

Abstract
Untangling the functional basis of divergence between closely related species is a step toward understanding species dynamics within communities at both the evolutionary and ecological scales. We investigated cellular (i.e., growth, domoic acid production, and nutrient consumption) and molecular (transcriptomic analyses) responses to varying nutrient concentrations across several strains belonging to three species of the toxic diatom genus Pseudo-nitzschia. Three main results were obtained. First, strains from the same species displayed similar transcriptomic, but not necessarily cellular, responses to the experimental conditions. It showed the importance of considering intraspecific diversity to investigate functional divergence between species. Second, a major exception to the first finding was a strain recently isolated from the natural environment and displaying contrasting gene expression patterns related to cell motility and domoic acid production. This result illustrated the profound modifications that may occur when transferring a cell from the natural to the in vitro environment and asks for future studies to better understand the influence of culture duration and life cycle on expression patterns. Third, transcriptomic responses were more similar between the two species displaying similar ecology in situ, irrespective of the genetic distance. This was especially true for molecular responses related to TCA cycle, photosynthesis, and nitrogen metabolism. However, transcripts related to phosphate uptake were variable between species. It highlighted the importance of considering both overall genetic distance and ecological divergence to explain functional divergence between species.
Keywords: phenotype, transcriptomics, Pseudo-nitzschia, divergence, nutrients
Introduction
The high number of microeukaryote species co-occurring in the highly dynamic marine pelagic environment and competing for a small set of resources, that is, the “Paradox of the plankton,” has long puzzled scientists (Hutchinson 1961). Understanding how cells from the various species diverge in terms of cell functions may help characterize the evolutionary and ecological processes at play during the divergence and coexistence of these species. This also has practical implications when considering species responsible for harmful algal blooms. This is for instance the case of the cosmopolite and common diatom genus Pseudo-nitzschia, producer of domoic acid (DA), the toxin responsible for amnesic shellfish poisoning worldwide (Lelong et al. 2012; Trainer et al. 2012; Bates et al. 2018). Although numerous species within the genus are considered as DA producers (Teng et al. 2016; Zabaglo et al. 2016; Lema et al. 2017), in the localities where toxicity events occur, only a few species are responsible for toxicity events. It is for example the case of Pseudo-nitzschiamultiseries and Pseudo-nitzschiaaustralis, two highly toxic species which often co-occur with less toxic species (Lelong et al. 2012; Trainer et al. 2012; Bates et al. 2018). Numerous studies have analyzed cellular phenotypes of different Pseudo-nitzschia species in response to different environmental parameters to understand phenotypic divergence among these closely related species (Lelong et al. 2012; Trainer et al. 2012; Bates et al. 2018). By monitoring simultaneously the expression of thousands of transcripts, the analysis of mRNA levels is a powerful tool to investigate the functional divergence of closely related organisms.
Overall, distantly related diatoms tend to display divergent gene expression patterns when placed in similar environmental conditions. For instance, <5% of the orthologous genes between Fragilariopsis cylindrus, P. multiseries, and Thalassiosira pseudonana displayed similar transcriptional responses when placed under nitrogen limiting conditions (Bender et al. 2014). After manipulating the environment of natural communities dominated by Pseudo-nitzschia sp. and Thalassiosira sp., these two taxa were shown to express different sets of genes under high and low Fe (Cohen et al. 2017). A study performed comparative transcriptomic approaches among three closely related Pseudo-nitzschia species (P.arenysensis, P. delicatissima, and P. multistriata; Di Dato et al. 2015). The three species expressed a comparable set of functions, with small differences in specific functions such as the ones involved in the biosynthesis of isoprenoids and related to the presence of nitric oxide synthase encoding transcripts. A recent study focusing on P. multiseries identified a cluster of four genes involved in DA biosynthesis from glutamate and geranyl pyrophosphate (Brunson et al. 2018). Interestingly, these genes were only identified in the reference transcriptome of two highly toxic DA producing species, P. multiseries and P. australis.
We recently compared inter- and intra-specific growth and DA production in relation to nutrient ratios and concentrations for several strains of three Pseudo-nitzschia species: P. australis, P. pungens, and P. fraudulenta (Lema et al. 2017). Two of these species (P. australis and P. pungens) are phylogenetically closely related, whereas the third one (P. fraudulenta) belongs to a divergent clade (Moschandreou et al. 2012; Ajani et al. 2018). In contrast, P. australis and P. fraudulenta may be considered as ecologically more similar, as they are found in mixed blooms during the spring and autumn, whereas P. pungens is present more regularly throughout the year and tends to bloom in early summer (Klein et al. 2010; Thorel et al. 2017). In order to improve our understanding of the functional divergence, both within and between Pseudo-nitzschia species, the present study investigated phenotypic responses at both the cellular and molecular levels. We investigated growth, DA production, nutrient consumption, as well as mRNA levels through transcriptomic analyses, in three experimental conditions. Based on our previous study (Lema et al. 2017), we chose three media with contrasting nutrient conditions differently affecting growth and DA production: 1) a low phosphate medium (X1); 2) a low nitrate medium (X2) and; 3) a nutrient replete environment (X3). To be able to appreciate whether the observed phenotypic variations are linked to difference between species, and not to any other kind of segregating polymorphism (i.e., physiological, intraspecific genetic polymorphism), two to three strains (i.e., clones) were assayed per species (two P. pungens and P. fraudulenta strains and three P. australis strains). Using this experimental setting, we investigate 1) the magnitude of functional divergence at the intra- and inter-specific levels; 2) whether interspecific divergence is more related to genetic relatedness or to ecological similarity; and 3) whether intraspecific divergence may be explained by intraspecific genetic diversity.
Materials and Methods
Cellular Phenotype
Pseudo-nitzschia Cultures
Seven strains of three Pseudo-nitzschia species (three strains of P. australis, two for P. fraudulenta, and two for P. pungens) were collected and isolated from different locations of the north coast of France (supplementary table 1, Supplementary Material online). To establish monoclonal strains, single cells were isolated using a sterile micropipette and washed thoroughly with filter-sterilized seawater. Cultures were maintained in sterile-filtered oligotrophic seawater amended with nutrients (K/2 + Si media; 100.8 μM Na2SiO3⋅5H2O, pH ∼8, and salinity ∼33.5; modified from Keller et al. 1987) at 16 °C, under a 12:12 L:D cycle and 80 μmol photons m−2 s−1, in algal incubators. Pseudo-nitzschia species were identified using transmission electron microscopy and/or genotyping.
Experimental Setup
Strains were grown in three modified K/2 + Si media, in which concentrations of NaNO3, NaH2PO4⋅H2O, and Na2SiO3⋅5H2O varied (at 12:12 L:D cycle, temperature ∼16 °C, and pH ∼8). From the results of a previous study on the same strains (Lema et al. 2017), we chose three media with contrasting effects on growth and DA production: 1) a low phosphate medium (X1: N = 48, μM, Si = 48μM, and P = 3μM); 2) a low nitrate medium (X2: N = 48μM, Si = 48μM, and P = 10μM); and 3) a nutrient replete environment (X3: N = 480μM, Si = 480μM, and P = 30μM).
All strains and species were grown in quadruplicates in each of the three experimental media, in 500 ml flasks (T175, Sarstedt AG & Co, Nümbrecht, Germany) containing 200 ml of media and a starting cell concentration of ∼1,500 cells ml−1 (making a total of 84 cultures). Preliminary experiments showed that the strains reached stationary phase 8 days after inoculation in X1 and X2, and 13 days after inoculation in X3. For the experiment inoculations in medium X3 were initiated 5 days earlier than in media X1 and X2. Growth was then followed for all treatments by subsampling 1 ml of homogenized culture every 2 days (for a total of 8 days for X2 and X1 media, and 13 days for X3 medium; supplementary fig. 1, Supplementary Material online) into a 48-well plate and quantifying in vivo chlorophyll fluorescence (ex: 440/40, em: 680/20) in a FLX800 fluorescence microplate reader (Biotek Inc., VT).
