

Abstract
Objective
Amyloid‐beta oligomers (Aßo) trigger the development of Alzheimer's disease (AD) pathophysiology. Cellular prion protein (PrPC) initiates synaptic damage as a high affinity receptor for Aßo. Here, we evaluated the preclinical therapeutic efficacy of a fully human monoclonal antibody against PrPC. This AZ59 antibody selectively targets the Aβo binding site in the amino‐terminal unstructured domain of PrPC to avoid any potential risk of direct toxicity.
Methods
Potency of AZ59 was evaluated by binding to PrPC, blockade of Aβo interaction and interruption of Aβo signaling. AZ59 was administered to mice by weekly intraperitoneal dosing and brain antibody measured. APP/PS1 transgenic mice were treated with AZ59 and assessed by memory tests, by brain biochemistry and by histochemistry for Aß, gliosis and synaptic density.
Results
AZ59 binds PrPC with 100 pmol/L affinity and blocks human brain Aßo binding to PrPC, as well as prevents synaptotoxic signaling. Weekly i.p. dosing of 20 mg/kg AZ59 in a murine form achieves trough brain antibody levels greater than 10 nmol/L. Aged symptomatic APP/PS1 transgenic mice treated with AZ59 for 5–7 weeks show a full rescue of behavioral and synaptic loss phenotypes. This recovery occurs without clearance of plaque pathology or elimination of gliosis. AZ59 treatment also normalizes synaptic signaling abnormalities in transgenic brain. These benefits are dose‐dependent and persist for at least 1 month after the last dose.
Interpretation
Preclinical data demonstrate that systemic AZ59 therapy rescues central synapses and memory function from transgenic Alzheimer's disease pathology, supporting a disease‐modifying therapeutic potential.
Introduction
Amyloid‐ß (Aβ) peptide accumulation as senile plaque defines Alzheimer's Disease (AD) pathologically and occurs as an early triggering step in Alzheimer's Disease (AD).1 Moreover, early onset familial AD is inherited with high penetrance through mutations that increase Aβ42 production.1 Multiple studies have demonstrated that soluble Aβ oligomers (Aßo), rather than the relatively inert plaque accumulation, are responsible for AD‐related dysfunction and initiate synaptotoxicity.2, 3, 4 Once triggered by Aßo‐induced synaptic damage, AD pathophysiology expands to include Tau pathology as well as inflammatory and gliotic amplification. Despite the triggering role of Aß, clinical efforts to reduce amyloid burden, either by inhibiting production or promoting clearance, have failed.5, 6
Cellular Prion Protein (PrPC), a glycol‐phosphatidyl‐inositol‐anchored protein highly expressed in neurons, acts as a high‐affinity receptor for Aβo and is required for learning, memory, and synaptic deficits in APP/PS1 and Tg2576 mice,4, 7, 8, 9 though not in all AD models10 (reviewed in11). Downstream components of the Aβo‐PrPC pathway have been elucidated and successfully targeted in APP/PS1 mice, including mGluR5,12, 13, 14, 15, 16 Fyn kinase,17, 18 and the human AD risk gene product, Pyk2 kinase.15, 17 Interventions targeting PrPC, Fyn, or mGluR5 do not alter Aβ levels, demonstrating that the Aβo‐PrPC pathway can be beneficially modified without altering plaque load per se. PrPC is an attractive therapeutic target for AD because neither selective CNS ablation nor constitutive ablation of PrPC expression in mice has detectable deleterious effects.19
Targeting PrPC, either by genetic deletion of Prnp 4, 7, 8, 9 or binding by anti‐PrPC monoclonal antibodies, has shown promise preclinically.20, 21, 22, 23, 24, 25 However, antibody studies have been limited to acute electrophysiological outcomes, to intracerebral doses or to large acute doses systemically. While potential benefit from therapeutically targeting PrPC for AD, or for transmissible spongiform encephalopathy, is recognized, there is debate over whether engagement of PrPC via monoclonal antibodies is neurotoxic.26, 27, 28, 29, 30, 31 Binding of PrPC by certain antibodies may mimic scrapie (PrPSc) pathology and cause cell death.30, 31 In particular, antibodies targeting the globular domain of PrPC are implicated and require the N‐terminal tail to transmit toxicity.30, 31 While targeting PrPC with antibodies might be damaging, antibody interaction with the Aβo binding domain near the N‐terminus7 avoids toxicity and is protective against any C‐terminally mediated effects.30, 31
Here, we develop a high potency fully human AZ59 antibody selectively targeting the Aßo binding site in the natively unstructured amino‐terminal sequence of PrPC. We confirm that a murine version of this antibody reaches the brain after chronic low dose systemic administration and displays no toxic effects in mice. Critically, weekly AZ59 dosing rescues learning, memory, and synaptic deficits in APP/PS1 mice, and beneficial effects are maintained after a one‐month washout. The beneficial action is specific to PrPC‐Aβo synaptic signaling, as astrocytosis, microgliosis, and plaque load are unaltered by AZ59 treatment. Thus, AZ59 provides a potential therapeutic for AD to protect synapses from Aßo damage, an approach which is fully separate from Aß‐lowering strategies.
Materials and Methods
AZ59 antibody
Identification and production of the anti‐PrPC antibody AZ59 has been described in published patent application WO2017178288 from AstraZeneca. In brief, anti‐PrPC antibodies were isolated from an M13‐based human single‐chain variable fragments (scFv)‐phage display library32 using an alternating series of selections against biotinylated human and cynomolgus PrPC(23‐111). Isolated ScFv clones were converted to null effector function IgG1‐TM whole immunoglobulin by subcloning into human VH and VL vectors.33 Vectors were transiently transfected into a CHO mammalian expression system and IgG purified using Protein A chromatography. Initial clones were affinity matured using a targeted CDR mutagenesis approach. The combination of a particular VH clone and a VL clone formed a new clone referred to as “AZ59”. Unless specifically stated as using human AZ59, all studies here utilized a murine version of AZ59 in which the variable regions of AZ59 were transferred to the murine IgG1 backbone. The control monoclonal IgG is isotype matched and is non‐reactive with mouse antigens.
Transgenic and control mouse strains
Mice were cared for by the Yale Animal Resource Center and all experiments were approved by Yale's Institutional Animal Care and Use Committee. Wild type and APP‐SWE/PSEN1∆E9 mice were purchased from Jackson Laboratory (Bar Harbor, ME) and maintained on a C57/Bl6J background as described previously.8, 16, 18 Mice were group housed (2–5 mice per cage) in a 12‐h light/dark cycle.
Aßo preparation
Aßo‐containing Tris‐buffered‐saline‐(TBS)‐soluble lysates from human AD autopsy brain were prepared as described.4, 18 All experiments utilized deidentified samples and were approved as exempt by Yale's Human Investigational Committee. Synthetic Aβ1‐42 peptide was obtained as lyophilized powder from Keck Large Scale Peptide Synthesis Facility (Yale University). Preparation and characterization of Aβ1‐42 oligomers (Aβo) have been described previously.18 Aβo was prepared in glutamate‐free F12 medium to avoid direct stimulation of glutamate receptors. Concentrations of Aβo are expressed in monomer equivalents, with 1 μmol/L total Aβ1‐42 peptide corresponding to approximately 10 nmol/L oligomeric species.7
Detection of anti‐PrPC antibody
MaxiSorp 384 well white microplates (ThermoFisher Scientific, 460372) were coated overnight with 20 μL/well of 250 nM full‐length PrPC(23‐230) in 30 mmol/L Na2CO3, 80 mmol/L NaHCO3, pH 9.6, at 4°C. After washing three times with PBST (PBS, 0.05% Tween 20), the plates were blocked with 100 μL/well protein‐free T20 PBS blocking buffer (Pierce, 37573) for 1 h at room temperature. After washing three times with PBST, 20 μL of TBS‐ and RIPA‐soluble brain homogenate was applied to microplates in triplicate and incubated overnight at 4°C. Serial dilutions of AZ59 were prepared in naïve TBS and RIPA extract as a standard curve. Plates were then washed three times with PBST and incubated with 20 μL/well of 1:8000 dilution of Eu‐N1 goat anti‐mouse IgG (PerkinElmer Life Sciences, AD0124) in DELFIA assay buffer (PerkinElmer Life Sciences) for 1 h. Finally, after washing three times in PBST, 20 μL of DELFIA Enhancement Solution (PerkinElmer Life Sciences) was applied to each well, and time‐resolved europium fluorescence was measured using a Victor 3V plate reader (PerkinElmer Life Sciences) as described.4, 18
Aßo and mGluR5 binding to immobilized PrPC
The binding of synthetic Aßo or human autopsy brain TBS‐soluble Aß to plate‐immobilized PrPC(23‐111) or PrPC(23‐230) was measured by Prion‐Linked ImmunoSorbent Assay (PLISA) as described4, 18 but with the addition of AZ59 immediately prior to Aß‐containing fractions. In order to measure mGluR5 soluble ectodomain binding to immobilized full length PrPC(23‐230), the same assay format was utilized with substitution of mGluR5 protein fragment in place of Aßo. The mGluR5 protein was collected from HEK‐293T cell medium after transfection with a secreted protein expression vector for C‐terminally Myc‐His–tagged ectodomain of rat mGluR5(aa21‐578). The vector encodes the signal peptide of IgG‐Kappa chain fused in frame with aa21‐578 of rat mGluR5 with Myc‐His fused at the C‐terminus. The mGluR5 protein was purified by Nickel‐resin affinity chromatography to greater than 95% homogeneity by Coommassie stain of SDS‐PAGE. After binding of mGluR5 ectodomain to full length PrPC(23‐230)‐coated plates with or without the addition of AZ59, bound mGluR5 was detected by rabbit anti‐Myc antibody.
Aßo level in mouse brain
The presence of Aßo in TBS‐soluble ultracentrifuged homogenates of mouse brain was measured by capture with plate‐immobilized PrPC(23‐111) followed by detection with rabbit anti‐Aß antibody, PLISA, as described.4, 18 The capture reagent is in greater than 100‐fold molar excess over Aßo and endogenous PrPC in this assay format.
Biolayer Interferometry
Full‐length PrPC(23‐230) and PrPC(23‐111) protein were biotinylated by incubation with N‐hydroxy‐succinimide‐activated biotin and free biotin was removed. The biotin‐PrPC was immobilized on streptavidin‐coated BioLayer Interferometry (BLI) probes (Pall ForteBio, 18‐5019). Interaction with different concentrations of AZ59 in PBS in 0.05% Tween 20 (Sigma, P7949) was detected and analyzed by an OctetRed instrument in 96 well format using ForteBio software.
