

Abstract
Phenotypic rather than genotypic tests remain the gold standard for diagnosing glucose-6-phosphate dehydrogenase (G6PD) deficiency. However, with increasing use of genomic arrays and whole exome or genome sequencing, G6PD genetic data are increasingly available. We examined the utility of G6PD genetic data in patients with hematologic malignancies and the association of G6PD genotype and phenotype with rasburicase-induced methemoglobinemia. We analyzed G6PD activity for 990 patients. Genotype data were available from the Affymetrix DMET array (n=379), whole exome sequencing (n=374), and/or the Illumina exome array (n=634) for 645 patients. Medical records of 341 patients with methemoglobin measures were assessed for the administration of rasburicase. We observed 5 non-synonymous SNPs, 4 of which were known to be associated with deficient G6PD activity (WHO Class I-III). Genotyping 367 males resulted in a positive predictive value of 81.8% (47.8–96.8%), and two males with a Class I-III allele having normal activity both received a red blood cell transfusion prior to the activity assay. However, genotyping males had only 39.1% (20.5–61.2%) sensitivity. Two of the 12 heterozygous females had deficient G6PD activity. Rasburicase-induced methemoglobinemia occurred in 6 patients, 5 of whom had at least one Class I-III allele, despite 2 of these having normal G6PD activity. We conclude that although an apparent nondeficient genotype does not necessarily imply a normal phenotype, a deficient genotype result indicates a deficient phenotype in those without transfusions, and may be a useful adjuct to phenotype to prevent adverse drug reactions.
Introduction
Glucose-6-phosphate-dehydrogenase (G6PD) deficiency is a well-recognized pharmacogenetic trait. (1) G6PD functions as the rate-limiting step of the hexose monophosphate shunt, which maintains the supply of NADPH available to replenish the cellular stores of reduced glutathione used for detoxifying reactive oxygen species. (1, 2) G6PD is the only source of NADPH in erythrocytes, leaving erythrocytes prone to oxidative stress from either endogenous or exogenous sources, including medications. Thus, erythrocytes are especially sensitive to G6PD deficiency, (1–4) and patients with G6PD deficiency are susceptible to hemolytic anemia and methemoglobinemia. (3–6) G6PD deficiency is estimated to affect almost 5% of the global population. (7)
The gold-standard method to assess clinical G6PD status is quantitating G6PD activity in whole red blood cells through spectrophotometry. (8, 9) Over 180 variant genetic alleles of G6PD have been reported, and uptake of genetic tests rather than phenotypic tests of activity has been slow. This is partly because genetic tests are thought to be poorly predictive in females, for whom X-linked mosaicism in red cells results in variable expression of G6PD activity, (10–13) and because G6PD status is often important to ascertain with short turn-around-time (14) and few point-of-care G6PD genetic tests have yet been developed. (15) Moreover, most low-activity G6PD alleles are rare, and the optimal set of alleles for genotyping has not been established (16) and may differ by ancestry. (7) The World Health Organization (WHO) classifies variant alleles into five different classes based on relative enzyme activity compared to the wildtype enzyme and the clinical presentation of deficiency. (16) The Clinical Pharmacogenetics Implementation Consortium (CPIC®) makes pharmacogenetic recommendations based on the assumption that genotypes may be available pre-emptively, and has assigned four different likely G6PD phenotypes based on the presence of WHO class alleles: normal (class IV), deficient (classes II and III), deficient with chronic nonspherocytic hemolytic anemia (CNSHA) (Class I), and variable (heterozygous females with one class IV and one deficient (class I-III) allele) (Supplemental Table S1). (6)
As array-based, whole exome and whole genome sequencing becomes more common, (6, 7, 17) clinicians will be faced with G6PD genetic information that is generated “incidentally.” These data will identify patients who carry class I-III low-function G6PD alleles, making it potentially possible to assign G6PD status to individuals based on genomic information alone. It is unknown how well genetic testing accurately predicts G6PD deficiency. We tested for this concordance of genotype with phenotype in an American pediatric population with hematological malignancies. We also tested how G6PD genotype and phenotype were associated with rasburicase-induced methemoglobinemia in pediatric patients receiving treatment for acute leukemia.
Methods
G6PD Activity
We retrospectively analyzed 990 pediatric patients with hematological malignancies (acute myeloid leukemia, acute lymphoblastic leukemia, chronic myeloid leukemia, or another hematological malignancy) enrolled in St. Jude Children’s Research Hospital from 1993–2013 on research protocols with institutional review board (IRB) approval who had G6PD activity measured in their blood as part of clinical care. Four different quantitative spectrophotometric G6PD assays were used over time by St. Jude’s Department of Pathology to measure G6PD activity for 990 patients, with different ranges defining “normal activity” by date: before December 1996, 4.6–13.5 units/g Hb; between December 1996 and August 2002, 7–20.5 units/g Hb; between August 2002 and September 2004, 10.8–16.2 units/g Hb; and after September 2004, 6.3–18.5 units/g Hb. For patients with leukocyte counts >100 X 103 cells/µl, a buffy-coat-free sample was used to measure G6PD activity; however, this could not be confirmed for samples prior to 1996. Twelve patients were excluded from analysis as described in the results section for a total of 978 patients in the cohort (Fig. 1). Patients were considered deficient if their activity fell below the lower normal limit for the quantitative spectrophotometric assay.
Figure 1. G6PD patient cohort.
