

Abstract
Over the last decade, a subfamily of G protein-coupled receptors that are agonized by endogenous and dietary free-fatty acids (FFA) has been discovered. These free-fatty acid receptors include FFA2 and FFA3, which are agonized by short-chained FFA, as well as FFA1 and FFA4, which are agonized by medium-to-long chained FFA. Ligands for FFA1 and FFA4 comprise the family of long chain polyunsaturated omega-3 fatty acids including α-linolenic acid (ALA), eicosapentaenoic acid (EPA), and docosahexaenoic acid (DHA), suggesting that many of the long-known beneficial effects of these fats may be receptor mediated. In this regard, FFA4 has gathered considerable interest due to its role in ameliorating inflammation, promoting insulin sensitization, and regulating energy metabolism in response to FFA ligands. The goal of this review is to summarize the body of evidence in regard to FFA4 signal transduction, its mechanisms of regulation, and its functional role in a variety of tissues. In addition, recent endeavors toward discovery of small molecules that modulate FFA4 activity are also presented.
1. Introduction
G protein-coupled receptors (GPCR) represent the largest and most diverse family of cell surface receptors and are known to regulate a variety of physiological processes. GPCRs also represent the largest family of drugged proteins, with some estimates suggesting that roughly 40% of marketed drugs target a member of the superfamily. Sequencing of the human genome revealed almost 900 distinct GPCR-encoding genes, with almost half of these classified as chemosensory receptors that transmit signals in response to olfaction or gustation, while approximately one-third have been characterized as ‘deorphanized’ receptors that bind a cognate endogenous ligand [1]. Currently, less than 100 GPCRs remain classified as ‘orphan’ receptors, lacking positive identification of an associated endogenous ligand [1]. Bioinformatic mining coupled with molecular biology based approaches in the early 2000’s revealed a subfamily of orphan GPCRs, including GPR40, GPR43, GPR41, and GPR120, which were subsequently shown to recognize and be agonized by endogenous and dietary free fatty acids (FFA) [2–3]. As a consequence of their deorphanization, these free-fatty acid receptors have recently been renamed by the International Union of Basic and Clinical Pharmacology as FFA1, FFA2, FFA3, and FFA4, respectively [4]. FFA2 and FFA3 recognize and are agonized by short-chain fatty acids such as acetate, propionate, and butyrate, which are the primary metabolic byproducts of anaerobic fermentation by intestinal microflora [5]. Meanwhile, though FFA1 and FFA4 share low sequence homology, they are both activated by medium and long-chain fatty acids, including the family of omega-3 (n-3) polyunsaturated fatty acids (PUFA), typified by α-linolenic acid (ALA), eicosapentaenoic acid (EPA), and docosahexaenoic acid (DHA) [4].
Recent studies have elucidated a profound role for FFA4 in modulation of metabolism and energy utilization as well as endocrine and immune function, and as a consequence, have thrust FFA4 to the forefront of drug discovery efforts. For example, activation of FFA4 by fatty acid or synthetic ligands has been shown to elicit incretin hormone release from the gastrointestinal tract, modulate anti-inflammatory effects in macrophages, enhance hepatic glucose uptake and decrease steatosis, and improve insulin resistance and sensitivity [6–10]. The relative importance of FFA4 in modulating metabolic homeostasis is highlighted in FFA4−/− animals, which develop obesity, glucose intolerance, hepatic steatosis, and demonstrate impaired glucose metabolism and heightened lipogenesis compared to control animals when fed high fat diets [11]. Moreover, FFA4 expression is elevated in obese humans compared to lean controls, and a non-synonymous mutation (R270H) of the human receptor inhibits its function, enhances inflammation and hepatic steatosis, and increases the risk of obesity in European populations, further underscoring the significance of the receptor in human metabolism and endocrine regulation [11]. The goal of this research update is to summarize the most recent and seminal advances in our understanding of the structure, function, and regulation of FFA4, and to highlight agents that are known to modulate its activity.
2. FFA4 gene, protein, and alternative splice variants
The gene for the human FFA4 was initially cloned from a genomic DNA fragment [6] and was shown to contain 1134 nucleotides encoding for a 377 amino acid protein with structural topology predictive of GPCRs [3,6,12]. Interestingly, in humans, two gene products arise due to alternative splicing of exon 3, an effect that can form both the longer 377 amino acid protein, commonly referred to as FFA4-’Long’ (FFA4-L), as well as a shorter transcript of 1086 nucleotides that encodes for a 361 amino acid protein, referred to as FFA4-’Short’ (FFA4-S) [12–13] (Fig. 1A). Notably, FFA4-L contains an additional 16 amino acid sequence within intracellular loop 3 (ICL3), a GPCR domain that is typically involved in protein interactions, downstream signaling, and desensitization. Cloning of the mouse and rat orthologs showed high sequence homology of 98% between the rodent amino acid sequences, and 85% and 86% between the rat and human and mouse and human FFA4-S proteins, respectively (Fig. 1B) [13]. An important species distinction has shown that FFA4-L exists only in humans as no evidence of the longer transcript has been demonstrated to date in rodents or non-human primates [13]. As will be discussed in detail below, the long isoform has signal transduction effects that are distinct from that of the short and its tissue distribution is sparse compared to FFA4-S.
Figure 1: (A) Snake Diagram of FFA4-S and FFA4-L.

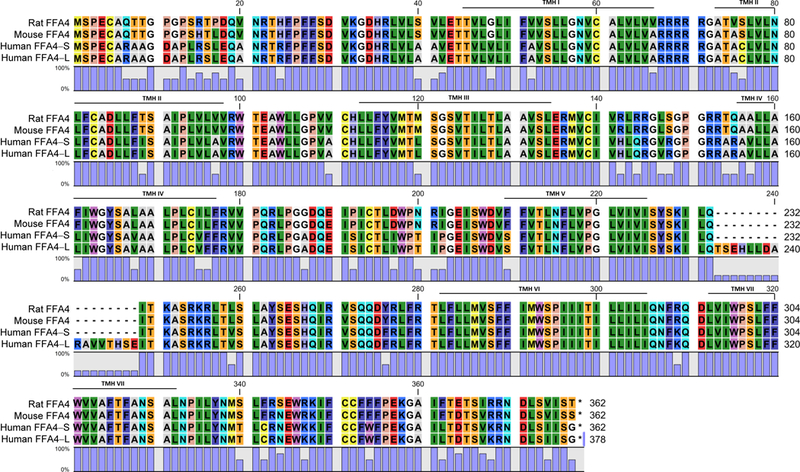
Numbers signify amino acid residues relative to initiating methionine. The insert shows the additional 16 amino acid gap within ICL3 of FFA4-L and corresponding numbering of FFA4-S/L residues after this point are noted. The amino acid residues shaded in red are implicated in ligand binding (R99, Y104, F115 F211, W207, W277, F304). The amino acids shaded in yellow are known phosphorylation sites (T347, T349, S350, S357, S361) of FFA4-S, which along with the noted acid residues (E341, D348, D355) shaded in green, create the β-arrestin phosphosensor. (B) Amino acid homology of rat, mouse, and human FFA4. Rat and mouse FFA4 share 98% sequence homology, while sequence homology of human FFA4-S with the rat and mouse protein is 85 and 86%, respectively. The percentage of homology (0–100%) at each amino acid residue across all four proteins is noted by the blue bar. Putative transmembrane helical (TMH) domains are noted.
3. FFA4 signal transduction
3.1. Ligand binding interactions
While no crystal structure of ligand bound or unbound FFA4 has been solved to date, mutational analysis and homology modeling has shed considerable insight into structural features that direct ligand binding to FFA4. Using the shorter isoform, Arg99 (2.64 in Ballesteros-Weinstein numbering), within extracellular loop 1 (ECL1), has been shown to coordinate binding of FFA4 agonists, putatively via the carboxylate pharmacophore common to the described fatty acid and synthetic ligands. Mutation of this residue abolishes functional activity of ALA as well as those induced by the synthetic FFA1/FFA4 agonist GW9508 and the FFA-4 selective agonist TUG-891 [14–15]. Using a mutagenesis-based approach, Hudson and colleagues developed an elaborate model of the orthosteric binding site of FFA4-S, revealing a role for five aromatic residues, F1153.29, F2115.42, W2075.38, W2776.48, and F3047.36 in formation of a hydrophobic binding pocket which likely contributes to π-stacking with aromatic moieties of the agonist ligands (Fig. 1A) [14]. Mutation of any of these to alanines effectively abolished agonist activity [14]. An additional aromatic residue located in ECL1, Trp104, was also predicted by the model to contribute a hydrogen-bond between its indole nitrogen to the ligand carboxylate, and mutation of the residue also abolished agonist activity (Fig. 1A) [14]. Given that these studies have utilized the shorter isoform, to date, no evidence exists to either claim or refute that agonist binding to FFA4-L would be distinct; and similarly, since these studies employ known functional agonists of the receptor, more work is required to establish the binding interactions of receptor antagonists (vide infra).
3.2. Intracellular signaling
FFA4-L was initially described to elicit intracellular Ca2+ mobilization in response to fatty acids, suggesting that it coupled to Gαq/11 or Gαi/o proteins, yet this effect required fusion to exogenous promiscuous G proteins [6], questioning the physiological relevance of this function. Following identification of the shorter isoform, it was reported that in response to agonism with the long-chain unsaturated fatty acid oleic acid, native FFA4-L is indeed unable to elicit Ca2+ responses, while FFA4-S produced robust Ca2+ mobilization [15]. The effect of FFA4-S was not sensitive to pertussis toxin (PTX), but was diminished by inhibition of IP3 receptors, suggesting that at least in transfected HEK293 cells, the short variant of the receptor couples to Ca2+ signaling via Gαq/11 [15]. Following agonism, both FFA4-L and FFA4-S elicited similar responses toward recruitment of β-arrestin partner proteins, and subsequently, both demonstrate robust internalization and are targeted to lysosomal compartments for degradation, rather than recycling [15]. These data shed the first important light on signaling differences between the two transcript variants and suggest that the insertion of the 16 amino acids into ICL3 of FFA4-L facilitates intrinsic bias toward β-arrestin signaling. While both Gαq/11 and β-arrestins have been implicated in temporally-regulated downstream signaling to phosphorylation of extracellular-signal regulated kinase 1/2 (ERK1/2) [16], Hudson and colleagues revealed that FFA4 activation by TUG-891 leads to rapid ERK1/2 phosphorylation via Gαq/11 signaling, followed by a sustained ERK1/2 effect dependent on both Gαq/11 and transactivation of epidermal growth factor receptor (EGFR) [17]. In addition, there have been tissue-specific reports of promiscuous FFA4 signaling through Gαi/o as well as GαS proteins. In this regard, both FFA4 mediated regulation of somatostatin secretion from murine pancreatic delta cells and ghrelin from primary murine gastric mucosal cell cultures were at least in large part inhibited by PTX, suggesting Gαi/o-dependent mechanisms [18–19]. Meanwhile, Tsukahara and colleagues reported that FFA4-mediated inhibition of GLP-2 secretion from rat enteroendocrine L-cells is sensitive to the protein kinase A (PKA) inhibitor H-89, suggesting that in these cells, FFA4 can modulate GαS/cAMP/PKA signaling [20]. The individual physiological contribution of the various signaling pathways within specific tissues is discussed further in section 4 below.
