

Abstract
Gastric cancer remains one of the leading cancers in the world with a high mortality, particularly in East Asia. Helicobacter pylori infection accounts for the majority of the noncardia gastric cancers by triggering gastric inflammation and subsequent neoplastic progression. Eradication of H. pylori can reduce, but not totally eliminate, subsequent risk of developing gastric cancer. Proton-pump inhibitors (PPIs) are one of the most widely prescribed medications worldwide. With their profound gastric-acid suppression, there are concerns about a possible carcinogenic role in gastric cancer, due to induced hypergastrinemia, gastric atrophy and bacterial overgrowth in the stomach. While randomized clinical trials to establish causality between long-term PPI use and gastric cancer are lacking, current evidence based on observational studies suggests PPIs are associated with an increased gastric cancer risk. However, opinions on causality remain divergent due to unmeasured and possible residual confounding in various studies. Our recent study has showed that even after H. pylori eradication, long-term PPI use is still associated with an increased risk of gastric cancer by more than twofold. Hence, long-term PPIs should be used judiciously after considering individual’s risk–benefit profile, particularly among those with history of H. pylori infection. Further well-designed prospective studies are warranted to confirm the potential role of PPIs in gastric cancer according to baseline gastric histology and its interaction with other chemopreventive agents like aspirin, statins and metformin.
Keywords: aspirin, enterochromaffin-like cells, gastrin, gastric adenocarcinoma, Helicobacter pylori, H. pylori, PPIs, stomach cancer
Introduction
Gastric cancer is the fifth commonest cancer and the third leading cause of cancer-related mortality worldwide. 1 In 2015, an estimated 1.3 million incident gastric cancer cases were diagnosed and there were 819,000 gastric-cancer-related deaths. Helicobacter pylori infection was classified by the World Health Organization (WHO) as a type I carcinogen in 1994. 2 Chronic H. pylori infection confers a more than threefold increase in risk of gastric cancer, 3 which accounts for 78% of all gastric cancer cases and 89% of noncardia cancers. 4 H. pylori-associated gastric carcinogenesis is generally believed to start from chronic gastritis, progressing to atrophic gastritis, intestinal metaplasia, dysplasia, and ultimately cancer. 5 Atrophic gastritis, intestinal metaplasia and dysplasia are therefore regarded as precancerous gastric lesions. In a prospective study, patients with corpus-predominant gastritis [relative risk (RR) 34.5; 95% confidence interval (CI) 7.1–166.7 versus antral-predominant gastritis], severe gastric atrophy (RR 4.9; 95% CI 2.8–19.2 versus absent/mild atrophy) and intestinal metaplasia (RR 6.4; 95% CI 2.6–16.1 versus absence of intestinal metaplasia) were all at higher risk of gastric cancer development. 6 The magnitude of risk was confirmed in another cohort study [atrophic gastritis: hazard ratio (HR) 4.5; 95% CI 3.5–5.8; intestinal metaplasia: HR 6.5; 95% CI 4.7–8.2; dysplasia: HR 10.9; 95% CI 7.7–15.4]. 7 In this regard, eradication of H. pylori has been shown to reduce the gastric cancer risk by 33–47%,8–10 but a considerable proportion of H. pylori-eradicated individuals still progress to develop gastric cancer.
Apart from chronic H. pylori infection, proton-pump-inhibitor (PPI) usage is another potential risk factor for the development of gastric atrophy. With the potent acid suppression, PPIs could induce changes in the gastric environment, including hypergastrinemia and enterochromaffin cells hyperplasia. 11 There is also evidence suggesting that PPIs could contribute to bacterial overgrowth in the stomach. 12 Intuitively, PPIs worsen gastric atrophy and hence could increase the risk of gastric cancer. 10
In this review, we will examine the latest literature to decipher the role of PPIs in gastric cancer development, particularly in relation to H. pylori infection.
Potential carcinogenic mechanisms of proton-pump inhibitors
Proton-pump inhibitors (PPIs) have become one of the most commonly prescribed medications worldwide since their introduction in 1980s, 13 and have been the cornerstone of the management of upper gastrointestinal diseases including peptic ulcer disease (PUD), H. pylori infection, dyspepsia, and gastroesophageal reflux disease (GERD). However, emerging data have shown that long-term PPIs are associated with a number of side effects, including bone fracture, 14 Clostridium difficile infection, 15 pneumonia, 16 myocardial infarction and stroke, 17 although a causality has not yet been confirmed.