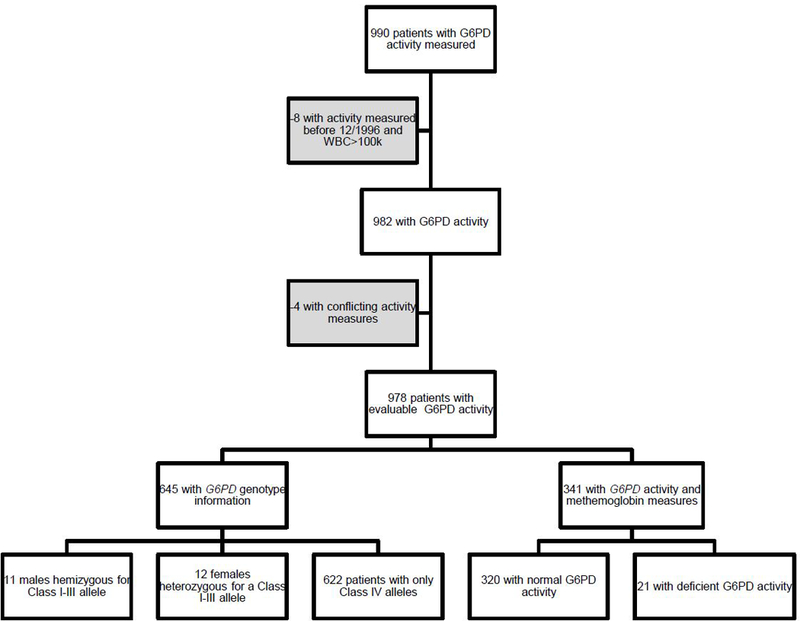
990 patients with hematological malignancies had at least one G6PD activity measurement. We removed 8 patients who had high leukocyte counts (>100 × 103 cells/µl) who were measured before December 1996 because we could not confirm that the buffy coat free method was performed. Four more patients were removed because they had two conflicting lab values (one normal and one deficient activity), for a total of 978 patients with evaluable G6PD activity. 645 of these patients had genotyping information and assigned G6PD genotype.341 patients with G6PD activity measurements had at least one methemoglobin lab value in their medical records. 320 of these patients had normal G6PD activity and 21 had deficient G6PD activity.
G6PD Genotype
DNA was extracted from peripheral blood after patients achieved clinical remission. 645 patients had their genotype assessed using one or more of the following platforms: Affymetrix Drug Metabolizing Enzymes and Transporters (DMET) array (Santa Clara, CA) (n=379) (18, 19), Illumina HumanExome BeadChip (Exomechip) (San Diego, CA) (n=634) (20), and WES (n=374) (21) (Supplemental Fig. S1). The Affymetrix DMET array interrogated 6 nonsynonymous single nucleotide polymorphisms (SNPs) (A (p.N126D), Canton (p.R459L), Chatham (p.A335T), Mediterranean (p.S188F), Sao Borja (p.D113N), 1 with unknown function (p.V77M)). The Illumina Exomechip interrogated 8 nonsynonymous SNPs (A-, 968 (p.L323P), Asahi (p.V68M), Mediterranean (p.S188F), Malaga (p.D181V), Sierra Leone (p.R104H), Seattle (p.D282H), Mira d’Aire (p.D350H), and one with unknown function (p.Q11H)) (Supplemental Fig. S2, Table S2). Nonsynonymous coding SNPs were classified according to the WHO classification, (16) and patients were assigned a phenotype based on the CPIC guidelines for rasburicase. (6)
G6PD genotype and phenotype concordance
We estimated the positive predictive value (PPV), negative predictive value (NPV), sensitivity, and specificity when using the patient’s genotype to predict G6PD phenotype, with 95% confidence intervals also estimated. Fisher’s exact test was used to compare the frequency of rasburicase-induced methemoglobinemia.
Rasburicase and methemoglobinemia
Of the 978 patients with evaluable G6PD activity, 341 had at least one methemoglobin level measured during therapy. Each medical record was assessed for administration of rasburicase, and we compared the dates of the methemoglobin level to rasburicase administration. Methemoglobinemia was defined as methemoglobin >3%, which was three times the standard deviation plus the mean for methemoglobin levels in our cohort. For patients with methemoglobinemia in the absence of rasburicase administration, we reviewed records to identify any other drugs known to cause methemoglobinemia, such as sulfamethoxazole/trimethoprim and nitric oxide.
Results
G6PD Activity
Of 990 patients with G6PD activity measures, 8 were excluded because we could not confirm if the buffy coat free method was performed, and their leukocyte counts were > 100 ×103 cells/µl (Fig. 1). Four patients had conflicting activity measurements and were excluded from the analysis (Supplemental Material [Concordance of multiple G6PD activity assays] and Fig. 1).
Of 978 patients with evaluable G6PD activity, 51 had deficient G6PD activity (5.2%). Twenty-three (45%) of the deficient patients were black males, and there were more than twice as many deficient males as deficient females. (Table 1) The mean (SD) age for patients with deficient G6PD activity was 8.5 ± 5.4 years, and the mean age for patients with normal G6PD activity was 8.0 ± 5.4 years.
Table 1.
Frequency of G6PD deficiency in pediatric patients with hematological malignancies by sex and race*
| Normal G6PD activity |
Deficient G6PD activity |
||||
|---|---|---|---|---|---|
| n | % | n | % | ||
| Male | White | 347 | 97.2% | 10 | 2.8% |
| Black | 131 | 85.1% | 23 | 14.9% | |
| Other^ | 49 | 96.1% | 2 | 3.9% | |
| Total | 527 | 93.8% | 35 | 6.2% | |
| Female | White | 262 | 98.1% | 5 | 1.9% |
| Black | 104 | 93.7% | 7 | 6.3% | |
| Other^ | 34 | 89.5% | 4 | 10.5% | |
| Total | 400 | 96.2% | 16 | 3.8% | |
|
Both genders |
Total | 927 | 94.8% | 51 | 5.2% |
Race self-reported; confirmed with genotype
Other includes Hispanics, Asians, and patients with mixed racial background.
G6PD Genotype
We observed variant genotypes at 5 nonsynonymous SNPs, 4 of which were Class II or III in 645 patients with genotype data: A variant (p.N126D), Asahi variant (p.V68M), Viangchan variant (p.V291M), A-,968 variant (p.L323P), and Pawnee variant (p.R439P) (Fig. 2, Supplemental Table S2). Two of the observed SNPs (Viangchan and Pawnee) were interrogated only by WES. Through WES, we found an additional 4 synonymous SNPs, 4 SNPs and 1 insertion/deletion (indel) in the 3’ untranslated region (UTR), 5 SNPs in the 5’ UTR, and 2 intronic SNPs (Supplemental Table S3). All variants were found in frequencies consistent with published allele frequencies (Supplemental Tables S2 and S3). All 645 patients were able to be assigned to a phenotype based on genotype (Fig. 1). Patients who had only the A variant were considered normal, as this is considered to be a Class IV allele.
