

Abstract
A 60‐year‐old man is presented with progressive involuntary muscle movements and neuropsychiatric symptoms who developed a variety of additional complaints over 2 years. Brain imaging revealed bilateral basal ganglia calcifications suggesting primary familial brain calcification. Analysis of the SLC20A2 gene revealed a missense mutation (c.541C>T, p.(Arg181Trp)), in silico predicted to be deleterious and not found in available databases. Segregation analysis confirmed his asymptomatic father to harbor the same mutation, though on brain imaging basal ganglia calcifications were found. This report illustrates the intrafamilial variability of the phenotype and generalized myoclonus as the presenting symptom.
Introduction
Primary familial brain calcification (PFBC, OMIM#213600) is an autosomal dominant neuropsychiatric disorder with clinical and molecular heterogeneity. Symptoms include migraine, mood swings, parkinsonism, cognitive decline and psychiatric manifestations with a variable age of onset between 30 and 60 years. Currently, four genes have been associated with PFBC: SLC20A2 (OMIM*158378), PDGFRB (OMIM*173410), PDGFB (OMIM*190040), and XPR1 (OMIM*605237), though no clear genotype–phenotype correlation could be established.1 However, SLC20A2 and XPR1 have been linked to phosphate metabolism while PDGFB and PDGFRB are connected to the blood–brain barrier integrity and pericyte maintenance.1 These four genes represent approximately 60% of all familial cases, with most mutations (40%) found in SLC20A2, encoding the inorganic phosphate transporter PIT‐2.2, 3 The diagnosis of PFBC is established with visualization of bilateral calcification of the basal ganglia on neuroimaging; presence of progressive neurologic dysfunction, in the absence of metabolic, infectious, toxic or traumatic cause. Identification of a heterozygous pathogenic variant by molecular genetic testing confirms the clinical diagnosis of PFBC. We report a family who presented with a variable presentation of PFBC, including progressive myoclonus, due to a likely pathogenic variant in the SLC20A2 gene.
Case Presentation
The proband is a 62‐year‐old man who has been suffering since 2 years from involuntary muscle movements and twitching of the lower limbs, which were initially thought to be segmental myokymias. Initially, he described these as brief electric shock sensations in the legs, predominantly occurring as 3–4 min episodes in the evening or while falling asleep, keeping him awake. Physical examination confirmed the muscle undulating twitches and a discrete positional tremor of the hands. Electromyography was performed but showed neither myokymic discharges nor myopathic or neuropathic changes. During the next months, involuntary movements – mainly muscle fasciculations – also arose in the arms and torso. The patient complained of neuropathic pain in the upper and lower limbs with a warm and burning sensation, and associated with muscle cramps and stiffness. The complaints were initially episodic and were relieved through exercise. However, the symptoms continued to worsen progressively over a period of 2 years, particularly in the legs. There were no seizures or epilepsy.
The medical history of the proband was further remarkable for a compression fracture of a cervical vertebra due to osteoporosis as well as mood swings at age 60 (mainly recurrent depressive moods, potentially as a result of the occurrence of the involuntary movements) for which he was treated with carbamazepine.
Brain imaging revealed bilateral basal ganglia calcifications, lenticulostriatal and in the globus pallidus (Fig. 1A and B). A small older lacunar infarction was noted in the pedunculus cerebri of the mesencephalon. Comprehensive neurological (electromyography, electroencephalogram), metabolic (serum electrolytes, thyroid and parathyroid hormones, calcitonin, albumin, billirubin, transaminases, gamma GT, cholesterol, triglyceride, vitamin B12, vitamin D, folate, urea, and creatinine; urine elektrolytes, urea and creatinine) and infectious (toxoplasmosis, cytomegalovirus virus) examinations were all normal. Family history was unremarkable for neurological problems, apart from unspecified psychiatric problems in his sister.
Figure 1.