Potent acid suppression has long been suspected a risk factor of gastric cancer by worsening gastric atrophy with ensuing hypergastrinemia and bacterial overgrowth in the stomach. Animal studies have shown that acid suppression by omeprazole 18 and the insurmountable histamine-2 receptor antagonist (H2RA) loxtidine 19 induce gastric mucosa neoplasia in rodents. However, evidence on human subjects remains controversial. Herein, we summarize the postulated mechanisms underlying the carcinogenic effects of PPIs on gastric cancer development (Figure 1).
Figure 1.
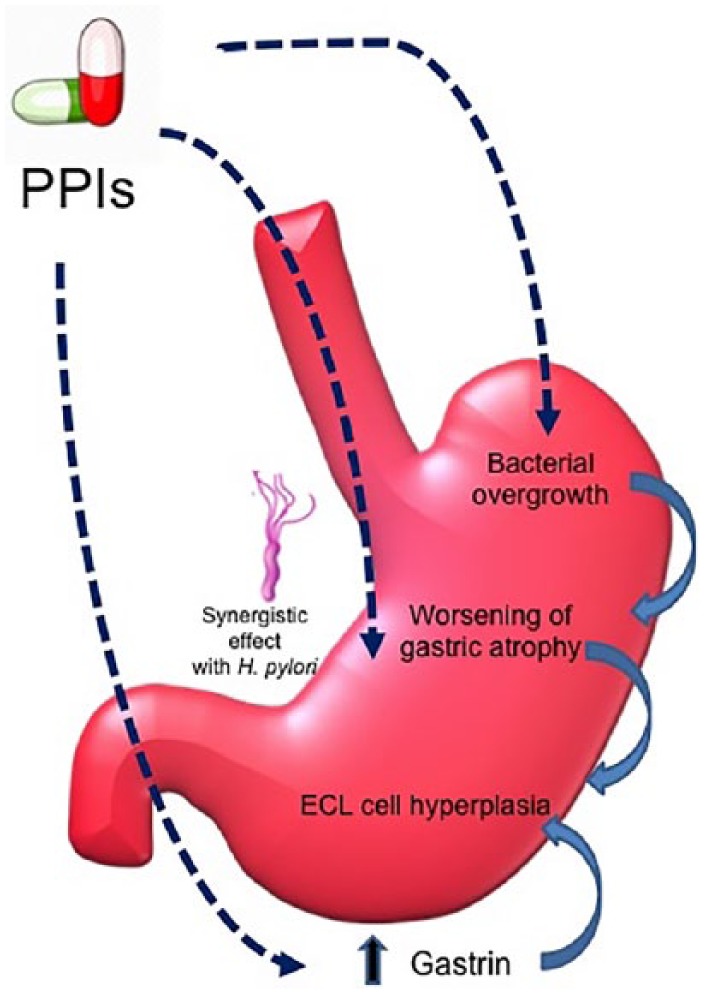
Postulated mechanisms underlying the carcinogenic effects of proton-pump inhibitors on gastric cancer development.
ECL, enterochromaffin like; H. pylori, Helicobacter pylori; PPIs, proton-pump inhibitors.
Interaction with Helicobacter pylori infection
H. pylori typically colonizes the gastric antrum, and cause an antrum-predominant gastritis in most infected subjects. 20 Antral mucosal inflammation in turn stimulates gastric secretion, maintaining a normal- or high-acidic environment. However, when the acid production is suppressed by PPIs, the pattern of gastritis shifts to a corpus-predominant gastritis with resultant impairment of parietal cell function; a phenomenon that does not occur in H. pylori-negative patients. 21 As such, the acid-suppressive effect of PPIs is further enhanced by H. pylori-induced corpus gastritis.22,23 In a cohort study of 177 patients, atrophic gastritis only developed in H. pylori-infected but not H. pylori-negative patients with long-term PPI use during a mean follow up of 5 years. 24 A systematic review also showed that the risk of corpus atrophy was more pronounced in H. pylori-infected than H. pylori-negative PPI users [odds ratio (OR) 11.45; 95% CI: 6.25–20.99]. 11
Hypergastrinemia
In response to suppression of gastric-acid production by PPIs, there is a compensatory increase in gastrin production as a negative feedback. 25 A recent systematic review by Lundell and colleagues 11 including 16 studies with 1920 patients showed that during long-term (>3 years) PPI use, the mean gastrin levels rose to one to three times the upper limit of normal. The progrowth effect of gastrin is suspected to play a role in gastric cancer development.26–28 Hypergastrinemia drives the hyperplasia of enterochromaffin-like (ECL) cells in the oxyntic mucosa, which occurs in 10–20% of patients with long-term PPI use. 29 It has been reported that the risk of ECL-cell hyperplasia is higher in H. pylori-infected than H. pylori-negative patients (OR 2.45; 95% CI 1.47–4.10). 11 Similar results were reported in two randomized clinical trials on acid reflux control by either antireflux surgery or long-term PPI therapy. 30 Analysis of the insulin–gastric (INS-GAS) transgenic mice raised the possibility that hypergastrinemia may promote gastric cancer through the gastrin receptor, cholecystokinin receptor-2 (CCK2R). 31 With the overexpression of amidated gastrin through the insulin promoter upstream of the human gastrin coding sequences, 32 these mice developed gastric intestinal metaplasia and dysplasia, and some even developed gastric corpus cancer, 33 which was accelerated by infection by Helicobacter felis or H. pylori. In another study, PPIs (omeprazole) led to a faster progression to dysplasia in H. felis-infected INS-GAS mice. 34
