies of Protein Phase Separation. J. Am. Chem. Soc 2018, 140, 2115–2126. [DOI] [PubMed] [Google Scholar]
- (16).Anthis NJ; Clore GM Sequence-Specific Determination of Protein and Peptide Concentrations by Absorbance at 205 nm. Protein Sci 2013, 22, 851–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Ceccon A; Tugarinov V; Bax A; Clore GM Global Dynamics and Exchange Kinetics of a Protein on the Surface of Nanoparticles Revealed by Relaxation-Based Solution NMR Spectroscopy. J. Am. Chem. Soc 2016, 138, 5789–5792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Delaglio F; Grzesiek S; Vuister GW; Zhu G; Pfeifer J; Bax A NMRPipe: a Multidimensional Spectral Processing System Based on UNIX Pipes. J. Biomol. NMR 1995, 6, 277–293. [DOI] [PubMed] [Google Scholar]
- (19).McConnell HM Reaction Rates by Nuclear Magnetic Resonance. J. Chem. Phys 1958, 28, 430–431. [Google Scholar]