3.3. Phosphoregulation of FFA4
Agonist occupation of GPCRs facilitates receptor phosphorylation via G protein-coupled receptor kinases (GRK), leading to high affinity recruitment of β-arrestins in a process termed homologous phosphorylation [21]. This regulatory mechanism serves to physically uncouple the GPCR from further G-protein interactions and effectively desensitizes downstream G-protein signaling [21]. Interaction between the phosphorylated receptor and β-arrestin partner proteins allows for the later to serve as an important signaling scaffold which can consequently facilitate β-arrestin-mediated signaling, as well as receptor internalization and trafficking [22]. GPCRs can also be heterologously phosphorylated by downstream signal transduction induced by activation of other GPCRs within the same cell, a process that typically involves second messenger activated serine/threonine kinases such as PKA or protein kinase C (PKC). Together, homologous and heterologous GPCR phosphorylation serves as a critical regulator of G-protein and β-arrestin signaling, internalization, as well as receptor internalization, degradation, and cell-surface recycling [22].
Due to the importance of β-arrestin signaling in FFA4 function, there has been much effort in characterizing phosphoregulation of FFA4. In 2010, Burns and Moniri showed that both ALA and DHA facilitate rapid phosphorylation of both FFA4 isoforms, and importantly, despite four additional phospholabile sites within ICL3 of FFA4-L, there is no significant difference in the rate or degree of agonist induced phosphorylation between the two isoforms [12]. Yet, it was revealed that FFA4-S was phosphorylated to a greater degree in the absence of agonist compared to the longer variant, suggesting that the additional portion of ICL3 in FFA4-L contributes to decreasing constitutive phosphorylation of the longer isoform [12]. Further efforts have focused on FFA4-S, where Burns and colleagues demonstrated that DHA-stimulated homologous phosphorylation was mediated by GRK6, whereas heterologous phosphorylation was mediated by agonism of other GPCRs that activate the Gαq/11/Ca2+/PKC pathway, or via direct activation of PKC with phorbol 12-myristate 13-acetate (PMA) [23]. Using mutational analysis, it was reported that GRK6 and PKC phosphorylate Thr347, Ser350, and Ser357 within the C-terminus of FFA4-S, and mutation of all three sites abrogated β-arrestin recruitment and increased agonist-mediated Ca2+ signals [23]. Using a more sensitive proteomic-based approach coupled to mutagenesis, Butcher and colleagues also showed that Thr347, Ser350, and Ser357 were phosphorylated upon agonism with TUG-891, and moreover, revealed additional contributions of nearby Thr349 and Ser360 to the TUG-891 induced phosphorylation (Fig. 1A) [24]. Perhaps most importantly, this groups’ work showed that mutation of all five phosphorylation sites decreased, but did not abolish β-arrestin-2 interactability, suggesting that other residues contribute to formation of the FFA4 β-arrestin phosphosensor [24]. Indeed, in the same body of work, Butcher and colleagues discovered three acidic residues within the C-terminus (Glu341, Asp348, and Asp355) that together with the five phosphorylation sites (Thr347, Thr349, Ser350, Ser357, and Ser360) form two distinct regions of negative charge that comprise the FFA4 β-arrestin-phosphosensor (Fig. 1A) [24]. The importance of phosphorylation in regulating β-arrestin signaling lends tightly to drug discovery efforts that must ensure adequate stimulus bias of novel drug agents for phosphorylation/arrestin activity versus more common screens based on FLIPR based Gαq/11/Ca2+. The tissue-specific physiological importance of β-arrestin biased FFA4 signaling, described in detail below, will underscore the need for such efforts in drug development.
4. FFA4 expression and tissue-specific physiological functions
Hirasawa and colleagues initially demonstrated dense expression of FFA4 by RT-PCR in human lung and gastrointestinal tract, as well as adrenal gland [6]. Since, FFA4 expression has been shown to be fairly ubiquitously distributed, with much of the literature focusing on expression of the receptor in tissues that function as dietary sensors, for example on the tongue or GI tract, or in tissues that modulate immune function or metabolic homeostasis (Table 1; Fig. 2).
Table 1:
Tissue specific expression and physiological/cellular function of FFA4.
| Tissue | Cell type or cell line | Physiological or cellular function | Reference |
|---|---|---|---|
| Intestinal | L-cells | Secretion of GLP-1 | [6] |
| L-cells | Inhibition of GLP-2 secretion | [20] | |
| K-cells | Secretion of GIP | [7], [25, 27] | |
| Murine STC1, GLUTag | Secretion of GLP-1 | [26] | |
| HCT116, HT-29, Caco-2 human colon epithelial cancer cells | Increase cytosolic Ca+2 Increase phosphorylation of ERK1/2 | [31] | |
| Gastric | Ghrelin secreting | Inhibition of ghrelin secretion | [33–35] |
| Tongue | Circumvallate papillae epithelia | Fat sensing, GLP-1 secretion | [40–42] |
| Type II taste cells | Taste perception | [40–42] | |
| Adipose | SQ, PR, Mes, Epi fat 3T3-L1 cells |
Adipocyte differentiation, GLUT4 translocation, glucose uptake VEGF-A induced angiogenesis |
[45–47], [52] |
| M2 and M1 macrophages, | Anti-inflammatory effects, insulin sensitization | [49–51] | |
| Macrophages | M2 and M1 macrophages, murine Raw264.7 cells, human THP-1 cells | Anti-inflammatory effects, reduced inflammasome assembly and activity, insulin sensitization | [49], [53–55] |
| Pancreatic Islets | Islet cells | Protection from lipotoxicity | [56] |
| Human and mouse α-cells | Glucagon synthesis and secretion Downstream hepatic glucagon sensitivity |
[18], [57] | |
| Mouse delta-cells | Inhibition of somatostatin secretion | [18] | |
| Murine αTC1–6 cells | [58] | ||
| Skeletal Muscle | Rat and murine gastrocnemius and soleus skeletal muscle | Reduced inflammation, improved insulin sensitivity | [51], [59] |
| Rat L6 and murine C2C12 myoblasts | GLUT4 translocation and increased glucose uptake | [60] | |
| Liver | Murine macrophage-like Kupffer cells | Reduced inflammation | [62] |
| Human hepatocytes, hepatic stem/progenitor cells, and hepatic Kupffer cells | Reduced inflammation, macrophage polarization | [63] | |
| Rat liver WB-F344 cells | Cell migration | [66] | |
| Bone | Osteoclasts | Reduced inflammation, inhibition of osteoclast development and bone resorption | [70–72] |
| Murine ST2 and MC3T3-E1 preosteoblasts | [70] |
Figure 2:
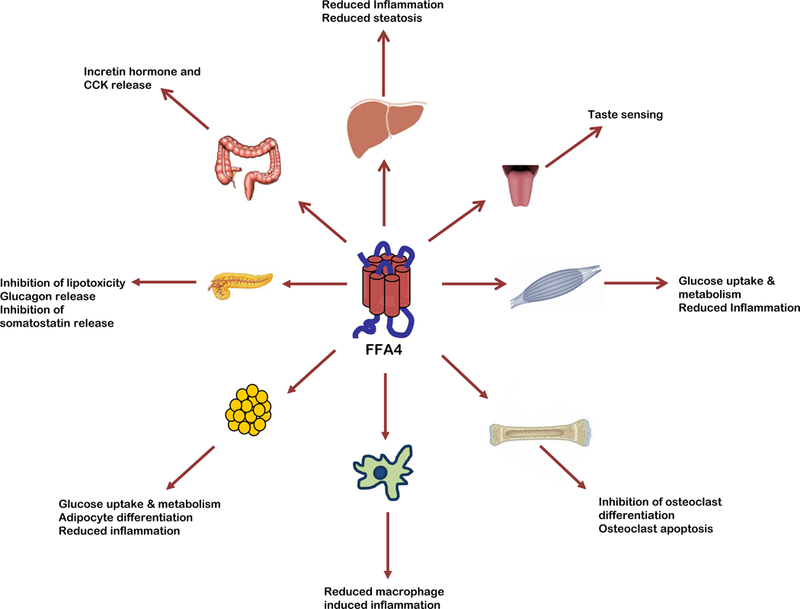
Known physiological effects of FFA4 agonism.
4.1. Hormone secretion and nutrient sensing from the Gastrointestinal tract
Initial studies showed that agonism of FFA4 increased glucagon-like peptide-1 (GLP-1) and cholecystokinin (CCK) secretion from gastrointestinal L- and K-cells, respectively, suggesting a role for the receptor in diet (i.e., fat) induced release of gastrointestinal hormones [6–7, 25]. In this regard, subsequent studies revealed that FFA4 is expressed on mouse STC1 and GLUTag enteroendocrine L-cells that secrete GLP-1 [6, 26], as well as mouse primary cultured K-cells, which secrete glucose-dependent insulinotropic peptide (GIP) [25, 27]. More recently, Paulsen and colleagues confirmed widespread expression of FFA4 in the ileal and colon epithelium; however, their in vivo results suggested that the role of FFA4 activation in GLP-1 secretion is minimal [28]. The role of fatty acid mediated GLP-1 secretion has also been suggested by results from other labs as being more dependent on FFA1 rather than FFA4 [29], signifying that further work is required to fully elucidate whether FFA4 plays a significant physiological role in fatty acid induced GLP-1 secretion in vivo.
To date, FFA4-L expression in humans has been demonstrated only within the colon and on human colonic cancer cell lines. Galindo and colleagues demonstrated expression of both FFA4 isoforms in colon from human tissue [30], while the immortalized human colon cancer cell lines HT-29 and HCT116 endogenously express both isoforms and only FFA4-L, respectively [31]. Importantly in regard to the colon, Wu and colleagues demonstrated that FFA4 expression is significantly upregulated in human colorectal cancer tissues and colon cancer cell lines [32], where interestingly, agonism of FFA4 activates PI3K/AKT/NF-κB signaling to promote cell migration as well as angiogenesis and tumorogenesis in vitro and in vivo [32].
