

Fast typing methods that can easily and accurately distinguish clonal groups and unrelated isolates are of particular interest for microbiologists confronted with outbreaks or performing epidemiological studies. Highly discriminatory universal methods, like PFGE, optical mapping, or WGS, are expensive and/or time-consuming. MLST is useful for phylogeny but is less discriminatory and requires sequencing facilities. PCR methods, which are fast and easy to perform, also have drawbacks. Random PCRs and REP-PCR are universal but lack reproducibility. Other PCR methods may lack the discriminatory power to differentiate isolates during outbreaks. MLVA combines the advantages of PCR methods with a high discriminatory power but in its standard form requires sequencing capillary electrophoresis. The method that we have developed combines the advantages of standard PCR (simple, fast, and inexpensive) with the high discriminatory power of MLVA and permits the typing of all E. coli isolates (either intestinal or extraintestinal pathogenic isolates as well as commensal isolates).
KEYWORDS: Escherichia coli, genotyping, MLVA
ABSTRACT
We developed a multiplex PCR method based on multiple-locus variable-number tandem-repeat (VNTR) analysis (MLVA) that was designed for the rapid typing of Escherichia coli and Shigella isolates. The method amplifies seven VNTRs and does not require a sequencing capillary or fluorescent dyes. The amplification products are simply loaded on a standard agarose gel for electrophoresis, and the banding patterns are analyzed visually. We evaluated the method on 220 strains belonging to different collections: the E. coli reference (ECOR) collection (n = 72), O1:K1 isolates causing neonatal meningitis (n = 38), extended-spectrum beta-lactamase-producing fecal isolates belonging to the worldwide sequence type 131 (ST131) clone (n = 38), Shiga toxin-producing E. coli (STEC) isolates of serogroups O157:H7 (n = 21) and O26 (n = 16, 8 of which belonged to an outbreak), 27 Shigella isolates (22 Shigella sonnei isolates, including 5 epidemic strains), and 8 reference strains. The performances were compared to those of multilocus sequence typing (MLST), the DiversiLab automated repetitive element palindromic PCR (REP-PCR), pulsed-field gel electrophoresis (PFGE), and whole-genome sequencing (WGS). We found 66 different profiles among the isolates in the ECOR collection. Among the clonal group O1:K1 isolates, 14 different profiles were identified. For the 37 STEC isolates, we found 23 profiles, with 1 corresponding to the 8 epidemic strains. We found 19 profiles among the 27 Shigella isolates, with 1 corresponding to the epidemic strain. The method was able to recognize strains of the ST131 clone and to distinguish the O16 and O25b serogroups and identified 15 different MLVA types among them. This method allows the simple, fast, and inexpensive typing of E. coli/Shigella isolates that can be carried out in any laboratory equipped for molecular biology and has a discriminatory power superior to that of MLST and DiversiLab REP-PCR but slightly lower than that of PFGE.
IMPORTANCE Fast typing methods that can easily and accurately distinguish clonal groups and unrelated isolates are of particular interest for microbiologists confronted with outbreaks or performing epidemiological studies. Highly discriminatory universal methods, like PFGE, optical mapping, or WGS, are expensive and/or time-consuming. MLST is useful for phylogeny but is less discriminatory and requires sequencing facilities. PCR methods, which are fast and easy to perform, also have drawbacks. Random PCRs and REP-PCR are universal but lack reproducibility. Other PCR methods may lack the discriminatory power to differentiate isolates during outbreaks. MLVA combines the advantages of PCR methods with a high discriminatory power but in its standard form requires sequencing capillary electrophoresis. The method that we have developed combines the advantages of standard PCR (simple, fast, and inexpensive) with the high discriminatory power of MLVA and permits the typing of all E. coli isolates (either intestinal or extraintestinal pathogenic isolates as well as commensal isolates).
INTRODUCTION
Escherichia coli is a major human commensal but also the most common cause of both nosocomial and community-acquired bacterial infections at all ages (1, 2). E. coli infections are categorized as intestinal or extraintestinal. Intestinal infections occur as diarrhea, which may eventually be complicated by hemolytic-uremic syndrome (HUS) when it is caused by Shiga toxin-producing E. coli (STEC) strains. Extraintestinal infections notably comprise urinary tract infections (UTI), bacteremia, and neonatal meningitis. Both STEC and extraintestinal pathogenic E. coli (ExPEC) strains harbor specific virulence factors and belong to a limited number of serotypes and clonal groups.
Although most E. coli infections are sporadic, large outbreaks of STEC infections have been described and investigated by use of different genotyping tools to identify the source of contamination (3). Beyond the investigation of numerous nosocomial outbreaks, the epidemiology of ExPEC strains, in particular, of those strains producing extended-spectrum beta-lactamases (ESBL) or carbapenemases, has also required the use of genotyping tools in order to understand the dissemination in human populations of clones with particular fitness and virulence, such as the sequence type 131 (ST131) E. coli clonal group (4).
Many genotyping tools have been applied to E. coli, and they all have advantages and drawbacks. Pulsed-field gel electrophoresis (PFGE), the gold standard, is highly discriminatory but time-consuming; sequencing methods ranging from multilocus sequence typing (MLST) to whole-genome sequencing (WGS) remain time-consuming and expensive; the available PCR methods either are poorly reliable (random PCRs) or expensive (automated repetitive element palindromic PCR [REP-PCR]) or lack discriminatory power (molecular serotyping, phylogrouping PCR, PCR of several virulence genes) (5).
Recently, multiple-locus variable-number tandem-repeat (VNTR) analysis (MLVA) has emerged as a rapid and highly discriminatory method for E. coli and Shigella genotyping (6–8). This method, based on the polymorphism of the variable-number tandem repeats (VNTRs) present in several loci dispersed on the bacterial chromosome, consists of PCRs with primers surrounding each locus, followed by electrophoresis. Some of these VNTRs display an important degree of polymorphisms, even among highly clonal strains, such as STEC strains belonging to the major serotype O157:H7. Studies analyzing STEC outbreaks have shown that MLVA can sometimes prove more discriminatory than PFGE, making it a promising genotyping tool (6, 9). However, in its standard form, MLVA requires specific equipment. Indeed, as tandem repeats (TRs) can be less than 10 bp long, to accurately differentiate the length of the amplification products, electrophoresis is performed on sequencing capillaries. Moreover, multiplexing in a single PCR tube requires the use of primers tagged with fluorescent dyes to identify each locus. Thus, although rapid and highly discriminatory, this method cannot be easily performed in most laboratories. Gorgé et al. have proposed a simplified version of the method adapted for Shigella isolates based on standard electrophoresis using VNTRs with TRs with long lengths, but each VNTR is amplified in a separate PCR (8). Several other combinations of VNTR have been proposed, and all of these require either fluorescent dyes or specific electrophoresis (capillary electrophoresis systems, polyacrylamide gels with silver staining) (10–16).
In this study, we modified the method in order, first, to combine in a single-tube multiplex PCR all VNTRs and, second, to discriminate both ExPEC and STEC strains on a standard agarose electrophoresis gel. We also validated its usefulness in situations requiring rapid E. coli genotyping.