Cell binding experiments
Cos‐7 cells were grown at 37˚C with 5% CO2 in DMEM (Gibco 11965) supplemented with 10% FBS and 1% Penicillin‐Streptomycin (Gibco 15140). Transfections were performed using Lipofectamine 3000 transfection reagent (Thermo L3000015). For binding studies, the cells were seeded and transfected in Lab‐tek II chamber slides (Nunc 154941). Transfected cells were washed twice with ice‐cold F‐12 (Atlanta biologicals M15350) and incubated with AZ59 antibody or mouse IgG antibody 100 nmol/L for 30 min. For Aßo antagonism experiments, cells were subsequently co‐incubated with biotin‐Aßo (1 μmol/L monomer equivalent) and AZ59 or IgG control for 1 h. Cells were subsequently washed three times with PBS, fixed with 3.7% formaldehyde (J.T. Baker 2106) in PBS, and blocked with 5% normal donkey serum (Jackson ImmunoResearch 017‐000‐121). For AZ59 binding experiments, hPrPC was detected with 1:1000 chicken anti‐PrPC (Aves PRN) and 1:1000 donkey anti‐chicken FITC (Invitrogen SA1‐72000). AZ59 and mouse IgG control were detected with 1:1000 donkey anti‐mouse alexa fluor 568 (Life technologies A10037). For Aßo antagonism experiments, myc‐tagged hPrP was detected with 1:1000 rabbit anti‐myc (CST 5605S), and 1:1000 donkey anti‐rabbit alexa fluor 488 (Life technologies A21206). Biotin‐Aßo was detected with 1:1000 streptavidin alexa fluor 568 (Life technologies S11226). After washing, vectashield with DAPI (Vector H‐1200) was added and the slide was covered with a coverslip (Fisher 12‐545‐E).
Co‐Immunoprecipitation of PrPC and Myc‐mGluR5
The co‐IP of PrPC and Myc‐mGluR5 was previously described.12 HEK‐293T cells were maintained in Dulbecco's Modified Eagle Medium (DMEM), supplied with 10% fetal calf serum (FCS), 1% L‐glutamine (2 mmol/L), 1% sodium pyruvate (1 mmol/L), and 1% penicillin/streptomycin (100 U/mL). Cells were transfected for 36 h using Lipofectamine 2000 transfection reagent (Invitrogen). AZ59, 6D11, and control IgG were applied to the cells for 3 h before 1 μmol/L Aβo was added to the plates for an additional hour; all treatment was done at 4˚C. To prepare detergent solubilized cell lysates, cells were rinsed with ice‐cold PBS and solubilized in RIPA buffer containing complete protease inhibitor cocktail (Roche) and phosSTOP phosphatase inhibitor cocktail (Roche). The insoluble fraction was removed by centrifugation at 20,000g and the supernatant was used for immunoprecipitation. One microgram of capture antibody was incubated overnight at 4°C with 1 mg of detergent solubilized lysate protein with continuous mixing. The antibodies used were anti‐Myc (SigmaAldrich, C3956) and SAF32 (Cayman, 189720) with capture by PureProteome Protein A/G Mix Magnetic Beads (Millipore, LSKMAGAG10). After incubation, beads were washed three times in wash buffer prior to elution of proteins in SDS‐PAGE sample loading buffer.
Immunoblots
Proteins were electrophoresed through precast 4–20% tris‐glycine gels (Bio‐Rad) and transferred with an iBlotTM Gel Transfer Device (Novex‐Life Technologies) onto nitrocellulose membranes (Invitrogen). Membranes were blocked in blocking buffer for fluorescent western blotting (Rockland MB‐070‐010) for 1 h at room temperature and incubated overnight in primary antibodies at 4°C. The following primary antibodies were used: anti‐actin (Sigma‐Aldrich A2066; 1:3,000), anti‐eEF2 (Cell Signaling Technology 2332; 1:1,000), anti‐Myc (Sigma‐Aldrich C3956; 1:1,000), PSD‐95 (Millipore MAB1598 1:1,000), β‐Actin (Cell Signaling Technology 3700, 1:10,000), anti‐phospho‐eEF2(T56) (Cell Signaling Technology 2331; 1:1,000), anti‐SAPK/JNK (Cell Signaling Technology 9252), anti‐phospho‐SAPK/JNK(T183/Y185) (Cell Signaling Technology 9251), and anti‐PrPC (Cayman 189720; 1:500). For blotting, AZ59 was used at 100 ng/mL. Appropriate secondary antibodies were applied for 1 hour at room temperature (Odyssey donkey anti‐mouse or donkey anti‐rabbit conjugated to IRDye 680 or IRDye 800, LI‐COR Biosciences) and proteins were visualized with a LI‐COR Odyssey infrared imaging system. Quantification of band intensities was performed within a linear range of exposure.
Treatment of mouse cohorts
Mice were cared for by the Yale Animal Resource Center and all experiments were approved by Yale's Institutional Animal Care and Use Committee. Wild type and APP/PS1 mice were purchased from Jackson Laboratory and maintained on a C57/Bl6J background as described previously.8 Animals were randomly assigned to treatment groups and the experimenter was blinded to both genotype and treatment status. All mice utilized in the experiments received either control IgG or AZ59 diluted in PBS and weekly doses were administered by i.p. injection. The first cohort that received the 100 and 20 mg/kg doses of AZ59 began treatment at 12–13 months of age and was 64% female. The second cohort that received 20, 4, or 0.8 mg/kg AZ59 began treatment at 10–11 months of age and was 54% female. The mice used for biochemical signaling analysis began treatment at 13 months of age and was 44% female. Treatment continued throughout the course of all behavioral testing except where specified in the cohort 2 washout phase.
Behavioral testing
Mice were tested for novel object recognition as described.9, 14, 16, 17, 34 Spatial learning and memory were analyzed using the Morris water maze as described.8, 9, 14, 16, 17 Throughout behavioral testing, the experimenter was unaware of genotype and treatment.
Immunohistochemistry of mouse brain slices
Mice were euthanized with CO2 and perfused with ice cold PBS before decapitation and brain dissection. The left hemispheres were drop‐fixed in 4% paraformaldehyde for 24 h at 4°C. Brains were sliced into 40 μm coronal brain sections using a Leica WT1000S vibratome. For PSD‐95, an antigen retrieval step was performed prior to exposure to primary antibody by incubating slices in 1x Reveal Decloaker buffer (RV 1000 M, Biocare Medical) for 15 min at 90°C in an incubator. Sections were blocked in 10% normal horse serum (Jackson ImmunoResarch Laboratories) in PBS + 0.1% Triton X‐100 for 1 h and then incubated with primary antibodies for 24 h at 4°C. The following primary antibodies were used: anti‐GFAP (glial fibrillary acidic protein; Abcam ab7260; 1:500), anti‐Iba1 (ionized calcium‐binding adapter molecule 1; Wako 019‐19741; 1:250), anti‐PSD95 (postsynaptic density protein 95; Invitrogen 51‐6900; 1:250), anti‐CD68 (cluster of differentiation 68; Bio‐Rad MCA1957; 1:1,000), and anti‐SV2A (synaptic vesicle glycoprotein 2A; Abcam 32942; 1:250). Sections were washed three times in PBS and incubated with secondary antibodies (donkey anti‐rabbit, donkey anti‐chicken, or donkey anti‐rat fluorescent antibodies; Invitrogen Alexa Fluor; 1:500) for 1 h at room temperature. After three washes in PBS, the sections were mounted onto glass slides (Superfrost Plus, Fisher Scientific) and coverslipped with Vectashield (Vector Laboratories H‐1200) antifade aqueous mounting medium. For Thioflavin S (Sigma T1892) staining, slices were washed twice with 70% ethanol, stained with 0.1% ThioS in 70% ethanol for 15 min at room temperature, and washed twice more with 70% ethanol before being mounted as previously described.
Imaging and analysis of immunohistochemistry
For imaging of synapse density stained by anti‐SV2A and anti‐PSD95 antibodies, a Zeiss 800 confocal microscope with a 63X 1.4 NA oil‐immersion lens was used. The area occupied by immunoreactive synaptic puncta from the molecular layer of the dentate gyrus was measured as described previously.8, 16, 18 For imaging and analysis of the tissue stained for Iba1, GFAP, or CD68, a Zeiss 800 confocal microscope with a 20X 0.8 air‐objective lens was used with tiling and a z‐stack through the full slice in the center of the hippocampus. β‐amyloid plaque load was imaged on a Zeiss AxioImager Z1 fluorescent microscope with a 4X air‐objective lens. The hippocampus was imaged for each mouse and the area occupied by synapses, plaques, or cell bodies was analyzed by quantification with ImageJ. Statistical analysis was based on separate mice, with three slices averaged per mouse.
Mouse brain tissue collection for biochemistry
For AZ59 antibody levels, cortex and hippocampus of left hemispheres were quickly dissected and homogenized in three times the brain tissue weight in TBS (Tris Buffered Saline) containing PhosSTOP (Roche) and cOmplete‐mini protease inhibitor cocktail (Roche) to extract cytosolic proteins. After centrifugation for 30 min at 100,000g and 4°C, the supernatants were collected as TBS‐soluble fractions. The remaining pellets were dissolved in three times the original brain tissue weight in RIPA lysis buffer. A centrifugation step for 30 min at 100,000g and 4°C separated the RIPA soluble from the RIPA insoluble fractions.
For assessment of synaptic PrPC expression after antibody treatment, synaptosomal fractions were obtained from whole brains as previously described.12
For signaling assays, the same procedure was followed except that that a volume six times the brain tissue weight was utilized and the TBS‐insoluble pellet was resuspended in 1% (w/v) Triton X‐100 lysis buffer with 125 mmol/L NaCl containing PhosSTOP (Roche) and cOmplete‐mini protease inhibitor cocktail (Roche).
Results
AZ59 binds specifically to PrPC with picomolar affinity and blocks Aβo binding
The isolated antibody AZ59 targets the N‐terminal 23‐111 Aβo‐binding region of PrPC and binds to full‐length PrPC with sub‐nanomolar affinity in different assays. Using substrate‐immobilized PrPC protein, the KD of AZ59 for full length PrPC(23‐230) was 87 pmol/L (Fig. 1A and B). Similarly, using biolayer interferometry (BLI), the KD of AZ59 binding to PrPC 23‐230 was 99 pmol/L (Fig. 1C) and for full‐length PrPC(23‐111) was 62 pmol/L (not shown). Thus, AZ59 binds the N‐terminus of PrPC with a KD of 50–100 pmol/L in two different binding formats. The murine and human versions of AZ59 exhibited indistinguishable affinities and bound to both human and murine PrPC antigen equally (murine AZ59 form binding to human PrPC antigen in Fig. 1A–C).
Figure 1.