Samples were collected on the same day in four successive batches (one batch per replicate). For each sample, 1) 5 ml of homogenized culture (cells and media) were collected and stored at −80 °C for DA analysis; 2) 50 ml of media (no cells) were collected and stored for nutrient analysis (i.e., nitrate, phosphate, and silicate); 3) 1 ml were fixed at a final concentration of 0.25% glutaraldehyde for flow-cytometry analysis; and 4) the remaining volume (∼130 ml) was used for immediate RNA extraction.
DA Quantification
Samples were thawed and centrifuged at ∼2,000 × g for 20 min to separate cells from media to analyze particulate DA and dissolved DA. DA was subsequently quantified as in Lema et al. (2017) using the DA ELISA kit (Mercury Science, Durham, NC).
Nutrient Analysis
Nutrient concentrations were determined after sample dilutions, using segmented flow analysis on a Seal Autoanalyseur AA3 and following classical methods detailed in Aminot et al. (2009).
Cell Quantification
Cell densities were quantified with a flow cytometer (FACSVerse, Becton Dickinson, San Jose, CA) equipped with three lasers (violet: 405 nm; blue: 488 nm; and red: 640 nm) and eight filters (527/32, 586/42, 700/54, and 783/56 nm for the blue laser; 448/45 and 528/45 nm for the violet laser; and 660/10 and 783/560 nm for the red laser).
Algal cells were discriminated by their natural red autofluorescence (linked to chlorophyll pigments) after excitation by the 488 nm laser detected on the red fluorescent detector of the flow cytometer (700/54 nm BP). Cell quantification was calculated based on the number of events, gated as algal cells, and the volume of sample recorded by the coupled Flow-Sensor device. Analyses were run for 180 s at medium flow rate (∼80 µl min).
Microscopy analyses of the samples revealed that the fixed cells were either isolated or forming two to three cell chains. As a result, the forward scatter (that can be considered as a proxy for size) of the flow cytometer displayed a bi- or three-modal distribution. Cell density estimates were corrected based on these multimodal distributions.
Statistical Analysis
ANOVA were used to investigate how each of cell density, DA level, silicate, phosphate, and nitrate concentrations, at the end of exponential phase, varied among strains and species in each of the three media. The models considered strains nested within species in interaction with media. Data were log transformed. Rather than putting emphases on P values (all of them being <0.0001), we focused on the effect sizes (η2) of the explanatory variables and of their interaction. Effect sizes were calculated as the sum of square of the focal explanatory variable (or interaction) divided by the total sum of square and therefore indicate the proportion of the variance explained by the focal explanatory variable (or interaction).
Molecular Phenotype
RNA Extraction, Library Preparation, and Sequencing
For total RNA extraction, cells were pelleted at 8,500 × g during 8 min at 4 °C. After addition of RLT buffer (Qiagen) supplemented with β-mercaptoethanol, samples were sonicated on ice, RNA was then purified using RNeasy Plus Minikit (Qiagen) following the manufacturer protocol. Extracted RNA was quantified using a Biotek Epoch spectrophotometer and the quality estimated on RNA 6000 nanochips using a Bioanalyzer (Agilent). RNA extraction failed for 15 samples (all, i.e., eight, P. fraudulenta samples in medium X1; four P. fraudulenta samples in medium X2; two P. pungens samples in medium X1; and one P. australis sample in medium X1). Starting from 0.5µg of total RNA, poly-A selection, reverse transcription and library preparation was performed using the Illumina Stranded Truseq Total RNA Library Preparation Kit. One library was generated per sample, representing a total of 69 libraries. Library quality was assessed on a Bioanalyzer (Agilent) using high sensitivity DNA analysis chips and quantified using Kappa Library Quantification Kit. Paired-end sequencing was performed in 2× 150 bp on three lanes of a Hiseq3000 (Illumina). To avoid batch effects, samples were randomized for RNA extraction, library preparation, and sequencing.
Reads Quality Assessment and Filtering
Adapter removal and quality filtering were initially performed on raw reads using Trimmomatic (v.0.32) (Bolger et al. 2014) with the following parameters: ILLUMINACLIP:2:30:10:8: TRUE, LEADING:3, TRAILING:3, MAXINFO:135:0.8, MINLEN:80.
Obtaining Reference Transcriptomes and Alignment
After initial quality filtering, for each species, the reads from the different samples were pooled, and de novo assembly of one reference transcriptome per species was obtained using Trinity (Haas et al. 2013). Only transcripts longer than 200 bp were retained. When several isoforms were identified, only the longest one was retained. Sequence homology of the transcripts with genes of identified function in the manually curated UniProt databank was investigated using BlastX with E-value <10−3. The transcripts were classified in various Gene Ontology (GO) categories (http://geneontology.org/, last accessed March 11, 2016) based on this result. Transcripts resulting from carry-over rRNA contaminants were identified and removed after performing BlastN against Silva database (Quast et al. 2013), corresponding to 37, 32, and 30 transcripts for P. australis, P. pungens, and P. fraudulenta, respectively. In addition, to specifically focus on the recently identified genes involved in DA biosynthesis (dabA, dabB, dabC, and dabD; Brunson et al. 2018), transcripts displaying nucleotide (blastn) homology (E-value <10−3) with these genes were searched for in each species-specific reference transcriptome.
The 69 samples were individually aligned on their corresponding species-specific reference transcriptome with Bowtie2 (Langmead and Salzberg 2012) using paired-end reads. Only reads with a mapping score >10 were retained. Pairs for which the two reads did not map concordantly on the same transcript were removed. Samtools was used to sort, index, and obtain raw read count tables from bam files (Li et al. 2009).
Intraspecific Differential Gene Expression
For all differential gene expression (DE) analyses, only transcripts with an average read count per sample higher than 10 were considered (supplementary table 2, Supplementary Material online).
Overall gene expression divergence between strains in each medium was calculated as the Euclidean distance among the average expression of each strain in each medium calculated as , where is the rlog transformation (as implemented in DESeq2; Love et al. 2014) of the number of reads mapping to a given transcript for the strain i in the medium j.
Within each species, pairwise DE was analyzed: 1) between media for each strain as well as 2) between strains in each media. DE was tested using: 1) negative binomial models on raw read counts (Wald tests as implemented in DESeq2; Love et al. 2014) with a false discovery rate (FDR) threshold set q-value =0.01 and 2. Linear models on voom normalized read counts (as implemented in limma; Ritchie et al. 2015) with a FDR threshold set q-value = 0.05. FDR thresholds were set based on preliminary analyses of the data sets. Only transcripts identified as significant using both approaches and displaying a log 2-fold change >2 were considered as differentially expressed. Considering the transcripts identified as significant in at least one pairwise comparison, the expression profiles across samples were clustered following negative binomial models as implemented in MBCluster.Seq, using expectation-maximization algorithm for estimating the model parameters and cluster membership (Si et al. 2014). After preliminary analyses, 20, 10, and 5 clusters were investigated for P. australis, P. pungens, and P. fraudulenta, respectively. The expression profiles of the selected clusters were subsequently visually inspected and clusters displaying similar profiles were merged (see Results). Expression is reported as the log 2-fold change of the median expression in the category of interest over the median expression considering all samples. It is calculated as where is the rlog transformation (as implemented in DESeq2) of the number of reads mapping to a given transcript for the strain i in the medium j and is the rlog transformation of the number of reads mapping to a given transcript for all strains in all media.
Overrepresentation of GO categories in the identified clusters were tested, for (GO) functional gene categories represented by at least five transcripts, using Fisher Exact tests followed by a FDR correction for multiple testing with a significance threshold set at q-value =0.05. Only GO categories containing more than five DE transcripts in a given cluster were considered. GO categories may be redundant (as similar set of genes belong to different GO categories). To mitigate such redundancy, GO categories displaying an overlap coefficient (where is the size of the GO category ) were clustered, and for each cluster only the GO category displaying the lowest q-value was reported.