RESULTS
After adjustment of the primer concentrations and annealing temperature, the multiplex MLVA PCR produced the expected bands on whole-genome-sequenced reference strains with molecular weights ranging from 102 bp to 1,023 bp by electrophoresis on agarose gels (Table 1; Fig. 1). Bands of higher molecular weights were either faint or not visible and were discarded from analysis.
TABLE 1.
Primers and variable-number tandem repeats selected for the MLVA scheme
| PCR | Primer | Sequence (5′-3′) | Coordinates on the E. coli strain K-12 DH10B genome (GenBank accession no. NC_010473) | Concn (μmol/liter) in PCR mix | VNTR name (gene) | Repeat unit size (bp) | Product size range (bp) on 17 reference genomesa | Origin of VNTR (source or reference) |
|---|---|---|---|---|---|---|---|---|
| ECMLV1 | ECMLV1-F | TCCCTGGACAAACCAGGACTG | 4191957–4191977 | 0.1 | RDB1 (rhaD) | 92 | 162–1,597 | This study |
| ECMLV1-R1 | CGTGCGGACTTATGAGAAAG | 4192485–4192466 | 0.1 | |||||
| ECMLV1-R2 | CGTGCGGGCTTATGAAAAAG | 4192485–4192466 | 0.5 | |||||
| ECMLV2 | ECMLV2-F | GAAACAGGCCCAGGCTACAC | 1797019–1797038 | 0.05 | ms11 (rsxC) | 96 | 575–869 | Gorgé et al. (8) |
| ECMLV2-R | CTGGCGCTGGTTATGGGTAT | 1797784–1797765 | 0.05 | |||||
| ECMLV3 | ECMLV3-F | TTCAGGAAATGGATAAAGTAGT | 2908149–2908170 | 0.8 | ms21 (tRNA-Arg) | 139 | 616–1,157 | Gorgé et al. (8) |
| ECMLV3-R | GGGAGTATGCGGTCAAAAGC | 2909170–2909151 | 0.8 | |||||
| ECMLV4 | ECMLV4-F | ACAACCGGCTGGGGCGAATCC | 988477–988497 | 0.05 | CNV001 (ftsK) | 39 | 413–539 | Lindstedt et al. (7) |
| ECMLV4-R | GTCAGCAAATCCAGAGAAGGCA | 988914–988893 | 0.05 | |||||
| ECMLV5 | ECMLV5-F | GCGGCGCTGAAGAAGAAAGC | 828727–828746 | 0.05 | CNV004 (tolA) | 48 | 375–438 | Lindstedt et al. (7) |
| ECMLV5-R | CTCCCGGCAGGCGAAGCATTGT | 829127–829106 | 0.05 | |||||
| ECMLV6 | ECMLV6-F | CAAAGAGCAATAACACTTTTAGCA | 4084801–4084824 | 0.1 | CNV014 (hemY) | 6 | 102–149 | Lindstedt et al. (7) |
| ECMLV6-R | GCAGCAGGGACAACGGAAGCTAA | 4084905–4084883 | 0.1 | |||||
| ECMLV7 | ECMLV7-F | GTGAAGGATAAGCTGCATTTGTCA | 4538101–4538124 | 0.1 | O157-33 (ytfL) | 17 | 176–211 | Keys et al. (6) |
| ECMLV7-R | GCCTGACGCTAAAGATAAAGAAGA | 4538293–4538270 | 0.1 |
The reference genomes were those of the following E. coli strains (GenBank accession numbers are given in parentheses): 536 (NC_008253), APECO1 (NC_008563), CFT073 (NC_004431), ED1a (NC_011745), IAI1 (NC_011741), IAI39 (NC_011750), K-12 DH10B (NC_010473), O103:H2 12009 (NC_013353), O104:H4 TY-2482 (AFVR01000000), O111:H− 11128 (NC_013364), O127:H6 E2348/69 (NC_011601), O157:H7 EDL933 (NC_002655), O26:H11 11368 (NC_013361), S88 (NC_011742), SMS-3-5 (NC_010498), UMN026 (NC_011751), and UTI89 (NC_007946).
FIG 1.
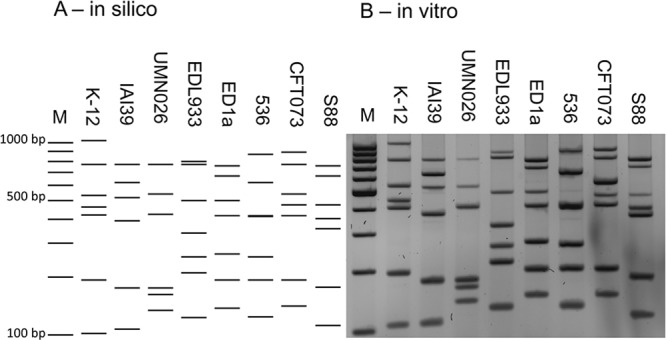
MLVA profiles of 8 reference E. coli strains, strains K-12 (GenBank accession number NC_010473), IAI39 (GenBank accession number NC_011750), UMN026 (GenBank accession number NC_011751), O157:H7 EDL933 (GenBank accession number NC_002655), ED1a (GenBank accession number NC_011745), 536 (GenBank accession number NC_008253), CFT073 (GenBank accession number NC_004431), and S88 (GenBank accession number NC_011742). Lanes M, molecular weight marker (100-bp DNA ladder). (A) In silico simulation of expected profiles based on whole-genome sequences. (B) In vitro actual banding pattern results for the same reference strains.
Evaluation of the MLVA method on the ECOR collection.
In order to assess the global discriminatory power of our MLVA method on unrelated E. coli strains from various origins, we first used strains from the E. coli reference (ECOR) collection (17). A total of 66 different MLVA types were recorded among the strains in the ECOR collection. Each pattern contained between 5 and 7 bands (Fig. 2). Precisely, 51.4% (37/72), 34.7% (25/72), and 13.9% (10/72) of the patterns possessed 7 bands, 6 bands, and 5 bands, respectively. In this collection, six pairs of isolates had identical MLVA types and 60 isolates each produced a unique pattern (Fig. 2). The Hunter and Gaston diversity index derived from Simpson’s index of diversity (D) (18) was calculated for the combined typing set and showed a value of 0.99 (95% confidence interval [CI], 0.99 to 1.0). The ECOR collection is composed of 49 different sequence types (STs) (Fig. 2). To assess the congruence between typing methods, the adjusted Rand coefficient (AR) and adjusted Wallace coefficient (W) were calculated. The overall congruence between MLVA and MLST, as determined by use of the Rand coefficient, was 0.024, indicating no agreement between MLST and MLVA typing. The directional congruence, as estimated by the adjusted Wallace coefficient going from MLVA to MLST, was 0.145, suggesting that isolates assigned to a cluster by MLVA had a low probability of being assigned to the same cluster when typed by MLST. Similarly, when examined in the other direction, the adjusted Wallace coefficient was 0.016, indicating a very low probability that isolates assigned to the same cluster by MLST would be assigned to the same cluster when typed by MLVA.
FIG 2.

