

Abstract
Objective
Parkin is the causative gene for autosomal recessive familial Parkinson's disease (PD), although it remains unclear how parkin dysfunction is involved with the general condition. Recently, serum and/or plasma metabolomics revealed alterations in metabolic pathways that might reflect pathomechanisms of idiopathic PD (iPD). Thus, we hypothesized that serum metabolomics of patients with homozygous or compound heterozygous parkin mutations (namely, PARK2) might reflect metabolic alterations due to parkin dysfunction.
Methods
We enrolled 15 PARK2 patients (52 ± 17.6 years) confirmed with homozygous (seven cases) and compound heterozygous (eight cases) parkin mutations, along with 19 healthy age‐matched controls (51 ± 11.5 years). We analyzed 830 metabolites from participants’ serum using well‐established metabolomics technologies, including ultra‐high performance liquid chromatography/tandem mass spectroscopy.
Results
Based on metabolic profiles, hierarchical matrix analysis can divide samples between control and PARK2 subjects. Profiles from PARK2 patients showed significantly higher levels of fatty acid (FA) metabolites and oxidized lipids, and significantly lower levels of antioxidant, caffeine, and benzoate‐related metabolites.
Interpretation
Metabolomics can identify specific metabolic alterations in PARK2 patients compared with controls. Alterations in FA metabolites suggest a relationship between parkin function and lipid metabolism. The elevation of oxidized lipids in combination with decreasing antioxidants may reflect general hyperoxidative stress. Decreasing benzoate‐related metabolites might be due to the alteration in gut microbiota. Consequently, caffeine and its metabolites may be decreased due to malabsorption. These findings are similar to metabolic alterations in iPD. Thus, serum/plasma metabolomics may reflect the association between parkin dysfunction and parkinsonism.
Introduction
Mutations in the gene encoding parkin are the most common causative gene mutations for autosomal recessive familial Parkinson's disease (ARPD).1 Characteristic features of parkin‐linked PD (PARK2) include early onset parkinsonism, dramatic response to treatment with levodopa, dystonia, and hyperreflexia. However, there are no specific clinical signs that distinguish patients with PARK2 from those with idiopathic PD (iPD).2, 3 Moreover, selective dopaminergic neuronal degeneration in the substantia nigra pars compacta without Lewy body (LB) formation is considered a characteristic pathological finding of PARK2, although LB‐positive cases have also been reported.4, 5 Parkin functions as an E3 ubiquitin ligase in the ubiquitin–proteasome system (UPS),6 and monoubiquitinates and polyubiquitinates target proteins.7, 8 UPS is a protein degradation system, and LBs are strongly stained for anti‐ubiquitin antibodies. Therefore, dysfunction in this system may be highly associated with pathomechanisms of iPD. Furthermore, collaboration between parkin and PTEN‐induced kinase 1 (PINK1), which is the causative protein of the second‐most frequent ARPD, may be associated with mitochondrial quality control systems.9 Mitochondrial dysfunction and oxidative stress are also highly linked to the pathological condition of iPD.10 In this context, although PARK2 and iPD show different phenotypes, parkin can be considered a key player in the pathogenesis of both diseases. Thus, biomarkers in PARK2 patients may provide understanding on the pathomechanisms of both PARK2 and iPD.
Recently, we and other groups revealed that serum and/or plasma metabolomics are useful for identifying alterations in metabolic pathways associated with the pathological condition of iPD.11, 12, 13, 14, 15, 16, 17, 18 In iPD, pathological alterations in the peripheral nervous system, such as peripheral vagus nerve degeneration with α‐synuclein deposition, are observed from an early stage. Thus, generalized metabolic changes may play an important role in disease onset and progression. Nonetheless, it remains unclear how association between genetics and environmental factors contribute to metabolic changes in PD. We hypothesized that serum metabolomics from PARK2 patients may impact on specific metabolic pathways associated with the pathomechanisms of PD due to parkin dysfunction. Hence, in this study, we aimed to examine metabolic alterations in PARK2 patients compared with subjects without any neurological disorder.
Methods
Participants
The target population was PARK2 patients. The participants consisted of 15 subjects with PARK2 (six men, age 52 ± 17.6 years) and 19 healthy age‐matched controls (nine men, age 51 ± 11.5 years; Table 1). No participant had a history of cancer, asthma, aspiration pneumonia, or collagen vascular diseases. Participants suffering from acute infectious diseases or acute/chronic renal failure at sample collection were also excluded. PARK2 and control participants’ characteristics are shown in Table 1. The information collected included age, consumption of caffeine status, body mass index (BMI), disease duration, motor function, levodopa equivalent dose, and parkin mutations. Levodopa equivalent dose was calculated based on a previously reported systematic review.19 All PARK2 patients had been treated as outpatients at Juntendo University, Tokyo, Japan. Control participants were the patients’ spouses and volunteers who were free of neurological and psychiatric illnesses. Three movement disorder specialists (A.O., T.H., and N.H.) performed clinical assessments, and all PARK2 patients met the Movement Disorder Society Clinical Diagnostic criteria for PD.20 Genetic analysis revealed that all PARK2 patients carried homozygous (seven cases) and compound heterozygous (eight cases) mutations in parkin. Screening was also performed for polymorphisms of cytochrome P450 family 1, subfamily A (CYP1A) and cytochrome P450 family 2, subfamily E member 1 (CYP2E1), which are involved in metabolism of xenobiotics including caffeine and its metabolites. The genetic analysis methods have been previously described.1, 21, 22 Data were collected between March 2014 and April 2016.
Table 1.