Furthermore, another study also suggested that promotion of growth of gastric carcinomas of ‘intestinal type’ might be due to the indirect effect of hypergastrinemia, via stimulation of the release of signal substances (e.g. histamine, regenerating-gene protein) from the ECL cells. 35 In line with these animal studies, clinical evidence from a case-control study nested within the all-male Alpha-Tocopherol, Beta-Carotene Cancer Prevention Study of 29,133 Finnish male smokers with more than 24 years of follow up, reported that a higher gastrin level (fourth quartile versus first quartile) was associated with an increased risk of noncardia gastric cancer (OR 1.92; 95% CI 1.21–3.05). 36
Although ECL cells are generally believed to play little role in human gastric carcinoma development, ECL-cell neuroendocrine tumors (NETs) 37 and adenocarcinomas 38 were observed in cases of pernicious anemia (autoimmune gastritis with corpus atrophy and thus low gastric-acid output). Early studies showed that the distinction between gastric NETs and adenocarcinomas may be difficult in both animals 39 and humans,40,41 as ECL cells may lose many of their neuroendocrine characteristics during neoplastic transformation. However, some studies later suggested that a proportion of the gastric adenocarcinomas, in particular, the signet ring subgroup of gastric carcinomas of ‘diffuse type,’ indeed develop from the ECL cells.42–44 With improved sensitivity of immunohistochemical methods for detecting neuroendocrine/ECL-cell makers, it was shown in one study that virtually all gastric adenocarcinomas in patients with severe hypergastrinemia were malignant NETs. 45
Non-Helicobacter pylori bacterial overgrowth
Acid suppression of PPIs can lead to both luminal and mucosal non-H. pylori bacterial overgrowth in the stomach that may exacerbate chronic inflammation. 12 It has been shown that non-H. pylori bacterial overgrowth is a risk factor for atrophic gastritis (antrum: RR 5.07; 95% CI 1.05–24.40; corpus: RR 6.38; 95% CI 0.78–53.89). 46 The simultaneous infection with H. pylori and non-H. pylori bacteria has a synergistic effect on inducing a higher level of serum cytokines [interleukin (IL)-1 beta and IL-8] and increasing the risk of atrophic gastritis. Moreover, there is an increase in the number of bacteria including nongastric microorganisms (mainly from oral flora) that possess nitrate reductase to produce N-nitroso compounds from food nitrates, known gastric carcinogens.47,48 Interestingly, it has been shown that gastric microbiota alteration is distinct in atrophic gastritis. 49 PPI users showed similarly high gastric microbial diversity as healthy subjects. A relatively high gastric microbial diversity, dominated by streptococcus, was observed in autoimmune gastritis, while a decreased microbial abundance and diversity was induced by H. pylori infection.
Clinical studies on the association between proton-pump inhibitors and gastric cancer
The incidence of gastric cancer indeed has been gradually declining in the past 50 years with an average estimated annual percentage decrease of 2.5% per year. 50 This observation has therefore been used as an argument against the potential carcinogenic role of PPIs, which has been available only since 1989. However, it could not be over-emphasized that reduction in the prevalence of H. pylori and improvement in food processing actually contribute to this observed decline in gastric cancer incidence, 51 hence masking the possible effect of PPIs on increasing gastric cancer incidence.