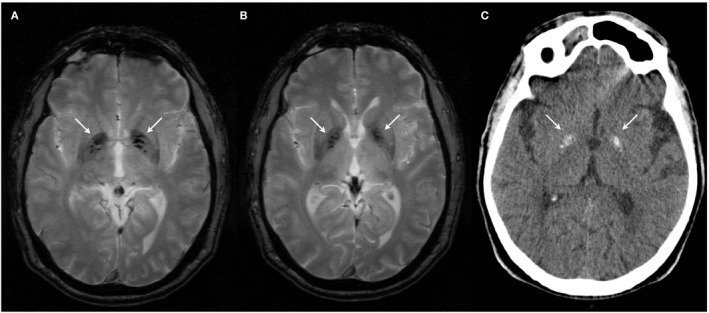
Brain imaging of the proband and his father. In the proband, T2* shows multiple hypointense lesions in the globus pallidus (A, B). There is no surrounding edema and no signs of leukoencefalopathy can be noted. In the proband's father, bilateral hyperintense calcifications in the basal ganglia were noted, without white matter involvement (C).
Within 2 years after the diagnosis, the patient showed a general worsening of the phenotype. The involuntary movements became generalized, occurring throughout the day but most severe in the evening. They could be provoked by yawning. Novel symptoms, which occurred during this period, included dizziness, ocular pain, headaches associated with photophonofobia and rustling sounds. The latter improved using a combined tricyclic anti‐depressive and anti‐psychotic drug (melitracen and flupentixol). Cognition remained normal at all times. Furthermore, the proband developed complaints of tinnitus as well as bilateral hearing loss for which hearing aids were necessary. Audiometry confirmed bilateral symmetrical neurosensorial hearing loss in the high tones (right: 45 dB at 8000 Hz; left 65 dB at 4000 Hz). Ophthalmological evaluation revealed mild cataract. The family history of the proband remained unchanged. At age 65, beginning of nuclear cataract was noted.
Molecular Analysis
Because of the brain imaging findings, PFBC was suspected and molecular analysis of the four known PFBC genes was performed. PCR amplification and subsequent Sanger sequencing of all protein‐coding exons and exon–intron boundaries of SLC20A2, PDGFB, PDGFRB, and XPR1 genes were performed. Novelty of the variants was investigated using the online databases: dbSNP142 (http://www.ncbi.nlm.nih.gov), 1000 Genomes Project (http://www.1000genomes.org), the Exome Variant Server (http://evs.gs.washington.edu/EVS), the Exome Aggregation Consortium (http://exac.broadinstitute.org) and the Genome Aggregation Consortium (http://gnomad.broadinstitute.org/). The potential effect of the variants at the protein level was predicted using PolyPhen‐2 (http://genetics.bwh.harvard.edu/pph2).
Sequencing revealed a missense variant, c.541C>T, p.(Arg181Trp), in exon 5 of SLC20A2, affecting a relatively well conserved nucleotide (Fig. S1), predicted to be deleterious by in silico tools and not found in European non‐Finnish control genomes (only found in one Finnish individual in the gnomAD database, MAF = 4.1e‐06). The SLC20A2 exon 5, encoding transmembrane helix 6 of the SLC20A2 protein, already harbors 4 other (likely) pathogenic variants (Fig. 2). This variant was therefore classified as likely pathogenic, according to the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (ACMG‐AMP) guidelines.4 Segregation analysis confirmed the father of the proband also carried the same variant. As he did not present any symptoms, brain imaging was done and demonstrated bilateral symmetric basal ganglia calcifications (Fig. 1C). The sibs of the proband were not available for molecular testing.
Figure 2.