FFA4 has also been shown to be expressed in the stomach and duodenum, where it inhibits ghrelin secretion in response to sensing of dietary fatty acids [19, 33–36]. These gastrointestinal effects have attracted much attention to the receptor due to the role of these hormones in modulation of metabolic processes such as insulin secretion, digestion, and satiety. Given the dense expression of FFA4 in the GI tract, it is of interest that studies in animals fed high fat diets reveal upregulation of FFA4 expression in variety of tissues, including within the GI tract. FFA4 transcript and protein expression in the intestines were increased in diet-induced obese rats compared to the diet-resistant genotype [37], while results from Cheshmehkani and colleagues confirm that FFA4 expression in the colon is also upregulated in diets supplemented with n-3 fatty acids [38]. Still, others have shown that FFA4 transcript and the number of FFA4-positive enteroendocrine cells in the mouse gastric corpus increased in as little as three weeks in animals fed a high fat diet, while the FFA4-positive cells in the gastric antrum was reduced, suggesting that dietary fat regulation of FFA4 expression is cell-type specific [39]. These data correspond to human studies that show chemosensory cells in the stomach express nearly 4-fold higher levels of FFA4 mRNA in morbidly obese individuals compared to controls [40]. There is also evidence of upregulation of receptor expression in inflammatory bowel diseases. In this case, the normal ileal epithelium has been shown to express only low levels of FFA4 in normal subjects, while mucosal inflammation of the ileum, as in the case of Crohn’s Disease, leads to significantly elevated FFA4 mRNA and protein in Crohn’s patients, an effect that also seems to link FFA4 to dysregulation of GLP-2 secretion [20].
4.2. Taste perception and nutrient sensing within the gustatory epithelium
Within the tongue, FFA4 has been shown to be expressed at significant levels in epithelia of rodent circumvallate papillae and type II taste cells that transmit gustatory signals, but not within in nonsensory papillae [41–43]. In man, FFA4 is present in both gustatory and nongustatory ligual epitheliua [44]. Within these cells, FFA4 is thought to play a role in sensing of dietary fatty acids and, along with FFA1, acts to transmit taste sensations and modulate taste preferences in response to the presence of fats [44]. Within the circumvallate papillae, FFA4 may also be involved in cross-talk to sweet taste preference through mechanisms that involve local secretion of lingual GLP-1, suggesting a role for the receptor in modulation of sweet taste preference in addition to that of fats [45].
4.3. Anti-inflammatory and metabolic effects within adipose
Consistent with the fundamental effects of fatty acids on energy storage and expenditure within adipose tissue, FFA4 seems to play a significant role in regulation of energy metabolism within adipocytes. High levels of FFA4, but not FFA1 mRNA, are expressed in human differentiated adipocytes, but not in human preadipocytes. FFA4 was found to be expressed in subcutaneous (SQ), perirenal (PR), mesenteric (Mes), and epididymal (Epid) adipose, as well as 3T3-L1 adipocytes, with demonstrated increases in its expression correlated to increased adipocyte differentiation [46–47]. Knockdown of FFA4 by siRNA resulted in inhibition of adipocyte differentiation and also decreased the expression of key adipocyte regulators, including PPAR-γ2, aP2, IRS, and GLUT4, further validating a role for the receptor in adipocyte differentiation [46, 48]. Meanwhile, animals fed high-fat diets expressed significantly heightened levels of FFA4 mRNA in SQ, Mes, and Epid adipose compared to control [46], and these data mirror those seen in obese humans, who express nearly 2−fold the amount of FFA4 mRNA in subcutaneous and omental adipose tissues compared to lean controls [49]. Seminal work by Oh and colleagues demonstrated that activation of adipose FFA4 by DHA or the synthetic agonist GW9508 significantly enhances glucose uptake by increasing GLUT4 translocation in a manner dependent on coupling to Gαq/11 and downstream activation of PI3-kinase (PI3K) [50]. Perhaps most importantly, this work also showed that n-3 fatty acids reduced in vivo migration of inflammatory macrophages into adipose tissue via activation of FFA4. Simultaneously, FFA4 agonism decreased expression of proinflammatory genes, including IL-6, TNF-α, MCP-1, IL-1β, iNOS, adiponectin and CD11c, while increasing expression of anti-inflammatory genes, including arginase-1, IL-10, MGL1, Ym-1, Clec7a, and MMR; effects that were lost in FFA4−/− mice [50]. Macrophage infiltrates within human adipose strongly inhibited FFA4 expression, further expanding the linkage between FFA4 expression and inflammation [51]. More recent evidence by Oliveira and colleagues showed that dietary n-3 and n-9 PUFA revert inflammation and insulin resistance in obese mice via mechanisms dependent on FFA4 expressed on adipose, as well as liver and muscle [52]. Together, the adipose effects of FFA4 serve to play major roles in regulation of weight, insulin sensitivity, and metabolic inflammation.
4.4. Anti-inflammatory effects in macrophages
Omega-3 PUFA have been historically linked to anti-inflammatory effects and recently, FFA4 has been noted to, at least in part, modulate many of these effects via effects on macrophages (summarized in Fig. 3). FFA4 expression has been demonstrated in primary intraperitoneally-derived macrophages, as well as on polarized M1 and M2 macrophages and the murine RAW264.7 macrophage cell line [50]. In addition to noted effects of macrophage infiltration of adipocytes above, Oh and colleagues reported that FFA4 agonism modulates reversal of LPS-induced phosphorylation of pro-inflammatory IKKβ and JNK, as well as downstream nuclear translocation of NF-κB within macrophages [50]. Moreover, FFA4 agonism inhibited release of pro-inflammatory mediators such as IL-6 and TNF-α from these cells [50]. These anti-inflammatory effects of FFA4 were shown to be fully dependent on the receptors interaction with β-arrestin partner proteins, which facilitate downstream inhibition of TAB1/TAK1/NF-κB signaling (Fig. 3) [50]. Using the human monocytotic THP-1 cell line and murine bone marrow derived macrophages, Williams-Bey and colleagues reported that FFA4 agonism by DHA inhibits NLRP3 inflammasome assembly and activity, inhibits the nuclear translocation of NF-κB, and results in significant reduction in proinflammatory IL-1β production and secretion, in a manner dependent on Gαq/11 as well as β-arrestin-2 signaling [54]. In 2013, Li and colleagues showed that FFA4 agonism with DHA reduced COX2, but not COX1, expression and activity, resulting in decreased prostaglandin E2 (PGE2) synthesis upon induction by LPS in macrophages [55]. Further, this group showed that TLR4 signaling through phosphorylation of AKT and JNK, as well as NF-κB translocation, was inhibited by DHA mediated agonism of FFA4 in a β-arrestin-2 dependent manner (Fig. 3) [55]. Subsequently, Liu and colleagues reported an additional mechanism whereby DHA and GW9508, via FFA4, activate cytosolic PLA2 and COX-2 to generate and release PGE2, which consequently acts on EP4 receptors to inhibit NF-κB-modulation of inflammation in RAW264.7 cells and human primary monocyte-derived macrophages [56]. This anti-inflammatory mechanism was again reported to be dependent on Gαq/11 and β-arrestin-2, and also relies on activation of ERK1/2 (Fig. 3) [56]. Given the critical role of macrophages in the pathophysiology of a multitude of human illnesses including insulin resistance, diabetes, obesity, cardiovascular disease, asthma and other pulmonary obstructive diseases, as well as tumorogenesis, it is very likely that modulation of FFA4 activity may play a wide-reaching role in future treatment of a variety of disease states and will be subject to significant research over the coming decade.
Figure 3: Anti-inflammatory FFA4 signaling pathways in macrophages.
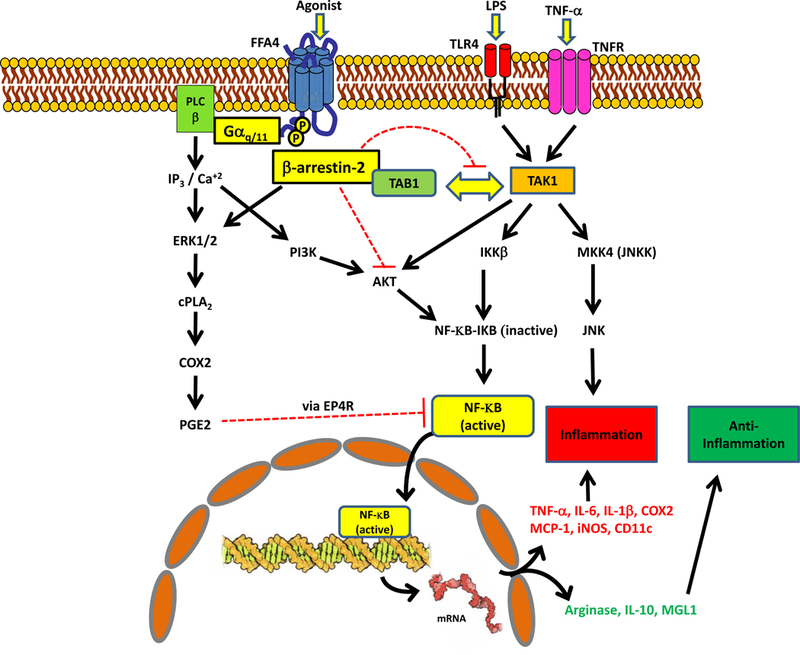
LPS and TNF-α are well known to promote inflammatory processes via activation of Toll-like receptor 4 (TLR4) and TNF-receptor (TNFR), respectively. Activation of either receptor causes the interaction of TAK1 kinase and its binding protein TAB1, activating the TAK1 complex. Active TAK1 phosphorylates MKK4 leading to phosphorylation of JNK. TAK1 can also phosphorylate IK-kinase-β which then phosphorylates IKB, releasing its ability to inhibit NF-κB. Phosphorylated IKB is ubiquitinated and degraded, thereby promoting further activity of NF-κB due to the loss of the inhibitor. NF-κB and phospho-JNK promote downstream inflammatory processes, including increased expression of TNF-α, IL-6, IL-1β, COX2, MCP-1, iNOS, and CD11c. Agonism of FFA4 leads to β-arrestin-2 recruitment, which sequesters TAB1, thereby, inhibiting its interaction with TAK1. Loss of this interaction inhibits downstream activation of NF-κB and JNK, and promotes anti-inflammatory effects, and can also facilitate expression of anti-inflammatory mediators such as Arginase-1, IL-10, and MGL1. Furthermore, FFA4-dependent β-arrestin recruitment inhibits AKT signaling, again, inhibiting the activation of NF-κB, thereby promoting anti-inflammatory effects. Finally, activation of FFA4 activation, via both Gαq/11 and β-arrestin-2, facilitates ERK1/2-dependent PGE2 synthesis, which can then act via prostaglandin EP4 receptors to inhibit NF-κB-modulated inflammatory effects.