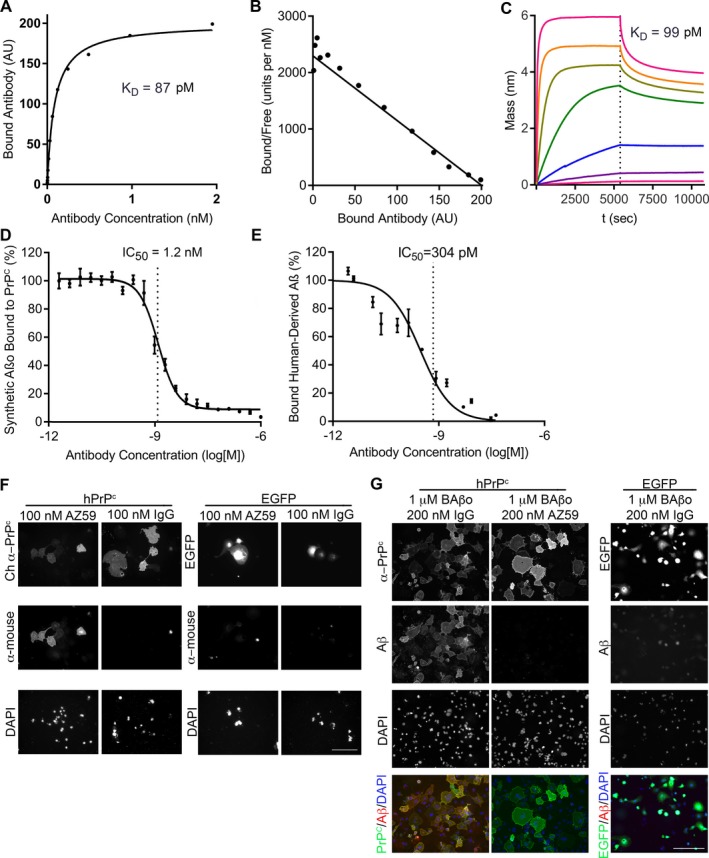
AZ59 binds to PrPC with high affinity and specificity and inhibits Aβo binding in vitro. (A) The binding of AZ59 to a 384‐well plate coated with PrPC(23‐230). Data are mean ± SEM, n = 3 replicates per sample. (B) Scatchard analysis of the data in (A). (C) Biolayer interferometric association (0–5400 sec) and dissociation (5401–10,800 sec) traces of AZ59 at concentrations of 0.024 nmol/L, 0.98 nmol/L, 0.39 nmol/L, 1.56 nmol/L, 6.25 nmol/L, 25 nmol/L, 100 nmol/L with PrPC(23‐230)‐coated sensor, indicating a dissociation constant of 99 pmol/L. (D) Prion‐Linked ImmunoSorbent Assay (PLISA) measurement of AZ59 inhibition of Aβo/PrPC(23‐230) interaction, indicating an IC50 of 1.2 nmol/L. Data are mean ± SEM, n = 3 replicates per sample. (E) PLISA measurement of AZ59 inhibition of human brain‐derived Aβo binding to PrPC, indicating an IC50 of 0.3 nmol/L. Data are mean ± SEM, n = 3 replicates per sample. (F) AZ59 binds to Cos‐7 cells expressing hPrPc but not EGFP. No binding of mouse IgG control was detected in either scenario. Scale bars = 200 μm. (G) AZ59 (100 nmol/L) completely blocks binding of biotin‐Aßo (1 μmol/L monomer equivalent) to Cos‐7 cells expressing myc‐tagged hPrPc. Scale bars = 200 μm.
Next, we sought to determine what concentration of AZ59 was required to block Aβo binding to PrPC 23‐230.7 After a 30‐min pretreatment with AZ59, the IC50 to inhibit binding of 3 nmol/L synthetic Aβo was 1.2 nmol/L in the plate binding format4, 18 (Fig. 1D). Similarly, the IC50 to inhibit binding of soluble Aβo derived from human AD autopsy brain extract4, 18 was 0.3 nmol/L (Fig. 1E).
To assess the selectivity of AZ59 for PrPC, we probed immunoblots of whole brain fractions with AZ59. In the RIPA‐resistant fraction of mouse brain, AZ59 recognizes PrPC from WT, but not Prnp −/− brains at the appropriate molecular weight (Fig. 1F). AZ59 also recognizes PrPC in human brain extract with a similar pattern to the mouse monoclonal anti‐PrPC antibody SAF32 (Fig. 1G). We conclude that the N‐terminally directed human anti‐PrPC AZ59 antibody provides high affinity and selectivity capable of blocking Aßo access to PrPC.
To evaluate AZ59 binding to PrPC and Aßo antagonism in a cellular context, we studied transfected Cos‐7 cells. AZ59 immunostained Cos‐7 cells transiently expressing human PrPC, as detected by co‐staining with a chicken polyclonal anti‐PrP antibody, but not non‐transfected cells in the same well or cells transfected with EGFP vector control (Fig. 1H). As described previously, biotin‐tagged Aßo bind to Cos‐7 cells expressing PrPC, but not control cells (Fig. 1I).7 Prereatment with 100 nmol/L AZ59 fully blocked Aßo binding to PrPC (Fig. 7I).
Figure 7.
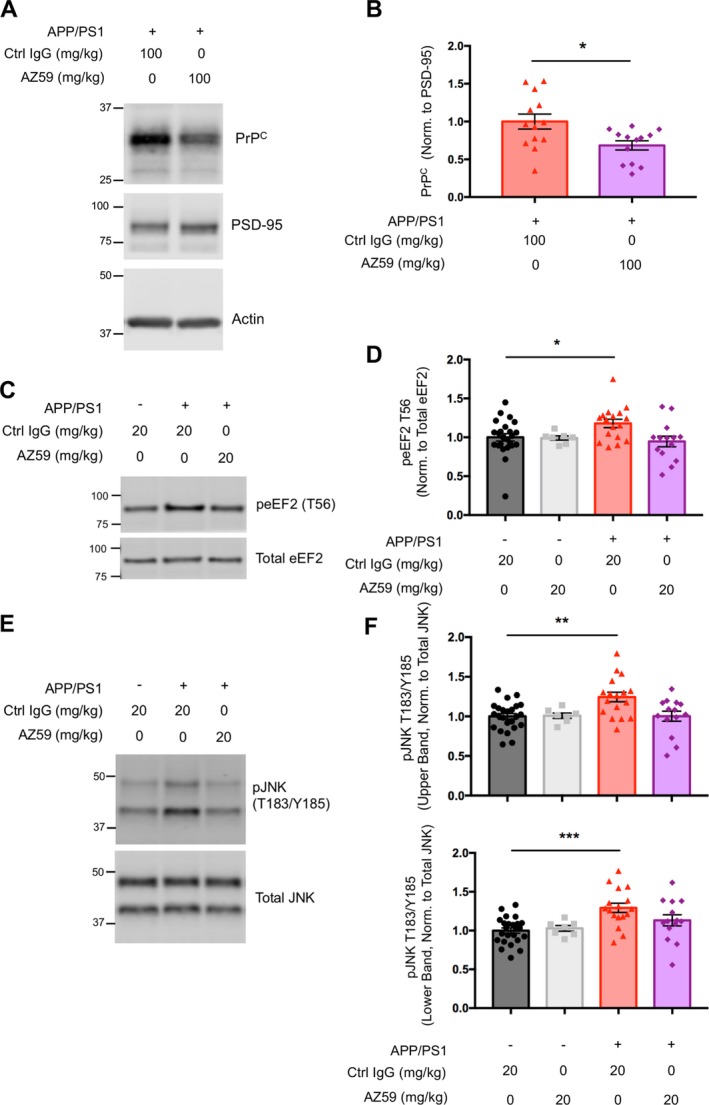
AZ59 reduces PrPC level and reverses aberrant eEF2 and JNK signaling in APP/PS1 mice. (A) Representative immunoblots of synaptosomal PrPC, PSD‐95 and β‐Actin from brains of 14‐16‐month APP/PS1 mice after 1 week of antibody treatment at 100 mg/kg. (B) Quantification of (A). One week (two doses) of treatment at 100 mg/kg significantly reduced levels of synaptic PrP relative to PSD‐95 (*,P < 0.05). Analysis by two‐tailed t‐test. n = 13 mice per group, data are mean ± SEM. (C) Representative western blots of TBS insoluble, Triton X‐100 soluble total, and phospho‐eEF2 (T56) from hippocampi of 14‐month wild type and APP/PS1 mice after 5 weeks of treatment. (D) Quantification of C. There was a significant increase in the level of Triton X‐100 soluble phospho‐eEF2 (T56) normalized to total eEF2 in hippocampi of 14‐month APP/PS1 mice treated with IgG control compared to wild type controls (*,P < 0.05). There was no significant difference in phospho‐eEF2 (T56) between 14‐month wild type controls and AZ59 treated APP/PS1 mice. Analysis by one‐way ANOVA with Dunnett's multiple comparisons test. n = 7–24 mice per group, data are mean ± SEM. (E) Representative western blots of TBS insoluble, Triton X‐100 soluble total, and phospho‐JNK (T183/Y185) from hippocampi of 14‐month wild type and APP/PS1 mice after 5 weeks of antibody treatment. (F) Quantification of (E). There was a significant increase in the level of Triton X‐100 soluble phospho‐JNK (T183/Y185) normalized to total JNK of the larger JNK isoform (upper band) in hippocampi of 14‐month APP/PS1 mice treated with IgG control compared to wild type controls (**,P < 0.01). There was no significant difference in phospho‐JNK (T183/Y185) of this isoform between wild type controls and APP/PS1 mice treated at 20 mg/kg. There was a significant increase in the level of phospho‐JNK (T183/Y185) normalized to total JNK of the smaller JNK isoform (lower band) in aged APP/PS1 mice treated with IgG control compared to wild type controls (***,P < 0.001). There was no significant difference in phospho‐JNK (T183/Y185) of this isoform between wild type controls and APP/PS1 animals treated with AZ59 at 20 mg/kg. Analysis by one‐way ANOVA with Dunnett's multiple comparisons test. n = 7–24 mice per group, data are mean ± SEM.
AZ59 disrupts the Aβo‐induced interaction of PrPC and mGluR5
The interaction of Aßo with PrPC engages mGluR5 to cause synaptic dysfunction and loss.13, 14, 15, 16 We sought to test whether AZ59 would prevent mGluR5 interactions driven by Aßo. We have previously mapped the mGluR5 binding site in PrPC to include the junction of the PrP(23‐111) N‐terminus and the C‐terminal globular domain.12 Using isolated proteins, the Venus fly trap ectodomain of mGluR5 exhibits high affinity for full length PrPC and the addition of AZ59 blocks the binding of the fly‐trap domain of mGluR5 to PrPC 23‐230) with an IC50 of 0.5 nmol/L (Fig. 2A).
Figure 2.