Demographic and clinical characteristics of study participants
| Control | PARK2 | P‐values | |
|---|---|---|---|
| Numbers (M:F) | 19 (9:10) | 15 (6:9) | 0.741 |
| Age, Mean (SD) | 51 (11.5) | 52 (17.6) | 0.832 |
| Total Caffeine Consumption (mg/day), Mean (SD) | 153.7 (110.6) | 130.6 (107.6) | 0.542 |
| Coffee Consumption, (cups/day; number) |
6+; 1 4–5; 4 1–3; 12 0; 2 |
6+; 0 4–5; 8 1–3; 2 0; 5 |
|
| Body Mass Index, Mean (SD) | 23.8 (4.4) | 22.7 (4.1) | 0.752 |
| Disease Duration (years), Mean (SD) | 23.5 (12) | ||
| UPDRS part III, Mean (SD) | 20.3 (21.6) | ||
| Hoehn & Yahr, Mean (SD) | 2.37 (0.69) | ||
| LED (mg), Mean (SD) | 762 (398) | ||
| Parkin Mutations |
exon 2‐4 deletion (homo) (2 patients) exon 3 deletion (homo) exon 4 deletion (homo) exon 5 deletion (homo) exon 6 duplication (homo) exon 6‐7 deletion (homo) exon 2 deletion (hetero)/exon 4 deletion (hetero) exon 2‐3 deletion (hetero)/c.1358G>A (p.W453X) (hetero) exon 2‐4 deletion (hetero) /c.535‐3A>G (p.G179RfsX10) (hetero) exon 2‐4 deletion (hetero)/exon 5‐6 deletion (hetero) exon 3 deletion (hetero)/exon 4 deletion (hetero) exon 3 deletion (hetero)/c.535‐3A>G (p.G179RfsX10) (hetero) exon 3‐5 deletion (hetero)/exon 3‐7 duplication (hetero) exon 6‐7 deletion (hetero)/c.97C>T (p.R33X) (hetero) |
LED, levodopa equivalent dose; PARK2, parkin‐linked Parkinson's disease; UPDRS, unified Parkinson's Disease rating scale.
P‐values obtained by 1Chi‐squared and 2Student's t‐test.
Ethics statement
Written informed consent was obtained from all subjects participating in this study, according to the declaration of Helsinki. The study was approved by the Ethics Committee of the Juntendo University School of Medicine (No. 2018095).
Collection procedure
Venous blood samples for laboratory analysis were collected between 9:00 and 11:00 AM, following overnight fasting. Serum samples were collected and stored at −80°C until processing at Metabolon, Inc., (Durham, NC, USA).
Metabolomics analysis
Metabolomics analysis was performed in a manner similar to our previous study.15 Briefly, individual serum samples were subjected to methanol extraction, then split into aliquots for analysis by ultra‐high‐performance liquid chromatography/mass spectrometry (UHPLC/MS). Global biochemical profiling analysis consisted of four unique arms composed of reverse phase chromatography with positive ionization methods optimized for hydrophilic compounds (liquid chromatography/mass spectrometry [LC/MS] Pos. Polar) and hydrophobic compounds (LC/MS Pos. Lipid), reverse phase chromatography with negative ionization conditions (LC/MS Neg.), as well as a HILIC chromatography method coupled to negative (LC/MS Polar).23 All methods alternated between full scan MS and data‐dependent MSn scans. The scan range varied slightly between methods but generally covered 70–1000 m/z. All the serum samples were extracted at the same time using a 48 well format and run on the HPLC instruments at the same time, which should minimize the intra and inter‐assay variability associated with sample extraction and HPLC analysis (instrument drift) at different time points.
Metabolites were identified by automated comparison of ion features in experimental samples to a reference library of chemical standard entries that included retention time, molecular weight (m/z), preferred adducts, in‐source fragments, and associated MS spectra. Metabolites were curated by visual inspection for quality control using software developed at Metabolon. The identification of known chemical entities was based on comparison to metabolomic library entries of purified standards.24
Statistical analysis
Statistical analyses of clinical characteristics of participants and correlation between caffeine consumption and scaled intensity were performed using JMP11 for Mac OS (SAS Institute Inc., NC, USA) and Prism 6 for Mac OS (GraphPad Software, Inc., San Diego, CA, USA). All demographic and clinical variables were normally distributed, as determined using the Kolmogorov–Smirnov test. Demographic and clinical data were analyzed by Student's t‐test for continuous variables, chi‐squared test for categorical variables, and Spearman correlation analysis and analysis of covariance (ANCOVA) for correlation between caffeine serum concentration and consumption. Statistical significance was set at P < 0.05.
For comparison between metabolomics profiling results in PARK2 and control subjects, two types of statistical analyses were performed: (1) significance tests and (2) classification analysis. Standard statistical analyses were performed using ArrayStudio (OmicSoft/QIAGEN Bioinformatics, https://omicsoftdocs.github.io/ArraySuiteDoc/) on log‐transformed data. For nonstandard ArrayStudio analyses, R program (http://cran.r-project.org/) was used. Following log transformation and imputation of any missing values with the minimum observed value for each compound, Welch's two sample t‐test was used to determine significance (P < 0.05) and identify biochemicals that differed significantly between experimental groups. Estimated false discovery rate (q‐value) was calculated to account for multiple comparisons that normally occur in metabolomic‐based studies. For classification analysis, hierarchical matrix analysis (HMA) was used. For scaled intensity graphics, each biochemical in original scale (raw area count) was rescaled to set the median across all cases and time points to 1.
Results
Table 1 shows characteristics of the participants in our cohort. There were no differences in age, sex, BMI, or caffeine consumption between PARK2 and control subjects. We analyzed a total of 830 metabolites using methods that have already been well‐established in PARK2 and in control subjects. Each compound was identified by reference to entries in the Metabolon chemical spectra library.
HMA can divide samples between control and PARK2 subjects based on their metabolic profiles. Indeed, participants were mainly divided into two clusters using HMA. The main difference between the two clusters was driven by the subject's age. Especially, in the older age cluster, PARK2 segregated to individual branches of the dendrogram, suggesting modest segregation of metabolic profiles between control and PARK2 groups (Fig. 1). In this study, PARK2 patients were distributed in young, middle, elder age (Figure S1).
Figure 1.
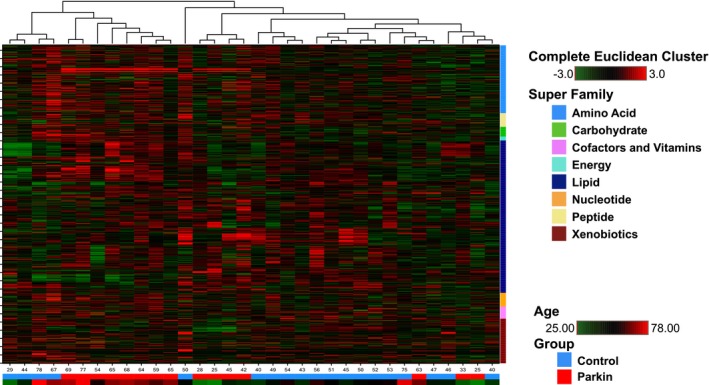
Hierarchical matrix dendrogram of metabolites in patients with parkin‐linked Parkinson's disease and normal subjects. Shades of red and green represent increased and decreased metabolite concentration, respectively, relative to median. Lower tables indicate participants group and age. Blue: normal control; and red: patients with homozygous or compound heterozygous parkin mutations and parameter of age. Shades of red and green (PARAM_AGE) indicate older and younger ages, respectively. Rows: metabolites; columns: patients.