Interspecific DE
Orthology of the transcripts considered for DE analyses was investigated using reciprocal BlastN with E-value <10−3. Only transcripts with an ortholog in each of the three species were considered (see supplementary table 2, Supplementary Material online). Overall gene expression divergence between strains of the different species in the three media was calculated as the Euclidean distance among the average expression of each strain in each medium (see above). DE was investigated as above: that is, investigating within each species, pairwise DE between media for each strain, as well as between strains in each medium. Transcripts displaying similar expression profiles across tested samples were clustered (10, 10, and 5 clusters were considered for P. australis, P. pungens, and P. fraudulenta, respectively). Interspecific overlap between the clusters was tested using Fisher Exact tests followed by FDR with a significance threshold set at q-value =0.01 and absolute log 2(odd ratio) >0.5. Overrepresentation of GO categories in the identified clusters was tested as above.
Intra- and Inter-Specific Genetic Divergence
For each strain, the fastq files obtained after sequencing were concatenated. The reads were quality filtered using Trimmomatic (as above). Reads were then aligned to their corresponding reference transcriptome (as above) using Bowtie2 (Langmead and Salzberg 2012). Single nucleotide polymorphisms (SNPs) were detected using FREEBAYES (Garrison and Marth 2012) and filtered using VCFTOOLS (Danecek et al. 2011). Positions were considered when displaying two alleles, a quality criterion higher than 40, and when found >20 times in each strain. Pairwise nucleotide divergence was calculated as the number of SNPs divided by two times (diploid organisms) the number of positions covered >20 times in each strain.
Interspecific genetic divergence was calculated as follows. Orthologous transcripts from each of the three species were aligned using MUSCLE with default parameters (Edgar 2004). Following this alignment, ambiguous regions were removed and conserved blocks of >100 bases were concatenated using Gblocks (Castresana 2000). Finally, Mega (Kumar et al. 2018) was used to compute the uncorrected-P genetic distances among the concatenated aligned sequences of the three species.
Results
Cellular Phenotype
Growth and DA
A total of seven strains from three Pseudo-nitzschia species (three P. australis, two P. pungens, and two P. fraudulenta) were grown in three media with variable nitrate, phosphate, and silicate initial concentrations. At the onset of the stationary phase, cell densities were measured using flow cytometry, DA was quantified, and nitrate, phosphate, and silicate concentrations were measured (fig. 1). Considering cell densities and DA, there were major and significant differences between species (fig. 1f: Species effect). More precisely, P. australis strains produced more DA (i.e., total DA: particulate and dissolved), whereas P. fraudulenta strains reached higher densities than the two other species (fig. 1a and b). Not surprisingly, for the three species, cell densities were higher in the medium with high initial nutrient concentrations (fig. 1a and f: media effect). As previously reported, the various strains produced more DA in the low phosphate medium X1 (Lema et al. 2017; fig. 1b and f: media effect). Intraspecific differences were smaller but nevertheless significant (fig. 1f, Strain effect). More complex interactions between strains and media or species and media could also be noted. One illustrative example being the strain Pa2 displaying much more variable cell density and DA production patterns across media than the other strains (fig. 1a and b).
Fig. 1.

—Cellular level phenotypes (a and b) and nutrient concentrations (c–e) for each of the seven strains (x axis) in the three media (color coded, see legend) at the time of RNA extraction. Each dot corresponds to a replicate. Cell densities (a) in cell ml−1, total DA (b) in ng cell−1, and nutrient concentration (c–e) are given in µmol l−1. (f) Effect size (percentage of the variance explained in the ANOVA) of the media, strain, species and their interactions on the cellular level phenotypes (cell density, and total DA) and nutrient concentration (silicate, phosphate, and nitrate) at the time of RNA extraction. The cumulated effect sizes do not reach 1, as a few percent of the variances (the residuals) are not explained by the explanatory variables of the models.
Nutrient Consumption
Not surprisingly, differences in nutrient concentrations at the end of stationary phase were strongly driven by initial nutrient concentration (fig. 1f). In medium X1, phosphate was almost completely depleted at stationary phase (average P in X1 = 0.06 µmol l−1 SE ± 0.01; fig. 1d) and therefore was potentially the nutrient limiting cell growth across all tested strains. In medium X2, the limiting nutrient seemed to be nitrate except for P.pungens (see below for interspecific difference; fig. 1e). In medium X3, all three nutrients were still present at the onset of stationary phase (fig. 1c–e). The second most important explanatory factor was the species × media interaction, indicating that species consumed nutrients in different ways in the three media (fig. 1f). For instance P. pungens consumed less phosphate in X2 and more silicate in X3 than the two other species. The most important difference, and probably with strong implications in terms of gene expression (see below), was related to nitrate consumption in medium X2. Although nitrate was entirely consumed by P. australis and P. fraudulenta, there was still a fair amount of nitrate left in P. pungens cultures (8–17 µmol l−1). Finally, to a lesser extent, there were also some strain × media interaction effects. This was apparent in the Si consumption in medium X2 that was quite different across different strains (e.g., Pa3 being the only strain that consumed all the silicate available in medium X2). Another example was the difference of phosphate consumption between Pf1 and Pf2 in X2.
Intra- and Inter-Specific Genetic Distance among Strains
Intraspecific pairwise genetic divergence based on SNPs was calculated within each species (supplementary table 3, Supplementary Material online). The total length of sequences considered for this analysis was similar for the three species (∼25 × 106 bases). Within P. pungens, the two strains displayed a divergence of 0.2%, corresponding to the presence of one SNP every ∼480 bases. Within P. fraudulenta, the two strains displayed a divergence of 0.05%, corresponding to the presence of one SNP every ∼2,100 bases. Within P. australis, the three strains displayed a pairwise divergence of ∼0.03%, corresponding to the presence of one SNP every ∼3,300 bases.
To be able to perform interspecific comparisons, only transcripts displaying orthologs in each of the three species were retained, reducing the number of transcripts to 6,557 (supplementary table 2, Supplementary Material online). Ortholog transcripts represented only ∼20%, ∼41%, and ∼32% of P.australis, P. fraudulenta, and P.pungens overall transcripts, respectively. Interspecific genetic divergence was also calculated based on unambiguous alignment of orthologous transcripts, corresponding to an alignment of 2,111,114 bases spanning 1,739 transcripts. Using this alignment, the genetic divergence between the pair of species was 23.9%, 24.9%, and 19.3% for P. australis and P. fraudulenta, P. fraudulenta and P. pungens, and P. australis and P. pungens, respectively.
Comparing Genetic Distance and Overall Expression Divergence among Strains
Gene expression distance among strains was computed for each strain in each medium. When considering all species-specific transcripts from reference transcriptomes, gene expression divergence among P. australis strains was extremely variable compared with the other two species, even though P. australis strains were genetically more similar (fig. 2a). This pattern was due to a single strain (Pa3) displaying extremely divergent gene expression in all three media, and particularly in X1. Though when restricting the analysis to the orthologous transcripts, this same strain only displayed slightly divergent gene expression (still especially in X1; fig. 2b), indicating that the divergent expression patterns were mainly due to species-specific transcripts (i.e., without orthologous genes in the other two species). Moreover, we observed that within the three species, the expression patterns of the strains were more similar in medium X3, than in X2, and more divergent in X1 (fig. 2b). Gene expression divergence among strains was ∼5 times higher when comparing strains from different species than strains from the same species (fig. 2b and c, y axis). In both intra- and inter-specific comparisons, gene expression divergence among strains did not necessarily increase with genetic distance (fig. 2). Indeed, in medium X2, gene expression was more similar among P. australis and P. fraudulenta strains (genetic distance =0.239) than between P. australis and P. pungens (genetic distance =0.193) or P. fraudulenta and P. pungens (genetic distance =0.249; fig. 2c).