Dendrogram of the MLVA results for the E. coli reference (ECOR) collection and E. coli K-12. The dendrogram was constructed by BioNumerics software using UPGMA and the Dice algorithm based on the band profiles on the electrophoresis gel. E. coli K-12 was integrated into each electrophoresis gel. The phylogenetic group and the ST (Warwick scheme) were reported by Clermont et al. (42), and the STc’s were obtained from http://pubmlst.org/bigsdb?b=pubmlst_mlst_seqdef. The dotted line corresponds to the 95% similarity cutoff.
MLVA typing and STEC isolates.
A collection of STEC isolates was further typed by MLVA. A set of strains from major STEC serogroups was previously typed. All these strains were typeable and showed multiple profiles (data not shown). After this verification assay, we focused on strains belonging to the worldwide predominant serotype O157:H7 and the emergent European serotype O26:H11. The two sets of strain were analyzed separately (Fig. 3 and 4).
FIG 3.

Dendrogram of the MLVA (A) and PFGE (B) results for E. coli O157:H7 strains. The dendrograms were constructed by BioNumerics software using UPGMA (A and B) and the Dice algorithm (A) based on the band profile on the electrophoresis gel. The PFGE profile was initially reported by aNRC-EC at the Robert Debré Hospital. The PFGE types C, C′, S, Sʺ, and Sʺʹ are profiles differentiated by less than three bands and considered similar according to the criteria of Tenover et al. (20). The dotted lines correspond to the 95% similarity cutoff.
FIG 4.

Dendrogram of the MLVA results for the E. coli O26 strain. The dendrogram was constructed by BioNumerics software using UPGMA and the Dice algorithm based on the band profile on the electrophoresis gel. The dotted line corresponds to the 95% similarity cutoff. Strains 41047, 41049 to 41051, 41054, and 41057 to 41059 were part of an epidemic outbreak and are indicated by a black box. Strains 9051, 9134, 9197, 9300, 9708, and 9821 were unrelated O26 strains from the Statens Serum Institut, Denmark (European External quality assessment STEC EQA-8 2017–2018).
All STEC isolates, whatever their serogroups, harbored 6 or 7 bands on gel electrophoresis. For the assay with O157:H7 strains, we selected 21 unrelated E. coli O157:H7 strains. Among the 21 isolates, 14 MLVA types were identified, and all O157 isolates possessed 7 bands on gel electrophoresis (Fig. 3). The Hunter and Gaston index of diversity (D) was 0.93 (95% CI, 0.88 to 0.99).
After analyzing the O157 isolate collection, we focused on the O26:H11 serotype, which is an emergent serotype of STEC in Europe. The aim of this assay was to evaluate our MLVA method in epidemic settings. We selected 16 isolates, including 8 isolates belonging to the same outbreak; the other 8 isolates were unrelated to the outbreak (19). Nine MLVA types were identified (Fig. 4). The same profile was identified for the eight isolates belonging to the outbreak (Fig. 4). The eight remaining strains unrelated to the outbreak harbored different profiles which differed from each other by at least one band (Fig. 4).
Comparison of PFGE and MLVA typing on O157 STEC isolates.
All the 21 O157:H7 E. coli isolates were also typed by the PFGE method by the Associated National Reference Center for E. coli (aNRC-EC) for epidemiological purposes. According to the criteria of Tenover et al., 17 PFGE profiles were distinguished among the 21 O157:H7 E. coli isolates, and 15 of the profiles were singletons (20). The remaining 2 clusters consisted of two and four isolates, respectively. The Hunter and Gaston diversity index (D) was 0.97 (95% CI, 092 to 1.0), which is slightly higher than the one obtained by MLVA. The adjusted Rand coefficient was 0.65, showing reasonable agreement between PFGE and MLVA typing, and the adjusted Wallace coefficient was 1.0, showing that PFGE was predictive of MLVA, whereas this was more limited in the reverse direction, with an adjusted Wallace coefficient of 0.483.
MLVA typing and Shigella isolates.
To test our MLVA method on Shigella isolates, we selected 27 isolates, including 22 Shigella sonnei isolates, 4 S. flexneri isolates, and 1 S. boydii isolate. Among the 22 S. sonnei strains, 5 strains were part of an outbreak in a religious (Jewish) school. All isolates presented between 5 and 7 bands on gel electrophoresis. Nineteen MLVA types were identified; of these, 15 were singletons, 2 were shared by 2 isolates, 1 corresponded to 3 isolates, and 1 was shared by 5 isolates (Fig. 5). Some profiles differed by only one band with minimal molecular weight variations. Furthermore, outbreak isolates from 5 children attending the same school presented an identical MLVA type. All S. sonnei isolates were grouped in the same cluster and apart from the S. boydii and S. flexneri isolates (Fig. 5).
FIG 5.

Dendrogram of the MLVA results for the Shigella isolates. The dendrogram was constructed by BioNumerics software using UPGMA and the Dice algorithm based on the band profile on the electrophoresis gel. The dotted line corresponds to the 95% similarity cutoff. The black box indicates the epidemic strains (SHIGA14, -22, -23, -24, -25) that were isolated from children attending the same school.
MLVA typing and WGS.
ExPEC strains, especially those belonging to ST clonal complex 95 (STc95), are responsible for invasive diseases, such as meningitis (21). We focused on serotype O1:K1, which belongs to STc95 and which is emergent among the predominant K1 serogroup in France. To evaluate the discriminatory power within a specific pathogenic clonal group, we used isolates previously characterized in a national study of unrelated neonatal meningitis cases (45). The 38 O1:K1 isolates had been isolated from the cerebrospinal fluid of neonatal meningitis cases in France. Each profile contained either 6 or 7 bands on gel electrophoresis. Fourteen MLVA types were identified; these were composed of 5 singletons, 6 pairs of isolates presenting the same profiles, 4 isolates presenting an identical profile, a group of 7 isolates harboring the same profile, and a group of 10 isolates harboring the same profile (Fig. 6). The Hunter and Gaston index of diversity (D) was calculated to be 0.89 (95% CI, 0.83 to 0.95), demonstrating a discriminatory capacity even in a highly clonal E. coli population. In comparison, MLST distinguished only 5 different STs, all belonging to STc95. This collection has been previously characterized by WGS, and the phylogeny was performed based on 19,547 single nucleotide polymorphisms. The clusters obtained were divided into subgroups as described by Gordon et al. (22). According to this method, 2 subgroups, namely, subgroups A and D, were obtained. Subgroup D was further subdivided into 3 clusters: D-1, D-2, and D-3. The isolates clustered together by MLVA typing in accordance with the WGS subgroups except for 3 D-1 isolates, which clustered with isolates of subgroup A (Fig. 6). This resulted in a poor adjusted Rand coefficient (AR = 0.10). The Wallace coefficient showed that MLVA can predict appropriately the WGS subgroup (W = 0.57) but that WGS subgroups are poorly predictive of the MLVA type (W = 0.06).
FIG 6.

Dendrogram of the MLVA results for the E. coli O1:K1 isolates. The dendrogram was constructed by BioNumerics software using UPGMA and the Dice algorithm based on the band profile on the electrophoresis gel. The WGS subgroups were reported by Geslain et al. (45). The dotted line corresponds to the 95% similarity cutoff. NR, not reported.
MLVA typing and E. coli ST131.