4.5. Protection from lipotoxicity and hormone secretion from endocrine pancreas
Taneera and colleagues reported that FFA4 expression in human islets is positively correlated with insulin content and secretion, as well as with lower glycated hemoglobin (HbA1c). Expression of FFA4 mRNA was substantially reduced in hyperglycemic donors with type 2 diabetes mellitus compared to non-diabetic or normoglycemic controls [57]. Treatment of human islets with EPA prevented lipotoxicity and increased cell viability, and these effects were reduced by 50% upon knockdown of FFA4, suggesting that islet FFA4 plays a major role in modulating protection from lipid-induced cytotoxicity [57]. While islet expression of FFA4 is evident, the specific cell types that modulate FFA4 responsiveness to fatty acid agonists remains somewhat controversial, particularly due to the functional feedback of GLP-1 on pancreatic islet function and the previously noted effects of FFA4 agonism on GLP-1 secretion. Suckow and colleagues revealed expression of FFA4 mRNA in human and mouse islets, and also reported the expression of the receptor on pancreatic α−cells [58]. Consistent with this observation, fatty acid induced glucagon secretion was significantly reduced in FFA4 knockout (KO) mice, and FFA4 and FFA1 were both shown to contribute to long-chain fatty acid mediated release of glucagon from these cells [58]. While islet glucagon levels were unaffected in FFA4 KO animals, islet insulin levels were reduced compared to wild-type controls, and importantly, FFA4 KO animals exhibited increased glucagon secretion and liver glucagon sensitivity compared to wild-type [58]. These results suggest that FFA4 modulation of the glucagon signaling axis also contributes to metabolic homeostasis. Stone and colleagues demonstrated dense localization of FFA4 within somatostatin secreting delta cells and more modest (>~15% of total) expression within the α-cells of mouse islets, with no detectable expression within β-cells [18]. Glucose-dependent somatostatin secretion from murine islets was dose-dependently inhibited by the synthetic FFA4 agonist Metabolex-36, and this effect was not seen in islets isolated from FFA4 KO mice [18]. Interestingly, the effects of this agonist on somatostatin secretion were at least in part attenuated by PTX, demonstrating FFA4 coupling to Gαi/o proteins in the delta cells. It is also noteworthy that although Metabolex-36 affected somatostatin secretion, the endogenous agonist DHA did not [18]. To date, there have been no reports in regard to the expression or role of FFA4 in the digestive exocrine pancreas.
4.6. Anti-inflammatory and metabolic effects within Skeletal muscle
FFA4 has been shown to be expressed in rat and mouse gastrocnemius and soleus (i.e., calf) as well as extensor digitorum longus (EDL) skeletal muscle [52, 60]. High fat diets increased FFA4 expression in EDL but not soleus skeletal muscle [60]. Meanwhile, diets enriched with 10% n-3 and n-9 fatty acids mediated improvement in muscle sensitivity to insulin as well as significant decreases in muscle inflammation as noted by reduced macrophage inflammation and increased IL-10, in a manner that was dependent on both FFA4 and FFA1 [52]. FFA4 is also expressed on murine C2C12 and rat L6 myoclasts, where it was shown to modulate DHA-mediated GLUT4 translocation and glucose uptake via a mechanism dependent on Ca2+/CaMKK/AMPK signaling [61]. These data are in contrast to those that show that FFA4 plays no role in fatty acid uptake and oxidation in muscle during endurance exercise [62]. Taken together, these results demonstrate that FFA4 influences inflammation of muscle as well as uptake and metabolism of glucose, but not of fatty acids.
4.7. Anti-inflammatory effects within liver
FFA4 has been shown to be expressed in the liver [60], and its extrahepatic activation, or lack thereof, has been shown to greatly influence hepatic function. In addition to becoming glucose intolerant and obese, FFA4 KO mice fed a high-fat diet demonstrate enhanced hepatic lipogenesis and develop fatty liver [11]. Immunohistochemistry of mouse liver sections revealed FFA4 expression in liver sinusoids, where it colocalizes exclusively with the macrophage marker F4/80, suggesting the restricted expression of the receptor in the resident macrophage-like Kupffer cells [63]. On these cells, FFA4 agonism serves to diminish hepatic inflammation via abrogation of NF-κB and JNK signaling [63]. On the contrary to this report and others [58], FFA4 was shown to be expressed in hepatocytes and hepatic stem/progenitor cells, in addition to hepatic Kupffer cells in biopsies from pediatric non-alcoholic fatty liver disease (NAFLD) patients [64]. Interestingly, the number of Kupffer cells expressing FFA4 was increased in these patients, and this elevation was strongly correlated with increased serum TNF-α, IL-6, and IL1-β. Treatment with DHA (250 mg/day) for 18-months caused a substantial increase in levels of receptor expressed in hepatocytes while inducing a significant reduction of FFA4-positive Kupffer cells, an effect which was again strongly correlated with significant reduction of phospho-NF-κB nuclear translocation and cytokine synthesis following the DHA treatment [64]. Together, these data show that FFA4-positive hepatic macrophages are responsible for proinflammatory cytokine production that can modulate steatosis, while activation of FFA4 could ameliorate this effect. This conclusion is validated by recent results showing that obese children and adolescents who heterozygously express the dysfunctional FFA4-R270H mutant variant are predisposed to hepatic steatosis and liver injury [65–66].
4.8. Anti-inflammatory effects and nutrient sensing in brain substructures
While FFA1 has been relatively well studied in the brain, the expression and function of FFA4 in various brain substructures and nuclei remains largely unstudied. Wellhauser and Belsham [68] demonstrated expression of FFA4 transcript in rat hypothalamus as well as on rat immortalized embryonic hypothalamic neurons, where its agonism reduces the inflammatory response to TNF-α in a TAB1/TAK1-dependent manner similar to that which was reported for the peripheral anti-inflammatory effects (Fig. 3) [8]. Expression of FFA4, but not FFA1 transcript has been shown in rat pituitary gland and pituitary primary cultures [69], while the immortalized LβT2 mouse gonadotrope cell line was shown to express both receptors [69]. Expression of FFA4 was also investigated in the intact mouse pituitary gland, where it was shown to be densely expressed on gonadotropes, but not thyrotropes, somatotropes, lactotropes, corticotropes, melanotropes, or within the posterior pituitary gland [70]. Colocalization of FFA4 with luteinizing and follicle stimulating hormones within the gonadotropes suggested that the receptor could play a role in synthesis and/or GnRH-dependent release of the gonadotrophs [70]. However, Garrel and colleagues showed that neither GW9508 nor the unsaturated fatty acid linoleate could induce LH secretion in LβT2 cells, implying that FFA1 and FFA4 are not involved in gonadotroph release [69]. Despite this, of particular interest, FFA4 mRNA was increased approximately 3−fold in mice that underwent a 24-hour fast, suggesting that the receptor could serve as a gonadotropic dietary fatty acid sensor sensitive to levels of serum fatty acids [70]. The function of FFA4 in this capacity, as well as within other brain structures remains to be elucidated.
4.9. Effects on bone resorption
FFA4 has been shown to be expressed in osteoblast-like cell lines including bone marrow derived ST2 cells and MC3T3-E1 preosteoblasts, as well as in chondrogenic ATDC5 cells [71–72], and the previously mentioned RAW264.7 cells, which can be differentiated to functional osteoclasts. In chondrocytes, DHA and EPA were shown to induce chondrocyte proliferation and differentiation and regulate expression of FFA4 [72]. Cornish and colleagues demonstrated a role for long chain fatty acid mediated inhibition of osteoclastogenesis, an effect that is reproduced with the FFA1/4 agonist GW9508, however, a clear delineation of the roles of either of these FFA receptors was not made here [71]. Seminal work in osteoclasts by Kim and colleagues showed that FFA4 expression is upregulated during osteoclast development, and agonism with GW9508 prevented maturation of murine bone marrow-derived macrophages to osteoclasts, via inhibition of RANKL-mediated NF-κB and JNK activation, as well as induction of nuclear factor of activated T cells c1 (NFATc1), a critical modulator osteoclastogesis [73]. Importantly, these effects were reversed in cells treated with FFA4-targeted shRNA and the net effect of FFA4 agonism in this cell type was acceleration of osteoclast apoptosis and inhibition of bone resorption [73], suggesting that FFA4 could be targeted to treat diseases involving bone degradation. Since bone density is modulated by constant remodeling due to osteoclasts and osteoblasts, further work to characterize the effect of FFA4 in osteoblast function and bone generation will be needed.
5. FFA4 ligands
5.1. FFA4 agonists
To date, there have been many endogenous fatty acid molecules that have been identified as FFA4 agonists, including ALA, DHA, EPA, as well as palmitoleic, myristic, and linoleic acids [6–15]. While these agonists are all long chain (e.g., 14–22 carbons) fatty acids, including saturated as well as unsaturated lipids, they are of low relative potency and are all non-selective towards agonism of both FFA4 and FFA1 (Table 2). Yore and colleagues recently identified a novel class of structurally distinct endogenous lipids containing branched fatty acid esters of hydroxy-fatty acids (FAHFA) that specifically agonize FFA4 and mimic the anti-inflammatory, insulin-sensitizing, and other metabolic events described above via engagement of FFA4 [74]. Distinct FAHFA isomers were identified with varying levels of tissue distribution and concentrations, with palmitic-acid-9-hydroxy-stearic-acid (9-PAHSA) (Table 2) being identified as the most abundant FAHFA in murine adipose tissue, while 13/12-, 11-, 10-, 9-, and 5-PAHSA isomers were all detected at low nanomolar concentrations, similar to other bioactive lipids, in serum [74]. Importantly, PAHSAs were shown to be components of various foods, were synthesized in vivo, and were susceptible to isomer-specific and tissue-specific regulation under fasting and high-fat feeding, as well as in insulin-resistant conditions in vivo. The signaling effects of various PAHSAs on Ca2+ mobilization and β-arrestin signaling were not studied. However, PAHSA levels strongly correlated with insulin sensitivity and were reduced in adipose and serum of insulin-resistant humans [74]. PAHSA administration lowered basal glycemia, improved glucose tolerance, and stimulated both GLP-1 and insulin secretion in vivo [74]. In addition, PAHSAs had an intense effect on reversing LPS-mediated inflammation and also increased GLUT-4 translocation and glucose uptakes in adipocytes, effects that were abolished in cells treated with FFA4 siRNA [74]. Together, these data convincingly show that PAHSAs are important endogenous modulators of FFA4 and its regulation of metabolism and inflammation. The structures of these various endogenous agonists are shown in Table 2, along with known relative potencies for both Gαq/11 and β-arrestin signaling at FFA1 versus FFA4.
Table 2.
