

Abstract
Sex chromosome constitution varies in the human population, both between the sexes (46,XX females and 46,XY males), and within the sexes (for example, 45,X and 46,XX females, and 47,XXY and 46,XY males). Coincident with this genetic variation are numerous phenotypic differences between males and females, and individuals with sex chromosome aneuploidy. However, the molecular mechanisms by which sex chromosome constitution impacts phenotypes at the cellular, tissue, and organismal levels remain largely unexplored. Thus emerges a fundamental question connecting the study of sex differences and sex chromosome aneuploidy syndromes: How does sex chromosome constitution influence phenotype? Here, we focus on Turner syndrome (TS), associated with the 45,X karyotype, and its synergies with the study of sex differences. We review findings from evolutionary studies of the sex chromosomes, which identified genes that are most likely to contribute to phenotypes as a result of variation in sex chromosome constitution. We then explore strategies for investigating the direct effects of the sex chromosomes, and the evidence for specific sex chromosome genes impacting phenotypes. In sum, we argue that integrating the study of TS with sex differences offers a mutually beneficial alliance to identify contributions of the sex chromosomes to human development, health, and disease.
Keywords: Turner syndrome, sex chromosome aneuploidy, sex differences, sex chromosome evolution
INTRODUCTION
The clinical features that define Turner syndrome (TS), including short stature, hypogonadism, and webbed neck, were first described in 1938 and TS was associated with the monosomy X karyotype (45,X) in 1959 (Ford, Jones, Polani, Dealmeida, & Briggs, 1959; Turner, 1938). In the 60 years since this association, questions regarding the causal role of the 45,X karyotype in TS phenotypes have accumulated. In parallel, another field has been considering the contributions of the sex chromosomes: the biology of sex differences. It is well-appreciated that males and females differ in sex chromosome constitution – most males are 46,XY, while most females are 46,XX – and that there are extensive phenotypic differences between males and females in health and disease (Wizemann & Pardue, 2001). For example, a variety of autoimmune diseases, including lupus, multiple sclerosis, and rheumatoid arthritis are more prevalent in females (Ngo, Steyn, & McCombe, 2014). In contrast, certain neurodevelopmental disorders, such as autism, attention-deficit hyperactivity disorder, and intellectual disability are male-biased (Boyle et al., 2011; Werling & Geschwind, 2013). Nonetheless, we know little about how the sex chromosomes contribute to many sex-biased phenotypes. Fundamentally, the TS and sex differences fields are united by the pursuit of connections between variation in sex chromosome constitution and phenotype. Thus, as we have previously argued (Page & Miller, 2016), it is time for a strategic alliance between TS and sex differences researchers.
A primary goal of this alliance should be to decipher the complex landscape of variables that contribute to TS and sex differences. These include sex chromosome constitution, gonadal hormone profiles, and disparate environmental exposures. In fact, the impact of sex chromosome constitution has long been minimized compared with the effects of gonadal hormones. In this regard, it was commonly thought that sex chromosomes modulate phenotypes throughout the body indirectly through their primary role in sex determination and the subsequent organizing effects of gonadal hormones. Progress in understanding the biology of the sex chromosomes has led to a significant conceptual departure from this idea: that the sex chromosomes have direct effects on cells and tissues outside of the reproductive tract, which are not mediated through gonadal hormones (Skaletsky et al., 2003). Indeed, many genes on the sex chromosomes are expressed throughout the body and participate in fundamental cellular processes such as transcription and translation (Bellott et al., 2014; Tukiainen et al., 2017). However, proving that sex chromosome constitution independently contributes to a given phenotype requires experimental models and strategies that can distinguish between effects of these co-occurring variables.
Here, we will review the evidence that sex chromosome constitution directly affects phenotypes in both TS and sex differences. We begin with a discussion of sex chromosome evolution, which can be considered a natural experiment that reveals promising candidate genes for mediating phenotypes throughout the body. Second, we consider experimental models that allow researchers to disentangle the effects of the sex chromosomes from other co-occurring variables. Third, we highlight two case studies of sex chromosome genes that have been convincingly implicated in mediating specific phenotypes in TS and may also contribute to sex differences. Finally, we suggest future approaches for increasing synergy between research in TS and sex differences.
STUDIES OF SEX CHROMOSOME EVOLUTION REVEAL CANDIDATE GENES FOR TS AND SEX DIFFERENCES
The investigation of sex chromosome evolution has been a successful strategy to identify genes that potentially contribute to TS and sex differences. Although research in TS typically focuses on the X chromosome, much of our understanding of sex chromosome evolution comes from the complementary study of the Y chromosome, as X and Y evolution occurred in parallel. The human sex chromosomes evolved from a pair of ordinary autosomes present in the common ancestor of mammals and birds approximately 300 million years ago (Lahn & Page, 1999; Ohno, 1967). X and Y chromosome differentiation began with the emergence of SRY, the male sex-determining gene, on the proto-Y chromosome before the evolutionary divergence of marsupials and placental mammals. Subsequently, a series of chromosomal inversions on the Y chromosome suppressed crossing-over with the X chromosome. In this region of suppressed X-Y crossing over, the X and Y chromosomes diverged, resulting in a male-specific region on the Y (MSY; Figure 1A). The X and Y chromosomes are identical in two pseudoautosomal regions (PARs) at the tips of the chromosomes, where X-Y crossing over occurs and is required for proper pairing and chromosome segregation in male meiosis (Cooke, Brown, & Rappold, 1985; Freije, Helms, Watson, & Donis-Keller, 1992). The loss of crossing-over with the X chromosome across the MSY led to genetic decay; only 3% of genes from the ancestral chromosome (17 of ~640 ancestral genes) survive on the human Y chromosome (Bellott et al., 2014; Skaletsky et al., 2003). In contrast, the X chromosome retained 98% of the ancestral genes (Bellott et al., 2014).
Figure 1. Evolution of the human sex chromosomes resulted in four gene classes on the X chromosome.