As gastric cancer is a relatively rare disease which requires a long-time lag to develop, randomized clinical trials (RCTs) demonstrating the potential carcinogenic effects of PPIs are both resource and labor intensive. More importantly, there may be an ethical issue in randomizing patients to receive intervention to observe primary adverse outcome. As such, the determination of the causal relationship could, at best, be determined by well-designed observational studies controlling for known confounding factors, with minimization of biases.
There are two meta-analyses based on RCTs assessing precancerous gastric lesions and PPIs.52,53 Not surprisingly, no association between PPIs and gastric cancer was found as the follow up in these studies was relatively short (maximum follow up was 36 months). On the other hand, a systemic review of three observational studies by Tran-Duy and colleagues showed that PPIs were associated with an increased gastric cancer risk (pooled OR 1.43; 95% CI: 1.23–1.66). 54 Inconsistent results were, however, observed when the effect of PPIs was studied according to duration of PPI use (<1 year, ⩾1 year and >3 years). If only patients with PPI use < 1 year were compared with non-PPI users, the pooled OR was 1.76 (95% CI 1.24–2.52). However, no statistically significant association was observed for patients using PPI ⩾ 1 year (pooled OR 1.31; 95% CI 0.79–2.19), but the greatest risk was noted for those using PPI > 3 years (pooled OR 2.45; 95% CI 1.41–2.45). The authors proposed that PPIs may be prescribed for a short period as part of the H. pylori eradication regimen for those using PPI < 1 year; and when sufficiently long enough time is allowed, PPIs and H. pylori infection act synergistically to increase the gastric cancer risk in those using PPI > 3 years.
On the other hand, previous reassuring studies on the long-term safety of PPIs neither had sufficient observation period nor adequate power with small sample size.29,55 Moreover, there are other limitations of previous studies. First, all except the study by Poulsen et al. 56 failed to adjust for H. pylori status or eradication. As such, the establishment of causal relationship between PPIs and gastric cancer is severely compromised by this most important confounding factor for gastric cancer development. Second, indication bias is possible as only the study by Garcia and colleagues 57 took the indication of PPIs (GERD, PUD and dyspepsia) into analysis. Third, protopathic bias (or reverse causality) exists, since the presence of prediagnosis cancer symptoms may warrant the prescription of PPIs. Fourth, most of the studies failed to take into account the potential role of other medications in gastric cancer development, including statins, 58 metformin, 59 aspirin, 60 nonsteroidal anti-inflammatory drugs (NSAIDs) and cyclooxygenase-2 (COX-2) inhibitors.
To address the confounding effect of H. pylori infection, we have recently carried out a territory-wide retrospective cohort study on H. pylori-eradicated patients (n = 63,397). During a median observation period of 7.6 years [interquartile range (IQR):5.1–10.3 years], 61 PPI use (defined as at least weekly use) was associated with an increased gastric cancer risk (HR 2.44; 95% CI 1.42–4.20). On the other hand, H2RAs, which served as negative control exposure, were not associated with an increased cancer risk (HR 0.72; 95% CI 0.48–1.07). With non-PPI use as the reference group, the risk of gastric cancer by PPI use increased with increasing frequency (HR 2.43 for weekly to less-than-daily use, and HR 4.55 for daily use) and duration (HR 5.04, 6.65 and 8.34 for ⩾1 year, ⩾2 years and ⩾3 years, respectively). The adjusted absolute risk difference was 4.29 more gastric cancer cases per 10,000 person-years in PPI users than non-PPI users. Furthermore, another matched cohort of PPI users who had not received H. pylori therapy (n = 142,460) was recruited for comparison. Of note, PPI users without prior H. pylori therapy had the lowest incidence rate of gastric cancer (0.8 cases per 10,000 person-years) among the three groups (non-PPI users with prior H. pylori therapy: 2.9 per 10,000 person-years; PPI users with prior H. pylori therapy: 8.1 per 10,000 person-years). Therefore, the presence of current or even prior H. pylori infection appears to be a more important determinant of gastric cancer risk than PPIs, and PPI-associated gastric cancers are more likely to occur in subjects with underlying H. pylori-associated gastric damage.
