

Abstract
Hereditary spastic paraplegia 73 (SPG73) was currently identified in only one family with variant in the neuronal isoform of carnitine palmitoyl‐transferase 1C (CPT1C) gene. We described a new family, in which affected individuals exhibited pure hereditary spastic paraplegia with benign clinical course. Exome sequencing revealed a novel nonsense variant in the CPT1C gene. The level of CPT1C mutant transcript significantly decreased compared to that of wild‐type transcript, and can be recovered after cycloheximide administration, which indicated that nonsense‐mediated mRNA decay was a mechanism that might be responsible for the phenotype. Our findings expanded the clinical and genetic spectrum of SPG73.
Introduction
Hereditary spastic paraplegia (HSP) is a clinically and genetically heterogeneous group of disorders characterized by progressive spasticity and weakness of the lower limbs resulting from a length dependent degeneration of the corticospinal tract, ascending sensory fibers, spinocerebellar fibers, and neuronal cell bodies.1 The phenotype of HSPs can be pure with spasticity of lower limbs alone or complicated with additional symptoms such as peripheral neuropathy, epilepsy, ataxia, optic neuropathy, retinopathy, deafness, dementia, mental retardation, ichthyosis, and bulbar dysfunction.2, 3
The inherited modes of HSPs include autosomal dominant (AD), autosomal recessive (AR), or X‐linked recessive.4 At least 80 different genetic loci for HSPs (SPG1‐80) have been identified, and 60 genes are currently cloned.5 Among pure and dominantly inherited HSPs, heterozygous variant in the neuronal isoform of carnitine palmitoyl‐transferase (CPT1C) gene as the genetic cause of SPG73 is a new locus added to the genetic list of SPGs recently.6 However, definitive proof about the pathogenicity of CPT1C will require identification of more heterozygote variants in new HSP patients. Herein, we report that three patients present with pure SPG73 accompanied with a benign clinical course in an autosomal dominant family.
Methods
Information regarding age of onset, progression of disease, and clinical manifestations was collected. Multiple laboratory tests were examined. Cerebral and spinal Magnetic Resonance imaging (MRI) were conducted. All patients were examined by at least two neurologists after written informed consent. The research was approved by ethics committee of the Peking University People's Hospital.
Genetic test was initially conducted in the index patient through exome next generation sequencing (NGS). Targeted exon enrichment was performed using SureSelect Human All Exon V5 (Agilent Technologies, Santa Clara, CA, USA). This enriched library was sequenced on a Hiseq 2500 platform (Illumina, San Diego, CA) for paired‐end reads of 100 bp. The reads were aligned against the human reference genome (hg19) using the SOAPaligner program. The single‐nucleotide polymorphisms (SNPs) were identified using the SOAPsnp program. Subsequently, the reads were realigned to the reference genome by the BWA program, and insertions or deletions (InDels) were identified with the GATK program. Allele frequency and reported pathogenicity were assessed using the dbSNP138, Human Gene Mutation Database (HGMD), 1000 Genomes Project, and Exome Sequencing Project (ESP6500). DNA from all available family members was directly sequenced for cosegregation analysis. In addition, the dynamic (CAG)n numbers for SCA1, SCA2, SCA3, SCA6, SCA7, SCA8, SCA12, SCA17, and DRPLA were directly detected by PCR amplified fragments. The copy number variation in exons in the SPAST, REEP1, and ATL1 gene were examined by multiplex ligation‐dependent probe amplification (MLPA).
Plasmids harboring wild‐type GFP‐CPT1C or mutant GFP‐CPT1C were transfected into HEK 293T cells for over‐expression. To explore the presence of nonsense mediated mRNA decay, cells were split into two subcultures. One subculture remained untreated, whereas the other was exposed to 150 mg/ml cycloheximide. The fluorescence of GFP was directly observed in these subcultures. Quantitative PCR (qPCR) for relative quantification of CPT1C transcript levels was performed on cDNA using primers specific for the CPT1C and GAPDH gene. Measurements were normalized against the GAPDH gene. The level of mutant transcript was relatively quantified to the level of wild‐type transcript. The protein level of CPT1C was evaluated by a monoclonal antibody (H00126129‐P01, Novus Biologicals, Centennial, CO, USA).