To evaluate our method on the E. coli ST131 clonal group disseminated worldwide, we used 38 strains from the study of Birgy et al. analyzing the fecal carriage of ESBL-producing E. coli among French children (23). Among these 38 strains, 29 belonged to the O25b serogroup and 9 belonged to the O16 serogroup. Fifteen profiles were identified; of these, 5 were singletons, 4 groups of 2 isolates each shared identical profiles, 4 clusters contained 3 isolates each, 1 cluster contained 5 isolates, and 1 cluster contained 8 isolates (Fig. 7). The Hunter and Gaston diversity index (D) was 0.92 (95% CI, 0.88 to 0.96). The profiles included 7 bands for 92.1% (35/37) of the strains and 6 bands for 7.9% (3/38) of the strains. All the profiles of the O25b subgroup harbored 7 bands. Moreover, 4 bands were identical in 97.4% (37/38) of the strains. The phylogenetic tree was organized into 2 main clusters, except for one isolate, named L49, which harbored an atypical profile. The profile patterns differed from each other by 3 bands between the two groups. Typical profiles could be established for the O25b and O16 subgroups.
FIG 7.
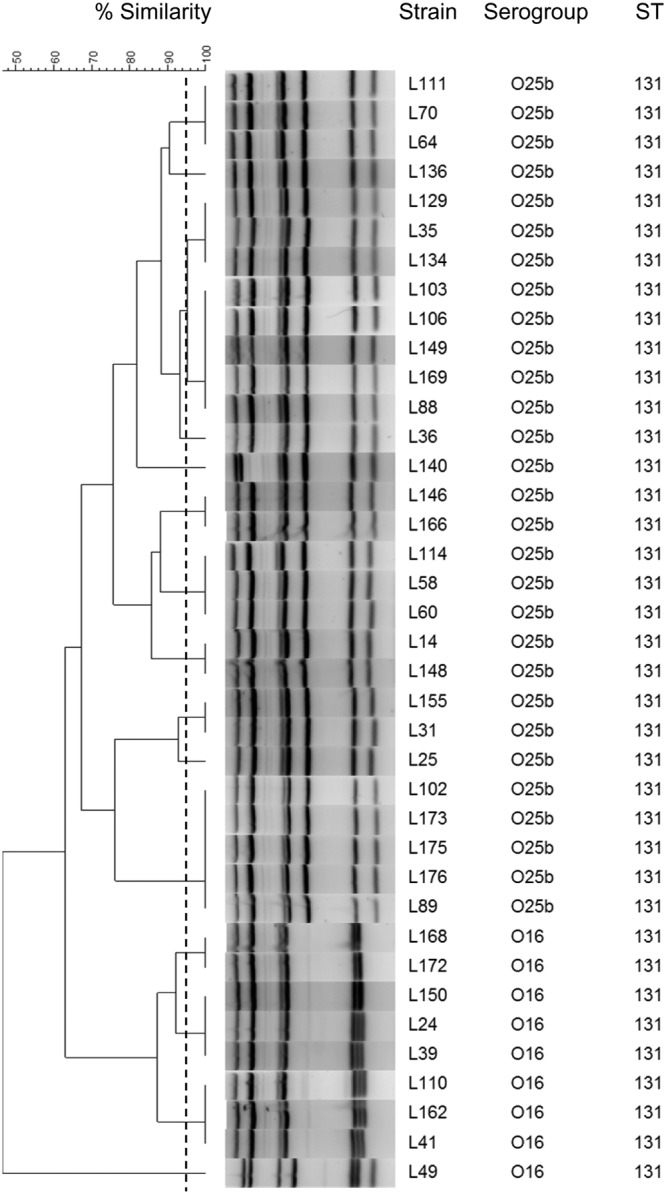
Dendrogram of the MLVA results for the E. coli ST131 isolates. The dendrogram was constructed by BioNumerics software using UPGMA and the Dice algorithm based on the band profile on the electrophoresis gel. The dotted line corresponds to the 95% similarity cutoff.
Repeatability and stability of MLVA typing.
Finally, to assess the stability of the VNTR loci during laboratory subcultures, 7 strains, including ECOR48, considered a hypermutator strain (24), were subcultured each day for 14 days and typed at the beginning and at the 7th and 14th days of subculture. No repeat number variation was observed during these passages at any of the 7 VNTR loci (data not shown).
DISCUSSION
In this study, we attempted to combine MLVA (which has the advantage of having a high discriminatory power) with standard agarose electrophoresis of single-tube multiplex PCR amplification products (which has the advantage of being fast and easy to perform). With this aim, we definitively abandoned the idea of determining the exact number of repeats for each VNTR analyzed. The results were analyzed in the same manner as those of other DNA fragment-generating genotyping methods based on either digestion (restriction fragment length polymorphism analysis, ribotyping, PFGE) (25–27) or PCR (randomly amplified polymorphic DNA analysis, arbitrarily primed PCR, enterobacterial repetitive intergenic consensus sequence-PCR, ribotyping PCR, REP-PCR) (28–31), that is, a simple visual comparison of band profiles similar to barcodes. The use of software like BioNumerics is in no case compulsory. If we did use this tool, it was only to facilitate the analysis of large collections of strains and to draw a visual representation of the genetic relationships among clonal groups through the use of dendrograms. The aim of this study was to check if the MLVA method could be a reliable screening tool for the identification of related/unrelated isolates.
This simplified MLVA assay designed for E. coli and Shigella typing is based on 6 loci described previously and another locus that we named RDB1 (6–8). Its discriminatory power and typing capacity were assessed on a large panel of strains responsible for various biological and medical concerns: intestinal and extraintestinal infections, epidemic outbreaks, multidrug resistance, and commensalism.
Thus, we evaluated our method first on strains from the ECOR collection, which is supposed to represent the global diversity of the species and which comprises both pathogenic and commensal isolates of various origins in terms of host and geography (17). The intestinal pathogens assayed were composed of Shigella and STEC strains that belonged to different serotypes and that were isolated from both sporadic and epidemic cases. For ExPEC strains, after having checked that reference strains representing major clonal groups produced different profiles, we specifically focused on an emergent group of O1:K1 strains causing neonatal meningitis to evaluate the discriminatory power of our method in a clone with restricted genetic diversity that we had previously characterized by WGS (45). Finally, we confronted our method with the worldwide emerging clonal group ST131 driving antimicrobial resistance in human microbiota (4, 23).
The MLVA approach managed to type all E. coli and Shigella strains that were part of this study; precisely, 76.5% (163/213), 18.3% (39/213), and 5.2% (11/213) of the strains harbored 7 bands, 6 bands, and 5 bands in their gel electrophoresis profiles, respectively. One or two of the seven loci of 23.5% (50/213) of the strains typed failed to be amplified. This may have been due to the fact that the corresponding locus was missing, that the VNTR was too long (>1,100 bp in the experiment with reference strains) to be easily amplified, or that sequence divergence resulted in mispriming (32). This uncertainty may reduce the discriminatory power and impedes determination of which VNTRs are most discriminating for E. coli in general or for a given E. coli group. However, among the 202 unrelated isolates, 136 different MLVA types were identified with a Hunter and Gaston index of diversity (D) of 0.99 (18). This calculated D value is considered highly discriminatory, even though highly clonal groups like ST131 E. coli or STc95 E. coli were being investigated.