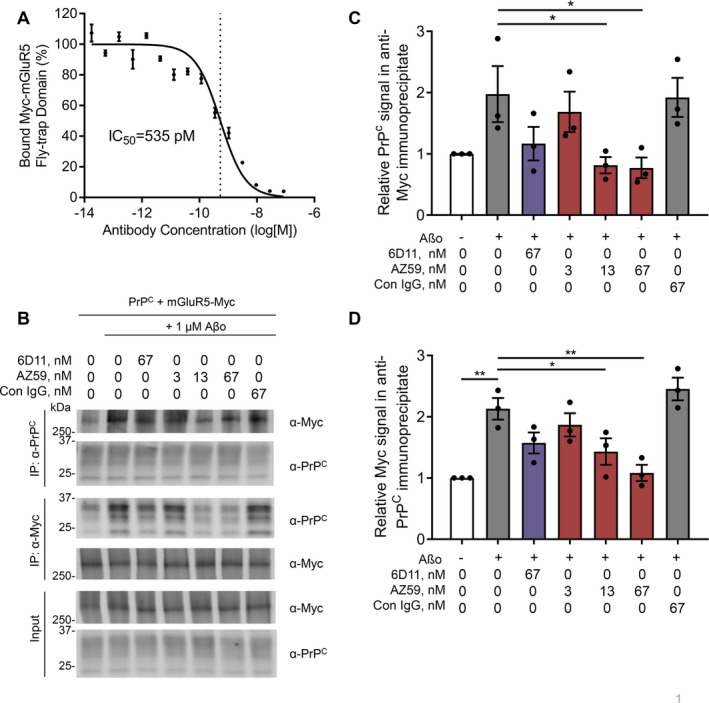
AZ59 disrupts the Aβo‐induced interaction of PrPC and mGluR5. (A) The binding of Myc‐tagged fly‐trap domain of mGluR5 to a 384‐well plate coated with PrPC(23‐230). Data are mean ± SEM, n = 3 replicates per sample. (B) Co‐immunoprecipitation of HEK cell lysates expressing PrPC and Myc‐mGluR5 pretreated with either vehicle, 6D11, AZ59, or control IgG before Aβo treatment. Membrane proteins were extracted with RIPA buffer and immunoprecipated with either α‐PrPC or α‐Myc. (C) Quantification of PrPC signal in (B) of α‐Myc immunoprecipitate. Data are mean ± SEM, n = 3 replicates per sample. (D) Quantification of Myc signal in (B) of α‐PrPC immunoprecipitate. Data are mean ± SEM, n = 3 replicates per sample.
Next, we considered AZ59 efficacy in a cellular environment. In HEK cells expressing both PrPC and mGluR5 and in neurons, we have previously shown that Aßo enhances co‐immunoprecipitation of the two proteins.12, 13 Pretreatment with AZ59 eliminates the Aβo‐induced association of PrPC and mGluR5 in a dose‐dependent manner, though it does not eliminate basal levels of association between the two full length proteins (Fig. 2B, C, and D). Thus, the ability of AZ59 to block the Aßo binding site on PrPC prevents engagement of downstream mGluR5 mediating synaptic damage.
AZ59 penetrates the blood‐brain barrier in a dose‐dependent manner
Previous studies of anti‐PrPC monoclonal antibodies in rodent AD models either did not quantify CNS drug levels25 or found that 0.1% of antibody in serum entered the CSF one hour after intracardiac injection.21 Here, we sought to determine AZ59 brain pharmacokinetic levels after a translationally relevant dosing paradigm. Mice were dosed i.p. with 0.8–100 mg/kg AZ59 weekly for 5 or 6 doses and then euthanized a week after their final dose to obtain trough levels after repeated dosing. A serum sample was obtained and then the blood was cleared by cardiac perfusion, and the brain was collected. After homogenizing the forebrain, TBS and RIPA fractions were plated into 384‐well plates coated with PrPC(23‐111) to capture PrPC‐interacting antibodies. Standard curves from untreated mouse brain samples from which lysate was spiked with known amounts of AZ59 were prepared for the TBS and RIPA fractions (Fig. 3A). In a WT naïve brain lysate with direct addition of AZ59 in amounts similar to those detected from dosed mice, approximately 25% of AZ59 activity was detected in each of the TBS and RIPA fractions (Fig. 3B). The 50% of AZ59 not recovered may be associated with the RIPA‐insoluble pellet and/or bound to endogenous antigen. The highest dose group, 100 mg/kg weekly, had an average trough antibody concentration of 11.4 nmol/L in brain TBS fraction, 24.9 nmol/L in brain RIPA fraction and 36.1 nmol/L in serum (Fig. 3C, D, and E). AZ59 levels for the other groups showed a direct dependence on dose level in all three compartments (Fig. 3F). We also measured brain concentrations 6 hours after a 20 mg/kg dose to obtain an estimate of peak brain concentration for that standard dose. For the 20 mg/kg weekly dose regimen, the sum of TBS and RIPA extractable brain AZ59 antibody varied from 19 nmol/L to 4 nmol/L during the dose period (Fig. 3F). This is consistent with a half‐life of AZ59 in the brain of approximately 3 days. Furthermore, the measurements indicate that antibody levels generated by 20 mg/kg doses remain an order of magnitude above the KD of the antibody for PrPC (Fig. 1) throughout the weekly dose period.
Figure 3.
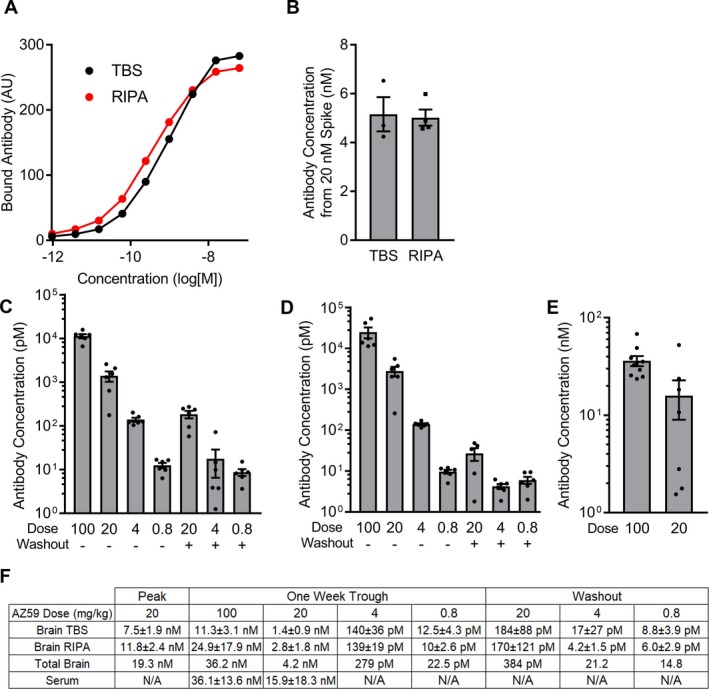
AZ59 penetrates the blood‐brain barrier at nanomolar concentrations. (A) Standard curves used to determine concentration of samples. 384‐well plates were coated with PrPC(23‐230) to capture AZ59 recovered from TBS and RIPA lysates from the cortex of treated mice. (B) AZ59 was added to total brain homogenate at a final concentration of 20 nmol/L and incubated overnight at 4C. TBS and RIPA fractions were collected and compared to a standard curve to determine the final concentration in each fraction. (C–D) AZ59 recovered from TBS (C) and RIPA (D) lysate from cortex of mice treated with the indicated mg/kg doses of AZ59 for either 5 or 6 weeks, and sacrificed 7 days after the last dose. Data are mean ± SEM, n = 5–6 per group, all samples were run in triplicate. (E) AZ59 recovered from serum of treated mice. Data are mean ± SEM, n = 7–10 per group, all samples were run in triplicate. (F) Table summarizing results in (C–E). Total brain concentration is the sum of TBS and RIPA fraction concentrations. Data are mean ± standard deviation. N/A, not assayed.
AZ59 reverses established learning and memory deficits in APP/PS1 mice
We sought to assess whether the AZ59 peripheral dosing would rescue AD‐related deficits in APP/PS1 mice. This strain first develops deficits in the Morris water maze (MWM) at 7 months in age.35 We began testing at 11 months of age, an age at which their memory deficits are firmly established. Before treatment began the mice completed MWM testing to verify the presence of a substantial learning and memory deficit (Fig. 4A). The APP/PS1 exhibited longer latency to learn the location of a hidden platform during six swim blocks of four trials each (Fig. 4B). After training, the AD transgenic mice spent less time in the quadrant previously containing the hidden target (Fig. 4C). In a first dose‐ranging trial, treatment groups were then randomized to receive once weekly i.p. injections of 100 mg/kg control IgG, 20 mg/kg AZ59, or 100 mg/kg AZ59. After three doses, the mice underwent behavioral tests in MWM and novel object recognition (NOR). The MWM paradigm was repeated over a week, with 24 forward swims followed by 24 reverse swims to opposite quadrants distinct from pretreatment target quadrant. During the acquisition phase of the MWM, the APP/PS1 mice receiving either 20 mg/kg or 100 mg/kg AZ59 learned to reach the second platform location significantly more quickly than did the APP/PS1 mice that received control IgG, and nearly as quickly as nontransgenic mice treated with control IgG (Fig. 4D). In WT mice, repeated treatment with 100 mg/kg AZ59 had no effect on learning. After the learning trials, a probe trial was completed to test memory for the trained platform location (Fig. 4E). The APP/PS1 mice treated with control IgG performed indistinguishably from chance (25%), and spent significantly less time in the target quadrant than did WT mice with either control IgG or AZ59. The APP/PS1 mice treated with either dose of AZ59 showed a nonsignificant trend to spend more time in the target quadrant than did APP/PS1 mice treated with control IgG and were not significantly different from WT mice.
Figure 4.