Figure 2. 5 nonsynonymous SNPs in the G6PD gene were observed.
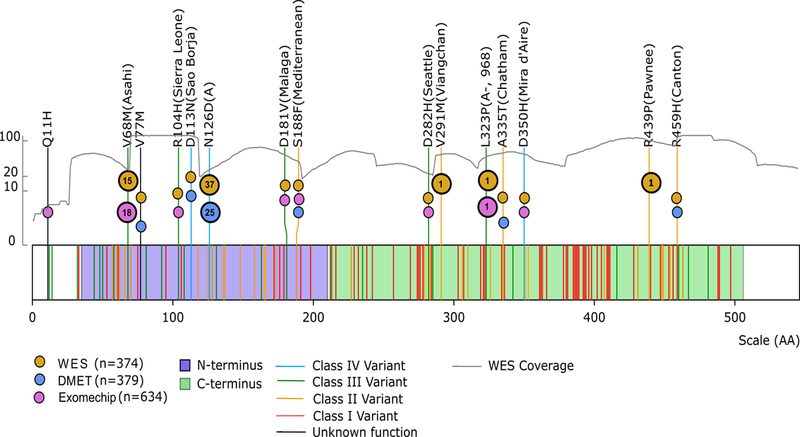
Lines represent G6PD variants reported in the CPIC guidelines (6), and color indicates the WHO classification. SNPs interrogated in our cohort are listed by their codon number, amino acid substitution, and common name. The colored circles indicate which platforms include each SNP. The number in the circles indicates how many patients interrogated on that platform harbored that variant SNP. The gray line represents the whole exome sequencing coverage plotted against the left y-axis. The purple shading indicates the N-terminus domain, and the green indicates the C-terminus domain. We observed four Class I-III variants and one Class IV variant.
Of 367 males, 11 were considered deficient based on genotype (hemizygous for a Class I-III allele). None of the 279 females were homozygous for a Class I-III allele, and thus none were considered deficient based on genotype. 12 females were heterozygous for a Class I-III allele and were assigned to have a “variable phenotype” (i.e. not assignable as normal or deficient) based on genotype.
G6PD Genotype and Phenotype Concordance
Of the 11 males deficient according to genotype, 9 had deficient G6PD activity (PPV=81.8% 47.8–96.8%)), and of the 23 genotyped males with deficient G6PD activity, 9 had a Class I-III allele (sensitivity=39.1% (20.5–61.2%)). Of the 356 males with a Class IV allele, 342 had normal G6PD activity (NPV=96.1% (93.3–97.7%)). 342 out of 344 males with normal activity had a Class IV allele (specificity=99.4% (97.7–99.9%)) (Fig. 3 and Supplemental Table S4). The two male patients with a Class I-III allele with normal activity had received a transfusion two days and 44 days prior to the G6PD activity assay respectively, which may have artificially increased the apparent G6PD activity (Fig. 3).
Figure 3. Genotyping G6PD has 81.8% positive predictive value and 39.1% sensitivity to predict G6PD phenotypic deficiency.
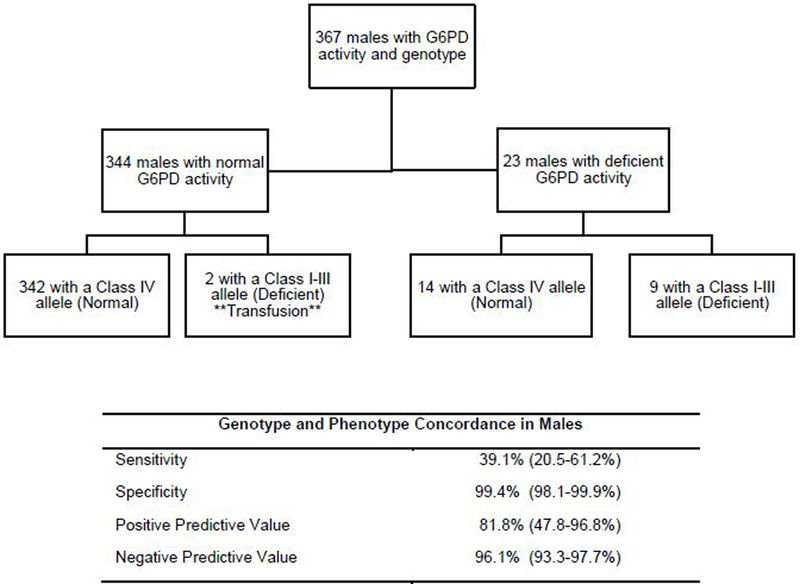
The assigned phenotype based on genotype according to the CPIC guidelines is in parentheses in the flowchart. Nine of the 23 males with deficient activity were also deficient according to genotype, resulting in a sensitivity of 39.1% (20.5–61.2%). Nine of the 11 males with a Class I-III allele had deficient G6PD activity, resulting in a positive predictive value of 81.8% (47.8–96.8%). The two patients with a Class I-III allele with normal G6PD activity had received a transfusion (**) prior to the activity measure. The 95% confidence interval is listed in parentheses.
Since there were no females homozygous for a Class I-III allele, a positive predictive value, sensitivity, and specificity could not be estimated for females. Of the 12 patients assigned as having a variable phenotype based on genotype, 2 (16.7%) had deficient G6PD activity. Genotyping females had a negative predictive value of 96.6% (93.5–98.3%): 258 out of 267 female patients with only Class IV alleles had normal G6PD activity (Fig. 4 and Supplemental Table S5).
Figure 4. G6PD genotype in females does not correspond well with phenotype.