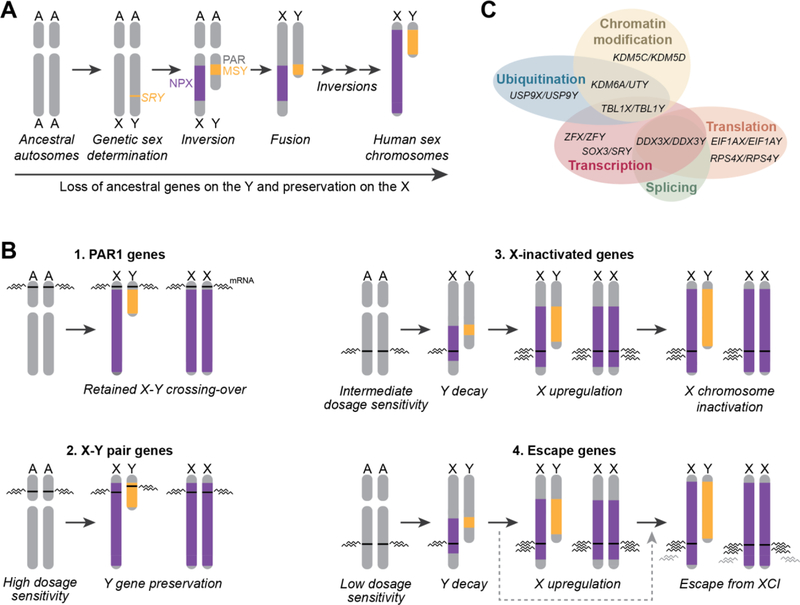
(A) Sex chromosome evolution began with ordinary autosomes. SRY emergence began Y chromosome differentiation, followed by a series of inversions that resulted in loss of crossing-over between the male-specific region of the Y (MSY) and the non-pseudoautosomal region of the X (NPX). The pseudoautosomal region (PAR) on the short arm retained X-Y crossing over. (B) Four classes of genes on the X chromosome underwent different evolutionary trajectories as a result of Y chromosome decay. The black wavy lines indicate the level of mRNA expressed from each gene. PAR1 genes (1) retained X-Y crossing-over and remain expressed from both the X and Y chromosomes. X-Y pair genes (2) reside in the NPX and MSY and do not cross-over, but due to high dosage sensitivity the Y gene was preserved and expression retained on both X chromosomes in 46,XX females. X-inactivated genes (3) had intermediate dosage sensitivity, which allowed for Y chromosome decay. This was followed by upregulation on the X chromosome to retain ancestral dosage in males, and subsequent inactivation of one allele in females. Escape genes (4) followed the same initial path as the X-inactivated genes, but did not become inactivated. Some escape genes may have avoided the step of X-upregulation, indicated by the gray dashed arrow bypassing this step. These genes are predicted to have lower levels of mRNA expression (gray wavy lines) compared to genes that underwent X-upregulation. (C) Functions of select human X-Y pair genes, adapted from (Bellott et al., 2014).
Although the X chromosome retained almost all of the ancestral genes, they did not follow a unified evolutionary path. Recent evidence shows that prior to sex chromosome differentiation, genes on the ancestral autosomes had inherent differences in sensitivity to gene dosage that resulted in four classes of divergent evolutionary trajectories on the X chromosome in the context of Y chromosome decay (Bellott et al., 2014; Bellott et al., 2017; Naqvi, Bellott, Lin, & Page, 2018). First are the PAR genes, which are distinguishable from the rest of the classes because this region never stopped crossing-over with the Y chromosome (Figure 1B). Here we specifically refer to the PAR1 genes (on the tip of the short arm), since PAR2 (on the end of the long arm) was not part of the ancestral chromosomes (Ciccodicola et al., 2000). The PAR genes remain expressed from both copies of the X chromosome in 46,XX females, and on the X and Y chromosomes in 46,XY males. TS and sex-biased phenotypes may be impacted by PAR genes that are dosage sensitive.
The second class is comprised of genes on the ancestral autosomes with a high inherent sensitivity to gene dosage. As a result, they could not tolerate transient dosage changes and resisted decay on the Y chromosome. These 14 genes remain as homologous pairs with a single copy on the X and Y chromosomes and continue to be expressed from both X chromosomes in 46,XX females, and from the X and Y chromosomes in 46,XY males (Lahn & Page, 1997) (Figure 1B). As the X-Y pair genes do not cross-over during meiosis, they have diverged and encode distinct protein isoforms; the X and Y copies retain 73–98% DNA and 71–99% protein identity (Skaletsky et al., 2003). Molecular functions of the X-Y pair genes have not been extensively compared; however, there is some evidence from mouse studies and in vitro experiments demonstrating either functional interchangeability or divergence, depending on the experimental context. For example, the histone lysine demethylase Uty is redundant with Kdm6a (Utx) in mouse development (Lee, Lee, & Lee, 2012; Shpargel, Sengoku, Yokoyama, & Magnuson, 2012), but the human UTY protein shows reduced demethylase activity in vitro compared to UTX (Hong et al., 2007; Lan et al., 2007).
Studies of independent sex chromosome systems reinforce the idea that survival of ancestral genes on sex-specific chromosomes is an important indicator of dosage sensitivity. Indeed, the same principle holds for the chicken ZW system, in which the W chromosome is present exclusively in females. Like the Y, the W chromosome has extensively degenerated over time, yet retains a set of genes that are highly dosage sensitive (Bellott et al., 2017). In the case of both the Y and W, the remaining ancestral genes are broadly expressed throughout the body and involved in fundamental cellular processes including transcription, epigenetic regulation, and translation (Figure 1C) (Bellott et al., 2014; Bellott et al., 2017). As we discuss below, these genes are some of the top candidates for contributing to TS and sex-biased phenotypes.
The third, and largest, class of X chromosome genes includes genes on the ancestral autosomes with intermediate dosage sensitivity. These genes went through a complex, three-step evolutionary process: first, decay of the Y-linked homolog; second, X chromosome upregulation; and third, X chromosome inactivation (Jegalian & Page, 1998; Ohno, 1967). Following loss of the Y homologs, the genes were transcriptionally upregulated on the X chromosome in both sexes to maintain the ancestral gene dosage in males. The increased expression would have been deleterious to females, therefore a mechanism of transcriptional dosage compensation, X chromosome inactivation (XCI), arose to silence one copy (Figure 1B). As these “X-inactivated” genes have equivalent expression in individuals with one or more X chromosomes, they are not likely to underlie phenotypes related to variation in sex chromosome constitution.