Lindstedt et al. have developed and validated an MLVA scheme for E. coli and Shigella genotyping based on only seven VNTRs (7). This method, which is able to genotype all intestinal pathogenic strains, was also validated on the reference ECOR collection representative of E. coli species diversity. The use of 7 VNTR-specific PCRs with fluorescent dyes and capillary electrophoresis permitted 63 genotypes to be distinguished among the 72 ECOR strains. This method was improved by adding 3 additional VNTR loci to increase the discriminatory power for ExPEC and intestinal pathogenic E. coli (InPEC) strain collections (14, 33). However, these results are similar to the 66 genotypes that we distinguished among the isolates in the ECOR reference collection with our simplified method (Fig. 2). None of the strains harboring an identical profile had the same multilocus enzyme electrophoresis (MLEE) profiles (17), but each of these clusters was composed of isolates belonging to the same phylogenetic group. This finding (17) is in agreement with the findings of the study of Lindstedt et al. (7) showing that the phylogenetic groups, based on MLEE data, are not very well conserved by MLVA.
Keys et al. developed the first MLVA scheme for E. coli, focusing on Shiga-toxin producing E. coli (STEC) isolates belonging to serotypes O157:H7 and O55:H7 (6). This scheme was based on an analysis of 30 VNTRs by 6 multiplex PCRs using fluorescent dye-labeled primers with electrophoresis on polyacrylamide gels. The method proved as discriminatory as PFGE. Nevertheless, our results for the STEC isolates, notably, for serotype O157:H7, showed that some identical MLVA types exhibited several PFGE profiles. The use of 7 VNTRs rather than 30 VNTRs may be the reason for the lower discriminatory power. Also, our MLVA results showed only partial agreement with the PFGE results, as reported in a previous study that used another MLVA scheme (33). The Hunter and Gaston index of diversity obtained (D = 0.93) can be considered a high discriminatory index value, especially in a clonal group such as serotype O157:H7. The results from the PFGE typing showed that two groups of isolates exhibited similar profiles according to the criteria of Tenover et al. (20). Interestingly, isolates within each of the groups also shared the same MLVA profiles (Fig. 3). The index of diversity based on PFGE data was 0.97, demonstrating a discriminatory power higher than that of MLVA for this set of isolates.
The studies of both Lindstedt et al. (7) and Keys et al. (6) have shown that MLVA has an efficient discriminatory power for genotyping E. coli/Shigella isolates. Their drawback was the requirement of fluorescent dyes and the need for sequencers to analyze the results, thus limiting the diffusion of this genotyping method to other laboratories devoid of these facilities.
Gorgé et al. have tried to make the MLVA method more accessible by use of simple agarose gel electrophoresis of the amplification products without fluorescent dyes (8). Their goal was to facilitate the genotyping of Shigella isolates using 15 VNTR-specific PCRs. As VNTRs with a repeat size of >9 bases were chosen, standard electrophoresis permitted the correct estimation of the number of repeats for each VNTR, and the genotype was analyzed using the standard numerical code of MLVA. Although it is simplified, this protocol has the drawback that each PCR must be performed independently, thus requiring 15 different PCRs to genotype the strains. In that study, they defined four main clusters, with one cluster including all S. sonnei isolates (8). Interestingly, all S. sonnei strains analyzed in our study belonged to the same cluster exclusively composed of these strains, since the five remaining strains, 4 S. flexneri strains and 1 S. boydii strain, had completely different profiles (Fig. 5). Moreover, 5 isolates yielding the same MLVA profile were isolated from 5 patients (Fig. 5), all of whom were schooled in the same Jewish institution, corresponding to an outbreak phenomenon, as was previously described for this community (34).
As shown by the results for the O1:K1 E. coli strains, the MLVA results showed agreement with the WGS data. The MLVA method can predict approximately the subgroup defined by WGS (22, 45). A study comparing WGS and MLVA in O157 E. coli isolates showed concordances for identifying epidemiologically related isolates but discordances in phylogenetic tree reconstruction for unrelated isolates (35).
Multiple MLVA profiles were also identified among the E. coli strains that exhibited an identical ST. Notably, in the ST131 E. coli subset, the MLVA analysis yielded multiple profiles organized into two main clusters: one corresponding to the O25b-ST131 isolates and the other corresponding to the O16-ST131 isolates (Fig. 7). Within the same cluster, discrimination of the profiles would be difficult to see directly from the electrophoresis gel unless both amplification products were loaded in contiguous wells and visualized next to each other. In this study, the visualization of the differences between the profiles was facilitated by the use of BioNumerics software. The identification of the ST131 clone can be determined by standard MLST, which requires Sanger sequencing; by multiplex PCR assay; and by high-resolution melting analysis (36–38). Besides showing a higher discriminatory power than MLST, MLVA could determine the serogroup of an ST131 E. coli strain. Indeed, when all the strains of this study (except O1:K1 isolates) were assembled in the same dendrogram, a cluster including exclusively the ST131 E. coli isolates was present (see Fig. S1 in the supplemental material). Moreover, inside this cluster, the O25b-ST131 and O16-ST131 isolates were separated into 2 distinct groups (Fig. 7; Fig. S1). These results confirm that our MLVA method can identify the ST131 clone and classify strains according to their serogroup.
Concerning the reproducibility of the method, the VNTRs appeared to be stable in our study. No change was observed among the eight strains from the O26 STEC outbreak or from those from the Shigella outbreak, indicating that genetic occurrence due to passaging through hosts has no impact on the genotype of the strain (Fig. 4). No change also occurred in the subculturing experiment that we conducted, even in the hypermutator ECOR48 strain (data not shown).
In conclusion, the simplified MLVA method developed in the study described here has multiple advantages. First, this technique was able to type all the strains used in this study. We recommend a cutoff of a minimum of 5 VNTRs by profile to accept strain typing, as used by Lindstedt et al. (7). The discriminatory power calculated for different sets of E. coli and Shigella strains has shown that this method can even distinguish isolates belonging to the same clonal complex (i.e., ST131 E. coli and O1:K1 STc95 E. coli isolates). MLVA necessitates a small amount of bacteria and standard molecular biology equipment, and analysis of profiles can be done by visualization of the band pattern directly from the gel used for electrophoresis. The results can be achieved within a day (∼4 h), which is faster than PFGE or WGS, and the cost of typing per strain (<$3) is lower than that by sequencing methods. These settings are suitable for use in all molecular biology-equipped laboratories.
The reliability and easiness of this method make it fit to a wide range of applications: rapid investigation of outbreaks, analysis of the diversity of an E. coli population in a microbiota, and identification of major clonal groups. It also proved a useful tool in our labs to easily distinguish transconjugants and mutants in conjugation experiments.
Nevertheless, in the E. coli O157:H7 set, PFGE showed a higher discriminatory power than MLVA. This finding may result in a false-positive signal suggestive of an outbreak. During utilization of the assay, we recommend that profiles presenting a similar band pattern be controlled by performing other highly discriminatory techniques, such as PFGE or WGS, to assess the similarity of strains.
MATERIALS AND METHODS
Bacterial isolates.