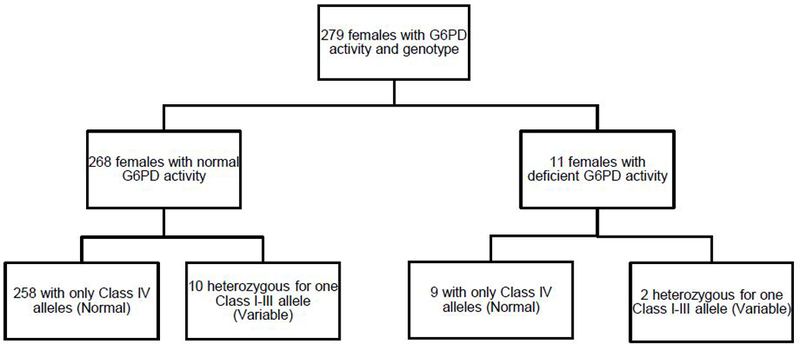
Of 279 females, 268 had normal and 11 had deficient activity. No female was homozygous for a Class I-III allele, and 10/268 (3.7%) of those with normal activity and 2/11 (18%) of those with deficient activity carried one Class I-III allele. Of the 12 “variable” females (those heterozygous for a Class I-III allele), only 2 had deficient activity (16.7%). 258 out of 267 patients with only Class IV alleles had normal G6PD activity, resulting in a negative predictive value of 96.6% (93.5–98.3%).
We expected false negatives to occur because of the possible existence of Class I-III alleles that were not interrogated. All 14 males who had deficient activity but did not have a class I-III allele were interrogated by whole exome sequencing of their genotypes, which failed to detect any nonsynonymous SNPs in 12, and detected only the A (p.N126D) variant (a WHO class IV allele) in the other 2 males (Supplemental Table S4). We also found false negatives among females. Of the 9 deficient females without a Class I-III allele, all were interrogated by WES, which did not detect any nonsynonymous variants in 8 females, and detected the A variant alone in 1 female (Supplemental Table S5).
Rasburicase and Methemoglobinemia
Of the 341 patients with at least one methemoglobin measurement in their medical records (196 males, 145 females), 320 had normal G6PD activity and 21 had deficient G6PD activity (Fig. 1). Sixty of the 341 patients received rasburicase (10 patients with deficient G6PD activity and 50 patients with normal G6PD activity), and 18 patients had methemoglobin measurements at the time of rasburicase administration (3 patients with deficient activity and 15 patients with normal activity) (Fig. 5) (Supplemental Fig. S3 for males and Supplemental Fig. S4 for females). All 3 of the patients with deficient G6PD activity who had an evaluable methemoglobin measurement after rasburicase administration developed rasburicase-induced methemoglobinemia, and each had a Class I-III allele. Three of the 15 patients with normal G6PD activity and with documented methemoglobin measurements at the time of rasburicase administration developed methemoglobinemia after rasburicase. Two of these patients had at least one Class I-III allele: a male hemizygous for a Class I-III allele who had received a transfusion 44 days prior to the activity assay and a female heterozygous for a Class I-III allele. The third patient was a black female who did not have a Class I-III allele by Exomechip array. The other 12 patients with normal G6PD activity who had only Class IV alleles or did not have any genotype information available did not develop methemoglobinemia after rasburicase administration (9 males and 3 females). In summary, 6 patients developed rasburicase-induced methemoglobinemia, and 5 of these 6 had a Class I-III allele (Fig. 5). Among patients with normal G6PD activity, methemoglobinemia after rasburicase administration was more common (2 of 2) in those with a Class I-III allele than in those without a Class I-III allele (1 of 13) (p = 0.029).
Figure 5. 60 patients with G6PD activity also had at least one methemoglobin measurement and received rasburicase at some point in therapy.
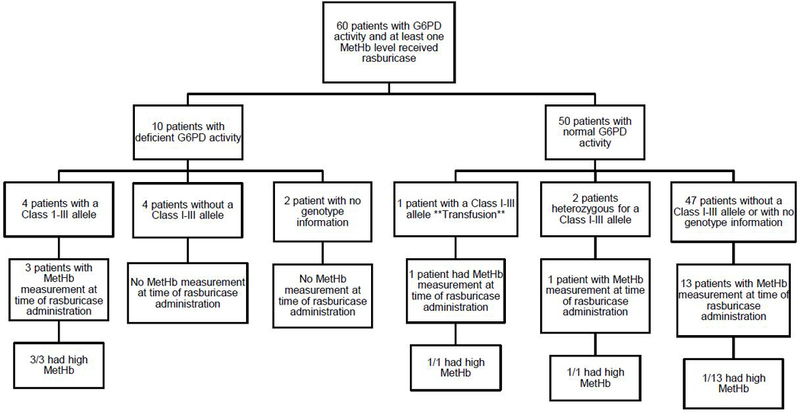
10 of these patients had deficient G6PD activity, and 50 had normal activity. The patients were then classified according to genotype. 18 patients had a methemoglobin level at the time of rasburicase administration: 3 patients with deficient activity and 15 patients with normal activity. The three patients with deficient activity all had a Class I-III allele and developed methemoglobinemia (>3%) after rasburicase administration. Of the 15 patients with normal activity who had evaluable methemoglobin after rasburicase, 3 developed methemoglobinemia: one male patient hemizygous for a Class I-III allele (this is the patient (**) who had received a transfusion prior to the G6PD activity assay); one was a female patient heterozygous for a Class I-III allele; and one was a female who did not have a Class I-III. Thus, of the 18 patients who had methemoglobin measurements at the time of rasburicase administration, 6 patients developed methemoglobinemia after rasburicase, and 5 of these 6 had a Class I-III allele.