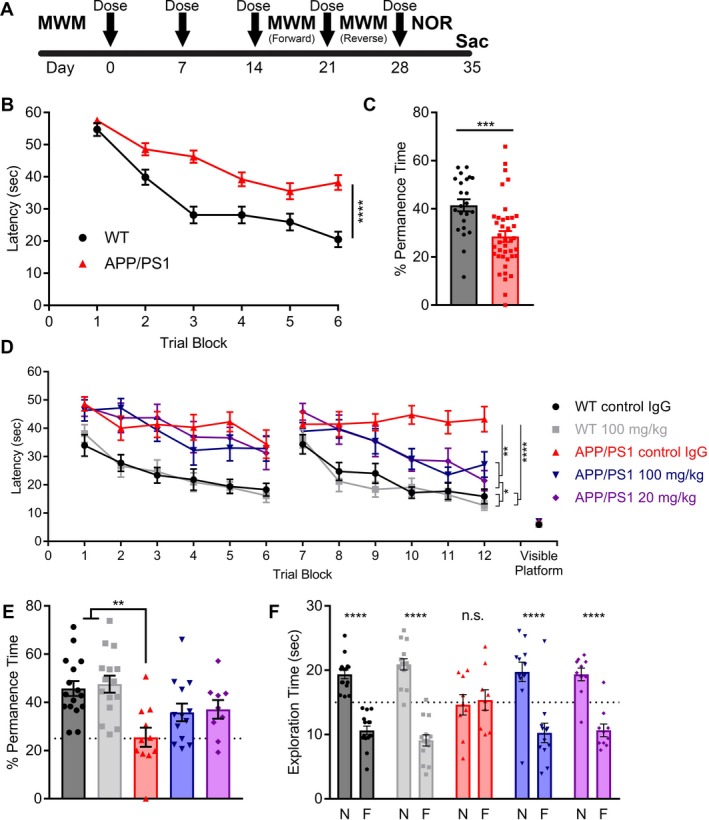
AZ59 reverses established learning and memory deficits in APP/PS1 mice. (A) Timeline of treatment and testing. Mice began treatment at 12–13 months, received five weekly doses via i.p. injection (arrows), and began behavior after 3 weeks of treatment. Tissue was collected 6 weeks after treatment began. (B) Prior to treatment, 12‐13‐month‐old WT and APP/PS1 completed the MWM to establish their baseline deficit. Data are mean ± SEM, n = 24 for WT and n = 42 for APP/PS1 mice. Performance was analyzed by two‐way analysis of variance with RM‐ANOVA over the last 12 trials (three blocks) and showed a significant effect of genotype (****,P < 0.0001). (C) 24 h after the learning trials, WT and APP/PS1 completed a probe trial. Time spent in the quadrant where the platform once was measured. Data are mean ± SEM, n = 24 for WT and n = 42 for APP/PS1 mice. Performance was analyzed by two‐tailed t test (***P < 0.001). (D) Mice were randomized into five groups receiving either 100 mg/kg of control IgG, 20 mg/kg AZ59, or 100 mg/kg AZ59 once weekly via i.p. injection. After three doses, they repeated the MWM twice with both a forward and reverse swim training set to new quadrants. The control IgG‐treated APP/PS1 group differed significantly from all other groups by one‐way RM‐ANOVA over the last 12 trials of the reverse swim with Tukey's post hoc multiple comparisons test (**P < 0.01; ****P < 0.0001). n = 9–16 per group, data are mean ± SEM. (E) 24 h after the reverse swim the mice completed a probe trial. WT mice spent significantly more time in the target quadrant than control APP/PS1 mice. APP/PS1 mice receiving AZ59 had intermediate performance, not differing significantly from either the WT or the control IgG APP/PS1 groups. n = 9–16 per group, data are mean ± SEM (***,P < 0.001), one‐way ANOVA with Dunnett's comparison to APP/PS1 control IgG. (F) After 3 weeks of treatment, mice completed the NOR test. APP/PS1 mice receiving AZ59 and WT mice preferred to interact with the novel object (****,P < 0.0001), while APP/PS1 mice receiving control IgG did not (P > 0.05). N, novel object; F, familiar object. Analysis by two‐way ANOVA with Sidak's multiple comparisons test. n = 10–16 per group, data are mean ± SEM. The dashed line indicates an equal amount of time spent with either object.
In a separate memory test for object recognition, the APP/PS1 mice that received control IgG injections failed to exhibit any preference for the novel object, while WT mice treated with control IgG or with AZ59 had a significant preference for the novel object. The APP/PS1 mice that received either 20 mg/kg or 100 mg/kg AZ59 showed the same significant novel object preference as WT mice (Fig. 4F). Thus, both the 20 mg/kg and 100 mg/kg weekly doses of AZ59 rescued learning and memory deficits in aged APP/PS1 mice, while the same AZ59 dose had no effect on WT mice.
AZ59 dose dependently and persistently rescues AD transgenic mice
To confirm and extend these initial data, we designed a second treatment study with larger cohorts and lower doses. Starting at 10.5 months, APP/PS1 mice were treated with 0.8 mg/kg, 4 mg/kg or 20 mg/kg weekly i.p. doses of AZ59 (Fig. 5A). The control WT and APP/PS1 groups received 20 mg/kg doses of control IgG on the same schedule. After the fifth dose, at 11.5 months age, mice underwent NOR and then MWM testing. For the MWM, the forward swim trial set was followed by a reverse swim to a target in the opposite quadrant. During the learning phase of the reverse swim, all three AZ59‐treated transgenic groups learned the platform location significantly faster than did the APP/PS1 control IgG group with latencies nearly identical to the WT group (Fig. 5B). In the probe trial for the reverse swim, the 4 mg/kg and 20 mg/kg groups showed a moderate, but non‐significant, improvement (Fig. 5C).
Figure 5.
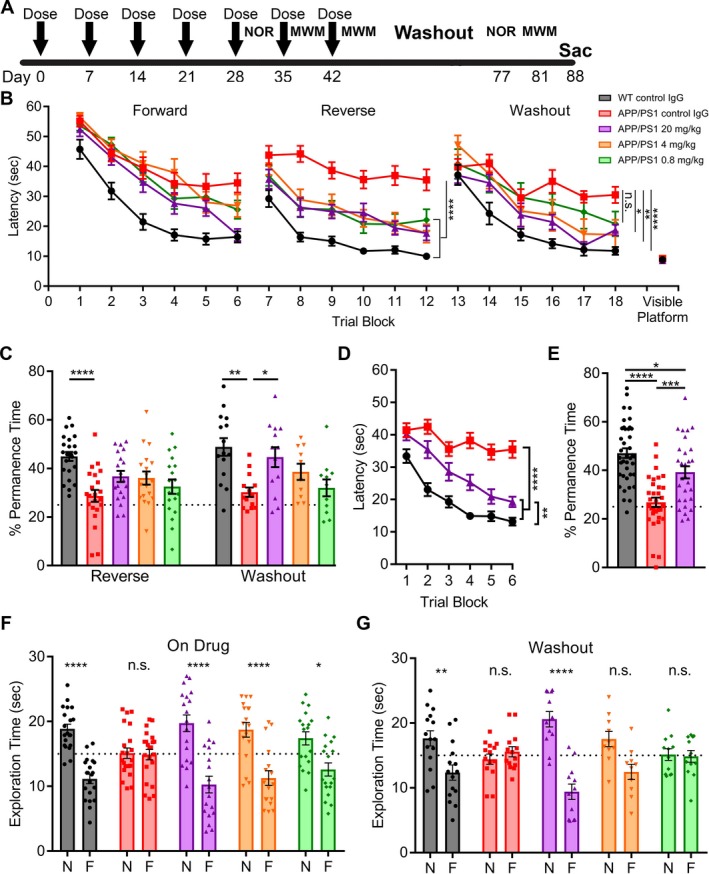
AZ59 reversal of learning and memory deficits in APP/PS1 mice is dose‐dependent and persists after washout. (A) Timeline of treatment and testing. WT and APP/PS1 mice (10–11 months old) were randomized into five groups receiving 20 mg/kg of control Ig, 0.8 mg/kg AZ59, 4 mg/kg AZ59 or 20 mg/kg AZ59 once weekly via i.p. injection. (B) After 5 weeks of treatment, mice completed the MWM twice with a forward and reverse set of 24 swims. The control IgG treated APP/PS1 group differed significantly from all other groups by one‐way RM‐ANOVA over the last twelve trials of the reverse swim with Tukey's post hoc multiple comparisons test (****,P < 0.0001). APP/PS1 mice receiving the two highest doses of AZ59 (4 mg/kg and 20 mg/kg) did not differ significantly from WT mice (P > 0.05), but those receiving the lowest dose (0.8 mg/kg) were slower than WT (*,P < 0.05). n = 18–26 per group. After 7 weeks of treatment, the mice entered a one‐month washout period. They then completed the MWM with the platform moved to a new quadrant. The control IgG treated APP/PS1 group differed significantly from all other groups except the 0.8 mg/kg group (P > 0.05) by one‐way RM‐ANOVA over the last twelve trials of the washout swim with Tukey's post hoc multiple comparisons test (*,P < 0.05; **, P < 0.01; ****, P < 0.0001). APP/PS1 mice receiving the two highest doses of AZ59 (4 mg/kg and 20 mg/kg) did not differ significantly from WT mice (P > 0.05), but those receiving the lowest dose (0.8 mg/kg) were slower than WT (*,P < 0.05). n = 10–15 per group. All data are mean ± SEM. (C) 24 h after the reverse and washout swims the mice completed a probe trial. WT mice spent significantly more time in the target quadrant than control APP/PS1 mice. APP/PS1 mice receiving 20 mg/kg AZ59 showed a trend to perform better than those receiving control IgG, but the difference was not significant (P = 0.069). (****,P < 0.0001), n = 18–26 per group. For the washout probe trial, APP/PS1 mice receiving 20 mg/kg AZ59 and WT mice spent significantly more time in the target quadrant than did control APP/PS1 mice (*,P < 0.05; **, P < 0.01). n = 10–15 per group. All data are mean ± SEM, one‐way ANOVA with Dunnett's comparison to APP/PS1 control IgG. (D) Meta‐analysis of the final block of swims each mouse receiving WT control IgG, APP/PS1 control IgG, or APP/PS1 20 mg/kg AZ59 performed. WT and APP/PS1 mice receiving 20 mg/kg AZ59 performed significantly better by one‐way RM‐ANOVA over the last twelve trials with Tukey's post hoc multiple comparisons test (****, P < 0.0001). WT mice also performed significantly better than APP/PS1 mice receiving 20 mg/kg AZ59 (**,P < 0.01). n = 32–39 per group, all data are mean ± SEM. (E) Meta‐analysis of the final probe trial for each mouse receiving WT control IgG, APP/PS1 control IgG, or APP/PS1 20 mg/kg AZ59 was performed. WT and APP/PS1 mice receiving 20 mg/kg AZ59 spent significantly more time in the target quadrant than APP/PS1 mice receiving control IgG (***,P < 0.001; ****, P < 0.0001). WT mice performed significantly better than APP/PS1 mice receiving 20 mg/kg AZ59 (*,P < 0.05). One‐way ANOVA with Tukey's post hoc multiple comparisons test, n = 32–39 per group, all data are mean ± SEM. (F) After 5 weeks of treatment, mice completed the NOR test. APP/PS1 mice receiving AZ59 and WT mice preferred to interact with the novel object (*,P < 0.05; ****, P < 0.0001), while APP/PS1 mice receiving control IgG did not (P > 0.05). N, novel object; F, familiar object. Analysis by two‐way ANOVA with Sidak's multiple comparisons test. n = 18–26 per group, data are mean ± SEM. The dashed line indicates an equal amount of time spent with either object. (G) After washout, APP/PS1 mice receiving 20 mg/kg AZ59 and WT mice preferred to interact with the novel object (**,P < 0.01; ****, P < 0.0001), while APP/PS1 mice receiving control IgG or 4 or 0.8 mg/kg AZ59 did not (P > 0.05). N, novel object; F, familiar object. Analysis by two‐way ANOVA with Sidak's multiple comparisons test. n = 10–15 per group, data are mean ± SEM. The dashed line indicates an even amount of time spent with either object.