Structures and relative FFA4 versus FFA1 potencies of various endogenous agonists expressed as nanomolar potency and (pEC50 ± S.D). Values are for human receptors and for FFA4-S unless noted. N.R. denotes data that are not reported.
| Endogenous Agonist | Ca+2 mobilization | β-arrestin BRET | References | ||
|---|---|---|---|---|---|
| FFA1 nM / (pEC50 ± SD) |
FFA4 nM / (pEC50 ± SD) |
FFA1 nM / (pEC50 ± SD) |
FFA4 nM / ( pEC50 ± SD) |
||
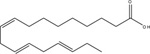 α-linolenic acid (ALA) |
1148 / (5.94 ± 0.03) | 550 / (6.26 ± 0.04) 5012 / (5.30 ± 0.07) 5623 / (5.25 ± 0.10) |
23,442 / (4.63 ± 0.15) 2884 / (5.54 ± 0.30) |
6918 / (5.16 ± 0.08) 7943 / (5.1 ± 0.1) 7762 / (5.11 ± 0.05) 3235 / (5.49 ± 0.14) |
[75] [14] [15] [17] [77] |
 Linoleic acid (LA) |
2238 / (5.65 ± 0.06) | 1288 / (5.89 ± 0.04) | N.R. | N.R. | [75] |
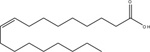 Oleic acid |
N.R. | 19,953 / (4.7 ± 0.3) | N.R. | 39,810 / (4.4 ± 0.1) | [15] |
|
Myristic acid |
N.R. | 63,095 / (4.2 ± 0.3) | N.R. | N.R. | [15] |
|
Palmitic acid |
1380 / (5.86 ± 0.05) | 759 / (6.12 ± 0.08) | N.R. | N.R. | [75] |
|
Eicosapentaenoic acid (EPA) |
1862 / (5.73 ± 0.04) | 2089 / (5.68 ± 0.06) | N.R. | N.R. | [75] |
|
Palmitic-acid-9-hydroxy-stearic-acid (9-PAHSA) |
N.R. | N.R. | N.R. | N.R. | [74] |
Due to the physiological relevance of FFA4 in modulating a myriad of metabolic processes, there have been significant efforts towards development of synthetic FFA4-selective small molecules. GlaxoSmithKline was the first to reveal a FFA4 acting agonist, however, this compound, 4-(3-Phenoxybenzylamino)phenylpropionic acid (GW9508), displayed higher potency towards FFA1 leading to an approximately 20−70-fold selectivity index for FFA1 over FFA4 (Table 3) [17,75]. Hence, studies that utilize GW9508 as an FFA4 agonist must be interpreted with caution unless lack of FFA1 expression is explicitly demonstrated within the tissue or cells of interest. Since FFA1, FFA4 and PPAR-γ share the same endogenous fatty acid agonists, Suzuki and colleagues utilized a known PPAR-γ pharmacophore coupled to homology modeling of FFA4 to synthesize 4-{4-[2[(Pheyl-2-pyridinylamino)ethoxy]phenyl}butyric acid (NCG21), which displays moderate-potency (EC50 = 1.2 μM) and full agonism at promoting FFA4-mediated Ca2+-mobilization, while retaining 16-fold selectivity for FFA4 versus FFA1 (Table 3) [76]. Despite this improved selectivity, the low relative potency of this agent means that FFA1 agonism is likely at micromolar concentrations required to elicit cell-based responses. More recently, Shimpukade and colleagues performed an elaborate structure-activity study on GW9508-like ortho-biphenyl propionic acids and reported on synthesis of the high potency FFA4 agonist 3-(4-((4-fluoro-4’-methyl-[1,1’-biphneyl]-2-yl)methoxy)phenyl)propionic acid (TUG-891) [77]. Importantly, TUG-891 behaves as a full agonist of human and murine FFA4 and exhibits high selectivity for FFA4 over FFA1 [17, 77] (Table 3). In regard to agonist-mediated β-arrestin-2 signaling, TUG-891 was 288-fold more selective for human FFA4 over FFA1, while ALA was non-selective, GW9508 showed 19-fold selectivity for FFA1, and NCG21 showed 12-fold selectivity for FFA4 [17]. When assessing agonist-induced Ca2+ mobilization, TUG-891 was 52-fold more selective for human FFA4 versus FFA1, while NCG21 showed 8-fold selectivity for FFA4 in this assay [17]. Taken together, these data reveal the utility of TUG-891 as the first high potency FFA4-selective agonist to date. Interestingly, Hudson and colleagues work revealed that the selectivity of TUG-891 diminishes appreciably at the murine FFA receptors. Here, TUG-891 displays improved potency toward murine FFA1, while the selectivity index for β-arrestin-2 signaling is reduced from 288-fold to 61-fold and that for Ca2+ mobilization is reduced from 52-fold to only 3-fold when assessing the effects at mouse FFA4 [17]. Given this context, Hudson and colleagues discussed potential challenges to the use of TUG-891 in assessing in vivo effects in rodent models or when using mouse-derived cells [17]. Nonetheless, TUG-891 remains an excellent high potency and highly selective tool for study of the human receptor and also serves as a promising structural template for discovery of further agonist ligands. In 2014, GlaxoSmithKline reported the synthesis and biological evaluation of a novel diarylsulfonamide compound, 4-methyoxy-N-(2,4,6-trimethylphenyl) benzenesulfonamide (GSK137647A) (Table 3) with approximately 500 nM potency at eliciting Ca2+ mobilization responses via FFA4 [78]. GSK137647A demonstrated greater than 50-fold selectivity for FFA4 versus the other three FFA receptors in regard to Ca2+ mobilization, a characteristic that was maintained with the mouse and rat orthologs [78]. This compound also elicited glucose-dependent insulin secretion from mouse MIN6 cells and displayed approximately 50% efficacy compared to linoleic acid in promoting GLP-1 secretion from NCI-H716 enteroendocrine cells [78]. Curiously, the arylsulfonamide moiety of GSK137647A is weakly acidic and hence, likely ionizable, and could conceivably form ionic interactions with Arg99. Alternatively, it is also feasible that the lack of a strongly acidic carboxylate isostere in this molecule could direct it to an allosteric binding site distinct from the orthosteric site centered around Arg99 described in section 3.1 above. Future work with these sulfonamide-containing agents should shed light on their mechanism of binding within the receptor binding pocket.
Table 3.
Structures and relative FFA4 versus FFA1 potencies of synthetic FFA4 agonists expressed as nanomolar potency and (pEC50 ± S.D). Values are for human receptors and for FFA4-S unless noted. N.R. denotes data that are not reported.
| Agonist | Ca+2 mobilization | β-arrestin BRET | References | ||
|---|---|---|---|---|---|
| FFA1 nM / (pEC50 ± SD) |
FFA4 nM / (pEC50 ± SD) |
FFA1 nM / (pEC50 ± SD) |
FFA4 nM / (pEC50 ± SD) |
||
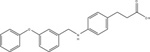 GW9508 |
48 / (7.32 ± 0.03) | 3467 / (5.46 ± 0.09) | [75] | ||
| 1950 / (5.71 ± 0.02) | 912 / (6.04 ± 0.04) | [14] | |||
| 851 / (6.07 ± 0.08) | 48 / (7.32 ± 0.20) | 933 / (6.03 ± 0.03) | [17] | ||
| 28 / (7.55 ± 0.15) | 1412 / (5.85 ± 0.05) | [77] | |||
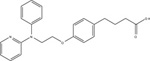 NCG21 |
19,054 / (4.72)* | 1260 / (5.9)* | [76] | ||
| 74,131 / (4.13 ± 0.13) | 9120 / (5.04 ± 0.10) | 23,442 / (4.63 ± 0.15) | 1905 / (5.72 ± 0.08) | [17] | |
| 9772 / (5.01 ± 0.23) | 2398 / (5.62 ± 0.10) | [77] | |||
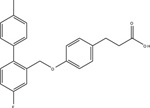 TUG-891 |
537 / ( 6.27 ± 0.02) | 72 / (7.14 ± 0.04) | [14] | ||
| 64,565 / (4.19 ± 0.97) | 44 / (7.36 ± 0.15) | [77] | |||
| 5011 / (5.30 ± 0.04) | 93 / (7.02 ± 0.09) | 65 / (7.19 ± 0.07) | [17] | ||
 GSK137647A |
< 32,000 / (< 4.5) | 501 / (6.3 ± 0.2) | N.R. | N.R. | [78] |
 cpdA |
Negligible effect | 24 / (7.62 ± 0.11) | N.R. | 347 / (6.46) | [9] |
Utilized Gα16 fused receptor construct, putatively FFA4-L
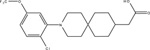
Recently, Oh and colleagues in collaboration with Merck Research Laboratories reported the discovery of a novel FFA4 agonist, cpdA, with high potency and selectivity for FFA4. This agent exhibits 24 nM EC50 at eliciting Gαq/11-mediated Ca2+ mobilization, and 350 nM EC50 at eliciting β-arrestin-2 recruitment [9]. There was negligible signaling effects of cpdA on FFA1 demonstrating full selectivity towards FFA4. Moreover, cpdA elicited similar signaling responses with the murine ortholog. Mice treated with cpdA for five weeks demonstrated improved glucose tolerance, lowered fasting blood insulin, and significantly improved insulin sensitivity [9]. Notably, cpdA mimicked the anti-inflammatory effects of DHA previously described by this group [8] including: decreased expression of proinflammatory genes and modulators and increased expression of anti-inflammatory genes; effects that were not seen in FFA4 KO mice.
Since the discovery of FFA receptors, nearly all major pharmaceutical companies have established FFA receptor drug discovery targeting programs and as a consequence, there are large numbers of putative FFA4 agonist structures reported within the patent literature. Given that these reports generally discuss hit compounds with moderate micromolar potency at eliciting Ca2+ mobilization in FLIPR-based screens, or alternatively, compounds that elicit broad-based desirable animal effects such as glucose-dependent-insulin secretion, and since these patents have been reviewed elsewhere [79–80], they are not discussed in further detail here.
5.2. FFA4 antagonists
Due to the noted beneficial metabolic effects of FFA4 agonism, development of FFA4 antagonists has lagged behind that of agonists. Hara and colleagues described that derivatives of the fungal product grifolin, including grifolic acid and its methyl ether (Fig. 4), promote Ca2+ mobilization and phosphorylation of ERK1/2 in HEK293 cells stably expressing a human FFA4-Gα16 fusion protein [81]. These compounds were also shown to induce GLP-1 secretion from mouse STC-1 cells. However, the efficacy of these compounds in these assays was 30–50% of that achieved by the fully efficacious fatty acid agonist ALA, suggesting that they behave as partial agonists of FFA4. True to this pharmacological character, grifolin acid and methyl ether dose-dependently inhibit ALA-induced ERK1/2 phosphorylation and Ca2+ mobilization, validating their partial agonist nature [81], and suggesting that these compounds would block the effect of the more efficacious fatty acid agonists in vivo.