A total of 220 isolates were included in this study. Eight reference E. coli strains whose whole genomes had been sequenced, strains K-12 (GenBank accession number NC_010473), IAI39 (GenBank accession number NC_011750), UMN026 (GenBank accession number NC_011751), O157:H7 EDL933 (GenBank accession number NC_002655), ED1a (GenBank accession number NC_011745), 536 (GenBank accession number NC_008253), CFT073 (GenBank accession number NC_004431), and S88 (GenBank accession number NC_011742), were used for in silico design and in vitro validation of the primers. E. coli strain K-12 was also used for quality control. The 72 strains of the ECOR collection were obtained from a previous study (39). Thirty-eight E. coli O1:K1 isolates that belonged to ST clonal complex 95 (STc95) and that were responsible for neonatal meningitis, including 38 isolates previously characterized in a study described elsewhere (45), were selected. Thirty-seven STEC isolates belonging to serogroup O157:H7 (n = 21) or O26:H11 (n = 16) were included; all of these isolates were from France, except for strains 9051, 9134, 9197, 9300, 9708, and 9821, which were unrelated O26 strains from the Statens Serum Institut, Denmark (European External quality assessment STEC EQA-8 2017-2018). These strains were not associated with a known outbreak, with the exception of eight O26 isolates from a child care facility outbreak occurring in southern France. Twenty-seven Shigella isolates were obtained from the strain collection of the Associated National Reference Center for E. coli (aNRC-EC) at the Robert Debré Hospital and the strain collection of the National Reference Center for Shigella (Institut Pasteur). The majority of these were sporadic isolates (n = 22), with five strains being associated with an outbreak at a single school. Thirty-eight unrelated E. coli ST131 isolates from the feces of healthy infants were described in a previous study (23).
Extraction of DNA.
The bacterial strains were grown overnight at 35°C on Trypticase soy agar (TSA). A 1-μl loopful of bacteria was suspended in 1 ml of sterile distilled water and boiled for 15 min at 100°C. The suspension was centrifuged at 3,500 rpm for 3 min. The supernatant was directly used in the PCRs.
VNTR.
The available genome sequences of reference E. coli strains K-12 (a commensal isolate, GenBank accession number NC_010473), CFT073 (a pyelonephritis isolate, GenBank accession number NC_004431), S88 (a meningitis isolate, GenBank accession number NC_011742), APECO1 (an avian pathogenic isolate, GenBank accession number NC_008563), UTI89 (a cystitis isolate, GenBank accession number NC_007946), and EDL933 and Sakai (both O157:H7 STEC strains, GenBank accession numbers NC_002655 and NC_002695, respectively) were used to search for VNTRs of interest among those previously published in the studies of Gorgé et al. (8), Lindstedt et al. (7), and Keys et al. (6), and VNTRs were identified using the research tool http://minisatellites.u-psud.fr (40). Each VNTR locus was selected (i) on the basis of its variability of repeats among the reference genomes and (ii) to have a weight range compatible with the weight ranges of the other VNTRs. A set of seven VNTRs was used for this study. Six VNTRs were previously described; notably, two were characterized by Gorgé et al. (8), three were from the study of Lindstedt et al. (7), and one was described by Keys et al. (6). In order to allow a correct visualization by maximizing the discrimination of the 7 bands on gel electrophoresis after amplification, we designed new primers for each VNTR locus using alignments of flanking sequences in E. coli reference genomes and the Primer3 software, available online (41). All the primers were selected so that the weight ranges allow a repartition of bands and minimize the risk of superposition on the electrophoresis gels. The main characteristics of the 7 selected VNTR loci are presented in Table 1.
MLVA typing.
All 7 VNTR regions were amplified by a single-tube multiplex PCR in a 50-μl volume using a Qiagen multiplex PCR kit (Qiagen, Valencia, CA) with Q solution, 5 μl of DNA extract (boiling lysis), and all primers at the concentrations indicated in Table 1. The PCR run conditions included a denaturation step of 95°C for 15 min, followed by 30 cycles of denaturation at 94°C for 30 s, elongation at 55°C for 90 s, and an extension step of 72°C for 90 s. A final extension of 72°C for 10 min was performed. The concentrations of the primers were adjusted to obtain the same intensities for all fragments on gel electrophoresis. Electrophoresis was performed in an ethidium bromide-stained 3% standard agarose gel for 75 min at 100 V. Each gel was flanked by two lanes loaded with a GeneRuler 100-bp DNA ladder (Thermo Scientific, Villebon-sur-Yvette, France) for normalization.
Stability of MLVA alleles.
Seven strains were subcultured on TSA plates 14 times over a period of 14 days. Every day, a colony was taken and streaked onto the next TSA plate. On the first day, the seventh day, and the last day, DNA from a bacterial sweep was extracted (see “Extraction of DNA” above) and subjected to the MLVA assay.
Phylogrouping, MLST, PFGE, and WGS.
The phylogrouping and MLST of the ECOR collection have been described elsewhere (42). PFGE analysis of XbaI-restricted total DNA of the isolates was performed according to a standardized protocol by the aNRC-EC at the Robert Debré Hospital (25). WGS of the 38 O1:K1 isolates was performed by the Illumina next-generation sequencing method (45).
Data analysis.
The PCR products obtained were subjected to agarose gel electrophoresis. The fingerprints of the PCR fragments were visualized using a Gel Doc 2000 system (Bio-Rad, Marnes-la-Coquette, France), and a TIFF image file of the gel used for electrophoresis was saved using QuantityOne software (Bio-Rad, Marnes-la-Coquette, France). In order to compare the fingerprints from different migrations, gel images were then loaded into a BioNumerics database (Applied Maths, Sint-Martens Latem, Belgium). The two molecular weight markers included in each gel (100-bp DNA ladder) enabled normalization between different migrations. The MLVA patterns were compared by use of a tolerance parameter of 1% and an optimization parameter of 0.5%. Dendrogram analysis by the unweighted pair group method using average linkages (UPGMA) and by use of the pairwise Dice similarity coefficient was performed with BioNumerics software (Applied Maths, Sint-Martens Latem, Belgium). Two profiles were considered identical when they displayed the same banding pattern (corresponding to >95% of similarity on the dendrograms). Discriminatory power was quantified by the Hunter and Gaston diversity index derived from Simpson’s index of diversity (D) (18). The concordance between typing methods was assessed by use of the adjusted Rand (AR) and Wallace (W) coefficients (43, 44). Statistical analysis (adjusted Rand and Wallace coefficients) was performed using the online tool of the Instituto de Medicina Molecular of the University of Lisbon (http://www.comparingpartitions.info/).
Supplementary Material
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AEM.02812-18.