Fig. 2.
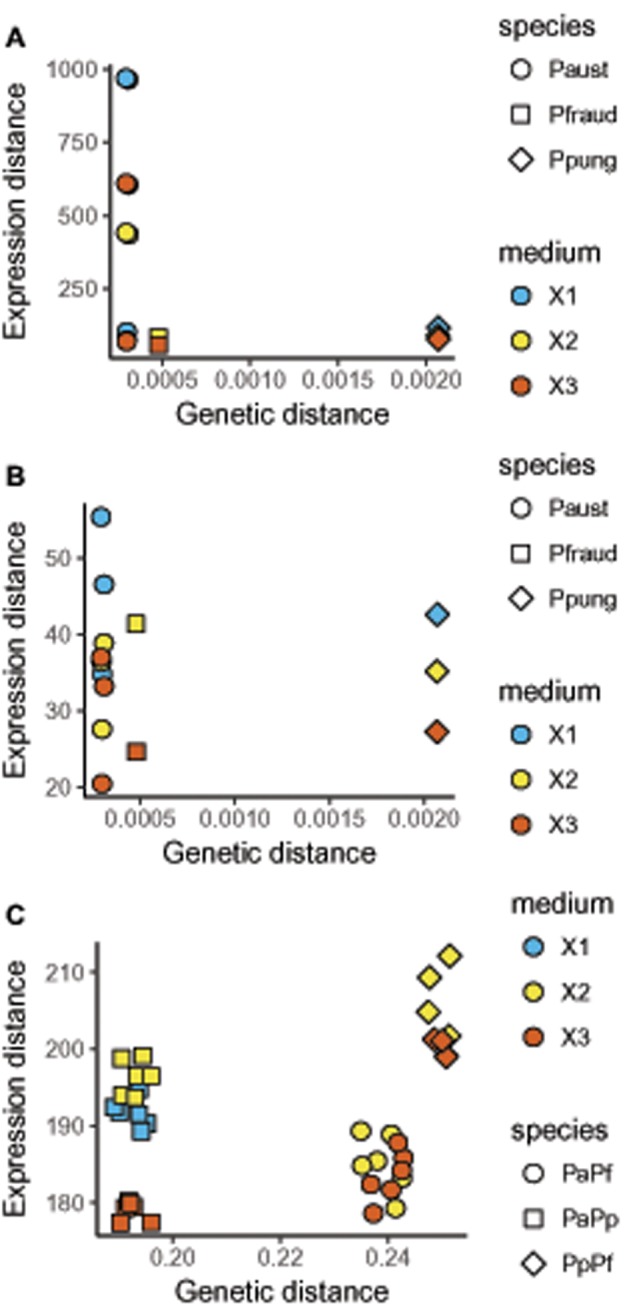
—Gene expression divergence as a function of genetic distance between strains at the intraspecific (a, b) and interspecific (c) levels. Overall gene expression divergence (y axis) between strains of the different species in the three media was calculated (see Materials and Methods), using all transcripts (a), or only orthologous transcripts (b and c). Overall genetic distance (x axis) between strains was calculated (see Materials and Methods) using SNP differences among strains (a and b) or conserved orthologous regions across species (c). Each point corresponds to a pair of strains in a given medium. The symbols indicate the species (intraspecific comparison: a and b) or pair of species (interspecific comparison: c), and the colors indicate the medium.
Intraspecific Gene Expression
Intraspecific DE was investigated in pairwise comparisons between media for each strain and between strains in each medium. As described in the methods section, clusters were retained to describe the expression profiles of these DE transcripts across the samples within each species. After observing expression patterns across clusters, two general trends could be recognized for the three studied species: 1) clusters reflecting a strain specific DE and 2) clusters reflecting a medium specific DE. The strain specific clusters contained transcripts barely expressed, if at all, in some strains but displaying variable expression levels in the other strains; whereas the medium specific clusters reflected variable DE in different media. Below, we develop these results for each species.
Pseudo-nitzschia australis
Within P. australis, DE was investigated in 18 pairwise comparisons (see Materials and Methods), resulting in the identification of 20,663 DE transcripts. Ten clusters were retained to describe the expression profiles of these DE transcripts across the samples (fig. 3, supplementary fig. 2, Supplementary Material online), five clusters reflected a strain specific DE (clusters Pa_C1, Pa_C7-10) and five clusters reflected a medium specific DE (Pa_C2-6).
Fig. 3.

—Pseudo-nitzschia australis expression profiles and overrepresented GO categories. Expression profiles, across strains and media, of the six main clusters of transcripts summarizing the changes in expression observed within P. australis. Expression is indicated using violin plots (rotated kernel density plots) based on the log 2-fold change of the median expression in the category of interest over the median expression considering all samples (see Materials and Methods). Black dots indicate median log 2-fold change. Blue, yellow, and red report to X1, X2, and X3 media, respectively. The number of transcripts belonging to each cluster is indicated. For each cluster, the ten GO categories displaying the highest overrepresentation of DE transcripts (based on q-values, see Materials and Methods) are indicated as rotated barplots. The lengths of the bars indicate the proportion of transcripts of each category in the clusters. The numbers of overrepresented GO categories is indicated above the barplots.
The main strain specific cluster (Pa_C1, fig. 3), in terms of both, fold change and number of transcripts, regrouped transcripts expressed in strain Pa3 and barely expressed, if at all, in the two other strains. More than 13,000 transcripts were found in this cluster (representing ∼40% of the transcripts considered for DE analysis in P. australis) and illustrating extensive divergence in terms of gene expression between Pa3 and the other two strains. More than 120 GO categories were overrepresented in this cluster. Of special interest were numerous functions related to cell motility (Pa_C1, fig. 3). The other four strain specific clusters were more anecdotal, as they only gathered a few tens of transcripts without any overrepresentation of GO category (Pa_C7-10, supplementary fig. 2, Supplementary Material online).
The five medium specific clusters were of similar size (from 951 to 2,512 transcripts). They regrouped transcripts that were more expressed in the medium X1 (Pa_C6), X2 (Pa_C2), and X3 (Pa_C5), as well as transcripts less expressed in X1 (Pa_C3) and X3 (Pa_C4). When looking at the GO categories overrepresented in the clusters, cluster Pa_C2 (fig. 3; supplementary table 5, Supplementary Material online) displayed an overrepresentation of transcripts involved not only in nitrate assimilation and nitrogen metabolism but also in the central metabolism (amino acid biosynthesis, glycolysis, and TCA cycle) (fig. 3). The cluster Pa_C3 displayed a slight overrepresentation of transcripts involved in photosynthesis and iron binding (fig. 3). The cluster Pa_C4 displayed an overrepresentation of transcripts that may be involved in cell communication (fig. 3). The cluster Pa_C5 displayed a strong overrepresentation of transcripts involved in photosynthesis, and to a lesser extent in glycolysis and amino acid synthesis (fig. 3). Finally, the cluster Pa_C6 displayed an overrepresentation of transcripts involved in photosynthesis, pentose phosphate shunt, and amino acid and fatty acid biosynthesis (fig. 3).
Pseudo-nitzschia fraudulenta
We were unable to extract enough RNA from P. fraudulenta strains grown in the medium X1 to perform the RNA sequencing therefore DE in P. fraudulenta was only investigated across two media (X2 and X3) and two strains. DE was investigated in two pairwise comparisons (between X2 and X3 for strain Pf2, and between strains Pf1 and Pf2 in medium X3, as only one replicate was sequenced for Pf1 in medium X2; no pairwise comparison could be done with Pf1 in X2), resulting in the identification of 2,057 DE transcripts. Only two clusters, displaying medium specific DE, were retained to describe the expression profiles of these DE transcripts across samples (fig. 4). The cluster Pf_C1 regrouped transcripts with higher expression in X2 than X3 and included an overrepresentation of transcripts related to the ribosomes and the transcription machinery in general (fig. 4; supplementary table 5, Supplementary Material online). Of special interest was also the overrepresentation of transcripts related to the nitrogen metabolism (fig. 4). The cluster Pf_C2 regrouped transcripts with higher expression in X3 than X2 and included an overrepresentation of transcripts related to photosynthesis and lipid metabolism (fig. 4).