Figure 4:
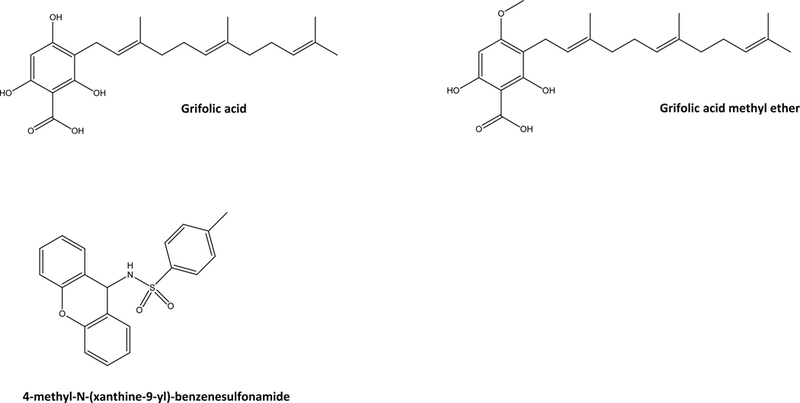
Structures of reported FFA4 antagonists.
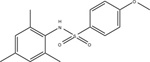
In seeking to develop novel arylsulfonamide agonists of FFA4, GlaxoSmithKline also reported the discovery of 4-methyl-N-(xanthen-9-yl)-benzenesulfonamide (Fig. 4), which has antagonistic effects on both ALA and GSK137647A-induced Ca2+ mobilization [78]. This tricyclic xanthenyl displays high potency for inhibition of human FFA4 function (pIC50 of 7.1 ± 0.35; 79 nM), and also demonstrates notable selectivity owed to its poor potency at inhibition of FFA1 responses (pIC50 of < 4.6; <25 μM) [78]. Interestingly, this agent was 10-fold more potent at rodent receptors (pIC50 of 8.1 for both rat and mouse FFA4; 8nM) compared to the human receptor [78]. To date, these are the only reports of FFA4 selective antagonists within the literature. The critical role of FFA4 activity in modulation of the metabolic and immunological homeostasis discussed throughout this report demonstrates that antagonism of the receptor in vivo would necessarily facilitate a high degree of metabolic and immunological dysfunction, which likely contributes to the overall scarcity of investigations into FFA4 antagonists. However, reports such as those by Wu and colleagues demonstrating that FFA4 activity can pose detrimental effects in certain cases, such as colon cancers [32], suggest that FFA4 antagonists may have therapeutic value in specialized instances. Additionally, their clinical use notwithstanding, FFA4 antagonists will also serve as important pharmacological tools that are greatly needed in order to add to the collective understanding of the receptors biochemistry and pharmacology from a basic science perspective.
6. Future Directions
Over the last decade, free-fatty acid receptors have emerged as important contributors to metabolic, gastrointestinal, and immune-system homeostasis and function, and have also been shown to serve in many cases, as dietary sensors that modulate such functional responses. These receptors have served to demonstrate that many of the effects of fatty acids that have long-been known to be beneficial in man are actually receptor-mediated. In this context, the nutriceutical value of FFA4 agonism by endogenous PUFA cannot be overstated, especially given the massive human public consumption of PUFA supplements, including fish oil and flax seed oil, which are enriched in the n-3 PUFA DHA/EPA and ALA, respectively. However, not all PUFA supplements are made equally and many are actually enriched with contaminating n-6 fatty acids, which can dysregulate the metabolic and in vivo beneficial effects of both n-3 and n-6 PUFA. The consequence of dietary n-3 : n-6 : n-9 ratios in man is well established, but the effect of dietary intake of these various fatty acids on activation and efficacy of FFA4 remains unstudied, as are the outcomes of chronic nutrient-based agonism of FFA4 on receptor desensitization, internalization, and cell-surface expression in vivo. Given that FFA4 is degraded from the cell surface fairly rapidly following its agonism in cells [15], it will be of interest to gauge what effect this effect may have on dietary supplementation of PUFA in vivo. This is an effect that should also be of concern for drug discovery efforts, as receptor desensitization following agonism with small molecules can circumvent effectiveness; yet, this lies in stark contrast to many published studies described here which show that long term administration of dietary PUFA actually increases FFA4 expression. Mechanisms of such regulation will need to be further characterized for better understanding on de novo synthesis, degradation, or recycling of FFA4 protein in a given tissue.
From a cell signaling perspective, given the importance of agonist-directed signaling and biased agonism, one goal of this research update was to review the known contributions of G protein-dependent versus β-arrestin-dependent signals toward promotion of beneficial functions that FFA4 facilitates. As described, many, but not all of the anti-inflammatory effects of FFA4 are modulated by β-arrestin-2 (Fig.3); yet, other beneficial effects (e.g., adipocyte GLUT-4 translocation and glucose uptake) are Gαq/11-mediated, while still others were noted to putatively involve Gαi/o or even Gαs. From the viewpoint of stimulus-bias, and given the importance of FFA4 phosphorylation and β-arrestin recruitment to receptor function, drug discovery efforts must ensure that proper high-throughput assays are utilized in screening of compound libraries to assess both Ca2+ mobilization, as has been historically performed for GPCRs, and also β-arrestin recruitment, as many are doing today.
In conclusion, FFA4 has been well established to play profound roles in mediating metabolic anti-inflammatory effects, and has also been shown to modulate important effects related to taste perception, gastrointestinal function, as well as endocrine balance, thrusting it to the forefront of drug discovery efforts. Since the discovery of FFA receptors, development of FFA1-acting agonists has been well ahead of that of FFA4 agonists, likely due to the role of the former in directly regulating pancreatic insulin secretion. However, as a consequence of its functional importance in reducing metabolic inflammation and its role in energy utilization, it is only a matter of time before clinically promising FFA4 agonists are progressed into phase III trials.
Acknowledgments
Portions of this work were supported fully or in part by the National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases Grant [DK098730] and three Diabetes Action Research and Educational Foundation grants to N.H.M.
The abbreviations used are:
- FFA
Free-fatty acid
- FFA4
Free-fatty acid receptor-4
- FFA1
Free-fatty acid receptor-1
- PUFA
polyunsaturated fatty acids
- ALA
α-linolenic acid
- EPA
eicosapentaenoic acid
- DHA
docosahexaenoic acid
REFERENCES
- [1].Civelli O, Reinscheid RK, Zhang Y, Wang Z, Fredriksson R, Schiöth HB. G protein-coupled receptor deorphanizations. Annu Rev Pharmacol Toxicol. 2013; 53:127–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Sawzdargo M, George SR, Nguyen T, Xu S, Kolakowski LF, O’Dowd BF. A cluster of four novel human G protein-coupled receptor genes occurring in close proximity to CD22 gene on chromosome 19q13.1. Biochem Biophys Res Commun. 1997; 239(2):543–7. [DOI] [PubMed] [Google Scholar]
- [3].Fredriksson R1, Höglund PJ, Gloriam DE, Lagerström MC, Schiöth HB. Seven evolutionarily conserved human rhodopsin G protein-coupled receptors lacking close relatives. FEBS Lett. 2003; 554(3):381–8. [DOI] [PubMed] [Google Scholar]
- [4].Davenport AP, Alexander SP, Sharman JL, Pawson AJ, Benson HE, Monaghan AE, Liew WC, Mpamhanga CP, Bonner TI, Neubig RR, Pin JP, Spedding M, Harmar AJ. International Union of Basic and Clinical Pharmacology. LXXXVIII. G protein-coupled receptor list: recommendations for new pairings with cognate ligands. Pharmacol Rev. 2013; 65(3):967–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Ulven T Short-chain free fatty acid receptors FFA2/GPR43 and FFA3/GPR41 as new potential therapeutic targets. Front Endocrinol (Lausanne). 2012; 3:111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Hirasawa A, Tsumaya K, Awaji T, Katsuma S, Adachi T, Yamada M, Sugimoto Y, Miyazaki S, Tsujimoto G. Free fatty acids regulate gut incretin glucagon-like peptide-1 secretion through GPR120. Nat Med. 2005; 11(1):90–4. [DOI] [PubMed] [Google Scholar]
- [7].Tanaka T, Katsuma S, Adachi T, Koshimizu TA, Hirasawa A, Tsujimoto G. Free fatty acids induce cholecystokinin secretion through GPR120. Naunyn Schmiedebergs Arch Pharmacol. 2008; 377(4–6):523–7. [DOI] [PubMed] [Google Scholar]
- [8].Oh DY, Talukdar S, Bae EJ, Imamura T, Morinaga H, Fan W, Li P, Lu WJ, Watkins SM, Olefsky JM. GPR120 is an omega-3 fatty acid receptor mediating potent anti-inflammatory and insulin-sensitizing effects. Cell. 2010; 142(5):687–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Oh DY, Walenta E, Akiyama TE, Lagakos WS, Lackey D, Pessentheiner AR, Sasik R, Hah N, Chi TJ, Cox JM, Powels MA, Di Salvo J, Sinz C, Watkins SM, Armando AM, Chung H, Evans RM, Quehenberger O, McNelis J, Bogner-Strauss JG, Olefsky JM. A Gpr120-selective agonist improves insulin resistance and chronic inflammation in obese mice. Nat Med. 2014; 20(8):942–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Williams-Bey Y, Boularan C, Vural A, Huang NN, Hwang IY, Shan-Shi C, Kehrl JH. Omega-3 free fatty acids suppress macrophage inflammasome activation by inhibiting NF-κB activation and enhancing autophagy. PLoS One. 2014; 9(6):e97957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Ichimura A, Hirasawa A, Poulain-Godefroy O, Bonnefond A, Hara T, Yengo L, Kimura I, Leloire A, Liu N, Iida K, Choquet H, Besnard P, Lecoeur C, Vivequin S, Ayukawa K, et al. Dysfunction of lipid sensor GPR120 leads to obesity in both mouse and human. Nature. 2012; 483(7389):350–4. [DOI] [PubMed] [Google Scholar]
- [12].Burns RN, Moniri NH. Agonism with the omega-3 fatty acids alpha-linolenic acid and docosahexaenoic acid mediates phosphorylation of both the short and long isoforms of the human GPR120 receptor. Biochem Biophys Res Commun. 2010; 396(4):1030–5. [DOI] [PubMed] [Google Scholar]