Our study is the first to demonstrate that long-term PPI use is still associated with an increased gastric cancer risk even after H. pylori eradication, with a dose- and time-response relationship. Several efforts of our study in minimizing the bias to determine a possible causal relationship should be noted. First, the large sample size and long observation time allows for an accurate estimation of the gastric cancer incidence among PPI users. Second, protopathic bias was avoided by excluding PPI prescriptions in the 6 months preceding gastric cancer diagnosis. Third, indication bias was unlikely a significant issue as a similar increase in gastric cancer risk was not observed for H2RAs, which are less potent than PPIs in terms of acid suppression. In addition, the lowest gastric cancer risk among PPI users without prior H. pylori therapy argues against a significant indication bias. Fourth, comorbid conditions and medications, which had not been adequately addressed in previous studies were included in the propensity score adjustment. Nevertheless, several limitations exist, including failure to capture some of the risk factors like lifestyle factors, family history and histology information from the electronic healthcare database system. 62
Four recent studies by other groups, published after our study, had findings consistent with ours. Among the four studies, one is a nationwide population-based study from Sweden, a country with a relatively low gastric cancer incidence. 63 It examined the gastric cancer risk in PPI users and H2RA users as compared with the Swedish background population of the same age, sex and calendar period (7.1–7.6 million adults). The standardized incidence ratio (SIR) of gastric cancer with PPI use was 3.38 (95% CI 3.25–3.53). Despite a sensitivity analysis to address protopathic bias (all gastric cancer cases diagnosed within 1 year of the study commencement were excluded), the risk remained albeit attenuated (SIR 1.6; 95% CI 1.51–1.71). Again, H2RA use was not a significant risk factor (SIR 0.57; 95% CI: 0.29–0.99).
The remaining three studies were from Asia, including two from Taiwan and one from Japan. The two Taiwanese studies were both case-control studies (controls were age- and sex-matched with cases in a 1:1 ratio). The first one (n = 2122) showed that PPIs were associated with an increased gastric cancer risk by 2.48 fold (95% CI 1.92–3.20) among GERD patients. 64 A higher risk was demonstrated by an increase in defined daily dose. The second study (n = 1298) also showed a similar result, with longer duration of PPI use conferring a higher risk as compared with non-PPI use [⩽6 month PPIs: OR 1.59 (95% CI 1.24–2.05) versus >6 month PPIs: OR 2.00 (95% CI 1.36–2.95)]. 65 The Japanese study was a retrospective cohort study of 571 H. pylori-eradicated patients, which addressed one important limitation of previous studies by adjusting for gastric precancerous lesions (corpus atrophy and intestinal metaplasia). 66 During a mean follow up of 6.9 years, PPI use was associated with an increased gastric cancer risk (HR 3.61; 95% CI 1.49–8.77). Such an association was only observed among patients with intestinal metaplasia, indicating PPIs potentially increase gastric cancer risk in patients with pre-existing gastric precancerous lesions. Notably, the risk of PPIs was higher among patients with mild intestinal metaplasia than those with severe changes [HR 16.0 (95% CI 1.90–134) versus HR 3.06 (95% CI 0.62–15.0)], which may be related to the small sample size from the subgroup analysis. Table 1 summarizes the characteristics and results of various observational studies.
Table 1.
Summary of observational studies on the association between proton-pump inhibitors and gastric cancer development.
| References | Study design | Sample size | Patient characteristics/ region | Factors considered (regression model or stratified analysis) |
* Results |
|---|---|---|---|---|---|
| Garcia Rodriguez et al. 57 | Nested case-control study (matched with age, sex, and calendar year) |
10,522 | United Kingdom | 1, 2, 4, 5, 13,14 | OR 1.75 (95% CI 1.10–2.79) |
| Tamim et al. 80 | Nested case-control study (matched with age and sex) |
8229 | Canada | 1,2, 17 | OR 1.46 (95% CI: 1.22–1.74) |
| Poulsen et al. 56 | Population-based cohort study | 280,872 | Denmark | 1, 3–5, 9, 10, 13, 17 | IRR 2.3 (95% CI 1.2–4.3; patients with ⩾5 years of follow up) |
| Cheung et al. 61 | Population-based cohort study | 63,397 | H. pylori-eradicated patients/Hong Kong | 1–12, 16, 17 | HR 2.44 (95% CI 1.42–4.20) |
| Brusselaers et al. 63 | Nationwide population-based cohort study | 843,003 PPI or H2RA users (versus general population of 7.1–7.6 million) | Sweden | 1–3, 6, 10, 13, 16, 17 | SIR 3.38 (95% CI 3.25–3.53) |
| Peng et al. 64 | Case-control study (matched with age, sex, and calendar year) | 2122 (1:1 ratio) |
GERD patients/Taiwan | 1, 2, 10, 13, 14 | OR 2.48 (95% CI 1.92–3.20) |
| Lai et al. 65 | Case-control study (matched with age, sex, and calendar year) | 1298 (1:1 ratio) |
Taiwan | 1–8, 10, 13, 17 | ⩽6-month PPI: OR 1.59 (95% CI 1.24– 2.05) >6-month PPI: OR 2.00 (95% CI 1.36–2.95) |
| Niikura et al. 66 | Retrospective cohort study | 571 | H. pylori-eradicated patients/Japan | 3, 10, 15 | HR 3.61; 95% CI 1.49–8.77 |
Results are presented after adjustment for covariates.