Finally, genes with the lowest dosage sensitivity followed a fourth path: they were lost from the Y chromosome but are not subject to XCI today. Instead they “escaped” XCI and are expressed from both X chromosomes in 46,XX females. The exact evolutionary trajectory they took to get there is not clear; they either were not upregulated following Y decay and did not require XCI, or they were upregulated but did not become inactivated (Figure 1B). Escape genes are expressed at higher levels in 46,XX females compared to 46,XY males across tissues (Tukiainen et al., 2017), and are plausible candidates for contributing to sex-biased phenotypes.
Since the phenotypes in TS are related to the loss of all or part of a sex chromosome, candidate genes should be sensitive to gene dosage and require expression from two alleles on the sex chromosomes (Ferguson-Smith, 1965; Held et al., 1992; Hook & Warburton, 1983). Given this, and because 46,XY males do not display TS phenotypes despite only having a single X chromosome, causal TS genes must be common to X and Y, and functionally interchangeable (Watanabe, Zinn, Page, & Nishimoto, 1993; Zinn, Page, & Fisher, 1993). There are two groups of genes that fit these criteria: 1) PAR genes and 2) X-Y pair genes (if they prove to be functionally interchangeable in specific phenotypic contexts relevant to TS; Figures 2 and 3).
Figure 2. Mechanisms of sex chromosome contributions to TS and sex differences.
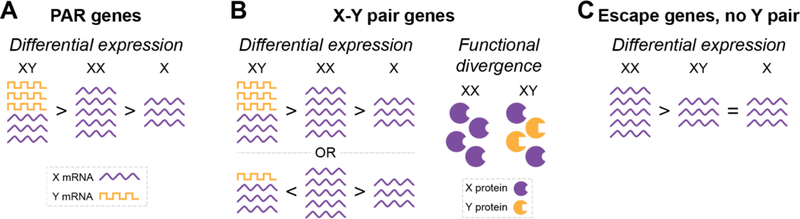
(A) PAR genes are typically more highly expressed in 46,XY males compared with 46,XX females, due to spreading of XCI, and expressed at the lowest levels 45,X individuals. (B) Left, X-Y pair genes may have differential mRNA expression levels from the X and Y homologs such that 46,XY or 46,XX individuals have unequal total expression. These genes escape XCI in 46,XX females so expression is lower in 45,X females. Right, X-Y pair genes may encode functionally divergent proteins that lead to molecular sex differences between 46,XX and 46,XY individuals. A combination of both mechanisms may also exist. (C) Genes that escape XCI without a Y homolog may contribute to sex differences due to differences in mRNA expression. Generally, expression of genes that escape XCI is higher in females.
Figure 3. X and Y genes that may contribute to Turner syndrome and sex differences.

This schematic includes two categories of genes that contribute to TS and sex differences phenotypes: X-Y pair genes and PAR1 genes (not shown are X escape genes without a Y homolog; these may also contribute to sex differences). PAR1 genes are listed in order from distal to proximal. Genes discussed in case studies are marked with an asterisk.
Sex-biased phenotypes must be mediated by genes that are 1) expressed at different levels based on sex chromosome dosage, and/or 2) encode Y chromosome proteins with unique functions. Escape genes and PAR genes fit the first criterion, while X-Y pairs fit both criteria. Genes that escape XCI are expressed higher in females (Carrel, Cottle, Goglin, & Willard, 1999); conversely, PAR genes tend to be more highly expressed in males due to spreading of XCI into the PAR in 46,XX females (Tukiainen et al., 2017). X-Y pair genes may have differences in the cumulative expression from both of the X chromosomes in 46,XX females or summed expression from the X and Y chromosomes in 46,XY males. Additionally, the X-Y pair genes may exhibit functional differences because they encode distinct protein isoforms (Skaletsky et al., 2003). This divergence allows for the intriguing possibility that the X and Y protein isoforms lead to distinct biochemical activities in males and females (Figures 2 and 3).
EXPERIMENTAL MODELS FOR IDENTIFYING PHENOTYPES MEDIATED BY SEX CHROMOSOME CONSTITUTION
At this point, we have a sufficient understanding of the types of genes that likely contribute to phenotypes due to variation in sex chromosome constitution. However, obtaining experimental evidence to prove the involvement of any specific gene or the sex chromosomes in general is not trivial. This is complicated by the co-occurrence of other variables with sex chromosome constitution, including gonadal hormone profiles and environmental exposures. For example, typical 46,XX females and 46,XY males differ in the levels of circulating estrogens and androgens; while women with TS have reduced estrogen levels compared to 46,XX females (Turner, 1938). There have been many strategies applied to this challenge, each with its pros and cons; we will evaluate some of them here and provide suggestions for moving forward.
One common strategy for deciphering genotype-phenotype relationships is to develop a mouse model, which has been applied in both TS and sex differences. For TS, there is a monosomy X mouse model (39,X karyotype), however it does not recapitulate the infertility, embryonic lethality, or the severity of phenotypes present in TS (Welshons & Russell, 1959). This is likely because the mouse and human sex chromosomes have significantly diverged in gene content and regulation. Seven of the 14 human X-Y pair genes are not present on the mouse Y chromosome: five were lost and two were retrotransposed onto autosomes (Hughes, Skaletsky, Koutseva, Pyntikova, & Page, 2015; Soh et al., 2014). Additionally, almost all of the orthologs of the human PAR genes are found on mouse autosomes or are missing in the mouse genome (Table 1) (Raudsepp & Chowdhary, 2015). Moreover, sex chromosome gene dosage regulation differs between mice and humans; only 3–7% of X-linked genes are expressed from both X alleles in the mouse, compared to at least 23% in humans (Berletch et al., 2015; Tukiainen et al., 2017).
Table 1.