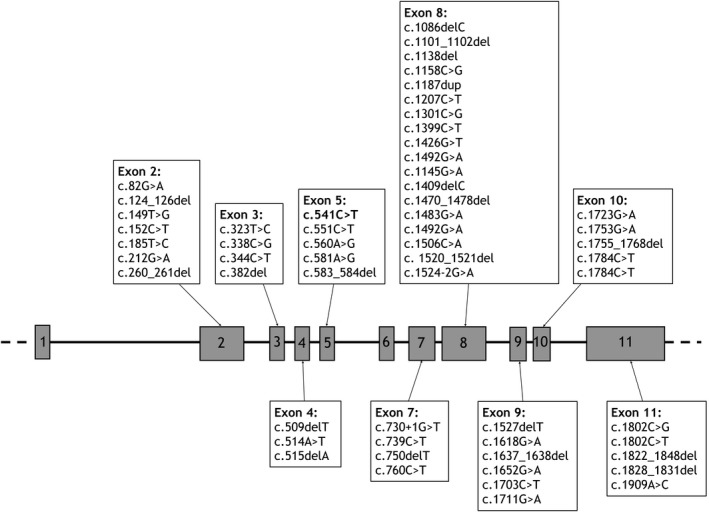
Reported (likely) pathogenic variants in the SLC20A2 gene. The exon structure of the SLC20A2 gene is shown, with the previously reported class 4 (likely pathogenic) and class 5 (pathogenic) variants. The likely pathogenic variant found in the proband is shown in bold in exon 5.
Discussion
Intracranial calcifications are common, often age‐dependent findings that do not lead to any medical concern. More rarely, pathological calcifications can be associated with infections (e.g., CMV) or be genetically determined. Among the latter, besides syndromic forms, PFBC is an important differential diagnosis, because of its severity and progressive nature. The phenotypic spectrum of PFBC is heterogeneous, including cognitive, psychiatric and movement disorders.5 Our proband initially presented with abnormal movements and neuropsychiatric symptoms but developed a wide variety of additional complaints over a two‐year period.
Movement disorders are a frequent symptom of SLC20A2‐related PFBC and seem more common compared to PDGFRB‐associated PFBC. Parkinsonism is the most common presenting movement disorder.6 To our knowledge, a presentation with myoclonus has been reported only once in brain calcification.7 Although rare, myoclonic attacks have been associated with (atypical) Aicardi–Goutières syndrome which also presents with basal ganglia calcifications 8 but have been considered to be mainly cortical in origin because of the positive response to piracetam.9 Our patient, however, did not show any cortical calcifications. More recently, myoclonic attacks and basal ganglia calcifications were described as an unusual presentation in a young patient with a progressive neurometabolic disorder due to mutations in the FOLR1 gene.10 Pathophysiologically, a role for the globus pallidus could be suggested. The internal globus pallidus inhibits the thalamocortical neurons via GABA and substance P. An increased thalamocortical drive has been previously suggested to be involved in the development of myoclonus.7
Pain has been associated with PFBC and, according to Manyam et al., occurred in as much as 16% of patients, though the precise nature of the pain sensation is not specified.11 Although it remains difficult to correlate this symptom with the anatomical location of the calcified lesions, in the absence of any neuropathic changes on the EMG, it cannot be excluded that the neuropathic pain sensations of the proband are part of the PFBC phenotype.
Other symptoms of the proband, which started after the PFBC diagnosis, such as tinnitus, osteoporosis, and sensorineural hearing loss are not known to be associated with PFBC. The basal ganglia have been reported to play a role in the pathophysiology of tinnitus, which can also be noted in patients with secondary basal ganglia calcifications. As the onset of the tinnitus co‐occurred with the abnormal movements, a relation with PFBC cannot be excluded.
Similarly, an inverse relation between soft tissue and bone mineralization is well known,12 though data on the presence or absence of a bone phenotype in PFBC are scarce. The proband had a compression fracture of a cervical vertebra due to osteoporosis. There were no known risk factors that would make the patient more susceptible. Blood results showed normal levels of calcitonin, magnesium, parathyroid hormone, phosphorus, and calcium (Table 1). Vitamin D levels were also within normal limits (53.8 pg/mL (19–95)). The risk of a fracture might have been increased by the dysregulation of the calcium and phosphate balance, known in PFBC. Vitamin D deficiency causes osteomalacia and poor bone mineralization.13 Hypovitaminosis D has been described in patients with primary brain calcifications and vitamin D treatment has been suggested to regulate SLC20A2 gene expression and reduce brain calcification.14 Phenotypic data of the Slc20a2 knockout mouse appears to correlate well with the loss of functions found in PFBC patients. This homozygous knockout model shows a variety of symptoms including general growth impairment, kyphosis, cataract and decreased bone mineral density,15 of which the latter two were also present in our proband.