1, age; 2, sex; 3, Helicobacter pylori status; 4, smoking; 5, alcohol; 6, history of PUD; 7, DM; 8, other comorbidities; 9, aspirin/NSAIDs/COX-2 inhibitors; 10, H2RAs; 11, statins; 12, metformin; 13, calendar period; 14, socioeconomic status; 15, gastric histology; 16, indication bias; 17, protopathic bias; CI, confidence interval; COX-2, cyclooxygenase-2; DM, diabetes mellitus; IRR, incidence rate ratio; GERD, gastroesophageal reflux disease; HR, hazard ratio; H2RAs, histamine-2 receptor antagonists; OR, odds ratio; PPIs, proton-pump inhibitors; PUD, peptic ulcer disease; NSAIDs, nonsteroidal anti-inflammatory drugs; SIR, standardized incidence ratio.
However, it is noteworthy that some important risk factors (e.g. diet, smoking, alcohol, family history of gastric cancer) were not taken into consideration in some of these observational studies. In addition, residual confounding is inherent in all observational studies. Therefore, a causal relationship between PPIs and gastric cancer still cannot be firmly established. Nevertheless, fulfillment of the Bradford Hill criteria may help to strengthen the possible causal relationship. 67
Effect of proton-pump inhibitors on gastric cancer in different subgroups
In a meta-analysis of seven studies, 68 Wan and colleagues found that the risk of PPIs was more prominent among Asians than Whites [OR 2.44 (95% CI 1.89–3.00) versus OR: 1.86 (95% CI 0.54–3.18)]. The effect of PPIs on gastric cancer is also site specific, with higher risk for noncardia than cardia gastric cancer [OR 2.45 (95% CI 1.44–3.45) versus OR 1.64 (95 % CI 0.23–3.51)].
In the nationwide Swedish study, 63 the risk of PPIs appears to be similar between males (SIR 3.65; 95% CI 3.45–3.85) and females (SIR 3.07; 95% CI 2.87–3.28). Importantly, the risk was most pronounced in younger age groups. The highest risk was observed among PPI users < 40 years when compared with non-PPI users (SIR 22.76; 95% CI 15.94–31.52), while the lowest risk was found among PPI users aged ⩾ 70 years (SIR 2.76; 95% CI 2.61–2.92). The authors provide several explanations, which included an increased prevalence of atrophic gastritis in relation to obesity 69 as well as acceleration of gastric carcinogenesis in younger age groups (with a higher likelihood of a positive family history 70 ), hence increasing the vulnerability to the potential carcinogenic effect of PPIs.
Stratified analysis according to PPI indications was also performed in this study. 63 The risk was much larger among H. pylori-infected (SIR 9.76; 95% CI 8.87–10.71) than H. pylori-negative patients (SIR 2.91; 95% CI 2.78–3.05). Among the common indications for PPIs, the risk was highest in patients with PUD (SIR 8.75; 95 CI 8.12–9.41), while the risk was similar among those with gastroduodenitis (SIR 3.68; 95% CI 3.31–4.09), dyspepsia (SIR 3.07; 95% CI 2.58–3.63) and GERD (SIR 3.04; 95% CI 2.80–3.31).
Effect of different proton-pump inhibitors on gastric cancer risk
Animal studies showed that longer-acting PPIs could produce a higher risk of gastric tumors due to greater systemic exposure from a higher concentration time (area under the curve), leading to more gastrin stimulation.26,27,71 However, a meta-analysis concludes that different PPIs can be used interchangeably based on potency as reflected by the percentage time pH > 4 over a 24 h period (omeprazole 30 mg is equivalent to lansoprazole 30 mg, esomeprazole 20 mg and rabeprazole 20 mg). 72 In a US Food and Drug Administration (FDA)-mandated long-term follow-up study of 61,864 PPI users, the risk of gastric cancer was comparable between the use of pantoprazole (a longer-acting PPI) and other shorter-acting PPIs (any combination of omeprazole, esomeprazole, lansoprazole or rabeprazole; HR 0.68; 95% CI 0.24–1.93) after adjusting for age, sex, H. pylori treatment, cumulative PPI dose and total years of PPI treatment. Although twice-daily PPI dosage suppresses gastric-acid production for a longer duration than once-daily dosage, 72 current data on risk of gastric cancer are lacking and further studies are required.