Mouse orthologs of human X-Y pair and PAR1 genes.
| Chromosomal location |
|||
|---|---|---|---|
| Human Gene | Human | Mousea | |
| Ancestral X-Y pair genes | SOX3/SRY | X/Y | X/Y |
| ZFX/ZFY | X/Y | X/Y | |
| DDX3X/Y | X/Y | X/Yb | |
| KDM6A/UTY | X/Y | X/Yb | |
| KDM5C/D | X/Y | X/Yb | |
| USP9X/Y | X/Y | X/Y | |
| EIF1AX/Y | X/Y | X/18c | |
| RPS4X/Y | X/Y | X/6c | |
| AMELX/Y | X/Y | X/- | |
| TXLNG/Y | X/Y | X/- | |
| TMSB4X/Y | X/Y | X/- | |
| NLGN4X/Y | X/Y | X/Y (PAR) | |
| TBL1X/Y | X/Y | X/- | |
| PRKX/Y | X/Y | X/- | |
| PAR genes | PLCXD1 | X/Y (PAR1) | 5 |
| GTPBP6 | X/Y (PAR1) | 5d | |
| PPP2R3B | X/Y (PAR1) | 9 | |
| SHOX | X/Y (PAR1) | -e | |
| CRLF2 | X/Y (PAR1) | 5 | |
| CSF2RA | X/Y (PAR1) | 19f | |
| IL3RA | X/Y (PAR1) | 14g | |
| SLC25A6 | X/Y (PAR1) | -g | |
| ASMTL | X/Y (PAR1) | - | |
| P2RY8 | X/Y (PAR1) | - | |
| AKAP17A | X/Y (PAR1) | X/Y (PAR) | |
| ASMT | X/Y (PAR1) | X/Y (PAR) | |
| DHRSX | X/Y (PAR1) | 4d | |
| ZBED1 | X/Y (PAR1) | -d | |
| CD99 | X/Y (PAR1) | 4h | |
PAR = pseudoautosomal region
For X-Y pairs, presence or absence of the Y copy in mouse is confirmed in (Soh et al., 2014).
X homolog expressed from both X chromosomes in human and mouse (Berletch et al., 2015; Yang, Babak, Shendure, & Disteche, 2010).
Human and mouse have a paralagous gene, SHOX2, on chr3 (Rao et al., 1997).
To study sex differences, a sex-reversed mouse model was developed to uncouple the effects of sex chromosome constitution and gonadal hormones. In this model, Sry, the Y-linked testis-determining gene, is deleted from the Y chromosome and relocated to an autosome as a transgene, resulting in the ability to generate gonadal males with either XY or XX chromosomes, and gonadal females with either XX or XY chromosomes. Therefore, this model allows for the identification of phenotypes independently associated with either gonadal sex or sex chromosome constitution. To date, several phenotypes dependent on sex chromosome constitution have been identified with this model, such as pathogenesis of autoimmune disease, addiction behaviors, and fat metabolism (Chen, McClusky, Itoh, Reue, & Arnold, 2013; Quinn, Hitchcott, Umeda, Arnold, & Taylor, 2007; Smith-Bouvier et al., 2008). Due to the divergence of the sex chromosomes, it will be useful to determine whether the genes mediating these phenotypes are conserved between mouse and human. The most informative genes in this regard are Kdm6a/Uty, Kdm5c/Kdm5d, and Ddx3x/Ddx3y, since, similar to humans, they remain as X-Y pairs and the X homologs are expressed from both X chromosomes in 40,XX female mice (Table 1).
Other strategies have been developed to study the effects of sex chromosome constitution directly in humans. The preimplantation embryo is a promising place to look at phenotypes mediated by the sex chromosomes prior to the initiation of gonadal development and hormone production. A single-cell study of human embryos from the 8-cell stage to embryonic day 7 found sex-biased expression of 58 autosomal genes, 13 Y-linked genes, and 105 X-linked genes (Petropoulos et al., 2016). It is likely that there were so many sex-biased X-linked genes observed because X chromosome inactivation is not yet complete during this developmental window. These results indicate that the sex chromosomes significantly shape the early transcriptome and may contribute to sex differences that include rate of growth to the blastocyst stage and glucose metabolism, which differ markedly between XX and XY human preimplantation embryos (Ray, Conaghan, Winston, & Handyside, 1995). Other groups have used human pluripotent stem cells (both embryonic and induced pluripotent stem cells) to model TS and sex differences in the preimplantation period (Ronen & Benvenisty, 2014; Syrett, Sierra, Berry, Beiting, & Anguera, 2018; Urbach & Benvenisty, 2009). These are promising model systems because they enable genetic manipulation and the potential to recapitulate tissue differentiation in vitro. However, many pluripotent cell lines have perturbed XCI status and must be carefully evaluated on a case-by-case basis prior to being used to address questions related to sex chromosome constitution.
Another human-centric approach is to study individuals with chromosomal mosaicism, in which cells with different sex chromosome constitutions are exposed to the same levels of gonadal hormones and environmental stimuli. Individuals with typical 46,XX or 46,XY karyotypes lose sex chromosomes in blood cells with increased age, which has stimulated interest in understanding the contributions of this phenomenon to disease phenotypes (Guttenbach, Koschorz, Bernthaler, Grimm, & Schmid, 1995; Jacobs, Doll, Goldstein, Brunton, & Courtbrown, 1963). Indeed, researchers have found that loss of the Y chromosome in men correlates with increased risk of Alzheimer’s disease and cancer (Dumanski et al., 2016; Forsberg et al., 2014). It is worth noting that general chromosomal instability predisposing to cancer may result in Y loss simply because this chromosome is easier to lose, so more research must be done to prove causality (Wright et al., 2017). Mosaicism is also a common phenomenon in women with TS; in fact, it is thought that all women with TS must be mosaic for cells of another karyotype, such as 46,XX, because ~99% of fetuses with a complete 45,X karyotype do not survive fetal development (Hook & Warburton, 1983, 2014). Mosaicism is associated with reduced severity of many phenotypes in TS, for example 45,X/46,XX mosaicism is associated with enhanced follicle development and fertility (Borgstrom et al., 2009). To most effectively harness mosaicism as a model in the future, we must learn more about its embryonic origins, dynamics throughout the lifespan, distribution across the body, and variability between individuals.
These experimental strategies have begun to help us understand the effects of the sex chromosomes in development, health, and disease, each with their strengths and weaknesses. Many of the approaches are used to investigate sex chromosome constitution as a whole, and not to study the influence of specific genes on the sex chromosomes on a given phenotype. We will now present two case studies that implicate one PAR gene and one X-Y pair in specific TS phenotypes. In addition, we speculate about how these genes may be relevant to sex differences.