Fig. 4.

—Pseudo-nitzschia fraudulenta expression profiles and overrepresented GO categories. Expression profiles, across strains and media, of the two clusters of transcripts summarizing the changes in expression observed within P. fraudulenta. Expression is indicated using violin plots (rotated kernel density plots) based on the log 2-fold change of the median expression in the category of interest over the median expression considering all samples (see Materials and Methods). Black dots indicate median log 2-fold change. Yellow and red report to X2, and X3 media, respectively. The number of transcripts belonging to each cluster is indicated. Note that for Pf1_X2 a single replicate was available, therefore the log 2-fold change of this replicate and not the median log 2-fold change is indicated. For each cluster, the ten GO categories displaying the highest overrepresentation of DE transcripts (based on q-values, see methods) are indicated as rotated barplots. The lengths of the bars indicate the proportion of transcripts of each category in the clusters. The numbers of overrepresented GO categories is indicated above the barplots.
Pseudo-nitzschia pungens
DE was investigated in nine pairwise comparisons (between media for each strain and between strains in each medium as for the other two species), resulting in the identification of 3,390 DE transcripts. Seven clusters were retained to describe the expression profiles of these DE transcripts across the samples (fig. 5, supplementary fig. 2, Supplementary Material online). Two of these clusters displayed strain specific DE patterns (Pp_C6-7) with a couple hundred transcripts in each cluster that were not related to any functional GO category (supplementary fig. 2, Supplementary Material online). The five other clusters were medium specific (Pp_C1-5). The medium specific cluster Pp_C1 harbored transcripts with a higher expression in medium X2 and displayed an overrepresentation of transcripts involved in photosynthesis (fig. 5; supplementary table 5, Supplementary Material online). The cluster Pp_C2 regrouped transcripts with higher expression in X1 than in the other two media and displayed an overrepresentation of transcripts involved in central metabolism processes (TCA cycle, amino acid biosynthesis, and oxido-reduction; fig. 5). The cluster Pp_C3 was characterized by transcripts displaying low expression in X3 compared with X1 and X2 (fig. 5). Among the overrepresented GO categories, we note the presence of kinase activity, phosphorylation, and lipid catabolism-related functions (fig. 5). The cluster Pp_C4 was characterized by transcripts displaying low expression in X2 compared with X1 and X3 (fig. 5), with an overrepresentation of transcripts involved in protein synthesis in general (fig. 5). The cluster Pp_C5 was characterized by transcripts displaying low expression in X1 compared with X2 and X3 with an overrepresentation of transcripts involved in photosynthesis (fig. 5).
Fig. 5.

—Pseudo-nitzschia pungens expression profiles and overrepresented GO categories. Expression profiles, across strains and media, of the five main clusters of transcripts summarizing the changes in expression observed within P. pungens. Expression is indicated using violin plots (rotated kernel density plots) based on the log 2-fold change of the median expression in the category of interest over the median expression considering all samples (see Materials and Methods). Black dots indicate median log 2-fold change. Blue, yellow, and red report to X1, X2, and X3 media, respectively. The number of transcripts belonging to each cluster is indicated. For each cluster, the ten GO categories displaying the highest overrepresentation of DE transcripts (based on q-values, see methods) are indicated as rotated barplots. The lengths of the bars indicate the proportion of transcripts of each category in the clusters. The numbers of overrepresented GO categories is indicated above the barplots.
DA Biosynthesis (dab) Gene Cluster
The four genes (dabABCD) recently identified as involved in the biosynthesis of DA in P. multiseries (Brunson et al. 2018) were identified in P. australis transcriptome (supplementary table 4, Supplementary Material online) and they displayed dynamic expression patterns across strains and media (fig. 6). The four genes displayed extremely different expression levels, with dabB and dabD displaying low (a few tens of reads per sample, maximum) and high expression levels (several thousands of reads per sample, maximum), respectively. Overall, the four genes displayed a similar expression pattern across media and were more expressed in medium X1, where total DA concentration was higher (fig. 6). However, the strain Pa3 displayed a distinct expression pattern with high expression in medium X3 where DA concentration was moderate and low expression in medium X2 where DA concentration was high (fig. 6). In P. pungens reference transcriptome, transcripts homologous to dabA and dabD were identified (supplementary table 4, Supplementary Material online), but only displayed extremely low expression levels (a handful of reads per sample, maximum). In P. fraudulenta reference transcriptome, a transcript homologous to dabD was found (supplementary table 4, Supplementary Material online) but displayed extremely low expression levels.
Fig. 6.

—Total DA concentration and DA biosynthesis gene expression in Pseudo-nitzschia australis. Total DA concentration at the time of RNA extraction (ng cell−1) is indicated as a function of the expression of the dabABCD gene clusters in P. australis strains. Shape and colors indicate strains and media, respectively. The average expression (y axes) and DA concentration (x axes) ±standard error is given for each strain in each medium.
Interspecific Comparisons
Pairwise DE analyses were performed as described above for the intraspecific comparisons (i.e., investigating, within each species, DE between media for each strain and between strains in each medium), but focusing on the 6,557 orthologs. Transcripts were clustered within each species to identify intraspecific expression profiles (fig. 7a). Overlaps between clusters of the different species were analyzed.
Fig. 7.

—Interspecific comparison of gene expression profiles. (a) Figures on the outer circle represent the expression profiles, across strains and media, of the clusters of transcripts summarizing the changes in expression observed for Pseudo-nitzschia fraudulenta (fc1-3), Pseudo-nitzschia pungens (pc1-5), and Pseudo-nitzschia australis (ac1-5), see legends of figure 4 for more details. Widths of the ribbons in the inner circle correspond to the number of transcripts shared between clusters. Ribbon colors indicate the odd ratio of the overlap, with purple and orange indicating excess and lack of shared transcripts compared with randomly expected, respectively (see color scale). The number of transcripts in each cluster is indicated. (b) Colored dots indicate the overrepresented GO categories in each of the clusters, with colored lines indicating overrepresented GO categories shared between clusters, the name of the shared categories are indicated.
A total of 2,325 transcripts were identified as DE in at least one pairwise comparison (supplementary table 2, Supplementary Material online). At the intraspecific level, contrary to the results obtained analyzing each species separately, no strain specific clusters could be identified. Even the extremely large cluster of transcripts highly expressed in the strain Pa1 (Pa_C1, fig. 3) was not identified, illustrating that the transcripts expressed in a strain specific manner had no homologs in all three species. In P. fraudulenta, transcripts of cluster fc3 (fig. 7a) displayed a more or less constant expression pattern across strains and media. Transcript clusters across the other two species were medium specific and often similar, in terms of DE profiles, to the ones identified when investigating intraspecific gene expression profiles. A notable exception were two clusters displaying low expression in medium X1 in P. australis (Pa_C3, fig. 3) and P. pungens (Pp_C5, fig. 5) when considering each species separately, but not identified when focusing the orthologs.
In M edia X2 and X3, P. australis and P. fraudulenta Display Similar Expression Patterns
Four hundred and twenty of the transcripts considered as DE (fig. 7a) displayed high expression levels in medium X2 in both P. fraudulenta (fig. 7a, fc2; 48% of the transcripts in fc2) and P. australis (fig. 7a, ac2; 62% of the transcripts in ac2), whereas 195 of these transcripts displayed low expression in P. pungens for that same medium (fig. 7a, pc4; 50% of the transcripts in pc4 shared with both fc2 and ac2). Transcripts shared between these three clusters were associated with several broad functional GO categories linked to the transcription and translation machineries, but also to two specific GO categories: nitrogen metabolism and TCA cycle (fig. 7b).