REFERENCES
- 1.Johnson JR, Russo TA. 2002. Extraintestinal pathogenic Escherichia coli: “the other bad E coli.” J Lab Clin Med 139:155–162. [DOI] [PubMed] [Google Scholar]
- 2.Kosek M, Bern C, Guerrant RL. 2003. The global burden of diarrhoeal disease, as estimated from studies published between 1992 and 2000. Bull World Health Organ 81:197–204. [PMC free article] [PubMed] [Google Scholar]
- 3.Grad YH, Lipsitch M, Feldgarden M, Arachchi HM, Cerqueira GC, FitzGerald M, Godfrey P, Haas BJ, Murphy CI, Russ C, Sykes S, Walker BJ, Wortman JR, Young S, Zeng Q, Abouelleil A, Bochicchio J, Chauvin S, DeSmet T, Gujja S, McCowan C, Montmayeur A, Steelman S, Frimodt-Moller J, Petersen AM, Struve C, Krogfelt KA, Bingen E, Weill F-X, Lander ES, Nusbaum C, Birren BW, Hung DT, Hanage WP. 2012. Genomic epidemiology of the Escherichia coli O104:H4 outbreaks in Europe, 2011. Proc Natl Acad Sci U S A 109:3065–3070. doi: 10.1073/pnas.1121491109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nicolas-Chanoine MH, Bertrand X, Madec JY. 2014. Escherichia coli ST131, an intriguing clonal group. Clin Microbiol Rev 27:543–574. doi: 10.1128/CMR.00125-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van Belkum A, Struelens M, de Visser A, Verbrugh H, Tibayrenc M. 2001. Role of genomic typing in taxonomy, evolutionary genetics, and microbial epidemiology. Clin Microbiol Rev 14:547–560. doi: 10.1128/CMR.14.3.547-560.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Keys C, Kemper S, Keim P. 2005. Highly diverse variable number tandem repeat loci in the E. coli O157:H7 and O55:H7 genomes for high-resolution molecular typing. J Appl Microbiol 98:928–940. doi: 10.1111/j.1365-2672.2004.02532.x. [DOI] [PubMed] [Google Scholar]
- 7.Lindstedt BA, Brandal LT, Aas L, Vardund T, Kapperud G. 2007. Study of polymorphic variable-number of tandem repeats loci in the ECOR collection and in a set of pathogenic Escherichia coli and Shigella isolates for use in a genotyping assay. J Microbiol Methods 69:197–205. doi: 10.1016/j.mimet.2007.01.001. [DOI] [PubMed] [Google Scholar]
- 8.Gorgé O, Lopez S, Hilaire V, Lisanti O, Ramisse V, Vergnaud G. 2008. Selection and validation of a multilocus variable-number tandem-repeat analysis panel for typing Shigella spp. J Clin Microbiol 46:1026–1036. doi: 10.1128/JCM.02027-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Noller AC, McEllistrem MC, Antonio GF, Boxrud DJ, Harrison LH, Pacheco AGF. 2003. Multilocus variable-number tandem repeat analysis distinguishes outbreak and sporadic Escherichia coli O157:H7 isolates. J Clin Microbiol 41:5389–5397. doi: 10.1128/JCM.41.12.5389-5397.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hyytiä-Trees E, Smole SC, Fields PA, Swaminathan B, Ribot EM. 2006. Second generation subtyping: a proposed PulseNet protocol for multiple-locus variable-number tandem repeat analysis of Shiga toxin-producing Escherichia coli O157 (STEC O157). Foodborne Pathog Dis 3:118–131. doi: 10.1089/fpd.2006.3.118. [DOI] [PubMed] [Google Scholar]
- 11.Kawamori F, Hiroi M, Harada T, Ohata K, Sugiyama K, Masuda T, Ohashi N. 2008. Molecular typing of Japanese Escherichia coli O157:H7 isolates from clinical specimens by multilocus variable-number tandem repeat analysis and PFGE. J Med Microbiol 57:58–63. doi: 10.1099/jmm.0.47213-0. [DOI] [PubMed] [Google Scholar]
- 12.Manges AR, Tellis PA, Vincent C, Lifeso K, Geneau G, Reid-Smith RJ, Boerlin P. 2009. Multi-locus variable number tandem repeat analysis for Escherichia coli causing extraintestinal infections. J Microbiol Methods 79:211–213. doi: 10.1016/j.mimet.2009.09.006. [DOI] [PubMed] [Google Scholar]
- 13.Izumiya H, Pei Y, Terajima J, Ohnishi M, Hayashi T, Iyoda S, Watanabe H. 2010. New system for multilocus variable-number tandem-repeat analysis of the enterohemorrhagic Escherichia coli strains belonging to three major serogroups: O157, O26, and O111. Microbiol Immunol 54:569–577. doi: 10.1111/j.1348-0421.2010.00252.x. [DOI] [PubMed] [Google Scholar]
- 14.Løbersli I, Haugum K, Lindstedt BA. 2012. Rapid and high resolution genotyping of all Escherichia coli serotypes using 10 genomic repeat-containing loci. J Microbiol Methods 88:134–139. doi: 10.1016/j.mimet.2011.11.003. [DOI] [PubMed] [Google Scholar]
- 15.Krüger A, Lucchesi PMA, Sanso AM, Etcheverría AI, Bustamante AV, Burgán J, Fernández L, Fernández D, Leotta G, Friedrich AW, Padola NL, Rossen JWA. 2015. Genetic characterization of Shiga toxin-producing Escherichia coli O26: H11 strains isolated from animal, food, and clinical samples. Front Cell Infect Microbiol 5:74. doi: 10.3389/fcimb.2015.00074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Timmons C, Trees E, Ribot EM, Gerner-Smidt P, LaFon P, Im S, Ma LM. 2016. Multiple-locus variable-number tandem repeat analysis for strain discrimination of non-O157 Shiga toxin-producing Escherichia coli. J Microbiol Methods 125:70–80. doi: 10.1016/j.mimet.2016.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ochman H, Selander RK. 1984. Standard reference strains of Escherichia coli from natural populations. J Bacteriol 157:690–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hunter PR, Gaston MA. 1988. Numerical index of the discriminatory ability of typing systems: an application of Simpson’s index of diversity. J Clin Microbiol 26:2465–2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Espié E, Grimont F, Mariani-Kurkdjian P, Bouvet P, Haeghebaert S, Filliol I, Loirat C, Decludt B, Minh NNT, Vaillant V, de Valk H. 2008. Surveillance of hemolytic uremic syndrome in children less than 15 years of age, a system to monitor O157 and non-O157 Shiga toxin-producing Escherichia coli infections in France, 1996-2006. Pediatr Infect Dis J 27:595–601. doi: 10.1097/INF.0b013e31816a062f. [DOI] [PubMed] [Google Scholar]
- 20.Tenover FC, Arbeit RD, Goering RV, Mickelsen PA, Murray BE, Persing DH, Swaminathan B. 1995. Interpreting chromosomal DNA restriction patterns produced by pulsed-field gel electrophoresis: criteria for bacterial strain typing. J Clin Microbiol 33:2233–2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Basmaci R, Bonacorsi S, Bidet P, Biran V, Aujard Y, Bingen E, Béchet S, Cohen R, Levy C. 2015. Escherichia coli meningitis features in 325 children from 2001 to 2013 in France. Clin Infect Dis 61:779–786. doi: 10.1093/cid/civ367. [DOI] [PubMed] [Google Scholar]