Table 1.
Biochemical measurements in the proband
| Proband values | Reference values | |
|---|---|---|
| Calcium | 2.37 | 2.12–2.62 mmol/L |
| Phosphorus | 1.13 | 0.80–1.45 mmol/L |
| Magnesium | 0.80 | 0.70–1.05 mmol/L |
| Parathyroid hormone | 22.9 | 15–65 ng/L |
| 1,25‐dihydroxy vitamin D | 53.8 | 19–95 pg/mL |
L, liter; mmol, millimol; mL, milliliter; ng, nanogram; pg, picogram.
Despite a fairly late onset of disease symptoms, the natural history of the proband was characterized by a rapid progression of disease and complicated by mood swings and depressive episodes, chronic headaches and vertigo – all previously associated with PFBC – as well as an increasing number of vague and specific complaints which are most probably of psychosomatic nature as they could be effectively treated with anti‐depressive drugs. The unspecified psychiatric problems reported in his sister may be due to PFBC but no brain imaging or molecular analysis could be obtained from this patient.
This family illustrates the important intrafamilial variability of the PFBC phenotype, though the penetrance of the basal ganglia calcifications seems higher. PFBC has an age‐dependent radiological penetrance, reaching 95% by age 50.2 The precise clinical penetrance has not been fully established for the different PFBC‐related genes and pathologic variants, but may be around 70% or even lower. According to Westenberger et al. only two‐thirds of mutation carriers develop neurologic and/or psychiatric symptoms, while the other third could remain asymptomatic throughout their lifespan.1, 16 In this case, the father of the proband, carrying the same likely pathogenic variant and having bilateral symmetric basal ganglia calcifications on brain imaging, remained asymptomatic. No correlation has been identified between age of onset, extent of calcium deposits, and neurologic deficits. Brain plasticity and resilience to calcification could explain why, in some cases, the calcification anticipates symptoms by various decades.17
In conclusion, we present a family with PFBC, harboring a likely pathogenic SLC20A2 variant and generalized myoclonus. Movement disorders and neuropsychiatric symptoms are the hallmark symptoms that – together with the typical basal ganglia imaging – should evoke the diagnosis of PFBC, with generalized nonepileptic myoclonus to be added to the list of presenting symptoms. The clinical significance of several other symptoms present in this family – some of which are recapitulated in the Slc20a2‐/‐ murine model – in relation to PFBC remains to be elucidated in a larger cohort of patients.
Author Contributions
Simon Lamquet contributed to Research project execution, data review and critique, and manuscript draft. Eliana Marisa Ramos and Andrea Legati also contributed to Research project execution, data review, and manuscript review. Giovanni Coppola contributed to Research project organization and execution, data review, and manuscript review. Dimitri Hemelsoet also contributed to Research project conception, organization and execution, data review and critique, and manuscript review. Olivier M. Vanakker also contributed to Research project conception, organization and execution, data review and critique, manuscript draft, and review.
Conflicts of Interest
The authors have no conflicts of interest to report.
Supporting information
Figure S1. Conservation of the affected nucleotide in the SLC20A2 gene.
Acknowledgments
This study was supported by a BOF research fellowship and a Methusalem grant (BOF08/01M01108) from the Ghent University. Olivier M. Vanakker is a Senior Clinical Investigator of the Research Foundation ‐ Flanders (Belgium).
Funding Information
This study was supported by a BOF research fellowship and a Methusalem grant (BOF08/01M01108) from the Ghent University. Olivier M. Vanakker is a Senior Clinical Investigator of the Research Foundation ‐ Flanders (Belgium).
Funding Statement
This work was funded by Ghent University grant BOF08/01M01108.