Transcripts associated with nitrogen uptake and metabolism (fig. 8, nar, nir, at, gas, gad) were often more expressed in the low nitrogen medium X2 within P. australis and P. fraudulenta, whereas the same transcripts displayed a constant lower expression across media within P. pungens. For example, P. pungens displayed a very low expression of nitrate reductase (a key enzyme of nitrogen metabolism) across the three media, whereas its expression was higher and highly dynamic across the three media in the two other species (fig. 8, nar). Urease, a key enzyme linking the urea cycle to the general nitrogen metabolism, tended to be much more expressed in all media in P. fraudulenta than in the other two species (fig. 8, ure).
Fig. 8.

—Central metabolism expression of the three Pseudo-nitzschia species in the three media. Median fold change (log 2) of the central metabolism transcripts for each of the three Pseudo-nitzschia species in the three media. Each heatmap corresponds to one transcript, transcript abbreviated names are in red, key intermediates are written in black.
A similar pattern was observed for transcripts associated in the TCA cycle (fig. 8, pdh, cs, aco, ldh, od, scs, ss, fum) and gluconeogenesis (fig. 8, gpd, pgk, pgm) which were more expressed in X2 within P. australis and P. fraudulenta, whereas the same transcripts displayed a constant lower expression across media for P. pungens. Other species-specific differences were observed. For example, malate dehydrogenase, the enzyme oxidizing malate into oxaloacetate during the TCA cycle, was barely expressed in P. australis, whereas it displayed a high expression levels in the other two species.
Transcripts encoding pigments (Fucoxanthin and Chlorophyll) and cytochromes, as well as transcripts associated with the photosystems I and II (figs. 7a and 8) displayed high expression levels in X3 within P. fraudulenta (fc1) and P. australis (fig. 7a, ac1, 403 shared transcripts, representing 52% of ac1 and 46% of fc1), and in contrast tended to be highly expressed in X2 in P. pungens (fig. 7a, ac1, 337 shared transcripts, representing 52% of pc1 and 43% of ac1).
In M edium X1, P. australis and P. pungens Globally Display Similar Expression Patterns, but Marked Differences Related to Phosphate Transporters
In the medium X1, where initial phosphate concentration was low, some similarities were noted between P. australis (fig. 7a, ac5) and P. pungens (pc5, 67 shared transcripts, representing 43% of ac5 and 20% of pc5) expression profiles though no clear functional roles were revealed. In the medium X1, transcripts linked to phosphate transport were a priori of specific interest. Phosphate transporters transcripts (identified in the three species) displayed expression profiles that seemed to be mainly driven by species-specific differences and to a lesser extent by the experimental environments. A transcript annotated as a sodium-dependent phosphate transport protein was highly expressed by P. australis, particularly in X1, whereas this same transcript displayed extremely low expression levels across all media for the two other species (fig. 8, sdpt). Pointing toward divergent management of phosphate intracellular pools, on the contrary, transcripts annotated as mitochondrial phosphate carriers were highly expressed by P. fraudulenta and P. pungens in all three media, but barely expressed by P. australis (fig. 8, mpc). Also, PEP/− translocator and 5′ bisphosphate nucleotidase displayed extremely different patterns of expression between species (fig. 8, bn, ppt).
Discussion
Intra- and inter-specific phenotypic divergence within diatoms of the genus Pseudo-nitzschia was investigated in three experimental conditions with contrasting initial concentrations of nitrate, phosphate and silicate. Phenotypic diversity was assessed at the cellular (DA, cell density), molecular (transcriptomic DE analysis) levels, as well as in terms of nutrient consumption for a total of seven strains from three Pseudo-nitzschia species (P.australis, P. fraudulenta, and P. pungens).
Intraspecific Variability
A major strength of our study was to investigate divergence at both the inter- and intra-specific levels. Strong intraspecific differences were observed at the cellular level but not at the molecular level, results that contrasted with the ones obtained when comparing the three species. At the cellular levels (DA, cell densities, as well as in terms of nutrient consumption), there were often contrasting responses between strains in different media. For instance, strain Pa3 produced as much DA in the low nitrate and low phosphate media, whereas strain Pa2 produced 500 times less DA in the low nitrate medium. Phosphate concentration remaining at the time of RNA extraction in the low nitrate medium was extremely different between the two P. fraudulenta strains (but not in the other two media). These results are in agreement with previous studies that highlighted major divergence in growth, DA production and nutrient usage by various strains of the same Pseudo-nitzschia species (Thessen et al. 2009; Lema et al. 2017). However, these intraspecific cellular differences did not translate into DE patterns, as the dynamics of expression across media were similar among strains belonging to the same species. This could reflect that the observed phenotypic differences among strains involve differential expression of a few specific genes rather than global changes in expression patterns. These phenotypic differences could also derive from dynamic gene expression during the course of the experiment but not captured by our end point analysis.
In contrast, at the cellular levels (DA, cell densities) as well as in terms of nutrient consumption strain specific effects preserved across media were moderate (i.e., no strain reached higher cell densities, produced more DA, or consumed more nutrients in all three media). However, at the molecular level, strain specific differential expression patterns (transcript DE in one of the strain irrespective of the experimental environment) were detected, even if they were usually extremely limited. The only major exception was one strain within P. australis (Pa3) that displayed an extremely divergent response (more than one third of the transcripts considered were almost exclusively expressed in this strain). We note that Pa3 was not necessarily divergent from the other strains when considering the cellular level phenotypes, the only exception being the exhaustion of silicate in the X2 environment. Moreover, aside from these numerous strain specific transcripts, the gene expression dynamics of Pa3 was rather similar to the expression patterns observed in the other two strains. This strongly suggests that the strain specific expression is related to a part of the cell metabolism that neither impacts the cellular phenotypes investigated, nor the nutrient consumption. The DE of this particular P. australis strain strongly affected specific cellular functions relating to cellular motility and chain formation. Although pennate diatoms do not carry cilia, they may glide across surfaces using a poorly characterized mechanism (Wang et al. 2013). Here, we identified numerous transcripts associated with motility and only expressed in the recently isolated P. australis strains. Motility-related genes have already been documented in the transcriptomes of numerous centric and pennate diatoms (Nanjappa et al. 2017). We may cite numerous transcripts related to dynein motor proteins (Roberts et al. 2013). The term “dynein” appeared in 70 out of the 224 transcripts only expressed in the recently isolated strain and associated with the GO category “cilium,” including 36 transcripts homologs to the dynein heavy chain of the axonemes. We also noted the presence of numerous homologs of the amoeba Dictyostelium discoideum and more precisely myosin homologs (18 out of 50 transcripts in the GO category “cell leading edge”: movement of plasma membrane at the cell front; Ridley 2011) and of a highly expressed homolog of Fimbrin (several thousand reads in the recently isolated Pa strain, 0 in the other strains). Microscopic observation of the strains showed that Pa3 displayed motile chains of cells whilst the other strains did not (data not shown). Environmental samples displaying long motile chains of Pseudo-nitzschia are often considered as indicative of healthy populations (Rines et al. 2002). We suggest that this strain specific DE pattern has a physiological rather than a genetic basis. Indeed, based on our transcriptome wide SNP analysis, Pa3 was not genetically divergent from the two other P. australis strains. Moreover, the genetic divergence between the two P. pungens strains was ten times higher than between the P. australis strains, without any strong strain specific differential expression pattern.
Interestingly, the strain Pa3 was isolated from the field <3 months before the experiments took place whereas all the other strains were cultivated for more than a year. We note that in our previous experiment, carried a few months before the present one, the same strain (Pa3) exhibited striking cellular level phenotypic differences (DA, cell density and size) that this time were not evidenced (Lema et al. 2017). Together, such observations strongly contribute to the idea of progressive physiological modifications in culture. We may speculate that motility could be progressively shut down because it is energetically costly and confer no advantage in culture.