CASE STUDIES: DIRECT EFFECTS OF SEX CHROMOSOME GENES ON PHENOTYPES IN TS AND SEX DIFFERENCES
The best characterized genetic explanation for a phenotype in TS is for the PAR gene short stature homeobox (SHOX), which, as the name describes, is associated with short stature. The SHOX gene was initially identified through studies of short-statured individuals with deletions on the tip of the short arm of the X chromosome (Rao et al., 1997). Since then, the molecular functions of SHOX have been comprehensively investigated (see (Marchini, Ogata, & Rappold, 2016) for an extensive review). Human studies show that SHOX is expressed in the developing long bones of the forearms and lower limbs, as well as the first and second pharyngeal arches, which contribute to the bones of the face (Clement-Jones et al., 2000). Thus, SHOX is also thought to be responsible for the skeletal phenotypes in TS, including cubitus valgus, short metacarpals, and a high arched palate. As a testament to the dosage sensitivity of SHOX in bone growth, individuals with an extra sex chromosome, and therefore an extra copy of SHOX, are taller than average (Ogata & Matsuo, 1993). It is also interesting to speculate about the possibility that SHOX may contribute to sex differences in height. Similar to other PAR genes, there is evidence that SHOX is more highly expressed in 46,XY males, compared to 46,XX females. This result comes from a gene expression survey of adult human tissues in which male-bias was observed in all six tissues where SHOX expression could be detected, but it remains to be seen whether this is also true in the context of the developing long bones (Tukiainen et al., 2017). Given the dosage sensitivity of SHOX demonstrated by studies of individuals with sex chromosome aneuploidies, a small difference in the expression of this gene may lead to non-trivial sex differences in height.
Recently, the X-Y pair transducin beta like 1 X and Y (TBL1X/Y) emerged as a compelling genetic explanation for the high prevalence of sensorineural deafness in TS (Elsheikh, Dunger, Conway, & Wass, 2002; Gravholt, 2004). Genetic sequencing of a family with X-linked inheritance of late-onset sensorineural deafness identified a causal loss-of-function mutation in TBL1X (Bassi et al., 1999). Another family, this time with a Y-linked inheritance pattern of hearing loss, was found to have a loss-of-function mutation in TBL1Y, the Y homolog of TBL1X (Di Stazio et al., 2018). This is in contrast to a different family in which Y-linked hearing loss was associated with transposition of a region of chromosome 1 previously implicated in hearing impairment to the Y chromosome (Wang et al., 2013). Available evidence points to the potential for functional redundancy of TBL1X and TBL1Y: both genes are expressed in the human cochlea, and in vitro studies show that they bind to the same transcriptional corepressor complex and have similar effects on gene expression (Di Stazio et al., 2018; Guenther et al., 2000). Previously, it was reported that a recurrent Y chromosome deletion encompassing TBL1Y and several other genes had “no major deleterious effects”, which cast doubt on whether TBL1X/TBL1Y could contribute to phenotypes in TS (Jobling et al., 2007). However, the authors state that they had no phenotypic information about the subjects with the Y deletion, therefore the results are not inconsistent with TBL1Y contributing to sensorineural deafness. There are also clues that TBL1X/Y may be involved in sex differences in hearing loss. TBL1Y is expressed three times higher than TBL1X in the human cochlea, raising the possibility of a sex difference in TBL1 activity in the inner ear (Di Stazio et al., 2018). Interestingly, several large studies have found that hearing loss, especially at high frequencies, is more prevalent in men compared to women (Gates, Cooper, Kannel, & Miller, 1990; Helzner et al., 2005; Moller, 1981). More research is required to definitively show that the TBL1X/Y pair is involved in hearing loss and to investigate its molecular functions in the inner ear. Nevertheless, these initial findings provide a promising avenue for a more thorough understanding of this prevalent phenotype in TS and may lead to novel therapeutic approaches
CONCLUSIONS AND FUTURE PROSPECTS
This is an exciting time to reinvigorate research into the sex chromosomes and apply our extensive evolutionary, structural, and functional understanding to new questions in the realms of Turner syndrome and sex differences. The tandem approach of combining the study of sex chromosome aneuploidy and sex differences is already yielding promising results. For example, it has been extensively applied towards understanding the contributions of the sex chromosomes to neurodevelopmental and psychiatric disorders (recently reviewed in (Green, Flash, & Reiss, 2019)). To make further progress, we issue the following recommendations: First, we must invest in resources such as large clinical datasets measuring phenotypes of interest in populations with different sex chromosome constitutions. We must also develop biorepositories to store cells and tissues from these individuals, as it is difficult for any single investigator to obtain enough samples for robust molecular studies of human tissues. Second, we must explore new methods for studying the sex chromosomes in the context of specific phenotypes, in a manner that is free of confounding variables. This includes improving our abilities to model sex differences and sex chromosome aneuploidies both in vivo, using engineered mouse models or exploring other model organisms, and in vitro through pluripotent stem cell cultures and directed differentiation into specific tissues of interest. Finally, we must engage multi-disciplinary teams of clinicians and scientists, the sex chromosome aneuploidy communities, and representatives from the biotechnology and pharmaceutical industries to join in these pursuits. Ultimately, biology has linked TS and sex differences through the shared molecular phenomenon of differences in X and Y chromosome dose. It is now up to researchers to achieve a broad understanding of the contributions of sex chromosome constitution to human development, health, and disease; and to use this knowledge to transform medical care for future generations.
ACKNOWLEDGEMENTS
Our work on Turner syndrome and sex differences has been supported by NIH grants F32HD091966 (AKSR) and U01HG0007587 (DCP); HHMI; a generous gift from Brit and Alexander† d’Arbeloff; and a philanthropic gift from Arthur W. Brill† and Carol Tobin Brill. We thank Paul Kruszka and Michael Silberbach for organizing the Turner Resource Network symposium; the National Institutes of Child Health and Development (NICHD R13 HD096857-01) and Patient-Centered Outcomes Research Institute (PCORI) Eugene Washington PCORI Engagement Award (#10460) for funding the Turner Resource Network symposium; Angela Lin for her mentorship; Kelly Ranallo and Cindy Scurlock for their support; and members of the Page laboratory for helpful comments on this manuscript.