We then examined all 341 patients with G6PD activity and at least one methemoglobin measurement, regardless of whether they ever received rasburicase (Fig. 6 and Supplemental Fig. S5 for males and Supplemental Fig. S6 for females). Three patients with deficient G6PD activity (all of whom also had class I-III alleles) developed rasburicase-induced methemoglobinemia. The other 18 deficient patients had only normal methemoglobin levels documented in their medical records and were unrelated to the administration of rasburicase. Of the 320 patients with normal G6PD activity, 296 patients had only normal methemoglobin levels recorded; 12 of these patients received rasburicase at the time of the methemoglobin level. Methemoglobinemia was documented in 24 patients with normal G6PD activity: 3 received rasburicase at the time methemoglobin was measured, and 21 did not. Of the 3 who received rasburicase, one was a male hemizygous for a Class I-III allele who had received a transfusion prior to the activity assay, one was a female heterozygous for a Class I-III allele, and one was a female who had only class IV alleles. The other 21 patients without a documented Class I-III allele were not administered rasburicase prior to the development of methemoglobinemia. Eleven of these 21 received another medication known to cause methemoglobinemia irrespective of G6PD status (nitric oxide (22, 23), dapsone (24, 25), phenazopyridine (26, 27), lidocaine (28, 29), and sulfamethoxazole/trimethoprim (30, 31)) (Fig. 6).
Figure 6. 341 patients had at least one methemoglobin measurement in their medical record, 21 of whom had deficient G6PD activity.
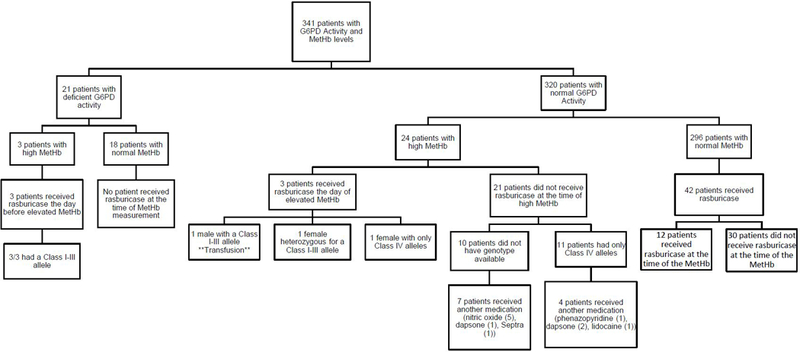
27 patients developed methemoglobinemia at some point during therapy (12 males and 15 females). All 3 patients with deficient G6PD activity developed elevated methemoglobinemia (>3%) after rasburicase administration, and all had a class I-III allele. Of the 24 patients with normal G6PD activity who developed methemoglobinemia at any time during therapy, 3 experienced methemoglobinemia after rasburicase administration. The one male was hemizygous for a Class I-III allele (he had received a transfusion prior to the activity assay**). One female was heterozygous for a Class I-III allele, and one female did not have a Class I-III allele (but was interrogated by the Exomechip array only). Of the other 21 patients with normal G6PD activity who experienced methemoglobinemia, 10 patients received another medication associated with methemoglobinemia.
Discussion
The prevalence of G6PD deficiency in 978 pediatric patients with hematologic malignancies was 5.2%, which is consistent with the prevalence in the general population. (7) In 367 males, genetic testing yielded a positive predictive value of 81.8% (47.8–96.8%), a negative predictive value of 96.1% (93.3–97.7%), sensitivity of 39.1% (20.5–61.2%), and specificity of 99.4% (98.1–99.9%). Genetic testing in 279 females yielded a negative predictive value of 96.6% (93.5–98.3%). No female in our patient population was homozygous for a Class I-III allele, and 2 of 12 (16.7%) heterozygous females had deficient activity. Rasburicase-induced methemoglobinemia occurred in 6 patients, 5 of whom had a Class I-III allele.
Despite extensive characterization of the G6PD gene, phenotypic enzyme activity is currently the gold standard for determining G6PD deficiency. However, various factors can influence activity assay results. Mature erythrocytes have approximately 50 times less G6PD activity than reticulocytes in the same individual; thus, G6PD activity measurements taken in the setting of reticulocytosis can be artificially high in G6PD-deficient patients (32–34). Although leukocytes usually contribute to a very small fraction of the measured G6PD activity, they could account for a substantial fraction in the setting of hyperleukocytosis (>100 X 103 cells/µL) and anemia, (35, 36) and for these cases, it is recommended to use a buffy coat free method. Red blood cell transfusions can also influence a G6PD activity measure. The lifespan of transfused red blood cells is approximately 60 days (37), and an activity measurement within this time frame could be reflective of the activity of the red blood cells from the donor instead of the patient. (6) In addition to patient factors, temperature and sample handling can also affect the results of activity assays. (9) A genetic test could avoid some of the artifacts inherent in measuring G6PD activity in blood.
Even if genetic variants are adequately interrogated, there are inherent limitations to genetic test for assigning G6PD status.It is difficult to predict activity in heterozygous females due to X-linked mosaicism, in which one copy of the gene is randomly inactivated in cells. (11–13) Most heterozygotes have more normal G6PD cells than deficient G6PD cells, suggesting a cell selection bias. (38) Age also plays a role in the distribution of normal and deficient red blood cells: with younger heterozygotes have more G6PD normal cells, whereas elderly patients have more G6PD deficient cells. (39, 40) In our cohort, 2 of 12 (16.7%) heterozygous females had deficient G6PD activity, which is consistent with reports in the literature (10, 41)
False positives in males (deficient genotype with normal activity) were not expected. The only two male patients in our cohort who had normal activity despite having a Class I-III allele had received red blood cell transfusions 44 days and 2 days prior to the activity assay, and thus activity may have reflected that of the red blood cells from the donor. One of these two patients developed methemoglobinemia after rasburicase administration, which also further suggests this patient was actually G6PD deficient. Hence our potential true positive predictive value for males could be 100%, given that both discordant patients may have had artificially elevated G6PD activity.
While a positive deficient genotype result should indicate a deficient phenotype, a negative deficient genotype (i.e. no detectable Class I-III alleles) does not necessarily indicate normal activity, because any genetic platform may fail to detect some important variants. (6) False negatives (G6PD deficient activity but without a Class I-III allele) were expected, especially in patients without whole exome sequencing. In our cohort, the sensitivity in males was only 39.1% (20.5–61.2%). There is the possibility that silent or noncoding variants may contribute to G6PD deficiency (Supplemental Material (Silent and noncoding variants in the G6PD gene)); however, we were unable to confirm that noncoding variants accounted for any of our cases of G6PD deficiency in the absence of Class I-III alleles (data not shown).