Welch's two sample t‐test (PARK2 vs. control subjects) identified 162 statistically significant (P < 0.05) metabolites (75 increasing), and 58 approaching statistically significant (0.05 ≤ P < 0.1) metabolites (33 increasing) in PARK2 patients compared with normal controls (Table S1). We also examined q‐values, which describe FDR, with a low q‐value (q < 0.10) an indication of high confidence in a result when examining a list of metabolites obtained simultaneously. Within the list of these compounds, we highlighted molecules involved in a common metabolic pathway and/or residing in a similar biochemical family with other significant compounds.
We revealed alterations in metabolic compounds including tyrosine metabolites, fatty acids (FAs) with increasing ketone bodies, polyunsaturated fatty acids (PUFAs), oxidative stress‐related markers, caffeine metabolism, and benzoate metabolism. In the tyrosine metabolic pathway, levodopa metabolites, including 3‐methoxytyrosine (fold of control: 115.98, P < 0.00001), 3‐methoxytyramine sulfate (76.90, P < 0.00001), dopamine 4‐sulfate (42.28, P < 0.00001), and homovanillate (11.28, P < 0.00001) were significantly higher in PARK2 patients than controls (Table S1). Whereas tyrosine metabolites were not associated with the levodopa metabolic pathway, including tyrosine (0.97, P = 0.54), phenol sulfate (1.27, P = 0.99), 3‐(4‐hydroxyphenyl), lactate (0.95, P = 0.64), and tyramine O‐sulfate (0.67, P = 0.32), and consequently not statistically significant different between groups (Table S1).
Fatty acid metabolites
PARK2 patients showed significantly different levels of serum FAs and PUFAs compared with control subjects. The uptake of FAs and PUFAs is facilitated by CD36, which is a known FA translocase (Fig . 2A),25, 26, 27 while the major FA degradation pathway is mitochondrial fatty acid β‐oxidation (FAO)28 (Fig. 2A). Serum levels of long‐chain FAs were significantly higher in PARK2 patients than control subjects (Fig. 2B). Additionally, medium‐chain FAs, including caprylate (8:0) (1.74, P < 0.05), caprate (10:0) (1.56, P < 0.05), laurate (12:0) (1.98, P < 0.0005), and 5‐dodecenoate (12:1n7) (2.14, P < 0.05) were significantly higher in serum from PARK2 than in control subjects. Increased serum FA might ultimately facilitate FAO, as suggested by elevated levels of carnitine‐conjugated lipids, including stearoylcarnitine (C18) (1.3, P < 0.01) and nervonoylcarnitine (C24:1) (1.48, P < 0.05), accompanied by accumulation of ketone bodies, such as acetoacetate (1.83, P = 0.07) and 3‐hydroxybutyrate (2.57, P < 0.05) (Table S1). Compared with control subjects, PARK2 patients also exhibited higher levels of several PUFAs, including stearidonate (18:4n3) (1.84, P < 0.005), docosapentaenoate (22:5n6) (1.24, P < 0.05), docosadienoate (n6 DPA; 22:2n6) (1.37, P < 0.05), and dihomo‐linoleate (20:2n6) (1.39, P < 0.05) (Fig. 2B). Additionally, endocannabinoids, including palmitoyl ethanolamide (1.30, P < 0.01), N‐oleoyltaurine (1.67, P < 0.05), N‐stearoyltaurine (1.43, P < 0.01), and linoleoyl ethanolamide (1.37, P < 0.05) were significantly elevated in serum from PARK2 patients (Fig. 2B).
Figure 2.
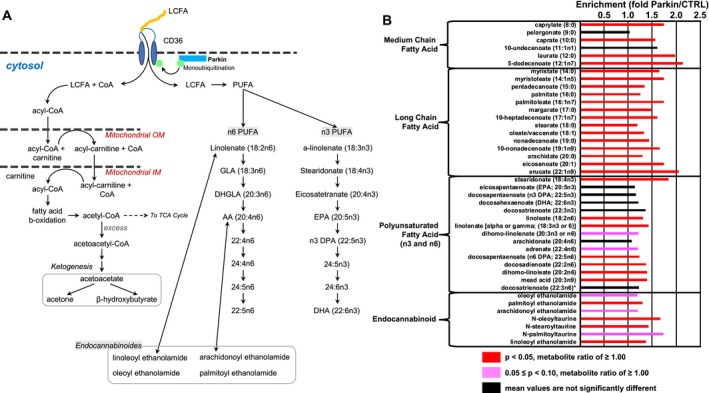
Alterations in fatty acids and related metabolites in patients with parkin‐linked Parkinson's disease. (A) CD36 is a long‐chain fatty acid (FA) transporter that is regulated by monoubiquitylation via parkin. The major degradation pathway of long‐ and medium‐chain FAs is mitochondrial fatty acid β‐oxidation. In the cytosol, FAs are activated to acyl‐coenzyme A (CoA) esters by acyl‐CoA synthetase. Acyl‐CoAs are imported to mitochondria via the carnitine shuttle, and degraded by β‐oxidation in mitochondria. Long‐chain FAs include polyunsaturated fatty acids (PUFAs), which can be classified into two families known as n3‐ and n6‐PUFAs, based on position of the double bond on the methyl terminal (n‐) end. n3‐ and n‐6 PUFAs are precursors for different classes of anti‐inflammatory or pro‐inflammatory eicosanoids, respectively. In addition, endocannabinoids are biosynthesized from PUFAs. (B) Several long‐ and medium‐chain FA, PUFAs, and their metabolites were significantly higher in Parkinson's disease due to parkin mutation. Red (P < 0.05) and pink (0.05 ≤ P < 0.10) columns indicate a significantly higher metabolite ratio and trending higher level of metabolites than controls, respectively. Black columns indicate no significant differences between patients and control subjects. AA, arachidonic acid; CTRL, control; DHA, docosahexaenoic acid; DHGLA, dihomo‐γ‐linolenic acid; DPA, docosapentaenoic acid; EPA, eicosapentaenoic acid; GLA, γ‐linolenic acid; IM, inner membrane; LCFA, long‐chain fatty acid; OM, outer membrane; Parkin, parkin‐linked Parkinson's disease; PUFA, polyunsaturated fatty acid; TCA cycle, tricarboxylic acid cycle.