We considered whether the benefits of AZ59 might persist for an extended period after dosing. Upon completion of the on‐drug MWM testing, the mice had received a total of seven weekly doses of drug and entered a one‐month washout, estimated to be nine half‐lives (Fig. 3). The mice then underwent MWM testing with the platform in a new third target quadrant. During the washout training phase, the 0.8 mg/kg, 4 mg/kg, 20 mg/kg groups performed with a dose responsive pattern, with the 20 mg/kg APP/PS1 group learning best and the 4 mg/kg and 0.8 mg/kg groups performing progressively worse (Fig. 5B). The same dose response pattern was observed in the washout probe trial, with the 20 mg/kg APP/PS1 showing a complete rescue in memory behavior while the 0.8 mg/kg APP/PS1 group did not differ from their transgenic peers receiving control IgG (Fig. 5C). Thus, the treatment benefit of weekly 20 mg/kg AZ50 persisted through the 39 days of washout and retesting.
Since the same 20 mg/kg dose was employed in both the Figures 3 and 4 experiments, we created a meta‐analysis of the WT control IgG, APP/PS1 control IgG, and APP/PS1 20 mg/kg AZ59 groups. This combination analysis reveals a significant improvement in APP/PS1 mice receiving 20 mg/kg AZ59 in both the learning and probe trials of the MWM, with close to WT performance levels (Fig. 5D and E).
Object memory was also performed on this cohort pre‐ and post‐washout by NOR. Before washout, the APP/PS1 mice receiving control IgG showed no preference for the novel object, while the 0.8 mg/kg, 4 mg/kg, and 20 mg/kg APP/PS1 groups all significantly preferred to interact with the novel object (Fig. 5F). Although the treated transgenic mice all showed an improvement in object recognition, there was a performance trend that correlated with the dose received (Fig. 5F). To repeat NOR after washout, new objects were chosen and WT mice were tested to confirm that there was no innate exploratory preference between the two (data not shown). After washout, the 0.8 mg/kg, 4 mg/kg, and 20 mg/kg APP/PS1 groups showed a significant dose response, with the 20 mg/kg group maintaining a full rescue in novel object preference, the 4 mg/kg group showing an intermediary phenotype (P = 0.13), and the 0.8 mg/kg group showing the same phenotype as the APP/PS1 control IgG group, with no preference for the novel object (Fig. 5G).
Thus, while on drug, all APP/PS1 groups receiving 0.8 mg/kg, 4 mg/kg, and 20 mg/kg of AZ59 showed improvements in memory during NOR and the training phase of the MWM. After a month‐long washout, the 0.8 mg/kg group behavioral improvement over control APP/PS1 was lost. In contrast, the 20 mg/kg APP/PS1 continued to perform as well as their WT counterparts. The 4 mg/kg APP/PS1 group performed at an intermediary level between the 20 and 0.8 mg/kg groups, demonstrating a dose response during the washout phase of treatment.
AZ59 treatment does not alter Aß accumulation or gliosis
The hypothesized role of PrPC is as a synaptic Aßo binding site, without a role in Aß production or Aß clearance or gliotic reaction to Aß. To assess the specificity of anti‐PrPC AZ59 action, we examined astrocyte, microglia, and plaque levels in the hippocampi of treated and non‐treated mice. The area of brain sections immunoreactive for two microglial markers, Iba1 and CD68, were significantly increased in APP/PS1 mice as expected, and their levels were not altered by AZ59 treatment (Fig. 6A–C). Similarly, immunoreactive area for GFAP, an astrocyte marker, was significantly increased in APP/PS1 mice but not altered by AZ59 treatment (Fig. 6D and E). Aß plaque load was assessed by Thioflavin‐S staining. AZ59 treatment had no effect on plaque accumulation in APP/PS1 mice (Fig. 6F and G), and no effect on PrP‐interacting soluble Aßo in TBS brain homogenate (Fig. 6H). Thus, the beneficial effects of AZ59 are PrPC‐mediated and independent of Aβo metabolism and glial activation.
Figure 6.
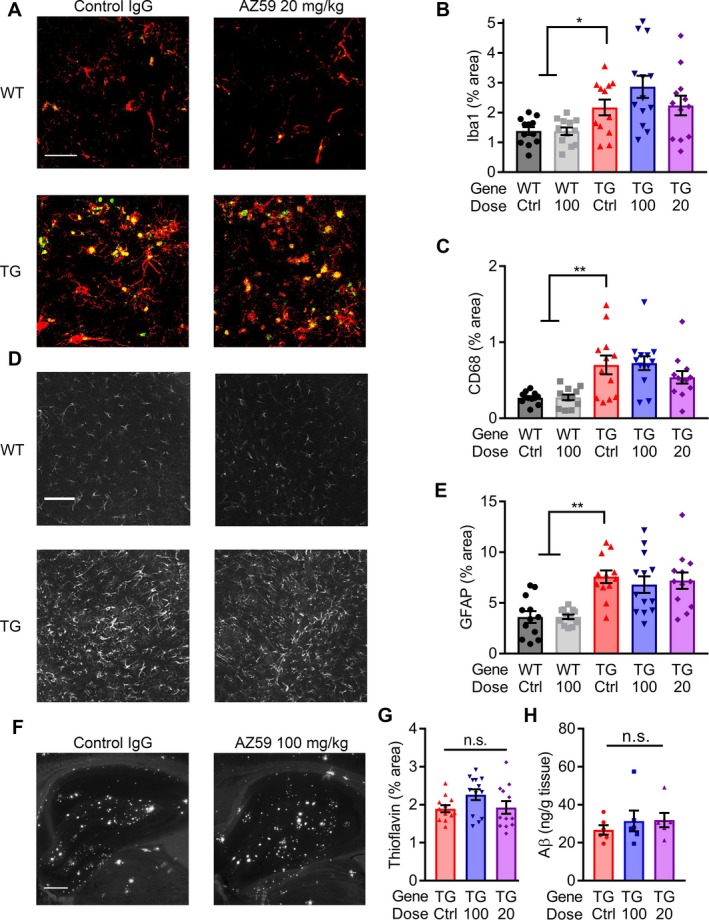
AZ59 does not alter glial activation or Aβ load. (A) Representative images of immunofluorescent staining for Iba1 (red) and CD68 (green) from the hippocampus of 14‐month‐old WT and APP/PS1 after 5 weeks of treatment. Scale bar = 60 μm. (B–C) Quantification of Iba1 (B) and CD68 (C) immunoreacted area. (*,P < 0.05; **, P < 0.01) n = 12–13 mice per group, data are mean ± SEM, one‐way ANOVA with Dunnett's comparison to APP/PS1 control IgG. (D) Representative images of anti‐GFAP immunostaining taken in the hippocampus of 14‐month‐old WT and APP/PS1 after 5 weeks of treatment. Scale bar = 60 μm. (E) Quantification of GFAP immunoreacted area. (**, P < 0.01) n = 12–13 mice per group, data are mean ± SEM, one‐way ANOVA with Dunnett's comparison to APP/PS1 control IgG. (F) Representative images of ThioS staining in the hippocampus of 14‐month‐old WT and APP/PS1 after 5 weeks of treatment. Scale bar = 200 μm. (G) Quantification of ThioS plaque area. No significance between the groups (P > 0.05) n = 12–13 mice per group, data are mean ± SEM, one‐way ANOVA with Dunnett's comparison to APP/PS1 control IgG. (H) Aβo from TBS homogenate of APP/PS1 mouse cortex captured by PrPC(23‐230) on a 384‐well plate. No significance between the groups (P > 0.05) n = 6 mice per group, data are mean ± SEM, one‐way ANOVA with Tukey's multiple comparison's test.
AZ59 reverses abnormal JNK and eEF2 signaling in APP/PS1 mice
We sought pharmacodynamic evidence for AZ59 engagement with PrPC and known synaptic signal transduction pathways in APP/PS1 mice. First, we assessed PrPC levels in aged transgenic mice after treatment with control or AZ59 antibody after two 100 mg/kg weekly doses. Synaptosomal levels of PrPC significantly decreased by 40% in AZ59 treated mice (Fig. 7A and B), indicating that a AZ59 antibody complex forms in vivo and promotes PrPC degradation.
Previous biochemical analyses of AD brains have revealed abnormal neuronal signaling patterns including increased phosphorylation of eEF2 and JNK (SAPK), which may contribute to synaptic loss.36, 37, 38 Increases in eEF2 and JNK phosphorylation are also observed in aged APP/PS1 mice.39, 40 More specifically, we have previously shown that eEF2 activation is immediately downstream of Aßo/PrPC‐induced activation of mGluR5.13, 16 Therefore, eEF2 activation is a known pharmacodynamic marker of PrPC signal transduction activity in APP/PS1 mice and JNK activation is a potential marker.
To determine whether AZ59 administration rescues abnormal eEF2 and JNK neuronal signaling, aged wild type and APP/PS1 mice treated for 5 weeks with either AZ59 or IgG control at 20 mg/kg weekly were sacrificed for biochemical analysis. Dissected hippocampi were homogenized and ultra‐centrifuged in TBS to obtain a TBS soluble protein fraction and the remaining pellet re‐homogenized and ultra‐centrifuged in Triton X‐100 to obtain a TBS insoluble, Triton X‐100 soluble protein fraction. Triton X‐100 soluble hippocampal fractions revealed a significant increase in hippocampal eEF2 phosphorylation at T56 in APP/PS1 samples, while no such increase was present in samples from APP/PS1 animals treated with AZ59 (Fig. 7C and D). The APP/PS1 brain fractions also showed a significant increase in the level of JNK phosphorylation at T183/Y185 for both the larger (54 kDa) and smaller (46 kDa) isoforms of JNK in APP/PS1 mice compared to WT controls. In contrast, the brain tissue from APP/PS1 mice administered AZ59 at 20 mg/kg weekly showed no increase of either phospho‐JNK isoform relative to WT samples, and a reduction relative to control antibody APP/PS1 samples (Fig. 7F). Together these results demonstrate that AZ59 administration successfully reverses abnormal neuronal signaling in APP/PS1 mice.
AZ59 reverses synaptic loss in APP/PS1 mice
The loss of synapses in AD parallels symptomatology and is likely to be a central pathophysiological mechanism. To determine whether the AZ59‐mediated behavioral rescue in APP/PS1 mice was correlated with a rescue of transgene‐dependent synaptic loss, we measured pre‐ and post‐synaptic markers in the dentate gyrus. The area immunopositive for PSD‐95, a postsynaptic marker, was reduced in APP/PS1 mice relate to WT controls, and was fully rescued in APP/PS1 mice by AZ59 treatment (Fig. 8C). AZ59 treatment had no effect of PSD‐95 area in WT mice. Similarly, SV2A, a presynaptic marker, was reduced in control APP/PS1 mice compared to their WT counterparts (Fig. 8G). Treatment with AZ59 at 100 mg/kg did not affect SV2A in WT mice but did rescue the transgene‐dependent deficits in APP/PS1 mice (Fig. 8G). After 7 weeks on treatment, AZ59 returned SV2A and PSD‐95 to WT levels in APP/PS1 mice at the 20 mg/kg dose, while the lower 4 and 0.8 mg/kg doses did not (Fig. 8D and H). A similar pattern is seen in both SV2A and PSD‐95 after a one‐month washout; synapse immunoreactivity was returned to WT levels in the 20 mg/kg APP/PS1 group, while the 4 and 0.8 mg/kg APP/PS1 groups did not differ significantly from the APP/PS1 control IgG group (Fig. 8E and I). A meta‐analysis of the WT control IgG, APP/PS1 control IgG, and APP/PS1 20 mg/kg AZ59 groups across cohorts revealed statistically robust rescue of both PSD‐95 and SV2A density in APP/PS1 mice with the 20 mg/kg AZ59 dose (Fig. 8F and J). Thus, AZ59 reverses APP/PS1 synaptic deficits in a dose‐dependent manner that persists after washout and does not alter WT levels.