REFERENCES
- Bassi MT, Ramesar RS, Caciotti B, Winship IM, De Grandi A, Riboni M, … Borsani G (1999). X-linked late-onset sensorineural deafness caused by a deletion involving OA1 and a novel gene containing WD-40 repeats. American Journal of Human Genetics, 64(6), 1604–1616. doi: 10.1086/302408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellott DW, Hughes JF, Skaletsky H, Brown LG, Pyntikova T, Cho TJ, … Page DC (2014). Mammalian Y chromosomes retain widely expressed dosage-sensitive regulators (vol 508, pg 494, 2014). Nature, 514(7520), 126–126. doi: 10.1038/nature13719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellott DW, Skaletsky H, Cho TJ, Brown L, Locke D, Chen N, … Page DC (2017). Avian W and mammalian Y chromosomes convergently retained dosage-sensitive regulators. Nature Genetics, 49(3), 387–394. doi: 10.1038/ng.3778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berletch JB, Ma WX, Yang F, Shendure J, Noble WS, Disteche CM, & Deng XX (2015). Escape from X Inactivation Varies in Mouse Tissues. Plos Genetics, 11(3). doi:ARTN e1005079 10.1371/journal.pgen.1005079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borgstrom B, Hreinsson J, Rasmussen C, Sheikhi M, Fried G, Keros V, … Hovatta O (2009). Fertility preservation in girls with turner syndrome: prognostic signs of the presence of ovarian follicles. J Clin Endocrinol Metab, 94(1), 74–80. doi: 10.1210/jc.2008-0708 [DOI] [PubMed] [Google Scholar]
- Boyle CA, Boulet S, Schieve LA, Cohen RA, Blumberg SJ, Yeargin-Allsopp M, … Kogan MD (2011). Trends in the prevalence of developmental disabilities in US children, 1997–2008. Pediatrics, 127(6), 1034–1042. doi: 10.1542/peds.2010-2989 [DOI] [PubMed] [Google Scholar]
- Carrel L, Cottle AA, Goglin KC, & Willard HF (1999). A first-generation X-inactivation profile of the human X chromosome. Proc Natl Acad Sci U S A, 96(25), 14440–14444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen XQ, McClusky R, Itoh Y, Reue K, & Arnold AP (2013). X and Y Chromosome Complement Influence Adiposity and Metabolism in Mice. Endocrinology, 154(3), 1092–1104. doi: 10.1210/en.2012-2098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccodicola A, D’Esposito M, Esposito T, Gianfrancesco F, Migliaccio C, Miano MG, … D’Urso M (2000). Differentially regulated and evolved genes in the fully sequenced Xq/Yq pseudoautosomal region. Human Molecular Genetics, 9(3), 395–401. doi: 10.1093/hmg/9.3.395 [DOI] [PubMed] [Google Scholar]
- Clement-Jones M, Schiller S, Rao E, Blaschke RJ, Zuniga A, Zeller R, … Rappold GA (2000). The short stature homeobox gene SHOX is involved in skeletal abnormalities in Turner syndrome. Human Molecular Genetics, 9(5), 695–702. [DOI] [PubMed] [Google Scholar]
- Cooke HJ, Brown WR, & Rappold GA (1985). Hypervariable telomeric sequences from the human sex chromosomes are pseudoautosomal. Nature, 317(6039), 687–692. [DOI] [PubMed] [Google Scholar]
- Di Stazio M, Collesi C, Vozzi D, Liu W, Myers M, Morgan A, … Gasparini P (2018). TBL1Y: a new gene involved in syndromic hearing loss. Eur J Hum Genet doi: 10.1038/s41431-018-0282-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Disteche CM, Brannan CI, Larsen A, Adler DA, Schorderet DF, Gearing D, … Park LS (1992). The human pseudoautosomal GM-CSF receptor alpha subunit gene is autosomal in mouse. Nature Genetics, 1(5), 333–336. doi: 10.1038/ng0892-333 [DOI] [PubMed] [Google Scholar]
- Dumanski JP, Lambert JC, Rasi C, Giedraitis V, Davies H, Grenier-Boley B, … Initiative, E. A. D. (2016). Mosaic Loss of Chromosome Y in Blood Is Associated with Alzheimer Disease. American Journal of Human Genetics, 98(6), 1208–1219. doi: 10.1016/j.ajhg.2016.05.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellison JW, Li X, Francke U, & Shapiro LJ (1996). Rapid evolution of human pseudoautosomal genes and their mouse homologs. Mamm Genome, 7(1), 25–30. [DOI] [PubMed] [Google Scholar]
- Elsheikh M, Dunger DB, Conway GS, & Wass JAH (2002). Turner’s syndrome in adulthood. Endocrine Reviews, 23(1), 120–140. doi: 10.1210/er.23.1.120 [DOI] [PubMed] [Google Scholar]
- Ferguson-Smith MA (1965). Karyotype-Phenotype Correlations in Gonadal Dysgenesis and Their Bearing on the Pathogenesis of Malformations. J Med Genet, 2(2), 142–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford CE, Jones KW, Polani PE, Dealmeida JC, & Briggs JH (1959). A Sex-Chromosome Anomaly in a Case of Gonadal Dysgenesis (Turners Syndrome). Lancet, 1(Apr4), 711–713. [DOI] [PubMed] [Google Scholar]
- Forsberg LA, Rasi C, Malmqvist N, Davies H, Pasupulati S, Pakalapati G, … Dumanski JP (2014). Mosaic loss of chromosome Y in peripheral blood is associated with shorter survival and higher risk of cancer. Nature Genetics, 46(6), 624–628. doi: 10.1038/ng.2966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freije D, Helms C, Watson MS, & Donis-Keller H (1992). Identification of a second pseudoautosomal region near the Xq and Yq telomeres. Science, 258(5089), 1784–1787. doi: 10.1126/science.1465614 [DOI] [PubMed] [Google Scholar]
- Gates GA, Cooper JC Jr., Kannel WB, & Miller NJ (1990). Hearing in the elderly: the Framingham cohort, 1983–1985. Part I. Basic audiometric test results. Ear Hear, 11(4), 247–256. [PubMed] [Google Scholar]
- Gianfrancesco F, Sanges R, Esposito T, Tempesta S, Rao E, Rappold G, … D’Urso M (2001). Differential divergence of three human pseudoautosomal genes and their mouse homologs: implications for sex chromosome evolution. Genome Res, 11(12), 2095–2100. doi: 10.1101/gr.197001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gravholt CH (2004). Epidemiological, endocrine and metabolic features in Turner syndrome. European Journal of Endocrinology, 151(6), 657–687. doi: 10.1530/eje.0.1510657 [DOI] [PubMed] [Google Scholar]