Oxidative stress markers
We previously identified alterations in oxidative stress markers, such as bilirubin and biliverdin, in iPD.15 Considering the association between mitophagy and parkin function, we hypothesized that oxidative stress markers might also be altered in PARK2 patients. Accordingly, we found increasing levels of oxidized lipids, such as 2‐hydroxypalmitate (1.18, P < 0.05), 9‐hydroxystearate (1.42, P < 0.05), 2‐hydroxydecanoate (1.24, P = 0.06), and 2‐hydroxystearate (1.14, P = 0.087), along with 13‐hydroxyoctadecadienoic acid (HODE) + 9‐HODE (1.31, P = 0.085) in PARK2 patients. This suggests an increasingly oxidizing environment associated with parkin dysfunction. Consistently, γ‐carboxyethyl hydroxychroman (CEHC) (0.61, P < 0.005), which is a metabolite of γ‐tocopherol, and α‐tocopherol (0.84, P < 0.05) was reduced in serum samples of PARK2 patients. Indeed, this indicates reduced antioxidative capacity or exposure to more oxidative stress (Fig. 3).
Figure 3.
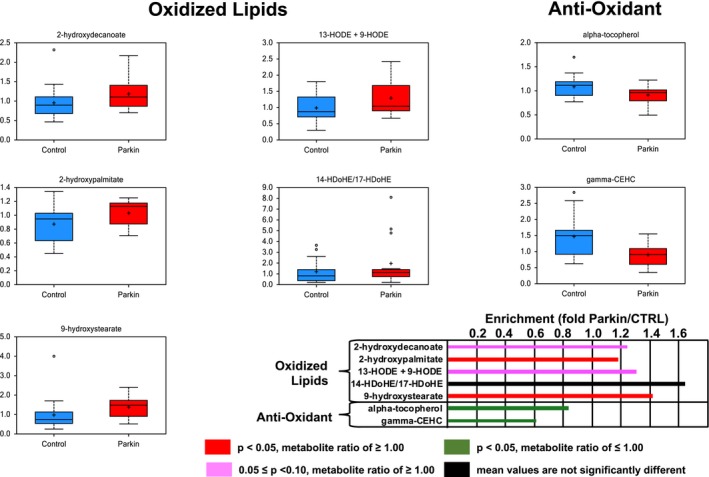
Alterations in redox‐related metabolites in patients with parkin‐linked Parkinson's disease. In patients, serum levels of oxidized lipids were higher, whereas antioxidant‐related metabolites, such as α‐tochopherol and γ‐CEHC, were lower than control subjects. Red (P < 0.05) and pink (0.05 ≤ P < 0.10) columns indicate a significantly higher metabolite ratio and trending higher level of metabolites than controls, respectively. Green (P < 0.05) columns indicate significantly lower metabolite ratio and a trending lower level of metabolites than controls, respectively. Black columns indicate no significant differences between patients and control subjects. γ‐CEHC, 2,7,8‐trimethyl‐2‐(2′‐carboxyethyl)‐6‐hydroxychroman; HDOHE, hydroxydocosahexaenoic acid; HODE, hydroxyoctadecadienoic acid; Parkin, parkin‐linked Parkinson's disease.
Hypoxanthine metabolites
Caffeine (0.25, P < 0.0005) and caffeine metabolites were significantly lower in PARK2 patients than in controls. Caffeine is metabolized into three primary metabolites: theophylline, theobromine, and paraxanthine (Fig. 4A). Although theobromine (0.94, P = 0.5), 3‐methylxanthine (0.8, P = 0.18), and 3,7‐dimethylurate (1.11, P = 0.98) were not significantly different between groups, theophylline and paraxanthine metabolites, including 1,3‐dimethylurate (0.43, P < 0.005), 1,7‐dimethylurate (0.4, P < 0.005), 1,3,7‐trimethylurate (0.26, P < 0.0005), 1‐methylxanthine (0.5, P < 0.0005), 5‐acetylamino‐6‐amino‐3‐methyluracil (0.45 P < 0.01), and 5‐acetylamino‐6‐formylamino‐3‐methyluracil (0.56, P < 0.05) were significantly lower in PARK2 patients (Fig. 4A and B). Meanwhile purine metabolites were not statistically different between groups (Table S1). Spearman correlation analysis and ANCOVA revealed a positive correlation between daily consumption and serum concentration of caffeine and its metabolites, including paraxanthine and theophylline, in control subjects (caffeine; ρ = 0.5462, P < 0.05, 95% confidence interval; 0.11–0.81, paraxanthine; ρ = −0.4622, P < 0.05, 95% confidence interval; 0–0.76, theophylline; ρ = 0.5206, P < 0.05, 95% confidence interval; 0.07–0.79), whereas no correlation was observed in PARK2 patients (caffeine; ρ = −0.302, P = 0.23, 95% confidence interval; −0.71–0.26, paraxanthine; ρ = −0.079, P = 0.71, 95% confidence interval; −0.58–0.46, theophylline; ρ = −0.201, P = 0.41, 95% confidence interval; −0.66–0.36) (Fig. 4C).
Figure 4.
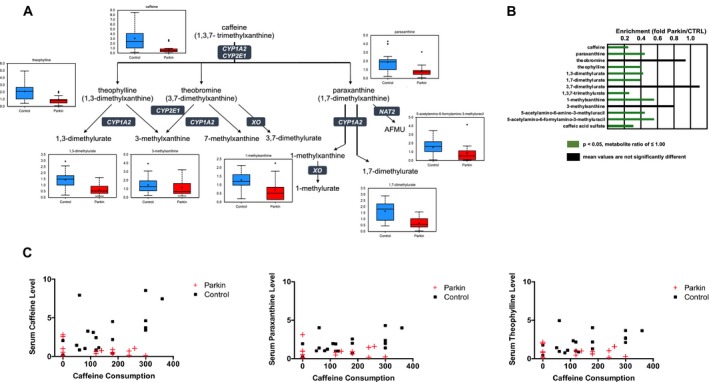
Alterations in xanthine metabolites in patients with parkin‐linked Parkinson's disease. (A) Caffeine is metabolized into three primary metabolites: theophylline, theobromine, and paraxanthine. (B) Significantly lower levels of caffeine and its metabolites were detected in serum from patients compared with controls. Green (P < 0.05) columns indicate a significantly lower metabolite ratio and trending lower level of metabolites than controls, respectively. Black columns indicate no significant differences between patients and control subjects. (C) Spearman correlation analysis revealed positive correlation between daily consumption and serum concentration of caffeine and its metabolites such as paraxanthine and theophylline in control subjects, whereas no correlation was observed in patients with parkin mutations. AFMU, 5‐acetylamino‐6‐formylamino‐3‐methyluracil; Parkin, parkin‐linked Parkinson's disease.