Potential chemopreventive agents to reduce proton-pump-inhibitor-associated gastric cancer risk
Meta-analyses have shown that aspirin reduces gastric cancer risk without stratification according to H. pylori status.73,74 To address this issue, we conducted another study on the effect of aspirin specifically on H. pylori-eradicated subjects. Aspirin was shown to be associated with a lower gastric cancer risk among H. pylori-eradicated subjects (HR 0.30; 95% CI 0.15–0.61), with a frequency–, duration–, and dose–response relationship being observed. 60 A subsequent post hoc analysis 75 showed that the harmful effect of PPIs was higher (HR 3.73; 95% CI 2.11– 6.60) among nonaspirin users, while PPIs were not associated with an increased risk of gastric cancer among aspirin users (HR 0.35; 95% CI 0.04–2.74). The potential detrimental effect of PPIs on gastric cancer appears to be negated by aspirin use, although this observation necessitates validation by studies from other centers. Nevertheless, our study finding prompts the investigation of the role of other potential chemopreventive agents, such as metformin 76 and statins, 58 in reducing gastric cancer risk among PPI users should be further explored.
Recommendations on proton-pump inhibitor use in clinical practice
Despite the potential harmful effects of PPIs, they are so far the most effective therapy for PUD, GERD and in preventing aspirin-/NSAID-related upper gastrointestinal bleeding (UGIB). Indeed, the rational use of PPIs should be promoted to minimize any potential side effects associated with long-term use, rather than irrational avoidance. Physicians should consider the lowest effective dose of PPIs with a finite treatment period, particularly for nonerosive GERD and nonulcer dyspepsia. A stepdown approach to less potent acid-suppressive agents such as H2RAs should also be considered in appropriate settings.
However, certain clinical conditions may mandate long-term PPI use. For instance, individuals who are at high risk of aspirin-/NSAID-related UGIB should be given long-term PPIs. 77 Barrett’s esophagus is another condition in which long-term use of PPIs is recommended. 78 If long-term PPI use is necessary, it is advisable to have H. pylori tested and eradicated if present so as to prevent the development of corpus atrophy, hence reducing gastric cancer risk. 79 It has also been proposed that nonfasting chromogranin A be used as a serological marker in long-term PPI users to monitor the degree of ECL-cell hyperplasia. 51 The effectiveness of this approach, however, has not yet been studied by prospective clinical studies and cost-effectiveness studies according to local gastric cancer incidence and different subgroups.
Conclusion
Emerging evidence from multiple observational studies suggests long-term use of PPIs is associated with a higher risk of gastric cancer development. However, the risk is likely limited to individuals with current or past history of H. pylori infection, particularly those with underlying precancerous gastric lesions. Physicians should prescribe PPIs according to individual’s risk–benefit profile rather than withholding PPIs from those with genuine indications, such as Barrett’s esophagus or high risk of UGIB. This is particularly important for aspirin users at high risk of UGIB, as aspirin may negate the potential harmful effects of PPIs on gastric cancer development. Further well-designed prospective studies are warranted to confirm the potential role of PPIs in gastric cancer according to baseline gastric histology and its interaction with other chemopreventive agents like aspirin, statins, and metformin.
Acknowledgments
All authors contributed equally to this paper with literature review and analysis, drafting and critical revision and editing, and approval of the final version of this article.
Footnotes
Funding: This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Conflict of interest statement: WKL has received honorarium for attending advisory board meetings of Takeda and Abbott Laboratories. There are no other competing interests.
Contributor Information
Ka Shing Cheung, Department of Medicine, The University of Hong Kong, Hong Kong.
Wai K. Leung, Department of Medicine, The University of Hong Kong, Queen Mary Hospital, 102 Pok Fu Lam Road, Hong Kong.