- [13].Moore K, Zhang Q, Murgolo N, Hosted T, Duffy R. Cloning, expression, and pharmacological characterization of the GPR120 free fatty acid receptor from cynomolgus monkey: comparison with human GPR120 splice variants. Comp Biochem Physiol B Biochem Mol Biol. 2009; 154(4):419–26. [DOI] [PubMed] [Google Scholar]
- [14].Hudson BD, Shimpukade B, Milligan G, Ulven T. The molecular basis of ligand interaction at free fatty acid receptor 4 (FFA4/GPR120). J Biol Chem. 2014; 289(29):20345–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Watson SJ, Brown AJ, Holliday ND. Differential signaling by splice variants of the human free fatty acid receptor GPR120. Mol Pharmacol. 2012; 81(5):631–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Shenoy SK, Drake MT, Nelson CD, Houtz DA, Xiao K, Madabushi S, Reiter E, Premont RT, Lichtarge O, Lefkowitz RJ. beta-arrestin-dependent, G protein-independent ERK1/2 activation by the beta2 adrenergic receptor. J Biol Chem. 2006; 281(2):1261–73. [DOI] [PubMed] [Google Scholar]
- [17].Hudson BD, Shimpukade B, Mackenzie AE, Butcher AJ, Pediani JD, Christiansen E, Heathcote H, Tobin AB, Ulven T, Milligan G. The pharmacology of TUG-891, a potent and selective agonist of the free fatty acid receptor 4 (FFA4/GPR120), demonstrates both potential opportunity and possible challenges to therapeutic agonism. Mol Pharmacol. 2013; 84(5):710–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Stone VM, Dhayal S, Brocklehurst KJ, Lenaghan C, Sörhede Winzell M, Hammar M, Xu X, Smith DM, Morgan NG. GPR120 (FFA4) is preferentially expressed in pancreatic delta cells and regulates somatostatin secretion from murine islets of Langerhans. Diabetologia. 2014; 57(6):1182–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Engelstoft MS, Park WM, Sakata I, Kristensen LV, Husted AS, Osborne-Lawrence S, Piper PK, et al. Seven transmembrane G protein-coupled receptor repertoire of gastric ghrelin cells. Mol Metab. 2013; 2(4):376–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Tsukahara T, Watanabe K, Watanabe T, Yamagami H, Sogawa M, Tanigawa T, Shiba M, Tominaga K, Fujiwara Y, Maeda K, Hirakawa K, Arakawa T. Tumor necrosis factor α decreases glucagon-like peptide-2 expression by up-regulating G-protein-coupled receptor 120 in Crohn disease. Am J Pathol. 2015; 185(1):185–96. [DOI] [PubMed] [Google Scholar]
- [21].Kohout TA and Lefkowitz RJ. Regulation of G protein-coupled receptor kinases and arrestins during receptor desensitization. Mol Pharmacol. 2003; 63:1, 9–18. [DOI] [PubMed] [Google Scholar]
- [22].Lefkowitz RJ and Shenoy SK. Transduction of receptor signals by beta-arrestins. Science. 2005; 280:24412–24419. [DOI] [PubMed] [Google Scholar]
- [23].Burns RN, Singh M, Senatorov IS, Moniri NH. Mechanisms of homologous and heterologous phosphorylation of FFA receptor 4 (GPR120): GRK6 and PKC mediate phosphorylation of Thr347, Ser350, and Ser357 in the C-terminal tail. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Butcher AJ, Hudson BD, Shimpukade B, Alvarez-Curto E, Prihandoko R, Ulven T, Milligan G, Tobin AB. Concomitant action of structural elements and receptor phosphorylation determines arrestin-3 interaction with the free fatty acid receptor FFA4. J Biol Chem. 2014; 289(26):18451–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Iwasaki K, Harada N, Sasaki K, Yamane S, Iida K, Suzuki K, Hamasaki A, Nasteska D, Shibue K, Joo E, Harada T, Hashimoto T, Asakawa Y, Hirasawa A, Inagaki N. Free fatty acid receptor GPR120 is highly expressed in enteroendocrine K cells of the upper small intestine and has a critical role in GIP secretion after fat ingestion. Endocrinology. 2015; 156(3):837–46. [DOI] [PubMed] [Google Scholar]
- [26].Iakoubov R, Izzo A, Yeung A, Whiteside CI, Brubaker PL. Protein kinase Czeta is required for oleic acid-induced secretion of glucagon-like peptide-1 by intestinal endocrine L cells. Endocrinology. 2007; 148(3):1089–98. [DOI] [PubMed] [Google Scholar]
- [27].Parker HE, Habib AM, Rogers GJ, Gribble FM, Reimann F. Nutrient-dependent secretion of glucose-dependent insulinotropic polypeptide from primary murine K cells. Diabetologia. 2009; 52(2):289–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Paulsen SJ, Larsen LK, Hansen G, Chelur S, Larsen PJ, Vrang N. Expression of the fatty acid receptor GPR120 in the gut of diet-induced-obese rats and its role in GLP-1 secretion. PLoS One. 2014; 10;9(2):e88227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Xiong Y, Swaminath G, Cao Q, Yang L, Guo Q, Salomonis H, Lu J, Houze JB, Dransfield PJ, Wang Y, Liu JJ, Wong S, Schwandner R, Steger F, Baribault H, Liu L, Coberly S, Miao L, Zhang J, Lin DC, Schwarz M. Activation of FFA1 mediates GLP-1 secretion in mice. Evidence for allosterism at FFA1. Mol Cell Endocrinol. 2013; 6369(1–2):119–29. [DOI] [PubMed] [Google Scholar]
- [30].Galindo MM, Voigt N, Stein J, van Lengerich J, Raguse JD, Hofmann T, Meyerhof W, Behrens M. G protein-coupled receptors in human fat taste perception. Chem Senses. 2012; 37(2):123–39. [DOI] [PubMed] [Google Scholar]
- [31].Kim JM, Lee KP, Park SJ, Kang S, Huang J, Lee JM, Sato K, Chung HY, Okajima F, Im DS. Omega-3 fatty acids induce Ca(2+) mobilization responses in human colon epithelial cell lines endogenously expressing FFA4. Acta Pharmacol Sin. 2015; ;36(7):813–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Wu Q, Wang H, Zhao X, Shi Y, Jin M, Wan B, Xu H, Cheng Y, Ge H, Zhang Y. Identification of G-protein-coupled receptor 120 as a tumor-promoting receptor that induces angiogenesis and migration in human colorectal carcinoma. Oncogene. 2013; 32(49):5541–50. [DOI] [PubMed] [Google Scholar]
- [33].Lu X, Zhao X, Feng J, Liou AP, Anthony S, Pechhold S, Sun Y, Lu H, Wank S. Postprandial inhibition of gastric ghrelin secretion by long-chain fatty acid through GPR120 in isolated gastric ghrelin cells and mice. Am J Physiol Gastrointest Liver Physiol. 2012; 303(3):G367–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Janssen S, Laermans J, Iwakura H, Tack J, Depoortere I. Sensing of fatty acids for octanoylation of ghrelin involves a gustatory G-protein. PLoS One. 2012; 7(6):e40168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Gong Z, Yoshimura M, Aizawa S, Kurotani R, Zigman JM, Sakai T, Sakata I. G protein-coupled receptor 120 signaling regulates ghrelin secretion in vivo and in vitro. Am J Physiol Endocrinol Metab. 2014; 306(1):E28–35. [DOI] [PubMed] [Google Scholar]
- [36].Egerod KL, Engelstoft MS, Lund ML, Grunddal KV, Zhao M, Barir-Jensen D, Nygaard EB, Petersen N, Holst JJ, Schwartz TW. Transcriptional and Functional Characterization of the G Protein-Coupled Receptor Repertoire of Gastric Somatostatin Cells. Endocrinology. 2015; 156(11):3909–23. [DOI] [PubMed] [Google Scholar]
- [37].Duca FA, Swartz TD, Sakar Y, Covasa M. Decreased intestinal nutrient response in diet-induced obese rats: role of gut peptides and nutrient receptors. Int J Obes (Lond). 37 (2013) 375–81. [DOI] [PubMed] [Google Scholar]
- [38].Cheshmehkani A, Senatorov IS, Kandi P, Singh M, Britt A, Hayslett R, Moniri NH. Fish oil and flax seed oil supplemented diets increase FFA4 expression in the rat colon. Inflamm Res. 2015; 64(10):809–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Widmayer P, Goldschmid H, Henkel H, Küper M, Königsrainer A, Breer H. High-fat feeding affects the number of GPR120 cells and enteroendocrine cells in the mouse stomach. Front Physiol. 6 (2015) 53:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Widmayer P, Küper M, Kramer M, Königsrainer A, Breer H. Altered expression of gustatory-signaling elements in gastric tissue of morbidly obese patients. Int J Obes (Lond). 2012; 36(10):1353–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Matsumura S, Mizushige T, Yoneda T, Iwanaga T, Tsuzuki S, Inoue K, Fushiki T. GPR expression in the rat taste bud relating to fatty acid sensing. Biomed Res. 2007; 28(1):49–55. [DOI] [PubMed] [Google Scholar]
- [42].Matsumura S, Eguchi A, Mizushige T, Kitabayashi N, Tsuzuki S, Inoue K, Fushiki T. Colocalization of GPR120 with phospholipase-Cbeta2 and alpha-gustducin in the taste bud cells in mice. Neurosci Lett. 2009; 450(2):186–90. [DOI] [PubMed] [Google Scholar]
- [43].Martin C, Passilly-Degrace P, Gaillard D, Merlin JF, Chevrot M, Besnard P. The lipid-sensor candidates CD36 and GPR120 are differentially regulated by dietary lipids in mouse taste buds: impact on spontaneous fat preference. PLoS One. 2011; 6(8):e24014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Cartoni C, Yasumatsu K, Ohkuri T, Shigemura N, Yoshida R, Godinot N, le Coutre J, Ninomiya Y, Damak S. Taste preference for fatty acids is mediated by GPR40 and GPR120. J Neurosci. 2010; 30(25):8376–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Martin C, Passilly-Degrace P, Chevrot M, Ancel D, Sparks SM, Drucker DJ, Besnard P. Lipid-mediated release of GLP-1 by mouse taste buds from circumvallate papillae: putative involvement of GPR120 and impact on taste sensitivity. J Lipid Res. 2012; 53(11):2256–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Gotoh C, Hong YH, Iga T, Hishikawa D, Suzuki Y, Song SH, Choi KC, Adachi T, Hirasawa A, Tsujimoto G, Sasaki S, Roh SG. The regulation of adipogenesis through GPR120. Biochem Biophys Res Commun. 2007; 354(2):591–7. [DOI] [PubMed] [Google Scholar]