Although PARK2 was associated with lower levels of caffeine and its metabolites, the prevalence of single‐nucleotide polymorphisms (SNPs) of CYP1A2 (rs2470890) and CYP2E1 (rs2515641) was not different between PARK2 and control subjects.
Benzoate metabolism
The gut microbiome is suspected to play important roles in the pathomechanisms of several neurological disorders, including iPD.29 In our previous study, we identified altered serum levels of catechol sulfate in iPD, which were associated with gut microbial metabolism.15 Benzoate is a simple carboxylic acid and produced from microbial degradation of dietary aromatic compounds in the intestine (Fig. 5). Several benzoate metabolites, including hippurate (0.52, P < 0.05), 3‐hydroxyhippurate (0.39, P < 0.005), catechol sulfate (0.62, P < 0.05), guaiacol sulfate (0.45, P < 0.0005), 3‐methyl catechol sulfate (0.34, P < 0.005), 3‐(3‐hydroxyphenyl)propionate sulfate (0.5, P < 0.01), and 3‐(3‐hydroxyphenyl)propionate (0.49, P < 0.005), were reduced in serum samples from PARK2 patients (Fig. 5), consistent with our previous reports on iPD.15, 17
Figure 5.
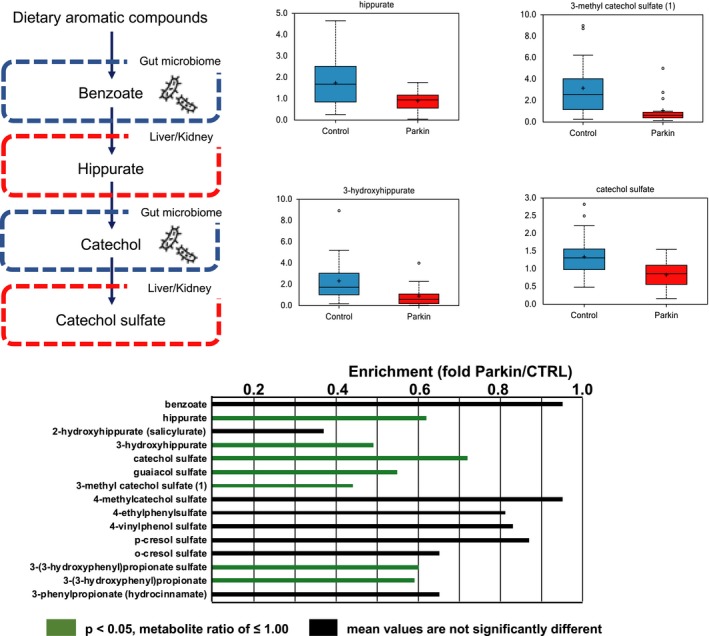
Alterations in benzoate and related metabolites in patients with parkin‐linked Parkinson's disease. Benzoate is produced from microbial degradation of dietary aromatic compounds in the intestine. The benzoate degradation pathway includes products of aromatic amino acids (phenylalanine, tyrosine, and tryptophan), as well as secondary bile acids, and is associated with microbial action in the gut. Significantly lower levels of benzoate and its metabolites were detected in patients than controls. Green (P < 0.05) columns indicate a significantly lower metabolite ratio and trending lower level of metabolites than controls, respectively. Black columns indicate no significant differences between patients and control subjects. Parkin, parkin‐linked Parkinson's disease.
Discussion
In this serum metabolomics study, we identified alterations in several metabolic pathways between PARK2 and control subjects. HMA analysis showed that the metabolic profiles of elderly PARK2 patients can be segregated into individual dendrogram branches. Aging and environmental factors may affect metabolic alterations associated with parkin dysfunction. In PARK2, FA metabolites, endocannabinoids, oxidative stress markers, caffeine metabolites, and derivatives of dietary aromatic compounds were significantly altered compared with normal subjects. Interestingly, metabolic alterations associated with PARK2 are similar to those of iPD.15, 17, 18 Considering the alterations in common metabolic pathways between PARK2 and iPD, it is still controversial whether the pathomechanisms of PARK2 are the same as those of iPD. Nonetheless, parkin dysfunction may play important roles in the pathomechanisms of not only PARK2 but also iPD.
We identified increasing levels of FA in serum from PARK2 patients compared with normal subjects. Recently, we and other groups have found that alterations in FA might be specifically associated with iPD progression.14, 16, 17, 18 Mitochondrial FAO is the major degradation pathway of free FA and an essential energy source under conditions of prolonged fasting, exercise, or metabolic stress. Impairment of FAO may reflect mitochondrial dysfunction in not only iPD,18 but also in PARK2. Additionally, we hypothesized that increasing FAs and PUFAs in PARK2 might be due to perturbation of membrane uptake of FA. CD36 is a candidate facilitator of very long‐chain FAs, including PUFAs. CD36 localizes on the plasma membrane of adipocytes, muscle cells, enterocytes, and hepatocytes, and delivers biologically active lipids to cells.30 Interestingly, Kim et al., reported that parkin ubiquitinates CD36 following the modulation of systemic fat uptake (Fig. 2A).31 CD36 is ubiquitinated by parkin and stabilized to promote FA uptake in the normal state. In contrast, parkin knockout mice and cultured cell models show a disabling of FA uptake and resistance to high‐fat diet‐induced fat accumulation in adipose and hepatic cells, which is attributable to decreasing levels of CD36. Moreover, parkin expression is not limited to the brain, but also expressed in diverse tissues in adult humans, including adipose tissue, skeletal muscle, and heart muscle (Expression Atlas EMBL‐EBI, Cambridge, UK; https://www.ebi.ac.uk/gxa/genes/ensg00000185345?bs=%7B%22homo%20sapiens%22%3A%5B%22ORGANISM_PART%22%5D%7D&ds=%7B%22kingdom%22%3A%5B%22animals%22%5D%7D#baseline). Therefore, the elevation of long‐chain FAs might be associated with lipid metabolic alternations due to parkin dysfunction in the adipose tissue, heart, and skeletal muscle. In our cohort, BMI was not significantly different between PARK2 and normal subjects. Despite the relatively small number of participants in our cohort, high serum FA levels might be related to difficulties in the uptake of FAs from serum into cells and tissues in PARK2.