Interspecific Divergence
Species-specific differences were observed at the cellular and molecular levels. For instance and as already reported in previous studies, P. australis produced more DA than the two other species (Trainer et al. 2012; Lema et al. 2017). The species P. fraudulenta reached higher cell densities than the other two species. Overall at the molecular level, the three species only displayed a moderate level of orthology (6,557 transcripts representing from 20% to 40% of the transcripts) indicating that distinct gene sets are used by the three species. This level of orthology is extremely similar to the one identified previously among three other Pseudo-nitzschia species (7,215 orthologs; Di Dato et al. 2015).
More specifically at the molecular level, the homologs of the four genes characterized as involved in DA biosynthesis in P. multiseries (Brunson et al. 2018) were only identified and expressed in P. australis. This is qualitatively in agreement with the higher DA production by P. australis. However, it seems important to note that the other two species also produced DA, albeit at lower concentration, but that the full dabABCD gene cluster was not identified as expressed in P. fraudulenta and P. pungens. This may indicate either that a very low expression of the dab gene cluster (too low to be detected in our analysis) is sufficient to enable the biosynthesis of DA, that the expression of the dab gene cluster and the biosynthesis of DA occurred before RNA was extracted in our experiment, or that other unknown genes are involved in DA biosynthesis. Investigating this would require genome sequencing and qPCR assays that are beyond the scope of the present study.
Notable differences between species were related to the expression of phosphate transporters. Indeed, each species expressed different sets of phosphate transporters and carriers. One low affinity phosphate transporter was among the eight most highly expressed transcripts in P. australis, but was barely expressed in P. pungens. The intracellular management of phosphate also seemed to differ between these two species as illustrated by the expression of transcripts linked to intracellular phosphate transport (PEP/− translocators and mitochondrial phosphate carriers), and to the recycling of phosphorus from nucleic acids (5′ bisphosphate nucleotidase). Such strong divergence in phosphate transporter expression in the field has also been highlighted between two distantly related diatoms (Alexander et al. 2015). Importantly, phosphate may be a strong limiting factor for phytoplankton growth in the field (Alexander et al. 2015; Grossman and Aksoy 2015; Lin et al. 2016). In the present study, the differences in the expression of phosphate transporters could not be directly linked to major differences in phosphate concentrations at the time of RNA extraction, but it did not preclude differences in terms of phosphate consumption dynamics during the time course of the experiment.
Turning toward the nitrate metabolism, both nitrate concentrations at the time of RNA expression and the expression of nitrate metabolism related transcripts were extremely different between species. This was particularly evident in the low nitrate medium, where P. australis and P. fraudulenta both exhausted nitrate, whereas around 10 µM of nitrate were still present in P. pungens cultures. At the molecular level, P. australis and P. fraudulenta expressed transcripts associated with the nitrogen metabolism, whereas P. pungens displayed constant expression for the same set of genes. These included the presence of nitrite reductase among the 11 most highly expressed transcripts in the low nitrate medium for both P. fraudulenta and P. australis. Moreover, P. pungens displayed a very low expression of nitrate reductase in the three media, whereas it was highly expressed in the other two species. The general pattern of over expression of nitrogen recycling transcripts under nitrogen stress (and specifically of nitrogen reductase as in the present study) has also been documented in recent studies specifically investigating transcriptional responses to nitrogen limitation in diatoms (Bender et al. 2014; Levitan et al. 2015). Altogether, and as already documented previously (Lelong et al. 2012), our results point toward major differences in nitrate usage by different Pseudo-nitzschia species. At the molecular level, a couple of studies also pointed toward divergence in nitrogen metabolism expression patterns among distantly related diatoms both in vitro (Bender et al. 2014) and in situ (Alexander et al. 2015). Our results suggest that such divergence also exists among closely related species.
Another interesting difference among Pseudo-nitzschia species was related to the expression of genes involved in photosynthesis such as pigments (Fucoxanthin and Chlorophyll) and photosystem I and II associated transcripts. These transcripts were more expressed in P. australis and P. fraudulenta in the high nutrient medium, but more expressed by P. pungens in the low nitrate medium. The effect of nutrient limitation on photosynthesis has been documented for a long time, but whether limitation increases or decreases photosynthetic activity tends to vary from one study to another (Cullen et al. 1992). In the present study, high expression of transcripts related to photosynthesis was detected when cells did not exhaust nutrients, but nevertheless reached stationary phase. One rather simple explanation for such pattern could be that self-shading among the cells induced light limitation that in turns induced increased expression of photosynthesis related genes (Flynn 2010). However, it seems unlikely that self-shading limitation is the sole explanation. Indeed, in P. pungens, cell densities are higher in the high nutrient medium but photosynthesis related genes are more expressed in the low nitrate medium.
Altogether, our results show similar responses of P. fraudulenta and P. australis across the experimental media. Although the link between in vitro and in situ environmental conditions is always difficult to draw, we note that in the area where the strains were isolated P. fraudulenta and P. australis display similar phenology (spring/summer blooms), whereas P. pungens is present more regularly throughout the year and tend to bloom in early summer when less nitrate is available (Klein et al. 2010, Thorel et al. 2017). It is interesting to note that this divergence may not simply be explained by the overall genetic divergence, as P. australis and P. pungens appear to be more closely related than P. fraudulenta.
Conclusion
In the present study, we investigated how closely related organisms diverged in their responses to contrasting experimental environments at both the intra- and inter-specific levels.
We highlighted that although cellular level responses across media may be rather different between strains belonging to the same species, global expression patterns tend to be preserved within species. It suggests that cellular phenotypic level differences may result from differential expression of a few specific genes rather than from global transcriptomic differences. As a result, in future work investigating functional divergence among species and coupling comparative transcriptomic with cellular level phenotypic approaches, it seems essential to investigate species divergence in the light of intraspecific diversity.
A major exception to the conclusion presented above was identified. A P. australis strain, displayed extreme DE compared with the other strains. As this strain was genetically similar to the others but was recently isolated from the natural environment, we argue that the strong DE reflects the progressive shut down of specific metabolic processes (in the present case, mainly associated with cell motility) following the transition from in situ to in vitro environmental conditions. Interestingly, this strong DE only affected precise metabolic processes without interfering with the species-specific expression patterns related to TCA cycle, photosynthesis, nitrogen metabolism or phosphate transport. An important exception was the contrasting expression pattern of the recently isolated strain regarding transcripts involved in motility and toxin biosynthesis. These results strongly advocate for future research efforts toward the understanding of the influence of culture duration and life cycle on the physiology and ecology of Pseudo-nitzschia species but also more generally on other microbial species.
The three species tended to display contrasting growth, toxin production and nutrient consumption that are associated with DE in genes related both to central (TCA cycle and photosynthesis) and specific (nitrogen metabolism, phosphate transport, and DA biosynthesis) metabolic processes. Interestingly, the expression patterns were more similar for the two species displaying similar phenology in the field (and thus probably occupying similar ecological niches) irrespective of their genetic divergence. In its present form, this result remains rather anecdotal. In order to be able to understand whether global transcriptomic, and more generally functional divergence, among closely related species is mainly driven by genetic distance between species and/or is influenced by ecology, we advocate for the systematic quantification of both genetic and ecological differences in comparative transcriptomic studies.
Data Availability
Raw reads and reference transcriptomes are accessible: https://doi.org/10.12770/78bba5d5-2112-467c-b35c-722992ded656.
Supplementary Material
Supplementary data are available at Genome Biology and Evolution online.
Supplementary Material
Acknowledgments
This work was supported by a “CNRS INSU EC2CO grant” (Pseudotox) to M.L.G., a postdoctoral fellowship from “Région Bretagne” and “Ifremer” to K.A.L., as well as a PhD fellowship from “Région Bretagne” and “Ifremer” to G.M. Sequencing has been done at the GeT-PlaGe Genotoul sequencing plateform (Toulouse, France). We thank the Ifremer RIC bio-informatic team for support.