Figure 8.
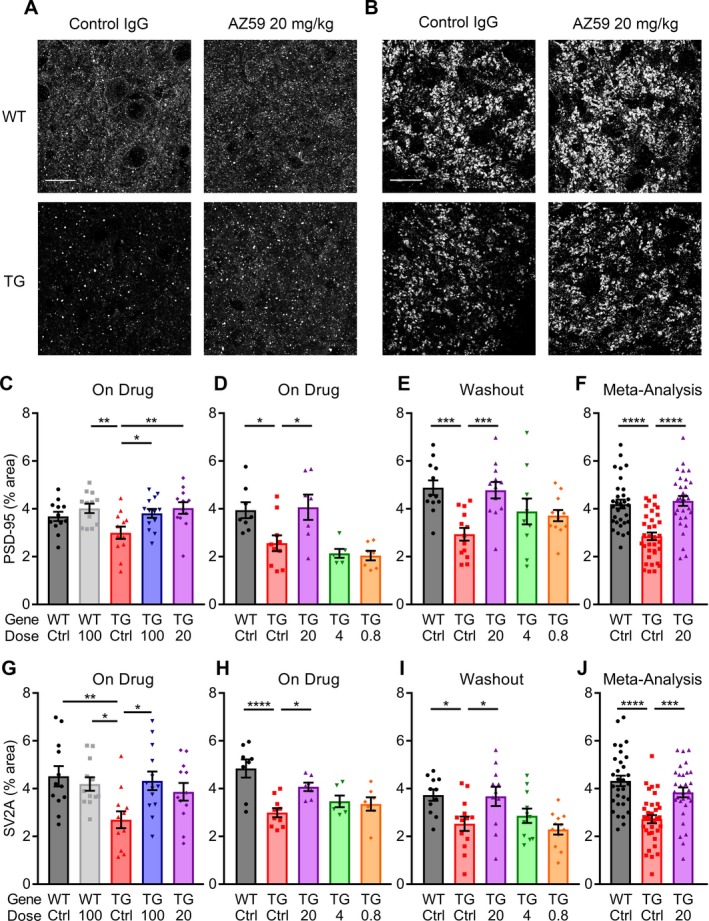
AZ59 reversal of synaptic deficits in APP/PS1 is dose‐dependent and persists after washout. (A–B) Representative images of PSD‐95 (A) and SV2A (B) taken in the dentate gyrus of 14‐month‐old WT and APP/PS1 mice after 5 weeks of treatment. Scale bar = 20 μm. (C–F) Quantification of PSD‐95 immunoreactive area in the dentate gyrus of treated mice. (C) 14‐month‐old mice after 5 weeks of treatment. (D) 12‐month‐old mice after 7 weeks of treatment. (E) 13‐month‐old mice after 7 weeks of treatment and a one‐month washout. (*, P < 0.05; **, P < 0.01; ***, P < 0.001) n = 6–13 per group. (F) Meta‐analysis of all PSD‐95 data for the WT control IgG, APP/PS1 control IgG, and APP/PS1 20 mg/kg AZ59. (****, P < 0.0001) n = 31–35 mice. All data are mean ± SEM, one‐way ANOVA with Dunnett's comparison to APP/PS1 control IgG. (G–J) Quantification of SV2A immunoreactivity in the dentate gyrus of treated mice. (C) 14‐month‐old mice after 5 weeks of treatment. (D) 12‐month‐old mice after 7 weeks of treatment. (E) 13‐month‐old mice after 7 weeks of treatment and a one‐month washout. (*, P < 0.05; **, P < 0.01; ****, P < 0.0001) n = 6–13 per group. (J) Meta‐analysis of all SV2A data for the WT control IgG, APP/PS1 control IgG, and APP/PS1 20 mg/kg AZ59. (***, P < 0.001; ****, P < 0.0001) n = 31–35 mice. All data are mean ± SEM, one‐way ANOVA with Dunnett's comparison to APP/PS1 control IgG.
Discussion
The major finding of the present study is that systemic administration of N‐terminally directed high‐affinity anti‐PrPC antibody provides safe and effective therapeutic benefit in preclinical AD models. Specifically, a monoclonal antibody binding with sub‐nanomolar affinity prevents access of Aßo derived from synthetic or human autopsy brain sources to the N‐terminus of PrPC. By blocking this binding step, the antibody protects against mGluR5 engagement. When a murine version of the fully human antibody is administered to mice at clinically feasible doses, it provides effective brain concentrations and reduces PrPC levels. Most importantly, in aged AD transgenic mice with Aß pathology and behavioral deficits, treatment with the antibody restores hippocampal synapse density and recovers learning and memory function. These benefits are dose‐dependent and persist for a month. The anti‐PrPC rescue occurs despite continued Aß plaque load and gliosis, separating this potential therapeutic approach from Aß‐lowering strategies. Overall, the data support development of anti‐PrPC antibodies as distinct disease‐modifying therapy for AD.
Previous studies have identified Aßo as key triggers for AD pathophysiology and PrPC as a high affinity binding site required for a range of deficits in preclinical models with cultured neurons, brain slices and mice.11, 41 For this reason, the ability of anti‐PrPC antibodies to abrogate AD‐related dysfunction has been investigated. A crucial step is demonstration of in vivo efficacy without toxicity. Initial studies showed that the mouse monoclonal 6D11 antibody eliminated a behavioral transgenic deficit at a very high 400 mg/kg dose delivered over 12 days. However, no dose responsiveness, pharmacokinetic or mechanistic information was available.25 Administration of other anti‐PrPC antibodies into the CNS or as single bolus injections has demonstrated benefit for long term potentiation deficits caused by either Aßo injection or AD‐related transgenes.20, 21, 22, 23, 24 However, these studies have not defined the utility or pharmacokinetics of human anti‐PrPC antibodies in an administration paradigm that is amenable to translation for clinical dosing. Another concern raised with anti‐PrPC administration has been the observation of neuronal toxicity in certain cases,27, 28, 31 though this has been controversial.26, 29 However, the reported anti‐PrPC antibody toxicity is blocked by the addition of a second antibody directed against the natively unfolded N‐terminus and overlapping with AZ59.31
In the current study, we administered the murine version of a human anti‐PrPC N‐terminally directed antibody to mice systemically, in a dose regimen consistent with clinical translation, and observed brain levels above the antibody affinity for PrPC. Critically, this dose regimen generated no evidence of toxicity as evaluated by extensive behavior testing, hippocampal synaptic densities, astrocytosis or microgliosis. The safety of this regimen may relate either to the N‐terminal epitope and/or the level of systemic dosing.
The AZ59 antibody exhibited predicted benefits both in vitro and in vivo. The antibody blocked binding of Aßo oligomers or mGluR5 ectodomain fragments to PrPC. Furthermore, Aßo enhancement of PrPC/mGluR5 association was prevented by AZ59. The ability of Aß accumulation driven by human mutant APP/PS1 transgenes to cause spatial and object learning and memory deficits was reversed in aged mice. Reductions of synaptic densities in the dentate gyrus of the hippocampus were recovered in the AZ59‐treated mice. Importantly, the behavioral tests show that even after dysfunction is well‐developed for months in the AD transgenic mice, AZ59 can lead to a reversal with restoration of function.
While memory and synapses are rescued to normal values by AZ59 in APP/PS1 mice, the levels of Aß plaque, astrocytic activation and microgliosis are not altered. The current findings are consistent with PrPC mediating the neuronal and synaptic effects of Aß accumulation, but not the accumulation or clearance of Aß. While we have observed no change in dense core plaque or soluble Aßo, future studies will be required to verify whether other APP metabolites might be altered by anti‐PrP treatment. The derangements of synaptic signaling pathways by Aß accumulation are corrected by AZ59 treatment. Previously we showed that eEF2 is activated downstream of Aßo/PrP/mGluR5,13, 16 and this is normalized by AZ59. In addition, activated JNK signaling has been linked to toxic signaling in AD transgenic mice37 and is normalized by AZ59. This is likely to occur through signaling driven by mGluR5 and Pyk213 since JNK is reported to be activated by Pyk2.42 The anti‐PrPC treatment also partially reduced PrPC levels in the brain and this clearly demonstrates target engagement. We hypothesize that efficacy is largely driven by near complete blockade of Aßo/PrPC binding and signaling, but reductions of PrPC level may contribute to efficacy. We conclude that AZ59 treatment prevents toxic Aßo/PrPC signaling at neuronal synapses to reverse memory dysfunction and synaptic loss in AD transgenic mice.
Gliosis can be driven by Aß accumulation, and microglia are prominent surrounding plaques. Human genetic studies have revealed contributions for Trem2 and other microglial genes in AD risk.43, 44 Microglia have been postulated to play a role in both encapsulating Aß plaque45 and in synaptic engulfment coupled with compliment fixation by damaged synapses.46 While AZ59 does not appear to alter microglia or astrocytes per se, it may abrogate their attack on synapses by preventing synaptic damage that leads to compliment fixation as a marker for engulfment. Future studies will be required to explore this potential pathway.
The benefits of A59 persist for over a month by memory testing and synaptic density measurements. Given the measured antibody levels, and an apparent half‐life of 3 days this is unlikely to be due to retained antibody. Instead, it demonstrates a disease modifying effect of AZ59 to restore synapse density. In the presence of APP/PS1 transgenes and Aß accumulation, we speculate that the disease will be reinstated slowly after the antibody is cleared, but the recrudescence of deficits may track their original onset occurring over months after the AZ59 falls below effective levels.
Because, the AZ59 anti‐PrPC approach does not alter Aß accumulation or gliosis, we hypothesize that its benefit would be synergistic with the development of any therapies successful in lessening these other steps in the AD process. Since it is doubtful that any single therapy will eradicate clinical AD symptoms, combination therapy may eventually provide the best hope for maximal efficacy in the clinic. Overall, the current preclinical work supports further development of AZ59 as disease‐modifying AD therapy.
Author Contributions
T.O.C., E.C.G., A.B. and S.M.S. designed the experiments. T.O.C. conducted mouse dosing behavior and histology; T.O.C. and E.C.G., binding assays; A.S., brain histology; A.H.B., M.T.C. and L.T.H., signaling assays; AstraZeneca personnel, antibody isolation and production. T.O.C., A.H.B., and S.M.S. analyzed data; and T.O.C. and S.M.S. wrote the paper data.