Furthermore, elevated serum PUFAs in PARK2 belong to the n‐6 (omega‐6) series, which are linked to precursors for generation of inflammatory lipid mediators such as eicosanoids and leukotrienes. PUFAs are essential for neuronal function, including neuronal survival, neurogenesis, synaptic function, and the regulation of brain inflammation.26 Brain PUFAs are mostly derived from serum, therefore serum PUFAs directly affect brain lipid composition.26 Further, n6‐PUFAs are precursors to endocannabinoids, which are also increased in PARK2 patients compared with controls.32 As agonists of nuclear receptors such as peroxisome proliferator‐activated receptor alpha (PPARα), endocannabinoids may play important roles in antioxidant pathways.32 Notably, palmitoyl ethanolamide was reported to exhibit anti‐inflammatory properties, and potentially contribute to recovery from neurological damage.32 Therefore, elevating levels of metabolites in the endocannabinoid pathway might be due to generalized inflammation.
Consistent with hyperinflammation in PARK2 patients, metabolomics revealed an increase in oxidative stress and decreased levels of antioxidative stress markers in PARK2. Correlation between inflammation and oxidative stress owing to mitochondrial dysfunction is well‐known in dopaminergic neurodegeneration in iPD.33, 34 Energy production via mitochondrial oxidative phosphorylation is associated with induction of reactive oxygen species (ROS). ROS can react with membrane lipids, resulting in oxidized lipids.35 In contrast, α‐ and γ‐tocopherol are lipophilic antioxidant compounds that scavenge lipid peroxyl radicals and quench other free radicals, such as singlet O2, superoxide, and hydroxyl radicals.36 The state of hyperoxidative stress might induce mitochondrial damage. Parkin is recruited from the cytosol to depolarized mitochondria to mediate selective autophagic removal of damaged organelles associated with PINK1, known as mitophagy.9 Both parkin and PINK1 are distributed in diverse tissues in adult humans. Therefore, in PARK2, disruption of the mitochondrial quality control system might induce generalized oxidative stress. We previously reported alteration of oxidative markers in iPD, such as decreased serum biliverdin and γ‐CEHC, and increased bilirubin.15 These findings suggest that alteration in oxidative stress markers might be a common etiology of dopaminergic neuronal degeneration in both PARK2 and iPD.
Meta‐analysis of epidemiological studies revealed that coffee drinkers have a significantly lower risk of iPD.37 Therefore, Berg et al., suggested that decreasing caffeine consumption was one of the risks of prodromal iPD.38 Furthermore, we revealed that serum levels of caffeine and its metabolites are significantly decreased in iPD, despite no difference in caffeine consumption between patients and controls.15, 22 Interestingly, similar to iPD, our results here also indicate decreased serum levels of caffeine and its metabolites in PARK2. Caffeine is a known antagonist of adenosine A2A receptors, and might function against dopaminergic nigrostriatal toxicity in 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine (MPTP)‐induced PD models.39 In addition, istradefylline is an adenosine A2A receptor antagonist, and available for treatment of wearing‐off in PD.40 Therefore, decreasing serum levels of caffeine might be associated with disease modification in PD. The CYP1A2 and CYP2E1 enzymes are involved in the caffeine metabolic pathway (Fig. 4A). It is well‐known that SNPs of these enzymes affect the efficiency of caffeine metabolism.22 However, the prevalence of CYP1A2 and CYP2E1 SNPs was not different between PARK2 and control subjects (Table 2). Considering the low levels of caffeine and its metabolites, our results may reflect lower intake and/or lower absorption of substances with caffeine. In this cohort, there were no statistical differences in caffeine consumption based on the questionnaire between control and PARK2 subjects. In control subjects, there was a positive correlation between caffeine consumption and measured serum levels, whereas in PARK2, there was no correlation between the two, indicating that absorption or catabolism of caffeine might be perturbed.22 Although it remains unclear whether parkin directly participates in absorption and/or metabolism of caffeine, decreasing levels of caffeine and its metabolites in PARK2 might reflect dietary and gut microbial influences, as seen in iPD.22, 41 Interestingly, PARK2 patients exhibited alterations in the benzoate degradation pathway, which includes products of the aromatic amino acids, phenylalanine, tyrosine, and tryptophan, as well as secondary bile acids, and is associated with microbial action in the gut. In this study, catechol sulfate, o‐methyl catechol sulfate, and 3‐methyl catechol sulfate (which are end products of aromatic compound metabolism generated by the combined activity of gut microbial metabolism and the liver and kidney function) were lower in PARK2. Furthermore, our previous study revealed that iPD also showed alterations in the benzoate degradation pathway, similar to PARK2.15 In this context, association between specific changes in the gut microbiota and common pathomechanisms of both PARK2 and iPD might induce decreased caffeine absorption efficiency due to malabsorption.
Table 2.
Single‐nucleotide polymorphisms of CYP1A2 and CYP2E1
| Gene and SNP | cDNA | Amino acid | PARK2 | Control | ExAC MAF | P‐values |
|---|---|---|---|---|---|---|
| CYP1A2 | ||||||
| rs2470890 | NM_000761.4:c.1548T>C | NP_000752.2:p.Asn516Ans | 0.667 | 0.579 | 0.4574 | 0.728 |
| CYP2E1 | ||||||
| rs2515641 | NM_000773.3:c.1263C>T | NP_000764.1:p.Phe421Phe | 0.133 | 0.158 | 0.8302 | 1 |
SNP, single‐nucleotide polymorphism; PARK2, parkin‐linked Parkinson's disease; ExAC, Exome Aggregation Consortium. P‐value obtained by Fisher's exact test.