This is one of few large-scale G6PD genotype-phenotype studies done in an American population and the first in pediatric patients undergoing treatment for hematological malignancies. A study of neonates in Pennsylvania which genotyped 5 variants (A, Asahi, Mediterranean, Kaiping, and Canton) in over 4000 patients had similar genotype/phenotype concordance rates as our study. (17) Most other studies have focused on specific populations and on the alleles most common to that population. In these studies, even if whole exome sequencing was performed, 6–28% of deficient patients did not have a missense variant detected. (42–45) Thus, it seems that factors other than G6PD coding variants may cause G6PD deficiency.
G6PD activity was measured without regard to transfusion history, a potential limitation of our retrospective study of clinical phenotypes. G6PD activity measurements were made based on clinical considerations (e.g. the need to consider rasburicase for tumor lysis syndrome) and so may not have been optimally timed relative to hematologic status. If a patient has normal G6PD activity and receives a transfusion from a G6PD-deficient donor, the transfusion should not be enough for the patient to present as deficient. However, a red blood cell transfusion from a donor with normal G6PD activity could artificially increase the G6PD activity of a G6PD deficient patient. Therefore, the activity assay may have missed some G6PD deficient patients, as we believe it did with the two males hemizygous for a Class I-III allele who had normal G6PD activity after a red blood cell transfusion. These were the only patients we expected to be G6PD deficient according to genotype who had normal G6PD activity. A related limitation of this study is that data were limited to what was in the medical record, which may not have accurately reflected all red blood cell transfusions given outside of St. Jude. Methemoglobin levels were also limited to those patients who had their methemoglobin levels monitored for a clinical reason, which was not clear from their medical record.
Rasburicase is a recombinant urate oxidase enzyme approved for the prevention and treatment of hyperuricemia of tumor lysis syndrome. As rasburicase breaks down uric acid to allantoin, it also produces hydrogen peroxide, an oxidizing agent, as a byproduct. (1, 46, 47) Rasburicase-induced methemoglobinemia is a potentially serious adverse effect that has been documented in several patients with G6PD deficiency, (48, 49) and rasburicase is thus contraindicated in patients with G6PD deficiency. A few patients in our cohort received rasburicase and were later determined to be G6PD deficient. We found that rasburicase-associated methemoglobinemia occurred in 6 patients, 5 of whom had at least one Class I-III allele. Importantly, performing only a phenotypic assay would have missed two of these patients: one hemizygous male who had received a blood transfusion prior to the activity assay and one heterozygous female with normal activity at the time of rasburicase administration. This indicates that having a fraction of deficient erythrocytes, even with a normal G6PD activity measure, could potentially be enough to predispose to methemoglobinemia after exposure to a strong oxidative trigger, like rasburicase. Similarly, in a retrospective analysis of a primaquine study, females heterozygous for the Mahidol variant, a class III allele, were more likely to require treatment for hemolytic anemia than wildtype females, even if the G6PD activity test was normal. (50) There have also been rare reports of methemoglobinemia after rasburicase administration in patients without G6PD deficiency. (51, 52) In our cohort, a black female with normal activity without a Class I-III allele also developed methemoglobinemia after rasburicase administration.
In conclusion, while genotyping G6PD has its limitations, it can provide valuable information, and may become more widely available as whole exome and whole genome sequencing increases. For drugs such as rasburicase, the turnaround time for a G6PD genetic test is currently too long, and an activity test will still be required for obtaining a patient’s G6PD status quickly. However, genetic tests are rapidly evolving to include point-of-care tests, and G6PD genotype can be useful for confirmation of G6PD status when phenotypic tests may be compromised. When a Class I-III allele is observed in male patients or two Class I-III alleles in females, medications known to cause hemolytic anemia or methemoglobinemia in G6PD deficiency, such as rasburicase, should be avoided. For heterozygous female patients, caution must be used with strong oxidative drugs, even if the G6PD activity measure is normal, due to the potential for hemolytic anemia or methemoglobinemia. Genotyping has the advantage that results are not altered by the hemolytic status of the patient, such as high white blood cell count, transfusion history, or anemic states. More work needs to be done to ensure greater sensitivity of DNA-based tests by increasing the alleles interrogated by commercially available genetic tests and to understand the mechanism of G6PD deficiency in the absence of a Class I-III allele.
Supplementary Material
Acknowledgments:
This study was supported by grants GM 115279 and CA 21765
Footnotes
Conflicts of interest: None declared
Supplementary information is available at The Pharmacogenomics Journal’s website.