Results
The index patient (II1, Table 1) was a 13‐year‐old girl who came from a nonconsanguineous family (Fig. 1A). She had a milestone delay of motor ability characterized as crawling at age 1, learning to walk at age 2, and walking independently at age 3.5. Before the age of 9, she gradually developed difficulty of walking steadily, weakness of lower limbs, and frequent falling‐down. Thereafter, she felt the aforementioned symptoms improved gradually, but still had muscle fatigue, stiffness, soreness, and clumsy gaits after walking with long‐distance. Her muscle strength was grade 4/5 (Medical Research Council Scale) in the foot dorsiflexion, grade 5‐/5 in the proximal lower limbs, and grade 5/5 in the upper limbs. The muscle tone of lower limbs increased. Deep tendon reflexes were brisk in the lower extremities. Babinski signs were elicited bilaterally. Sensation of vibration in the feet slightly decreased, but the other sensations were normal. No oculomotor disturbance, slow saccades, dysarthria, and ataxia signs were identified. Bilateral pes cavus was obvious. Neuropsychological battery tests revealed a normal cognitive status. Cerebral and spinal MRI were normal. Somatosensory evoked potentials indicated a delay latency and poor waveform differentiation, but peripheral nerve conduction velocities (NCV) were normal.
Table 1.
Clinical characteristics of affected individuals of the family
| Clinical variables | Patient II1 | Patient II2 | Patient I2 |
|---|---|---|---|
| Age at onset (years) | infancy | infancy | uncertainty |
| Age at walking (years) | 3.5 | 3 | 1 |
| Age at examinations (years) | 13 | 12 | 45 |
| Gender | female | male | female |
| Spasticity | + | + | − |
| Hyperreflexia | + | + | + |
| Plantar reflex | + | + | + |
| Muscle weakness | + | + | − |
| Muscle wasting | − | − | − |
| Vibration sensation | + | + | − |
| Urinary dysfunction | − | − | − |
| Pes cavus | + | + | − |
| Ataxia | − | − | − |
| Brain and spinal MRI | − | − | − |
| Neurophysiology | − | − | − |
| Progression | progression‐reversible | progression‐reversible | unnoticed |
Figure 1.
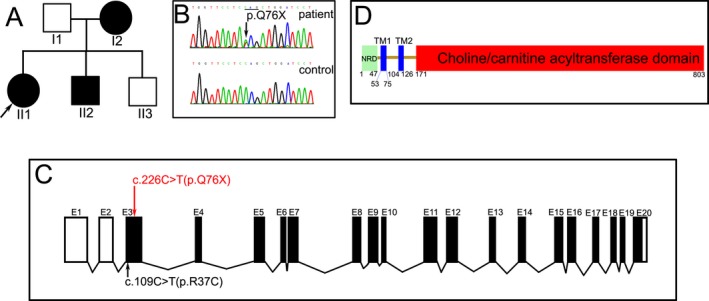
The clinical and genetic data in the SPG73 family. The family tree indicated an autosomal dominant inherited pattern (A, arrow indicates index patient). The chromagram revealed a variant (c.226C>T) in the CPT1C gene (B). The novel variant (red font) caused a premature stop in exon 3 of the CPT1C gene, the reported variant (black font) was also located in the exon 3 (C). CPT1C protein is composed of a small N‐terminal regulatory domain (NRD, 1‐47) and a large catalytic‐terminal domain (171‐803), separated by two transmembrane (TM) domains and a short connecting loop (D).