We need to consider several limitations in this study. First, patients with PARK2 were administered antiparkinsonian drugs, and samples from nonmedicated patients were not available for nontargeted metabolomics analysis. Therefore, we must consider the influence of drugs on metabolites and drug‐induced changes in metabolism. However, previous studies suggested medication effects for parkinsonism may not significantly impact the profiling of metabolism, with the exception of the tyrosine metabolic pathway.11, 15, 18 Further studies are needed to determine if dopaminergic treatments are potentially confounding factors. To minimize the effect of medications, the next step for investigating metabolic biomarkers of PARK2 would be to investigate differences between drug‐naïve PARK2 patients and nonmedicated control participants. A second limitation of our study was the small sample size for a case–control study, which was because of low prevalence of PARK2. Furthermore, metabolomics should be confirmed for intra and inter‐assay variability by investigation of two different cohorts. In our study, we could not compare variability between the different groups or different serum samples collected from same individuals. However, significantly altered metabolites in PARK2 often cluster within the same metabolic pathway. Additionally, our results are consistent with previous findings on metabolomic analyses or epidemiological investigations of iPD. Therefore, dopaminergic neuronal dysfunction might be associated with alterations in common metabolic pathways. Hence, despite these limitations, we believe that our results reflect alterations in metabolic pathways related to the pathomechanisms of PARK2.
Conclusions
This study provides an important insight and has identified several metabolic pathways altered in patients with PARK2 compared with controls. Although the results should be confirmed by other procedures such as cellular and animal models, these alterations in metabolic pathways are quite similar to those of iPD subjects, which were revealed by previous investigations. Therefore, these findings suggest that parkin dysfunction may perturb several metabolic pathways, signifying common pathomechanisms in PARK2 and iPD subjects. Our results will be a useful resource for the research field and further studies are required to determine specific mechanisms that link metabolic pathways to dopaminergic neurodegeneration in PD.
Author Contributions
T.H., S.Sai, and N.H. contributed to the conception and design of the study; A.O., T.H., S‐I.U., T.O., S.Sai, A.M., T.K., Y.O., K‐I.I., M.F., S.Sat, S.R., and R.P.M. contributed to the acquisition and analysis of data. All the authors contributed to drafting of the manuscript text.
Conflicts of Interest
Nothing to report.
Supporting information
Figure S1. Hierarchical matrix analysis sorted by age (on X‐axis).
Table S1. Heat map of statistically significant biochemicals profiled in this study.
Acknowledgments
We thank Drs. Yuuki Yonekawa, Yosuke Matsumoto (Summit Pharmaceuticals International Corporation), Dr. Hiroyo Yoshino, Ms. Yoko Imamichi (Juntendo University, School of Medicine), and all the participants in this study. This study was supported by the Brain Mapping by Integrated Neurotechnologies for Disease Studies Project (to N.H.); a Japan Science and Technology Agency grant for the creation of innovative technology for medical applications based on the global analyses and regulation of disease‐related metabolites (to N.H.); the Japan Agency for Medical Research and Development under Grant Number JP18ek0109393 (to N.H.); and Grants‐in Aid from the Research Committee of CNS Degenerative Disease, Research on Policy Planning and Evaluation for Rare and Intractable Diseases, Health, Labour and Welfare Sciences Research Grants, the Ministry of Health, Labour and Welfare, Japan (to N.H.).
Funding Information
This study was supported by the Brain Mapping by Integrated Neurotechnologies for Disease Studies Project (to N.H.); a Japan Science and Technology Agency grant for the creation of innovative technology for medical applications based on the global analyses and regulation of disease‐related metabolites (to N.H.); the Japan Agency for Medical Research and Development under Grant Number JP18ek0109393 (to N.H.); and Grants‐in Aid from the Research Committee of CNS Degenerative Disease, Research on Policy Planning and Evaluation for Rare and Intractable Diseases, Health, Labour and Welfare Sciences Research Grants, the Ministry of Health, Labour and Welfare, Japan (to N.H.).
Funding Statement
This work was funded by Japan Science and Technology Agency grant ; Japan Agency for Medical Research and Development grant JP18ek0109393; Ministry of Health, Labour and Welfare, Japan grant ; Brain Mapping by Integrated Neurotechnologies for Disease Studies Project grant ; Intractable Diseases, Health, Labour and Welfare Sciences Research grant ; Research Committee of CNS Degenerative Disease, Research on Policy Planning and Evaluation grant .
Contributor Information
Taku Hatano, Email: thatano@juntendo.ac.jp.
Nobutaka Hattori, Email: nhattori@juntendo.ac.jp.
References
- 1. Kitada T, Asakawa S, Hattori N, et al. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998;392:605–608. [DOI] [PubMed] [Google Scholar]
- 2. Doherty KM, Silveira‐Moriyama L, Parkkinen L, et al. Parkin disease: a clinicopathologic entity? JAMA Neurol 2013;70:571–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yamamura Y, Sobue I, Ando K, et al. Paralysis agitans of early onset with marked diurnal fluctuation of symptoms. Neurology 1973;23:239–244. [DOI] [PubMed] [Google Scholar]
- 4. Mori H, Kondo T, Yokochi M, et al. Pathologic and biochemical studies of juvenile parkinsonism linked to chromosome 6q. Neurology 1998;51:890–892. [DOI] [PubMed] [Google Scholar]
- 5. Miyakawa S, Ogino M, Funabe S, et al. Lewy body pathology in a patient with a homozygous parkin deletion. Mov Disord 2013;28:388–391. [DOI] [PubMed] [Google Scholar]
- 6. Shimura H, Hattori N, Kubo S, et al. Familial Parkinson disease gene product, parkin, is a ubiquitin‐protein ligase. Nat Genet 2000;25:302–305. [DOI] [PubMed] [Google Scholar]
- 7. Hatano T, Hattori N. Parkinson disease. ELS 2011. 10.1002/9780470015902.a0006026.pub2. [DOI] [Google Scholar]
- 8. Hattori N, Mizuno Y. Twenty years since the discovery of the parkin gene. J Neural Transm (Vienna) 2017;124:1037–1054. [DOI] [PubMed] [Google Scholar]