- 22.Gordon DM, Geyik S, Clermont O, O’Brien CL, Huang S, Abayasekara C, Rajesh A, Kennedy K, Collignon P, Pavli P, Rodriguez C, Johnston BD, Johnson JR, Decousser J, Denamur E. 2017. Fine-scale structure analysis shows epidemic patterns of clonal complex 95, a cosmopolitan Escherichia coli lineage responsible for extraintestinal infection. mSphere 2:e00168-17. doi: 10.1128/mSphere.00168-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Birgy A, Levy C, Bidet P, Thollot F, Derkx V, Béchet S, Mariani-Kurkdjian P, Cohen R, Bonacorsi S. 2016. ESBL-producing Escherichia coli ST131 versus non-ST131: evolution and risk factors of carriage among French children in the community between 2010 and 2015. J Antimicrob Chemother 71:2949–2956. doi: 10.1093/jac/dkw219. [DOI] [PubMed] [Google Scholar]
- 24.LeClerc JE, Li B, Payne WL, Cebula TA. 1996. High mutation frequencies among Escherichia coli and Salmonella pathogens. Science 274:1208–1211. [DOI] [PubMed] [Google Scholar]
- 25.Bidet P, Mariani KP, Grimont F, Brahimi N, Courroux C, Grimont P, Bingen E. 2005. Characterization of Escherichia coli O157:H7 isolates causing haemolytic uraemic syndrome in France. J Med Microbiol 54:71–75. doi: 10.1099/jmm.0.45841-0. [DOI] [PubMed] [Google Scholar]
- 26.Shima K, Terajima J, Sato T, Nishimura K, Tamura K, Watanabe H, Takeda Y, Yamasaki S. 2004. Development of a PCR-restriction fragment length polymorphism assay for the epidemiological analysis of Shiga toxin-producing Escherichia coli. J Clin Microbiol 42:5205–5213. doi: 10.1128/JCM.42.11.5205-5213.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Martin IE, Tyler SD, Tyler KD, Khakhria R, Johnson WM. 1996. Evaluation of ribotyping as epidemiologic tool for typing Escherichia coli serogroup O157 isolates. J Clin Microbiol 34:720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dalla-Costa LM, Irino K, Rodrigues J, Rivera ING, Trabulsi LR. 1998. Characterisation of diarrhoeagenic Escherichia coli clones by ribotyping and ERIC-PCR. J Med Microbiol 47:227–234. doi: 10.1099/00222615-47-3-227. [DOI] [PubMed] [Google Scholar]
- 29.Munday CJ, Whitehead GM, Todd NJ, Campbell M, Hawkey PM. 2004. Predominance and genetic diversity of community- and hospital-acquired CTX-M extended-spectrum beta-lactamases in York, UK. J Antimicrob Chemother 54:628–633. doi: 10.1093/jac/dkh397. [DOI] [PubMed] [Google Scholar]
- 30.Bonacorsi S, Clermont O, Houdouin V, Cordevant C, Brahimi N, Marecat A, Tinsley C, Nassif X, Lange M, Bingen E. 2003. Molecular analysis and experimental virulence of French and North American Escherichia coli neonatal meningitis isolates: identification of a new virulent clone. J Infect Dis 187:1895–1906. doi: 10.1086/375347. [DOI] [PubMed] [Google Scholar]
- 31.Dombek PE, Johnson LK, Zimmerley ST, Sadowsky MJ. 2000. Use of repetitive DNA sequences and the PCR to differentiate Escherichia coli isolates from human and animal sources. Appl Environ Microbiol 66:2572–2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Onteniente L, Brisse S, Tassios PT, Vergnaud G. 2003. Evaluation of the polymorphisms associated with tandem repeats for Pseudomonas aeruginosa strain typing. J Clin Microbiol 41:4991–4997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Helldal L, Karami N, Welinder-Olsson C, Moore ERB, Åhren C. 2017. Evaluation of MLVA for epidemiological typing and outbreak detection of ESBL-producing Escherichia coli in Sweden. BMC Microbiol 17:8. doi: 10.1186/s12866-016-0922-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Baker KS, Dallman TJ, Behar A, Weill F-X, Gouali M, Sobel J, Fookes M, Valinsky L, Gal-Mor O, Connor TR, Nissan I, Bertrand S, Parkhill J, Jenkins C, Cohen D, Thomson NR. 2016. Travel- and community-based transmission of multidrug-resistant Shigella sonnei lineage among international orthodox Jewish communities. Emerg Infect Dis 22:1545–1553. doi: 10.3201/eid2209.151953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Holmes A, Allison L, Ward M, Dallman TJ, Clark R, Fawkes A, Murphy L, Hanson M. 2015. Utility of whole-genome sequencing of Escherichia coli O157 for outbreak detection and epidemiological surveillance. J Clin Microbiol 53:3565–3573. doi: 10.1128/JCM.01066-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Matsumura Y, Pitout JDD, Peirano G, DeVinney R, Noguchi T, Yamamoto M, Gomi R, Matsuda T, Nakano S, Nagao M, Tanaka M, Ichiyama S. 2017. Rapid identification of different Escherichia coli sequence type 131 clades. Antimicrob Agents Chemother 61:e00179-17. doi: 10.1128/AAC.00179-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.François P, Bonetti E-J, Fankhauser C, Baud D, Cherkaoui A, Schrenzel J, Harbarth S. 2017. Rapid identification of ST131 Escherichia coli by a novel multiplex real-time allelic discrimination assay. J Microbiol Methods 140:12–14. doi: 10.1016/j.mimet.2017.06.018. [DOI] [PubMed] [Google Scholar]
- 38.Harrison LB, Hanson ND. 2017. High-resolution melting analysis for rapid detection of sequence type 131 Escherichia coli. Antimicrob Agents Chemother 61:e00265-17. doi: 10.1128/AAC.00265-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Clermont O, Bonacorsi S, Bingen E. 2000. Rapid and simple determination of the Escherichia coli phylogenetic group. Appl Environ Microbiol 66:4555–4558. doi: 10.1128/AEM.66.10.4555-4558.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Denoeud F, Vergnaud G. 2004. Identification of polymorphic tandem repeats by direct comparison of genome sequence from different bacterial strains: a web-based resource. BMC Bioinformatics 5:4. doi: 10.1186/1471-2105-5-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Untergasser A, Cutcutache I, Koressaar T, Ye J, Faircloth BC, Remm M, Rozen SG. 2012. Primer3—new capabilities and interfaces. Nucleic Acids Res 40:e115. doi: 10.1093/nar/gks596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Clermont O, Gordon D, Denamur E. 2015. Guide to the various phylogenetic classification schemes for Escherichia coli and the correspondence among schemes. Microbiology 161:980–988. doi: 10.1099/mic.0.000063. [DOI] [PubMed] [Google Scholar]
- 43.Carriço JA, Silva-Costa C, Melo-Cristino J, Pinto FR, de Lencastre H, Almeida JS, Ramirez M. 2006. Illustration of a common framework for relating multiple typing methods by application to macrolide-resistant Streptococcus pyogenes. J Clin Microbiol 44:2524–2532. doi: 10.1128/JCM.02536-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Severiano A, Pinto FR, Ramirez M, Carriço JA. 2011. Adjusted Wallace coefficient as a measure of congruence between typing methods. J Clin Microbiol 49:3997–4000. doi: 10.1128/JCM.00624-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Geslain G, Birgy A, Adiba S, Magnan M, Courroux C, Levy C, Cohen R, Bidet P, Bonacorsi S. 17 January 2019. Genome sequencing of strains of the most prevalent clonal group of O1:K1:H7 Escherichia coli that causes neonatal meningitis in France. BMC Microbiol 19:17. doi: 10.1186/s12866-018-1376-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.