The younger brother (II2) of index patient was 12 years old. He presented with clinical features similar to that of the index patient. The symptoms insidiously progressed before 10 years old, then gradually improved in the recent 2 years, despite the fact that spasticity of lower limbs still affected his gaits. Physical examination revealed spasticity and hyperreflexia in lower limbs, mild weakness of foot dorsi/plantar flexion, and bilateral pes cavus. Babinski signs were elicited bilaterally. No oculomotor disturbance, slow saccades, dysarthria, and ataxia signs were identified. Cerebral and spinal MRI were normal. Somatosensory evoked potentials indicated a delay latency and poor waveform differentiation, but NCV was normal. Before the index patient was diagnosed as SPG73, the 45‐year‐old mother (I2) felt no neurological symptoms. Physical examinations revealed high‐brisk reflexes in both knees and a positive response of the right Oppenheimer sign, but no other extensor plantar responses, spasticity, gait abnormalities, and sensory loss were identified. The father and little younger brother (II3) were normal in clinical evaluations. No other family members were available to examine clinically.
A novel heterozygous variant (c.226C>T) resulting in a premature stop (p.Q76X, Fig. 1B) was detected in exon 3 of the CPT1C gene by exome NGS (Fig. 1C). The same variant was identified in her mother and younger brother, but not in her father and healthy little brother, which suggested the variant was closely segregated with the phenotype. The variant was not found in 200 healthy Chinese controls, 1000 genomes database, ExAC database, and gnomAD database. No other causative variants associated with other SPGs were found in the NGS data; no abnormal expansion of nucleotide repeats was identified in SCAs. No copy number variation in exons was found in the SPAST, REEP1, or ATL1 gene.
The GFP fluorescence showed an increased expression of control (Fig. 2A) and wild‐type CPT1C (Fig. 2B) in the 293T cells, but the expression of mutant CPT1C significantly decreased (Fig. 2C). After administration of cycloheximide, the expression of mutant CPT1C was recovered (Fig. 2D). Premature stop variant might induce mRNA degradation via nonsense‐mediated mRNA decay. 293T cells carrying the p.Q76X variant indeed contained only about 5% CPT1C mRNA transcript compared to that of wild‐type cells (Fig. 2E). Transcript levels could be increased after treatment with the translation inhibitor cycloheximide that blocks nonsense‐mediated mRNA decay. No full‐length or truncated CPT1C proteins were detected by immunoblot in the 293T cell homogenates (Fig. 2F).
Figure 2.
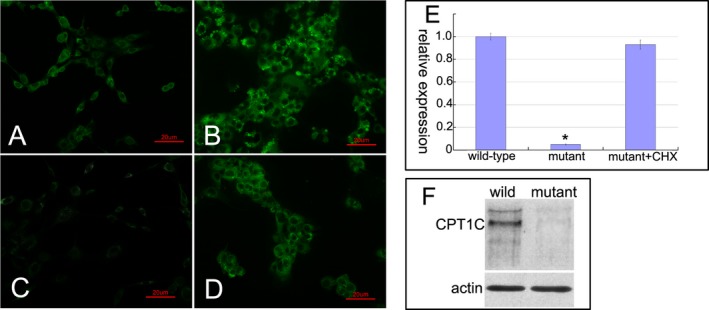
The pathogenic investigations of mutant CPT1C. The GFP fluorescence showed a normal expression in 293T cells transfected with pEGFP‐N1 vectors (A), an increased expression in 293T cells transfected with wild‐type pEGFP‐N1‐CPT1C vectors (B), and a significantly decreased expression in 293T cells transfected with c.226C>T mutant pEGFP‐N1‐CPT1C vectors (C). After administration of cycloheximide (CHX), the expression of mutant CPT1C was recovered (D). Quantitative PCR measurements showed a significant reduction in the mutant CPT1C transcript expression compared to the level of wild‐type transcript. Repeated three times for every test. Data were analyzed by one‐way ANOVA test; Error bars are SEM; *P < 0.01(E). Immunoblot showed that no full‐length or truncated CPT1C proteins were detected in the mutant cells (F).