- 9. Pickrell AM, Youle RJ. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson's disease. Neuron 2015;85:257–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bose A, Beal MF. Mitochondrial dysfunction in Parkinson's disease. J Neurochem 2016;139(Suppl. 1):216–231. [DOI] [PubMed] [Google Scholar]
- 11. Bogdanov M, Matson WR, Wang L, et al. Metabolomic profiling to develop blood biomarkers for Parkinson's disease. Brain 2008;131:389–396. [DOI] [PubMed] [Google Scholar]
- 12. Ahmed SS, Santosh W, Kumar S, Christlet HT. Metabolic profiling of Parkinson's disease: evidence of biomarker from gene expression analysis and rapid neural network detection. J Biomed Sci 2009;16:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Johansen KK, Wang L, Aasly JO, et al. Metabolomic profiling in LRRK2‐related Parkinson's disease. PLoS ONE 2009;4:e7551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Roede JR, Uppal K, Park Y, et al. Serum metabolomics of slow vs. rapid motor progression Parkinson's disease: a pilot study. PLoS ONE 2013;8:e77629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hatano T, Saiki S, Okuzumi A, et al. Identification of novel biomarkers for Parkinson's disease by metabolomic technologies. J Neurol Neurosurg Psychiatry 2016;87:295–301. [DOI] [PubMed] [Google Scholar]
- 16. Burte F, Houghton D, Lowes H, et al. metabolic profiling of Parkinson's disease and mild cognitive impairment. Mov Disord 2017;32:927–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. LeWitt PA, Li J, Lu M, et al., Parkinson Study Group DI . Metabolomic biomarkers as strong correlates of Parkinson disease progression. Neurology 2017;88:862–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Saiki S, Hatano T, Fujimaki M, et al. Decreased long‐chain acylcarnitines from insufficient beta‐oxidation as potential early diagnostic markers for Parkinson's disease. Sci Rep 2017;7:7328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tomlinson CL, Stowe R, Patel S, et al. Systematic review of levodopa dose equivalency reporting in Parkinson's disease. Mov Disord 2010;25:2649–2653. [DOI] [PubMed] [Google Scholar]
- 20. Postuma RB, Berg D, Stern M, et al. MDS clinical diagnostic criteria for Parkinson's disease. Mov Disord 2015;30:1591–1601. [DOI] [PubMed] [Google Scholar]
- 21. Hatano T, Okuzumi A, Kamagata K, et al. Neuromelanin MRI is useful for monitoring motor complications in Parkinson's and PARK2 disease. J Neural Transm (Vienna) 2017;124:407–415. [DOI] [PubMed] [Google Scholar]
- 22. Fujimaki M, Saiki S, Li Y, et al. Serum caffeine and metabolites are reliable biomarkers of early Parkinson disease. Neurology 2018;90:e404–e411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Evans AM, Bridgewater BR, Liu Q, et al. High resolution mass spectrometry improves data quantity and quality as compared to unit mass resolution mass spectrometry in high‐throughput profiling metabolomics. Metabolomics 2014;4:132 10.4172/2153-0769.1000132. [DOI] [Google Scholar]
- 24. Dehaven CD, Evans AM, Dai H, Lawton KA. Organization of GC/MS and LC/MS metabolomics data into chemical libraries. J ChemInform 2010;2:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. .23w?>Su X , Abumrad NA. Cellular fatty acid uptake: a pathway under construction. Trends Endocrinol Metab 2009;20:72–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bazinet RP, Laye S. Polyunsaturated fatty acids and their metabolites in brain function and disease. Nat Rev Neurosci 2014;15:771–785. [DOI] [PubMed] [Google Scholar]
- 27. Liu JJ, Green P, John Mann J, et al. Pathways of polyunsaturated fatty acid utilization: implications for brain function in neuropsychiatric health and disease. Brain Res 2015;1597:220–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Houten SM, Violante S, Ventura FV, Wanders RJ. The biochemistry and physiology of mitochondrial fatty acid beta‐oxidation and its genetic disorders. Annu Rev Physiol 2016;78:23–44. [DOI] [PubMed] [Google Scholar]
- 29. Tremlett H, Bauer KC, Appel‐Cresswell S, et al. The gut microbiome in human neurological disease: a review. Ann Neurol 2017;81:369–382. [DOI] [PubMed] [Google Scholar]
- 30. Silverstein RL, Febbraio M. CD36, a scavenger receptor involved in immunity, metabolism, angiogenesis, and behavior. Sci Signal 2009;2:re3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kim KY, Stevens MV, Akter MH, et al. Parkin is a lipid‐responsive regulator of fat uptake in mice and mutant human cells. J Clin Invest 2011;121:3701–3712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Piomelli D, Sasso O. Peripheral gating of pain signals by endogenous lipid mediators. Nat Neurosci 2014;17:164–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hattori N, Tanaka M, Ozawa T, Mizuno Y. Immunohistochemical studies on complexes I, II, III, and IV of mitochondria in Parkinson's disease. Ann Neurol 1991;30:563–571. [DOI] [PubMed] [Google Scholar]
- 34. Yoritaka A, Hattori N, Uchida K, et al. Immunohistochemical detection of 4‐hydroxynonenal protein adducts in Parkinson disease. Proc Natl Acad Sci USA 1996;93:2696–2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Navarro A, Boveris A. The mitochondrial energy transduction system and the aging process. Am J Physiol Cell Physiol 2007;292:C670–C686. [DOI] [PubMed] [Google Scholar]
- 36. Bostanci MO, Bas O, Bagirici F. Alpha‐tocopherol decreases iron‐induced hippocampal and nigral neuron loss. Cell Mol Neurobiol 2010;30:389–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hernan MA, Takkouche B, Caamano‐Isorna F, Gestal‐Otero JJ. A meta‐analysis of coffee drinking, cigarette smoking, and the risk of Parkinson's disease. Ann Neurol 2002;52:276–284. [DOI] [PubMed] [Google Scholar]
- 38. Berg D, Postuma RB, Adler CH, et al. MDS research criteria for prodromal Parkinson's disease. Mov Disord 2015;30:1600–1611. [DOI] [PubMed] [Google Scholar]
- 39. Chen JF, Xu K, Petzer JP, et al. Neuroprotection by caffeine and A(2A) adenosine receptor inactivation in a model of Parkinson's disease. J Neurosci 2001;21:RC143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mizuno Y, Hasegawa K, Kondo T, et al., Japanese Istradefylline Study G . Clinical efficacy of istradefylline (KW‐6002) in Parkinson's disease: a randomized, controlled study. Mov Disord 2010;25:1437–1443. [DOI] [PubMed] [Google Scholar]
- 41. Sampson TR, Debelius JW, Thron T, et al. Gut microbiota regulate motor deficits and neuroinflammation in a model of Parkinson's disease. Cell 2016;167:1469–1480 e1412 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Hierarchical matrix analysis sorted by age (on X‐axis).
Table S1. Heat map of statistically significant biochemicals profiled in this study.