References:
- 1.Luzzatto L, Nannelli C, Notaro R. Glucose-6-Phosphate Dehydrogenase Deficiency. Hematol Oncol Clin North Am 2016;30(2):373–93. [DOI] [PubMed] [Google Scholar]
- 2.Pandolfi PP, Sonati F, Rivi R, Mason P, Grosveld F, Luzzatto L. Targeted disruption of the housekeeping gene encoding glucose 6-phosphate dehydrogenase (G6PD): G6PD is dispensable for pentose synthesis but essential for defense against oxidative stress. The EMBO journal 1995;14(21):5209–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sivilotti ML. Oxidant stress and haemolysis of the human erythrocyte. Toxicological reviews 2004;23(3):169–88. [DOI] [PubMed] [Google Scholar]
- 4.Mason PJ, Bautista JM, Gilsanz F. G6PD deficiency: the genotype-phenotype association. Blood reviews 2007;21(5):267–83. [DOI] [PubMed] [Google Scholar]
- 5.McDonagh EM, Bautista JM, Youngster I, Altman RB, Klein TE. PharmGKB summary: methylene blue pathway. Pharmacogenetics and genomics 2013;23(9):498–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Relling MV, McDonagh EM, Chang T, Caudle KE, McLeod HL, Haidar CE, et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) guidelines for rasburicase therapy in the context of G6PD deficiency genotype. Clinical pharmacology and therapeutics 2014;96(2):169–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nkhoma ET, Poole C, Vannappagari V, Hall SA, Beutler E. The global prevalence of glucose-6-phosphate dehydrogenase deficiency: a systematic review and meta-analysis. Blood cells, molecules & diseases 2009;42(3):267–78. [DOI] [PubMed] [Google Scholar]
- 8.Standardization of procedures for the study of glucose-6-phosphate dehydrogenase. Report of a WHO Scientific Group. World Health Organization technical report series 1967;366:1–53. [PubMed] [Google Scholar]
- 9.Domingo GJ, Satyagraha AW, Anvikar A, Baird K, Bancone G, Bansil P, et al. G6PD testing in support of treatment and elimination of malaria: recommendations for evaluation of G6PD tests. Malar J 2013;12:391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Abdulrazzaq YM, Micallef R, Qureshi M, Dawodu A, Ahmed I, Khidr A, et al. Diversity in expression of glucose-6-phosphate dehydrogenase deficiency in females. Clin Genet 1999;55(1):13–9. [DOI] [PubMed] [Google Scholar]
- 11.Beutler E, Yeh M, Fairbanks VF. The normal human female as a mosaic of X-chromosome activity: studies using the gene for C-6-PD-deficiency as a marker. Proceedings of the National Academy of Sciences of the United States of America 1962;48:9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nance WE. GENETIC TESTS WITH A SEX-LINKED MARKER: GLUCOSE-6-PHOSPHATE DEHYDROGENASE. Cold Spring Harbor symposia on quantitative biology 1964;29:415–25. [DOI] [PubMed] [Google Scholar]
- 13.Rinaldi A, Filippi G, Siniscalco M. Variability of red cell phenotypes between and within individuals in an unbiased sample of 77 heterozygotes for G6PD deficiency in Sardinia. American journal of human genetics 1976;28(5):496–505. [PMC free article] [PubMed] [Google Scholar]
- 14.Luzzatto L, Seneca E. G6PD deficiency: a classic example of pharmacogenetics with on-going clinical implications. Br J Haematol 2014;164(4):469–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xia Z, Chen P, Tang N, Yan T, Zhou Y, Xiao Q, et al. Rapid detection of G6PD mutations by multicolor melting curve analysis. Mol Genet Metab 2016;119(1–2):168–73. [DOI] [PubMed] [Google Scholar]
- 16.Yoshida A, Beutler E, Motulsky AG. Human glucose-6-phosphate dehydrogenase variants. Bulletin of the World Health Organization 1971;45(2):243–53. [PMC free article] [PubMed] [Google Scholar]
- 17.Lin Z, Fontaine JM, Freer DE, Naylor EW. Alternative DNA-based newborn screening for glucose-6-phosphate dehydrogenase deficiency. Mol Genet Metab 2005;86(1–2):212–9. [DOI] [PubMed] [Google Scholar]
- 18.Fernandez CA, Smith C, Yang W, Lorier R, Crews KR, Kornegay N, et al. Concordance of DMET plus genotyping results with those of orthogonal genotyping methods. Clinical pharmacology and therapeutics 2012;92(3):360–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Burmester JK, Sedova M, Shapero MH, Mansfield E. DMET microarray technology for pharmacogenomics-based personalized medicine. Methods in molecular biology 2010;632:99–124. [DOI] [PubMed] [Google Scholar]
- 20.Grove ML, Yu B, Cochran BJ, Haritunians T, Bis JC, Taylor KD, et al. Best practices and joint calling of the HumanExome BeadChip: the CHARGE Consortium. PLoS One 2013;8(7):e68095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang W, Wu G, Broeckel U, Smith CA, Turner V, Haidar CE, et al. Comparison of genome sequencing and clinical genotyping for pharmacogenes. Clinical pharmacology and therapeutics 2016;100(4):380–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Davidson D, Barefield ES, Kattwinkel J, Dudell G, Damask M, Straube R, et al. Inhaled nitric oxide for the early treatment of persistent pulmonary hypertension of the term newborn: a randomized, double-masked, placebo-controlled, dose-response, multicenter study. The I-NO/PPHN Study Group. Pediatrics 1998;101(3 Pt 1):325–34. [DOI] [PubMed] [Google Scholar]
- 23.Dellinger RP, Zimmerman JL, Taylor RW, Straube RC, Hauser DL, Criner GJ, et al. Effects of inhaled nitric oxide in patients with acute respiratory distress syndrome: results of a randomized phase II trial. Inhaled Nitric Oxide in ARDS Study Group. Critical care medicine 1998;26(1):15–23. [DOI] [PubMed] [Google Scholar]
- 24.Mandrell BN, McCormick JN. Dapsone-induced methemoglobinemia in pediatric oncology patients: case examples. J Pediatr Oncol Nurs 2001;18(5):224–8. [DOI] [PubMed] [Google Scholar]
- 25.Plotkin JS, Buell JF, Njoku MJ, Wilson S, Kuo PC, Bartlett ST, et al. Methemoglobinemia associated with dapsone treatment in solid organ transplant recipients: a two-case report and review. Liver Transpl Surg 1997;3(2):149–52. [DOI] [PubMed] [Google Scholar]
- 26.Shahani L, Sattovia S. Acquired methaemoglobinaemia related to phenazopyridine ingestion. BMJ Case Rep 2012;2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Green ED, Zimmerman RC, Ghurabi WH, Colohan DP. Phenazopyridine hydrochloride toxicity: a cause of drug-induced methemoglobinemia. Jacep 1979;8(10):426–31. [DOI] [PubMed] [Google Scholar]