- [47].Miyauchi S, Hirasawa A, Iga T, Liu N, Itsubo C, Sadakane K, Hara T, Tsujimoto G. Distribution and regulation of protein expression of the free fatty acid receptor GPR120. Naunyn Schmiedebergs Arch Pharmacol. 2009; 379(4):427–34. [DOI] [PubMed] [Google Scholar]
- [48].Liu D, Wang L, Meng Q, Kuang H, Liu X. G-protein coupled receptor 120 is involved in glucose metabolism in fat cells. Cell Mol Biol (Noisy-le-grand). 2012; Suppl.58:OL1757–62. [PubMed] [Google Scholar]
- [49].Ichimura A, Hirasawa A, Poulain-Godefroy O, Bonnefond A, Hara T, Yengo L, Kimura I, Leloire A, Liu, et al. Dysfunction of lipid sensor GPR120 leads to obesity in both mouse and human. Nature. 2012; 483(7389):350–4. [DOI] [PubMed] [Google Scholar]
- [50].Oh DY1, Talukdar S, Bae EJ, Imamura T, Morinaga H, Fan W, Li P, Lu WJ, Watkins SM, Olefsky JM GPR120 is an omega-3 fatty acid receptor mediating potent anti-inflammatory and insulin-sensitizing effects. Cell. 2010; 142(5):687–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Trayhurn P, Denyer G. Mining microarray datasets in nutrition: expression of the GPR120 (n-3 fatty acid receptor/sensor) gene is down-regulated in human adipocytes by macrophage secretions. J Nutr Sci. 2012; 1:e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Oliveira V, Marinho R, Vitorino D, Santos GA, Moraes JC, Dragano N. Diets containing alpha-linolenic (omega 3) or oleic (omega 9) fatty acids rescues obese mice from insulin resistance. Endocrinology. 2015; in press. [DOI] [PubMed] [Google Scholar]
- [53].Hasan AU, Ohmori K, Konishi K, Igarashi J, Hashimoto T, Kamitori K, Yamaguchi F, et al. Eicosapentaenoic acid upregulates VEGF-A through both GPR120 and PPARγ mediated pathways in 3T3-L1 adipocytes. Mol Cell Endocrinol. 2015; 406:10–8. [DOI] [PubMed] [Google Scholar]
- [54].Williams-Bey Y, Boularan C, Vural A, Huang NN, Hwang IY, Shan-Shi C, Kehrl JH. Omega-3 free fatty acids suppress macrophage inflammasome activation by inhibiting NF-κB activation and enhancing autophagy. PLoS One. 2014. June 9;9(6):e97957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Li X, Yu Y, Funk CD. Cyclooxygenase-2 induction in macrophages is modulated by docosahexaenoic acid via interactions with free fatty acid receptor 4 (FFA4). FASEB J. 2013; 27(12):4987–97. [DOI] [PubMed] [Google Scholar]
- [56].Liu Y, Chen LY, Sokolowska M, Eberlein M, Alsaaty S, Martinez-Anton A, Logun C, Qi HY, Shelhamer JH. The fish oil ingredient, docosahexaenoic acid, activates cytosolic phospholipase A₂ via GPR120 receptor to produce prostaglandin E₂ and plays an anti-inflammatory role in macrophages. Immunology. 2014; 143(1):81–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Taneera J, Lang S, Sharma A, Fadista J, Zhou Y, Ahlqvist E, Jonsson A, Lyssenko V, Vikman P, et al. A systems genetics approach identifies genes and pathways for type 2 diabetes in human islets. Cell Metab. 2012; 16(1):122–34. [DOI] [PubMed] [Google Scholar]
- [58].Suckow AT, Polidori D, Yan W, Chon S, Ma JY, Leonard J, Briscoe CP. Alteration of the glucagon axis in GPR120 (FFA4) knockout mice: a role for GPR120 in glucagon secretion. J Biol Chem. 2014; 289(22):15751–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Whalley NM, Pritchard LE, Smith DM, White A. Processing of proglucagon to GLP-1 in pancreatic α-cells: is this a paracrine mechanism enabling GLP-1 to act on β-cells? J Endocrinol. 2011; 211(1):99–106. [DOI] [PubMed] [Google Scholar]
- [60].Cornall LM, Mathai ML, Hryciw DH, McAinch AJ. Diet-induced obesity up-regulates the abundance of GPR43 and GPR120 in a tissue specific manner. Cell Physiol Biochem. 2011; 28(5):949–58. [DOI] [PubMed] [Google Scholar]
- [61].Kim N, Lee JO, Lee HJ, Kim HI, Kim JK, Lee YW, Lee SK, Kim SJ, Park SH, Kim HS. Endogenous Ligand for GPR120, Docosahexaenoic Acid, Exerts Benign Metabolic Effects on the Skeletal Muscles via AMP-activated Protein Kinase Pathway. J Biol Chem. 2015; 290(33): 20438–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Fujitani M1, Matsumura S, Masuda D, Yamashita S, Fushiki T, Inoue K CD36, but not GPR120, is required for efficient fatty acid utilization during endurance exercise. Biosci Biotechnol Biochem. 2014; 78(11):1871–8. [DOI] [PubMed] [Google Scholar]
- [63].Raptis DA, Limani P, Jang JH, Ungethüm U, Tschuor C, Graf R, Humar B, Clavien PA. GPR120 on Kupffer cells mediates hepatoprotective effects of ω3-fatty acids. J Hepatol. 2014; 60(3):625–32. [DOI] [PubMed] [Google Scholar]
- [64].Nobili V, Carpino G, Alisi A, De Vito R, Franchitto A, Alpini G, Onori P, Gaudio E. Role of docosahexaenoic acid treatment in improving liver histology in pediatric nonalcoholic fatty liver disease. PLoS One. 2014; 9(2):e88005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Marzuillo P, Grandone A, Conte M, Capuano F, Cirillo G, Di Sessa A, Umano GR, Romano R, Perrone L, del Giudice EM. Novel association between a nonsynonymous variant (R270H) of the G-protein-coupled receptor 120 and liver injury in children and adolescents with obesity. J Pediatr Gastroenterol Nutr. 2014; 59(4):472–5. [DOI] [PubMed] [Google Scholar]
- [66].Marzuillo P, Grandone A, Perrone L, Miraglia Del Giudice E. Understanding the pathophysiological mechanisms in the pediatric non-alcoholic fatty liver disease: The role of genetics. World J Hepatol. 2015; 7(11):1439–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Ishii S, Hirane M, Kitamura Y, Mori S, Fukushima N, Honoki K, Tsujiuchi T. Role of GPR120 in cell motile activity induced by 12-O-tetradecanoylphorbol-13-acetate in liver epithelial WB-F344 cells. Mol Cell Biochem. 2015; 400(1–2):145–51. [DOI] [PubMed] [Google Scholar]
- [68].Wellhauser L, Belsham DD. Activation of the omega-3 fatty acid receptor GPR120 mediates anti-inflammatory actions in immortalized hypothalamic neurons. J Neuroinflammation. 2014; 11:60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Garrel G, Simon V, Denoyelle C, Cruciani-Guglielmacci C, Migrenne S, Counis R, Magnan C, Cohen-Tannoudji J. Unsaturated fatty acids stimulate LH secretion via novel PKCepsilon and -theta in gonadotrope cells and inhibit GnRH-induced LH release. Endocrinology. 2011; 152(10):3905–16. [DOI] [PubMed] [Google Scholar]
- [70].Moriyama R, Deura C, Imoto S, Nose K, Fukushima N. Expression of the long-chain fatty acid receptor GPR120 in the gonadotropes of the mouse anterior pituitary gland. Histochem Cell Biol. 2015; 143(1):21–7. [DOI] [PubMed] [Google Scholar]
- [71].Cornish J, MacGibbon A, Lin JM, Watson M, Callon KE, Tong PC, Dunford JE, van der Does Y, Williams GA, Grey AB, Naot D, Reid IR. Modulation of osteoclastogenesis by fatty acids. Endocrinology. 2008; 149(11):5688–95. [DOI] [PubMed] [Google Scholar]
- [72].Koren N, Simsa-Maziel S, Shahar R, Schwartz B, Monsonego-Ornan E. Exposure to omega-3 fatty acids at early age accelerate bone growth and improve bone quality. J Nutr Biochem. 2014; 25(6):623–33. [DOI] [PubMed] [Google Scholar]
- [73].Kim HJ, Yoon HJ, Kim BK, Kang WY, Seong SJ, Lim MS, Kim SY, Yoon YR. G Protein-Coupled Receptor 120 Signaling Negatively Regulates Osteoclast Differentiation, Survival, and Function. J Cell Physiol. 2015; In Press. [DOI] [PubMed] [Google Scholar]
- [74].Yore MM, Syed I, Moraes-Vieira PM, Zhang T, Herman MA, Homan EA, et al. Discovery of a class of endogenous mammalian lipids with anti-diabetic and anti-inflammatory effects. Cell. 2014; 159(2):318–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Briscoe CP, Peat AJ, McKeown SC, Corbett DF, Goetz AS, Littleton TR, et al. Pharmacological regulation of insulin secretion in MIN6 cells through the fatty acid receptor GPR40: identification of agonist and antagonist small molecules. Br J Pharmacol. 2006; 148(5):619–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Suzuki T, Igari S, Hirasawa A, Hata M, Ishiguro M, Fujieda H,, et al. Identification of G protein-coupled receptor 120-selective agonists derived from PPARgamma agonists. J Med Chem. 2008; 51(23):7640–4. [DOI] [PubMed] [Google Scholar]
- [77].Shimpukade B, Hudson BD, Hovgaard CK, Milligan G, Ulven T. Discovery of a potent and selective GPR120 agonist. J Med Chem. 2012; 55(9):4511–5. [DOI] [PubMed] [Google Scholar]
- [78].Sparks SM, Chen G, Collins JL, Danger D, Dock ST, Jayawickreme C, Jenkinson S, Laudeman C, et al. Identification of diarylsulfonamides as agonists of the free fatty acid receptor 4 (FFA4/GPR120). Bioorg Med Chem Lett. 2014; 24(14):3100–3. [DOI] [PubMed] [Google Scholar]
- [79].Halder S, Kumar S, Sharma R. The therapeutic potential of GPR120: a patent review. Expert Opin Ther Pat. 2013; 23(12):1581–90. [DOI] [PubMed] [Google Scholar]
- [80].Formicola R, Pevarello P, Kuhn C, Liberati C, Piscitelli F, Sodano M. FFA4/GPR120 agonists: a survey of the recent patent literature. Pharm Pat Anal. 2015; 4(6):443–51. [DOI] [PubMed] [Google Scholar]
- [81].Hara T, Hirasawa A, Sun Q, Sadakane K, Itsubo C, Iga T, Adachi T, Koshimizu TA, Hashimoto T, Asakawa Y, Tsujimoto G. Novel selective ligands for free fatty acid receptors GPR120 and GPR40. Naunyn Schmiedebergs Arch Pharmacol. 2009; 380(3):247–55. [DOI] [PubMed] [Google Scholar]