- Green T, Flash S, & Reiss AL (2019). Sex differences in psychiatric disorders: what we can learn from sex chromosome aneuploidies. Neuropsychopharmacology, 44(1), 9–21. doi: 10.1038/s41386-018-0153-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guenther MG, Lane WS, Fischle W, Verdin E, Lazar MA, & Shiekhattar R (2000). A core SMRT corepressor complex containing HDAC3 and TBL1, a WD40-repeat protein linked to deafness. Genes & Development, 14(9), 1048–1057. [PMC free article] [PubMed] [Google Scholar]
- Guttenbach M, Koschorz B, Bernthaler U, Grimm T, & Schmid M (1995). Sex-Chromosome Loss and Aging - in-Situ Hybridization Studies on Human Interphase Nuclei. American Journal of Human Genetics, 57(5), 1143–1150. [PMC free article] [PubMed] [Google Scholar]
- Held KR, Kerber S, Kaminsky E, Singh S, Goetz P, Seemanova E, & Goedde HW (1992). Mosaicism in 45,X Turner Syndrome - Does Survival in Early-Pregnancy Depend on the Presence of 2 Sex-Chromosomes. Human Genetics, 88(3), 288–294. [DOI] [PubMed] [Google Scholar]
- Helzner EP, Cauley JA, Pratt SR, Wisniewski SR, Zmuda JM, Talbott EO, … Newman AB (2005). Race and sex differences in age-related hearing loss: the Health, Aging and Body Composition Study. J Am Geriatr Soc, 53(12), 2119–2127. doi: 10.1111/j.1532-5415.2005.00525.x [DOI] [PubMed] [Google Scholar]
- Hong S, Cho YW, Yu LR, Yu H, Veenstra TD, & Ge K (2007). Identification of JmjC domain-containing UTX and JMJD3 as histone H3 lysine 27 demethylases. Proc Natl Acad Sci U S A, 104(47), 18439–18444. doi: 10.1073/pnas.0707292104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hook EB, & Warburton D (1983). The Distribution of Chromosomal Genotypes Associated with Turners Syndrome - Livebirth Prevalence Rates and Evidence for Diminished Fetal Mortality and Severity in Genotypes Associated with Structural-X Abnormalities or Mosaicism. Human Genetics, 64(1), 24–27. doi: 10.1007/Bf00289473 [DOI] [PubMed] [Google Scholar]
- Hook EB, & Warburton D (2014). Turner syndrome revisited: review of new data supports the hypothesis that all viable 45,X cases are cryptic mosaics with a rescue cell line, implying an origin by mitotic loss. Human Genetics, 133(4), 417–424. doi: 10.1007/s00439-014-1420-x [DOI] [PubMed] [Google Scholar]
- Hughes JF, Skaletsky H, Koutseva N, Pyntikova T, & Page DC (2015). Sex chromosome-to-autosome transposition events counter Y-chromosome gene loss in mammals. Genome Biology, 16. doi:ARTN 104 10.1186/s13059-015-0667-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs PA, Doll R, Goldstein H, Brunton M, & Courtbrown WM (1963). Change of Human Chromosome Count Distributions with Age: Evidence for a Sex Difference. Nature, 197(487), 1080-&. doi: 10.1038/1971080a0 [DOI] [PubMed] [Google Scholar]
- Jegalian K, & Page DC (1998). A proposed path by which genes common to mammalian X and Y chromosomes evolve to become X inactivated. Nature, 394(6695), 776–780. doi: 10.1038/29522 [DOI] [PubMed] [Google Scholar]
- Jobling MA, Lo IC, Turner DJ, Bowden GR, Lee AC, Xue Y, … Parkin EJ (2007). Structural variation on the short arm of the human Y chromosome: recurrent multigene deletions encompassing Amelogenin Y. Human Molecular Genetics, 16(3), 307–316. doi: 10.1093/hmg/ddl465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahn BT, & Page DC (1997). Functional coherence of the human Y chromosome. Science, 278(5338), 675–680. doi: 10.1126/science.278.5338.675 [DOI] [PubMed] [Google Scholar]
- Lahn BT, & Page DC (1999). Four evolutionary strata on the human X chromosome. Science, 286(5441), 964–967. doi: 10.1126/science.286.5441.964 [DOI] [PubMed] [Google Scholar]
- Lan F, Bayliss PE, Rinn JL, Whetstine JR, Wang JK, Chen S, … Shi Y (2007). A histone H3 lysine 27 demethylase regulates animal posterior development. Nature, 449(7163), 689–694. doi: 10.1038/nature06192 [DOI] [PubMed] [Google Scholar]
- Lee S, Lee JW, & Lee SK (2012). UTX, a histone H3-lysine 27 demethylase, acts as a critical switch to activate the cardiac developmental program. Dev Cell, 22(1), 25–37. doi: 10.1016/j.devcel.2011.11.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchini A, Ogata T, & Rappold GA (2016). A Track Record on SHOX: From Basic Research to Complex Models and Therapy. Endocrine Reviews, 37(4), 417–448. doi: 10.1210/er.2016-1036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moller MB (1981). Hearing in 70 and 75 year old people: results from a cross sectional and longitudinal population study. Am J Otolaryngol, 2(1), 22–29. [DOI] [PubMed] [Google Scholar]
- Naqvi S, Bellott DW, Lin KS, & Page DC (2018). Conserved microRNA targeting reveals preexisting gene dosage sensitivities that shaped amniote sex chromosome evolution. Genome Res, 28(4), 474–483. doi: 10.1101/gr.230433.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngo ST, Steyn FJ, & McCombe PA (2014). Gender differences in autoimmune disease. Frontiers in Neuroendocrinology, 35(3), 347–369. doi: 10.1016/j.yfrne.2014.04.004 [DOI] [PubMed] [Google Scholar]
- Ogata T, & Matsuo N (1993). Sex chromosome aberrations and stature: deduction of the principal factors involved in the determination of adult height. Human Genetics, 91(6), 551–562. [DOI] [PubMed] [Google Scholar]
- Ohno S (1967). Sex chromosomes and sex-linked genes Berlin, New York etc.: Springer-Verlag. [Google Scholar]
- Page DC, & Miller DE (2016). Turner syndrome as a model for understanding sex biases in human disease. In Silberbach M & Quigley CA (Eds.), Turner Syndrome Health and Wellness in the 21st Century: The crossroads of health care delivery and health research (pp. 28–32): Llumina Press, Plantation, Florida. [Google Scholar]