Conflict of Interest
J.H., G.R., B.D., M.G., M.G., C.D., T.V., I.C., and A.B. are full time employees of AstraZeneca MedImmune and have filed a patent application describing AZ59 antibody. S.M.S. is Inventor for an issued patent claiming use of PrPC inhibition for Alzheimer's disease.
Acknowledgments
We thank Stefano Sodi for expert technical assistance. This work was supported by grants from the National Institutes of Health (NIH) and from the Falk Medical Research Trust to S.M.S.
Funding Information
This work was supported by grants from the National Institutes of Health (NIH) and from the Falk Medical Research Trust to S.M.S.
Funding Statement
This work was funded by National Institutes of Health (NIH) grants P50AG047270, R01AG034924, R35NS097283, and RF1AG053000.
References
- 1. Citron M, Westaway D, Xia W, et al. Mutant presenilins of Alzheimer's disease increase production of 42‐residue amyloid beta‐protein in both transfected cells and transgenic mice. Nat Med 1997;3:67–72. [DOI] [PubMed] [Google Scholar]
- 2. Cleary JP, Walsh DM, Hofmeister JJ, et al. Natural oligomers of the amyloid‐beta protein specifically disrupt cognitive function. Nat Neurosci 2005;8:79–84. [DOI] [PubMed] [Google Scholar]
- 3. Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 2002;297:353–356. [DOI] [PubMed] [Google Scholar]
- 4. Kostylev MA, Kaufman AC, Nygaard HB, et al. Prion‐protein‐interacting Amyloid‐beta oligomers of high molecular weight are tightly correlated with memory impairment in multiple Alzheimer mouse models. J Biol Chem 2015;290(28):17415–17438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Giacobini E, Gold G. Alzheimer disease therapy–moving from amyloid‐beta to tau. Nat Rev Neurol 2013;9:677–686. [DOI] [PubMed] [Google Scholar]
- 6. Schneider LS, Mangialasche F, Andreasen N, et al. Clinical trials and late‐stage drug development for Alzheimer's disease: an appraisal from 1984 to 2014. J Intern Med 2014;275:251–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lauren J, Gimbel DA, Nygaard HB, et al. Cellular prion protein mediates impairment of synaptic plasticity by amyloid‐beta oligomers. Nature 2009;457:1128–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gimbel DA, Nygaard HB, Coffey EE, et al. Memory impairment in transgenic Alzheimer mice requires cellular prion protein. J Neurosci 2010;30:6367–6374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Salazar SV, Gallardo C, Kaufman AC, et al. Conditional deletion of prnp rescues behavioral and synaptic deficits after disease onset in transgenic Alzheimer's disease. J Neurosci 2017;37:9207–9221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cisse M, Sanchez PE, Kim DH, et al. Ablation of cellular prion protein does not ameliorate abnormal neural network activity or cognitive dysfunction in the J20 line of human amyloid precursor protein transgenic mice. J Neurosci 2011;31:10427–10431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Purro SA, Nicoll AJ, Collinge J. Prion protein as a toxic acceptor of amyloid‐beta oligomers. Biol Psychiatry 2018;83:358–368. [DOI] [PubMed] [Google Scholar]
- 12. Haas LT, Kostylev MA, Strittmatter SM. Therapeutic molecules and endogenous ligands regulate the interaction between brain cellular prion protein (PrPC) and metabotropic glutamate receptor 5 (mGluR5). J Biol Chem 2014;289:28460–28477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Haas LT, Salazar SV, Kostylev MA, et al. Metabotropic glutamate receptor 5 couples cellular prion protein to intracellular signalling in Alzheimer's disease. Brain 2016;139(Pt 2):526–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Haas LT, Salazar SV, Smith LM, et al. Silent allosteric modulation of mGluR5 maintains glutamate signaling while rescuing Alzheimer's mouse phenotypes. Cell Rep 2017;20:76–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Haas LT, Strittmatter SM. Oligomers of amyloid beta prevent physiological activation of the cellular prion protein‐metabotropic glutamate receptor 5 complex by glutamate in Alzheimer disease. J Biol Chem 2016;291:17112–17121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Um JW, Kaufman AC, Kostylev M, et al. Metabotropic glutamate receptor 5 is a coreceptor for Alzheimer abeta oligomer bound to cellular prion protein. Neuron 2013;79:887–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kaufman AC, Salazar SV, Haas LT, et al. Fyn inhibition rescues established memory and synapse loss in Alzheimer mice. Ann Neurol 2015;77:953–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Um JW, Nygaard HB, Heiss JK, et al. Alzheimer amyloid‐beta oligomer bound to postsynaptic prion protein activates Fyn to impair neurons. Nat Neurosci 2012;15:1227–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nicoll AJ, Collinge J. Preventing prion pathogenicity by targeting the cellular prion protein. Infect Disord Drug Targets 2009;9(1):48–57. [DOI] [PubMed] [Google Scholar]
- 20. Zhang D, Qi Y, Klyubin I, et al. Targeting glutamatergic and cellular prion protein mechanisms of amyloid beta‐mediated persistent synaptic plasticity disruption: longitudinal studies. Neuropharmacology 2017;121:231–246. [DOI] [PubMed] [Google Scholar]
- 21. Klyubin I, Nicoll AJ, Khalili‐Shirazi A, et al. Peripheral administration of a humanized anti‐PrP antibody blocks Alzheimer's disease Abeta synaptotoxicity. J Neurosci 2014;34:6140–6145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hu NW, Nicoll AJ, Zhang D, et al. mGlu5 receptors and cellular prion protein mediate amyloid‐beta‐facilitated synaptic long‐term depression in vivo. Nat Commun 2014;5:3374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Freir DB, Nicoll AJ, Klyubin I, et al. Interaction between prion protein and toxic amyloid beta assemblies can be therapeutically targeted at multiple sites. Nat Commun 2011;2:336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Barry AE, Klyubin I, Mc Donald JM, et al. Alzheimer's disease brain‐derived amyloid‐beta‐mediated inhibition of LTP in vivo is prevented by immunotargeting cellular prion protein. J Neurosci 2011;31:7259–7263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chung E, Ji Y, Sun Y, et al. Anti‐PrPC monoclonal antibody infusion as a novel treatment for cognitive deficits in an Alzheimer's disease model mouse. BMC Neurosci 2010;11:130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Klohn PC, Farmer M, Linehan JM, et al. PrP antibodies do not trigger mouse hippocampal neuron apoptosis. Science 2012;335:52. [DOI] [PubMed] [Google Scholar]
- 27. Solforosi L, Criado JR, McGavern DB, et al. Cross‐linking cellular prion protein triggers neuronal apoptosis in vivo. Science 2004;303:1514–1516. [DOI] [PubMed] [Google Scholar]
- 28. Reimann RR, Aguzzi A. Intrinsic toxicity of Antibodies to the globular domain of the prion protein. Biol Psychiatry 2018;84:e51–e52. [DOI] [PubMed] [Google Scholar]
- 29. Purro SA, Mead S, Khalili‐Shirazi A, et al. Reply to: intrinsic toxicity of antibodies to the globular domain of the prion protein. Biol Psychiatry 2018;84:e53–e54. [DOI] [PubMed] [Google Scholar]
- 30. Herrmann US, Sonati T, Falsig J, et al. Prion infections and anti‐PrP antibodies trigger converging neurotoxic pathways. PLoS Pathog 2015;11:e1004662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sonati T, Reimann RR, Falsig J, et al. The toxicity of antiprion antibodies is mediated by the flexible tail of the prion protein. Nature 2013;501:102–106. [DOI] [PubMed] [Google Scholar]
- 32. Lloyd C, Lowe D, Edwards B, et al. Modelling the human immune response: performance of a 1011 human antibody repertoire against a broad panel of therapeutically relevant antigens. Protein Eng Des Sel 2009;22:159–168. [DOI] [PubMed] [Google Scholar]
- 33. Douthwaite JA, Sridharan S, Huntington C, et al. Affinity maturation of a novel antagonistic human monoclonal antibody with a long VH CDR3 targeting the Class A GPCR formyl‐peptide receptor 1. MAbs. 2015;7:152–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhang R, Xue G, Wang S, et al. Novel object recognition as a facile behavior test for evaluating drug effects in AbetaPP/PS1 Alzheimer's disease mouse model. J Alzheimers Dis 2012;31:801–812. [DOI] [PubMed] [Google Scholar]
- 35. Serneels L, Van Biervliet J, Craessaerts K, et al. gamma‐secretase heterogeneity in the Aph1 subunit: relevance for Alzheimer's disease. Science 2009;324:639–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhu X, Raina AK, Rottkamp CA, et al. Activation and redistribution of c‐jun N‐terminal kinase/stress activated protein kinase in degenerating neurons in Alzheimer's disease. J Neurochem 2001;76:435–441. [DOI] [PubMed] [Google Scholar]
- 37. Le Pichon CE, Meilandt WJ, Dominguez S, et al. Loss of dual leucine zipper kinase signaling is protective in animal models of neurodegenerative disease. Sci Transl Med 2017;9:eaag0394. [DOI] [PubMed] [Google Scholar]
- 38. Li X, Alafuzoff I, Soininen H, et al. Levels of mTOR and its downstream targets 4E‐BP1, eEF2, and eEF2 kinase in relationships with tau in Alzheimer's disease brain. FEBS J 2005;272:4211–4220. [DOI] [PubMed] [Google Scholar]
- 39. Kim HY, Kim HV, Jo S, et al. EPPS rescues hippocampus‐dependent cognitive deficits in APP/PS1 mice by disaggregation of amyloid‐beta oligomers and plaques. Nat Commun 2015;6:8997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ma T, Chen Y, Vingtdeux V, et al. Inhibition of AMP‐activated protein kinase signaling alleviates impairments in hippocampal synaptic plasticity induced by amyloid beta. J Neurosci 2014;34:12230–12238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Salazar SV, Strittmatter SM. Cellular prion protein as a receptor for amyloid‐beta oligomers in Alzheimer's disease. Biochem Biophys Res Commun 2017;483:1143–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tokiwa G, Dikic I, Lev S, Schlessinger J. Activation of Pyk2 by stress signals and coupling with JNK signaling pathway. Science 1996;273:792–794. [DOI] [PubMed] [Google Scholar]
- 43. Jonsson T, Stefansson H, Steinberg S, et al. Variant of TREM2 associated with the risk of Alzheimer's disease. N Engl J Med 2013;368:107–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Guerreiro R, Wojtas A, Bras J, et al. TREM2 variants in Alzheimer's disease. N Engl J Med 2013;368:117–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Condello C, Yuan P, Grutzendler J. Microglia‐mediated neuroprotection, TREM2, and Alzheimer's Disease: evidence from optical imaging. Biol Psychiatry 2018;83:377–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hong S, Beja‐Glasser VF, Nfonoyim BM, et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 2016;352:712–716. [DOI] [PMC free article] [PubMed] [Google Scholar]