References
- 1. Westenberger A, Klein C. The genetics of primary familial brain calcifications. Curr Neurol Neurosci Rep 2014;14:490. [DOI] [PubMed] [Google Scholar]
- 2. Lemos RR, Ferreira JBMM, Keasey MP, Oliveira JRM. Chapter Fourteen ‐ An Update on Primary Familial Brain Calcification In: Bhatia K. P., Schneider S. A., eds. International review of neurobiology. 2013;110:349–371. [DOI] [PubMed] [Google Scholar]
- 3. Ramos EM, Oliveira J, Sobrido MJ. Primary Familial Brain Calcification In: Adams M. P., Ardinger H. H., Pagon R. A., Wallace S. E., Bean L. J. H., Stephens K., et al. eds Genereviews. Seattle, MA:University of Washington, 2017. [PubMed] [Google Scholar]
- 4. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and genomics and the association for molecular pathology. Genet Med 2015;17:405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nicolas G, Pottier C, Charbonnier C, et al. Phenotypic spectrum of probable and genetically‐confirmed idiopathic basal ganglia calcification. Brain 2013;136:3395–3407. [DOI] [PubMed] [Google Scholar]
- 6. Nicolas G, Charbonnier C, de Lemos RR, et al. Brain calcification process and phenotypes according to age and sex: lessons from SLC20A2, PDGFB, and PDGFRB mutation carriers. Am J Med Genet B Neuropsychiatr Genet 2015;168:586–594. [DOI] [PubMed] [Google Scholar]
- 7. Lauterbach EC, Spears TE, Prewett MJ, et al. Neuropsychiatric disorders, myoclonus, and dystonia in calcification of basal ganglia pathways. Biol Psychiatry 1994;35:345–351. [DOI] [PubMed] [Google Scholar]
- 8. Berger A, Schroeter C, Wiemer‐Kruel A, et al. Atypical case of Aicardi‐Goutieres syndrome with late‐onset myoclonic status. Epileptic Disord 2007;9:140–144. [DOI] [PubMed] [Google Scholar]
- 9. Genton P, Guerrini R, Remy C. Piracetam in the treatment of cortical myoclonus. Pharmacopsychiatry 1999;32:49–53. [DOI] [PubMed] [Google Scholar]
- 10. Toelle SP, Wille D, Schmitt B, et al. Sensory stimulus‐sensitive drop attacks and basal ganglia calcification: new findings in a patient with FOLR1 deficiency. Epileptic Disord 2014;16:88–92. [DOI] [PubMed] [Google Scholar]
- 11. Manyam BV, Walters AS, Narla KR. Bilateral striopallidodentate calcinosis: clinical characteristics of patients seen in a registry. Mov Disord 2001;16:258–264. [DOI] [PubMed] [Google Scholar]
- 12. Cannata‐Andia JB, Roman‐Garcia P, Hruska K. The connections between vascular calcification and bone health. Nephrol Dial Transplant 2011;26:3429–3436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bhan A, Rao AD, Rao DS. Osteomalacia as a result of vitamin D deficiency. Endocrinol Metab Clin North Am 2010;39:321–331. [DOI] [PubMed] [Google Scholar]
- 14. Keasey MP, Lemos RR, Hagg T, Oliveira JRM. Vitamin‐D receptor agonist calcitriol reduces calcification in vitro through selective upregulation of SLC20A2 but not SLC20A1 or XPR1. Sci Rep 2016;6:25802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bezerra DP, Oliveira JR. New studies on knockout mouse for the SLC20A2 Gene show much more than brain calcifications. J Mol Neurosci 2016;59:565–566. [DOI] [PubMed] [Google Scholar]
- 16. Gagliardi M, Morelli M, Iannello G, et al. A SLC20A2 mutation identified in an asymptomatic patient with brain calcification. J Neurol Sci 2017;372:70–72. [DOI] [PubMed] [Google Scholar]
- 17. Oliveira JR, Lima Filho JL, Zatz M. Identical twins with idiopathic basal ganglia calcification (“Fahr disease”) presenting with a remarkable similar pattern of neuroimaging findings. Parkinsonism Relat Disord 2009;15:396. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Conservation of the affected nucleotide in the SLC20A2 gene.