Discussion
SPG73 has been considered a pure form of AD‐HSP characterized by adult‐onset progressive weakness and spasticity of the lower limbs, but the disease progression is relentless and will lead to loss of ambulation at 10 to 15 years after the onset of symptoms.6 Currently the clinical features of SPG73 were only described in an Italian family.6 Herein we reported the second SPG73 family with genetic confirmation. The affected patients presented with a typical phenotype of pure HSP characterized by spasticity of lower limbs and pes cavus, but they simultaneously showed some distinct characteristics. The index patient and her brother showed a motor milestone delay from infant period, and then exhibited a reversible course at the age of around 10. Conversely, the mother was only identified to have hyperreflexia and mild extensor plantar response without any other symptoms, which represented a putatively asymptomatic carrier. The phenotype of our patients indicated that the clinical course of SPG73 was relatively benign, despite the fact that the patients had congenital onset.
The list of genes responsible for HSPs is quickly expanding, mostly owing to the convenient availability of NGS method.7 The CPT1C gene responsible for SPG73 was reported in autosomal dominant HSP. Only a missense (p.R37C) in the CPT1C gene had been reported in an Italian family with pure HSP currently.6 In this study, we found a CPT1C variant in a Chinese family through NGS method. The nonsense mutation was a novel variant that was predicted to cause a premature stop, and closely segregated in the family. Therefore, the variant can be qualified as pathogenic according to the American College Medical Genetics and Genomics (ACMG) criteria.8
CPT1C encodes the neuronal isoform of the carnitine palmitoyl‐transferase enzyme that is mainly expressed in the soma and dendritic projections of motor neurons, and localizes at the endoplasmic reticulum (ER).9 CPT1C protein is composed of a small N‐terminal domain and a large catalytic‐terminal domain, separated by two transmembrane domains and a short connecting loop (Fig. 1D).10 Rinaldi et al. found that the missense mutation (p.R37C) would impair the function of N‐terminal regulatory domain, and markedly reduced the number and size of lipid droplets in the cortical neurons through a dominant negative mechanism.6 However, we found that the level of mutant CPT1C transcript significantly decreased compared to the level of wild‐type transcript, and can be recovered after treatment with cycloheximide that blocks nonsense‐mediated mRNA decay. The immunoblot study revealed that no full or truncated CPT1C proteins were detected. These findings indicated that the nonsense‐mediated mRNA decay might be responsible for the pathogenesis of SPG73 in our patients.11 The stop codon Pro76 is located at the last residue of the first transmembrane helix, and will lose the whole catalytic‐terminal domain. The C‐terminal choline/carnitine acyltransferase domain of CPT1C can directly interact with atlastin‐1 (SPG3A) in the network of ER, and assists in the formation and maintenance of long neuron processes, which suggest that abnormalities of ER morphogenesis may be a common pathogenesis of HSPs.12, 13
Carrasco et al. found that CPT1C‐deficient mice developed early onset progressive motor disturbances, including impaired gait and coordination, severe muscle weakness, and reduced locomotor activity.14 During mouse development, CPT1C levels were low in the cerebellum, striatum, and motor cortex from birth to postnatal day 10. Levels then increased gradually and peaked on postnatal day 21. In adulthood, CPT1C expression was substantially reduced in the striatum and cerebellum, but remained high in the motor cortex. The experimental evidence suggested that the availability of CPT1C protein was important to the development of neurons in the young individual, but the insufficiency of CPT1C protein function might be tolerated in the adult. The pattern of dynamic expression might partially explain the phenotype of motor milestone delay and reversible course with age in our patients.15
In conclusion, our findings provided additional patients to confirm that autosomal dominant SPG73 could be caused by the CPT1C variant. The clinical features exhibited pure form of HSP with benign clinical course. Nonsense‐mediated mRNA decay was a mechanism that might be responsible for the phenotype in our patients.
Author Contribution
H.D. contributed to research execution and manuscript composition; C. L. contributed to genetic evaluation. Z.S. and L.L contributed to cell and RNA experiments. X.Y. contributed to critical review and revision of manuscript; Z.J. contributed to design, conception, and manuscript composition.