- Park SH, Shin YK, Suh YH, Park WS, Ban YL, Choi HS, … Jung KC (2005). Rapid divergency of rodent CD99 orthologs: Implications for the evolution of the pseudoautosomal region. Gene, 353(2), 177–188. doi: 10.1016/j.gene.2005.04.023 [DOI] [PubMed] [Google Scholar]
- Petropoulos S, Edsgard D, Reinius B, Deng Q, Panula SP, Codeluppi S, … Lanner F (2016). Single-Cell RNA-Seq Reveals Lineage and X Chromosome Dynamics in Human Preimplantation Embryos. Cell, 165(4), 1012–1026. doi: 10.1016/j.cell.2016.03.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinn JJ, Hitchcott PK, Umeda EA, Arnold AP, & Taylor JR (2007). Sex chromosome complement regulates habit formation. Nat Neurosci, 10(11), 1398–1400. doi: 10.1038/nn1994 [DOI] [PubMed] [Google Scholar]
- Rao E, Weiss B, Fukami M, Rump A, Niesler B, Mertz A, … Rappold GA (1997). Pseudoautosomal deletions encompassing a novel homeobox gene cause growth failure in idiopathic short stature and Turner syndrome. Nature Genetics, 16(1), 54–63. doi: 10.1038/ng0597-54 [DOI] [PubMed] [Google Scholar]
- Raudsepp T, & Chowdhary BP (2015). The Eutherian Pseudoautosomal Region. Cytogenet Genome Res, 147(2–3), 81–94. doi: 10.1159/000443157 [DOI] [PubMed] [Google Scholar]
- Ray PF, Conaghan J, Winston RM, & Handyside AH (1995). Increased number of cells and metabolic activity in male human preimplantation embryos following in vitro fertilization. J Reprod Fertil, 104(1), 165–171. [DOI] [PubMed] [Google Scholar]
- Ronen D, & Benvenisty N (2014). Sex-Dependent Gene Expression in Human Pluripotent Stem Cells. Cell Reports, 8(4), 923–932. doi: 10.1016/j.celrep.2014.07.013 [DOI] [PubMed] [Google Scholar]
- Shpargel KB, Sengoku T, Yokoyama S, & Magnuson T (2012). UTX and UTY demonstrate histone demethylase-independent function in mouse embryonic development. Plos Genetics, 8(9), e1002964. doi: 10.1371/journal.pgen.1002964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skaletsky H, Kuroda-Kawaguchi T, Minx PJ, Cordum HS, Hillier L, Brown LG, … Page DC (2003). The male-specific region of the human Y chromosome is a mosaic of discrete sequence classes. Nature, 423(6942), 825–U822. doi: 10.1038/nature01722 [DOI] [PubMed] [Google Scholar]
- Smith-Bouvier DL, Divekar AA, Sasidhar M, Du S, Tiwari-Woodruff SK, King JK, … Voskuhl RR (2008). A role for sex chromosome complement in the female bias in autoimmune disease. J Exp Med, 205(5), 1099–1108. doi: 10.1084/jem.20070850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soh YQS, Alfoldi J, Pyntikova T, Brown LG, Graves T, Minx PJ, … Page DC (2014). Sequencing the Mouse Y Chromosome Reveals Convergent Gene Acquisition and Amplification on Both Sex Chromosomes. Cell, 159(4), 800–813. doi: 10.1016/j.cell.2014.09.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syrett CM, Sierra I, Berry CL, Beiting D, & Anguera MC (2018). Sex-Specific Gene Expression Differences Are Evident in Human Embryonic Stem Cells and During In Vitro Differentiation of Human Placental Progenitor Cells. Stem Cells Dev, 27(19), 1360–1375. doi: 10.1089/scd.2018.0081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tukiainen T, Villani AC, Yen A, Rivas MA, Marshall JL, Satija R, … MacArthur DG (2017). Landscape of X chromosome inactivation across human tissues. Nature, 550(7675), 244–248. doi: 10.1038/nature24265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner HH (1938). A syndrome of infantilism, congenital webbed neck, and cubitus valgus. Endocrinology, 23(5), 566–574. doi: 10.1210/endo-23-5-566 [DOI] [PubMed] [Google Scholar]
- Urbach A, & Benvenisty N (2009). Studying Early Lethality of 45,XO (Turner’s Syndrome) Embryos Using Human Embryonic Stem Cells. Plos One, 4(1). doi:ARTN e4175 10.1371/journal.pone.0004175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Xue Y, Zhang Y, Long Q, Asan, Yang F, … Tyler-Smith C (2013). Genetic basis of Y-linked hearing impairment. American Journal of Human Genetics, 92(2), 301–306. doi: 10.1016/j.ajhg.2012.12.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe M, Zinn AR, Page DC, & Nishimoto T (1993). Functional Equivalence of Human X-Encoded and Y-Encoded Isoforms of Ribosomal Protein-S4 Consistent with a Role in Turner Syndrome. Nature Genetics, 4(3), 268–271. doi: 10.1038/ng0793-268 [DOI] [PubMed] [Google Scholar]
- Welshons WJ, & Russell LB (1959). The Y-Chromosome as the Bearer of Male Determining Factors in the Mouse. Proc Natl Acad Sci U S A, 45(4), 560–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werling DM, & Geschwind DH (2013). Sex differences in autism spectrum disorders. Current Opinion in Neurology, 26(2), 146–153. doi: 10.1097/WCO.0b013e32835ee548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wizemann TM, & Pardue ML (2001). Exploring the biological contributions to human health: Does sex matter? (pp. 267). Retrieved from http://purl.oclc.org/DLF/benchrepro0212 [PubMed]
- http://www.nap.edu/catalog.php?record_id=10028.
- Wright DJ, Day FR, Kerrison ND, Zink F, Cardona A, Sulem P, … Perry JRB (2017). Genetic variants associated with mosaic Y chromosome loss highlight cell cycle genes and overlap with cancer susceptibility. Nature Genetics, 49(5), 674–679. doi: 10.1038/ng.3821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang F, Babak T, Shendure J, & Disteche CM (2010). Global survey of escape from X inactivation by RNA-sequencing in mouse. Genome Res, 20(5), 614–622. doi: 10.1101/gr.103200.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinn AR, Page DC, & Fisher EM (1993). Turner syndrome: the case of the missing sex chromosome. Trends Genet, 9(3), 90–93. [DOI] [PubMed] [Google Scholar]