- 28.Bayat A, Kosinski RW. Methemoglobinemia in a newborn: a case report. Pediatr Dent 2011;33(3):252–4. [PubMed] [Google Scholar]
- 29.Karim A, Ahmed S, Siddiqui R, Mattana J. Methemoglobinemia complicating topical lidocaine used during endoscopic procedures. Am J Med 2001;111(2):150–3. [DOI] [PubMed] [Google Scholar]
- 30.Kawasumi H, Tanaka E, Hoshi D, Kawaguchi Y, Yamanaka H. Methemoglobinemia induced by trimethoprim-sulfamethoxazole in a patient with systemic lupus erythematosus. Intern Med 2013;52(15):1741–3. [DOI] [PubMed] [Google Scholar]
- 31.Carroll TG, Carroll MG. Methemoglobinemia in a Pediatric Oncology Patient Receiving Sulfamethoxazole/Trimethoprim Prophylaxis. Am J Case Rep 2016;17:499–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marks PA, Johnson AB. Relationship between the age of human erythrocytes and their osmotic resistance: a basis for separating young and old erythrocytes. The Journal of clinical investigation 1958;37(11):1542–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Morelli A, Benatti U, Gaetani GF, De Flora A. Biochemical mechanisms of glucose-6-phosphate dehydrogenase deficiency. Proceedings of the National Academy of Sciences of the United States of America 1978;75(4):1979–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Beutler E Glucose-6-phosphate dehydrogenase deficiency. Diagnosis, clinical and genetic implications. American journal of clinical pathology 1967;47(3):303–11. [DOI] [PubMed] [Google Scholar]
- 35.Echler G Determination of glucose-6-phosphate dehydrogenase levels in red cell preparations. The American journal of medical technology 1983;49(4):259–62. [PubMed] [Google Scholar]
- 36.Morelli A, Benatti U, Lenzerini L, Sparatore B, Salomino F, Melloni E, et al. The interference of leukocytes and platelets with measurement of clucose-6-phosphate dehydrogenase activity of erythrocytes with low activity variants of the enzyme. Blood 1981;58(3):642–4. [PubMed] [Google Scholar]
- 37.Liumbruno G, Bennardello F, Lattanzio A, Piccoli P, Rossetti G. Recommendations for the transfusion of red blood cells. Blood transfusion = Trasfusione del sangue 2009;7(1):49–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Filosa S, Giacometti N, Wangwei C, De Mattia D, Pagnini D, Alfinito F, et al. Somatic-cell selection is a major determinant of the blood-cell phenotype in heterozygotes for glucose-6-phosphate dehydrogenase mutations causing severe enzyme deficiency. American journal of human genetics 1996;59(4):887–95. [PMC free article] [PubMed] [Google Scholar]
- 39.Sanna G, Frau F, De Virgiliis S, Piu P, Bertolino F, Cao A. Glucose-6-phosphate dehydrogenase red blood cell phenotype in GdMediterranean heterozygous females and hemizygous males at birth. Pediatric research 1981;15(11):1443–6. [DOI] [PubMed] [Google Scholar]
- 40.Au WY, Ma ES, Lam VM, Chan JL, Pang A, Kwong YL. Glucose 6-phosphate dehydrogenase (G6PD) deficiency in elderly Chinese women heterozygous for G6PD variants. American journal of medical genetics Part A 2004;129a(2): 208–11. [DOI] [PubMed] [Google Scholar]
- 41.Johnson MK, Clark TD, Njama-Meya D, Rosenthal PJ, Parikh S. Impact of the method of G6PD deficiency assessment on genetic association studies of malaria susceptibility. PLoS One 2009;4(9):e7246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sirdah M, Reading NS, Vankayalapati H, Perkins SL, Shubair ME, Aboud L, et al. Molecular heterogeneity of glucose-6-phosphate dehydrogenase deficiency in Gaza Strip Palestinians. Blood cells, molecules & diseases 2012;49(3–4):152–8. [DOI] [PubMed] [Google Scholar]
- 43.Laouini N, Bibi A, Ammar H, Kazdaghli K, Ouali F, Othmani R, et al. Glucose-6-phosphate dehydrogenase deficiency in Tunisia: molecular data and phenotype-genotype association. Mol Biol Rep 2013;40(2):851–6. [DOI] [PubMed] [Google Scholar]
- 44.Sirdah MM, Shubair ME, Al-Kahlout MS, Al-Tayeb JM, Prchal JT, Reading NS. Possible association of 3’ UTR +357 A>G, IVS11-nt 93 T>C, c.1311 C>T polymorphism with G6PD deficiency. Hematology 2017;22(6):370–4. [DOI] [PubMed] [Google Scholar]
- 45.Dallol A, Banni H, Gari MA, Al-Qahtani MH, Abuzenadeh AM, Al-Sayes F, et al. Five novel glucose-6-phosphate dehydrogenase deficiency haplotypes correlating with disease severity. J Transl Med 2012;10:199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Navolanic PM, Pui CH, Larson RA, Bishop MR, Pearce TE, Cairo MS, et al. Elitek-rasburicase: an effective means to prevent and treat hyperuricemia associated with tumor lysis syndrome, a Meeting Report, Dallas, Texas, January 2002. Leukemia 2003;17(3):499–514. [DOI] [PubMed] [Google Scholar]
- 47.Pui CH. Rasburicase: a potent uricolytic agent. Expert opinion on pharmacotherapy 2002;3(4):433–42. [DOI] [PubMed] [Google Scholar]
- 48.Bontant T, Le Garrec S, Avran D, Dauger S. Methaemoglobinaemia in a G6PD-deficient child treated with rasburicase. BMJ Case Rep 2014;2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sonbol MB, Yadav H, Vaidya R, Rana V, Witzig TE. Methemoglobinemia and hemolysis in a patient with G6PD deficiency treated with rasburicase. American journal of hematology 2013;88(2):152–4. [DOI] [PubMed] [Google Scholar]
- 50.Chu CS, Bancone G, Moore KA, Win HH, Thitipanawan N, Po C, et al. Haemolysis in G6PD Heterozygous Females Treated with Primaquine for Plasmodium vivax Malaria: A Nested Cohort in a Trial of Radical Curative Regimens. PLoS Med 2017;14(2):e1002224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bauters T, Mondelaers V, Robays H, De Wilde H, Benoit Y, De Moerloose B. Methemoglobinemia and hemolytic anemia after rasburicase administration in a child with leukemia. International journal of clinical pharmacy 2011;33(1):58–60. [DOI] [PubMed] [Google Scholar]
- 52.Kizer N, Martinez E, Powell M. Report of two cases of rasburicase-induced methemoglobinemia. Leukemia & lymphoma 2006;47(12):2648–50. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.