Conflicts of Interest
None.
Acknowledgments
We thank the family and control individuals for their cooperation. This work was supported by the National Natural Science Foundation of China (81460199 and 81870996) and Peking University People's Hospital Research and Development Funds (RDX2018‐08).
Funding Information
This work was supported by the National Natural Science Foundation of China (81460199 and 81870996) and Peking University People's Hospital Research and Development Funds (RDX2018‐08).
Funding Statement
This work was funded by National Natural Science Foundation of China grants 81460199 and 81870996; Peking University People's Hospital Research and Development Funds grant RDX2018‐08.
References
- 1. Lo Giudice T, Lombardi F, Santorelli FM, et al. Hereditary spastic paraplegia: clinical‐genetic characteristics and evolving molecular mechanisms. Exp Neurol 2014;261:518–539. [DOI] [PubMed] [Google Scholar]
- 2. Kara E, Tucci A, Manzoni C, et al. Genetic and phenotypic characterization of complex hereditary spastic paraplegia. Brain 2016;139:1904–1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Blackstone C. Hereditary spastic paraplegia. Handb Clin Neurol 2018;148:633–652. [DOI] [PubMed] [Google Scholar]
- 4. Finsterer J, Löscher W, Quasthoff S, et al. Hereditary spastic paraplegias with autosomal dominant, recessive, X‐linked, or maternal trait of inheritance. J Neurol Sci 2012;318:1–18. [DOI] [PubMed] [Google Scholar]
- 5. Faber I, Pereira ER, Martinez ARM, et al. Hereditary spastic paraplegia from 1880 to 2017: an historical review. Arq Neuropsiquiatr 2017;75:813–818. [DOI] [PubMed] [Google Scholar]
- 6. Rinaldi C, Schmidt T, Situ AJ, et al. Mutation in CPT1C associated with pure autosomal dominant spastic paraplegia. JAMA Neurol 2015;72:561–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Qiu Y, Zhong S, Cong L, et al. A novel KIF5A gene variant causes spastic paraplegia and cerebellar ataxia. Ann Clin Transl Neurol 2018; 5:1415–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bush LW, Beck AE, Biesecker LG, et al. Professional responsibilities regarding the provision, publication, and dissemination of patient phenotypes in the context of clinical genetic and genomic testing: points to consider‐a statement of the American College of Medical Genetics and Genomics (ACMG). Genet Med 2018;20:169–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Price N, van der Leij F, Jackson V, et al. A novel brain‐expressed protein related to carnitine palmitoyltransferase I. Genomics 2002;80:433–442. [DOI] [PubMed] [Google Scholar]
- 10. Gratacòs‐Batlle E, Olivella M, Sánchez‐Fernández N, et al. Mechanisms of CPT1C‐dependent AMPAR trafficking enhancement. Front Mol Neurosci 2018;11:275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Baker KE, Parker R. Nonsense‐mediated mRNA decay: terminating erroneous gene expression. Curr Opin Cell Biol 2004;16:293–299. [DOI] [PubMed] [Google Scholar]
- 12. Blackstone C, O'Kane CJ, Reid E. Hereditary spastic paraplegias: membrane traffic and the motor pathway. Nat Rev Neurosci 2011;12:31–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hu J, Shibata Y, Zhu PP, et al. A class of dynamin‐like GTPases involved in the generation of the tubular ER network. Cell 2009;138:549–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Carrasco P, Jacas J, Sahún I, et al. Carnitine palmitoyltransferase 1C deficiency causes motor impairment and hypoactivity. Behav Brain Res 2013;256:291–297. [DOI] [PubMed] [Google Scholar]
- 15. Salinas S, Proukakis C, Crosby A, Warner TT. Hereditary spastic paraplegia: clinical features and pathogenetic mechanisms. Lancet Neurol 2008;7:1127–1138. [DOI] [PubMed] [Google Scholar]