Literature Cited
- Ajani PA, Verma A, Lassudrie M, Doblin MA, Murray SA.. 2018. A new diatom species P. hallegraeffii sp. nov. belonging to the toxic genus Pseudo-nitzschia (Bacillariophyceae) from the East Australian Current. PLoS One 13(4):e0195622.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander H, Jenkins BD, Rynearson TA, Dyhrman ST.. 2015. Metatranscriptome analyses indicate resource partitioning between diatoms in the field. Proc Natl Acad Sci U S A. 112(17):E2182–E2190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aminot A, Kerouel R, Coverly SC.. 2009. Nutrients in seawater using segmented flow analysis In: Practical guidelines for the analysis of seawater. Boca Raton: CRC Press; p. 143–178. [Google Scholar]
- Bates SS, Hubbard KA, Lundholm N, Montresor M, Leaw CP.. 2018. Pseudo-nitzschia, Nitzschia, and domoic acid: new research since 2011. Harmful Algae 79:3–43. [DOI] [PubMed] [Google Scholar]
- Bender SJ, Durkin CA, Berthiaume CT, Morales RL, Armbrust EV.. 2014. Transcriptional responses of three model diatoms to nitrate limitation of growth. Front Mar Sci. 1: 1–15. [Google Scholar]
- Bolger AM, Lohse M, Usadel B.. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30(15):2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunson JK, et al. 2018. Biosynthesis of the neurotoxin domoic acid in a bloom-forming diatom. Science 361(6409):1356–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castresana J. 2000. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol. 17(4):540–552. [DOI] [PubMed] [Google Scholar]
- Cohen NR, et al. 2017. Diatom transcriptional and physiological responses to changes in iron bioavailability across ocean provinces. Front Mar Sci. 4: 360. [Google Scholar]
- Cullen JJ, Yang X, MacIntyre HL.. 1992. Nutrient limitation of marine photosynthesis In: Primary productivity and biogeochemical cycles in the sea. Environmental science research. Boston (MA: ): Springer; p. 69–88. [Google Scholar]
- Danecek P, et al. 2011. The variant call format and VCFtools. Bioinformatics 27(15):2156–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Dato V, et al. 2015. Transcriptome sequencing of three Pseudo-nitzschia species reveals comparable gene sets and the presence of Nitric Oxide Synthase genes in diatoms. Sci Rep. 5:12329.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32(5):1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn KJ. 2010. Do external resource ratios matter? J Mar Syst. 3–4:170–180. [Google Scholar]
- Garrison E, Marth G.. 2012. Haplotype-based variant detection from short-read sequencing. ArXiv12073907 Q-Bio. Available from: http://arxiv.org/abs/1207.3907 (accessed February 26, 2018).
- Grossman AR, Aksoy M.. 2015. Algae in a phosphorus-limited landscape In: Plaxton WC, Lambers H, editors. Annual plant reviews. Vol. 48 Hoboken: John Wiley & Sons, Inc; p. 337–374. [Google Scholar]
- Haas BJ, et al. 2013. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat Protoc. 8(8):1494–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson GE. 1961. The paradox of the plankton. Am Nat. 95(882):137–145. [Google Scholar]
- Keller MD, Selvin RC, Claus W, Guillard RRL.. 1987. Media for the culture of oceanic ultraphytoplankton. J Phycol. 23(4):633–638. [Google Scholar]
- Klein C, Claquin P, Bouchart V, Le Roy B, Véron B.. 2010. Dynamics of Pseudo-nitzschia spp. and domoic acid production in a macrotidal ecosystem of the Eastern English Channel (Normandy, France). Harmful Algae 9(2):218–226. [Google Scholar]
- Kumar S, Stecher G, Li M, Knyaz C, Tamura K.. 2018. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. 35(6):1547–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL.. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods. 9(4):357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lelong A, Hégaret H, Soudant P, Bates SS.. 2012. Pseudo-nitzschia (Bacillariophyceae) species, domoic acid and amnesic shellfish poisoning: revisiting previous paradigms. Phycologia 51(2):168–216. [Google Scholar]
- Lema KA, Latimier M, Nézan É, Fauchot J, Le Gac M.. 2017. Inter and intra-specific growth and domoic acid production in relation to nutrient ratios and concentrations in Pseudo-nitzschia: phosphate an important factor. Harmful Algae 64:11–19. [DOI] [PubMed] [Google Scholar]
- Levitan O, et al. 2015. Remodeling of intermediate metabolism in the diatom Phaeodactylum tricornutum under nitrogen stress. Proc Natl Acad Sci U S A. 112(2):412–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, et al. 2009. The sequence alignment/map format and SAMtools. Bioinformatics 25(16):2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S, Litaker RW, Sunda WG.. 2016. Phosphorus physiological ecology and molecular mechanisms in marine phytoplankton. J Phycol. 52(1):10–36. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S.. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15(12):550.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moschandreou KK, et al. 2012. Inter- and intra-specific diversity of Pseudo-nitzschia (Bacillariophyceae) in the northeastern Mediterranean. Eur J Phycol. 47(3):321–339. [Google Scholar]
- Nanjappa D, Sanges R, Ferrante MI, Zingone A.. 2017. Diatom flagellar genes and their expression during sexual reproduction in Leptocylindrus danicus. BMC Genomics. 18:813.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quast C, et al. 2013. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41(D1):D590–D596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridley AJ. 2011. Life at the leading edge. Cell 145(7):1012–1022. [DOI] [PubMed] [Google Scholar]
- Rines JEB, Donaghay PL, Dekshenieks MM, Sullivan JM, Twardowski MS.. 2002. Thin layers and camouflage: hidden Pseudo-nitzschia spp. (Bacillariophyceae) populations in a fjord in the San Juan Islands, Washington, USA. Mar Ecol Prog Ser. 225:123–137. [Google Scholar]
- Ritchie ME, et al. 2015. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43(7):e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts AJ, Kon T, Knight PJ, Sutoh K, Burgess SA.. 2013. Functions and mechanics of dynein motor proteins. Nat Rev Mol Cell Biol. 14(11):713–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Si Y, Liu P, Li P, Brutnell TP.. 2014. Model-based clustering for RNA-seq data. Bioinformatics 30(2):197–205. [DOI] [PubMed] [Google Scholar]
- Teng ST, et al. 2016. High diversity of Pseudo-nitzschia along the northern coast of Sarawak (Malaysian Borneo), with descriptions of P. bipertita sp. nov. and P. limii sp. nov. (Bacillariophyceae). J Phycol. 52(6):973–989. [DOI] [PubMed] [Google Scholar]
- Thessen AE, Bowers HA, Stoecker DK.. 2009. Intra- and interspecies differences in growth and toxicity of Pseudo-nitzschia while using different nitrogen sources. Harmful Algae 8:792–810. [Google Scholar]
- Thorel M, et al. 2017. Nutrient ratios influence variability in Pseudo-nitzschia species diversity and particulate domoic acid production in the Bay of Seine (France). Harmful Algae 68:192–205. [DOI] [PubMed] [Google Scholar]
- Trainer VL, et al. 2012. Pseudo-nitzschia physiological ecology, phylogeny, toxicity, monitoring and impacts on ecosystem health. Harmful Algae 14:271–300. [Google Scholar]
- Wang J, Cao S, Du C, Chen D.. 2013. Underwater locomotion strategy by a benthic pennate diatom Navicula sp. Protoplasma 250(5):1203–1212. [DOI] [PubMed] [Google Scholar]
- Zabaglo K, et al. 2016. Environmental roles and biological activity of domoic acid: a review. Algal Res. 13:94–101. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw reads and reference transcriptomes are accessible: https://doi.org/10.12770/78bba5d5-2112-467c-b35c-722992ded656.