References
- 1. Fitzmaurice C, Allen C, Barber RM, et al. Global, regional, and national cancer incidence, mortality, years of life lost, years lived with disability, and disability-adjusted life-years for 32 cancer groups, 1990 to 2015: a systematic analysis for the global burden of disease study. JAMA Oncol 2017; 3: 524–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Anwar W, Armstrong BK, Correa P. Infection with Helicobacter pylori. IARC Monogr Eval Carcinog Risks Hum 1994; 61: 177–240. [PMC free article] [PubMed] [Google Scholar]
- 3. Helicobacter and Cancer Collaborative Group. Gastric cancer and Helicobacter pylori: a combined analysis of 12 case control studies nested within prospective cohorts. Gut 2001; 49: 347–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. International Agency for Research on Cancer. Helicobacter pylori eradication as a strategy for preventing gastric cancer. IARC Working Group Reports. Vol. 8, WHO Press, World Health Organization, Geneva, Switzerland, 2014. [Google Scholar]
- 5. Correa P. Human gastric carcinogenesis: a multistep and multifactorial process–first American Cancer Society Award lecture on cancer epidemiology and prevention. Cancer Res 1992; 52: 6735–6740. [PubMed] [Google Scholar]
- 6. Uemura N, Okamoto S, Yamamoto S, et al. Helicobacter pylori infection and the development of gastric cancer. N Engl J Med 2001; 345: 784–789. [DOI] [PubMed] [Google Scholar]
- 7. Song H, Ekheden IG, Zheng Z, et al. Incidence of gastric cancer among patients with gastric precancerous lesions: observational cohort study in a low risk Western population. BMJ 2015; 351: h3867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lee YC, Chiang TH, Chou CK, et al. Association between Helicobacter pylori eradication and gastric cancer incidence: a systematic review and meta-analysis. Gastroenterology 2016; 150: 1113–1124.e5. [DOI] [PubMed] [Google Scholar]
- 9. Ford AC, Forman D, Hunt RH, et al. Helicobacter pylori eradication therapy to prevent gastric cancer in healthy asymptomatic infected individuals: systematic review and meta-analysis of randomised controlled trials. BMJ 2014; 348: g3174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cheung KS, Leung WK. Risk of gastric cancer development after eradication of Helicobacter pylori. World J Gastrointest Oncol 2018; 10: 115–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lundell L, Vieth M, Gibson F, et al. Systematic review: the effects of long-term proton pump inhibitor use on serum gastrin levels and gastric histology. Aliment Pharmacol Ther 2015; 42: 649–663. [DOI] [PubMed] [Google Scholar]
- 12. Sanduleanu S, Jonkers D, De Bruine A, et al. Non-Helicobacter pylori bacterial flora during acid-suppressive therapy: differential findings in gastric juice and gastric mucosa. Aliment Pharmacol Ther 2001; 15: 379–388. [DOI] [PubMed] [Google Scholar]
- 13. Forgacs I, Loganayagam A. Overprescribing proton pump inhibitors. BMJ 2008; 336: 2–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yang YX, Lewis JD, Epstein S, et al. Long-term proton pump inhibitor therapy and risk of hip fracture. JAMA 2006; 296: 2947–2953. [DOI] [PubMed] [Google Scholar]
- 15. Janarthanan S, Ditah I, Adler DG, et al. Clostridium difficile-associated diarrhea and proton pump inhibitor therapy: a meta-analysis. Am J Gastroenterol 2012; 107: 1001–1010. [DOI] [PubMed] [Google Scholar]
- 16. Laheij RJ, Sturkenboom MC, Hassing RJ, et al. Risk of community-acquired pneumonia and use of gastric acid-suppressive drugs. JAMA 2004; 292: 1955–1960. [DOI] [PubMed] [Google Scholar]
- 17. Sherwood MW, Melloni C, Jones WS, et al. Individual proton pump inhibitors and outcomes in patients with coronary artery disease on dual antiplatelet therapy: a systematic review. J Am Heart Assoc 2015; 4: e002245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Havu N. Enterochromaffin-like cell carcinoids of gastric mucosa in rats after life-long inhibition of gastric secretion. Digestion 1986; 35(Suppl. 1): 42–55. [DOI] [PubMed] [Google Scholar]
- 19. Poynter D, Pick CR, Harcourt RA, et al. Association of long lasting unsurmountable histamine H2 blockade and gastric carcinoid tumours in the rat. Gut 1985; 26: 1284–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Weeks DL, Eskandari S, Scott DR, et al. A H+-gated urea channel: the link between Helicobacter pylori urease and gastric colonization. Science 2000; 287: 482–485. [DOI] [PubMed] [Google Scholar]
- 21. Kuipers EJ, Uyterlinde AM, Pena AS, et al. Increase of Helicobacter pylori-associated corpus gastritis during acid suppressive therapy: implications for long-term safety. Am J Gastroenterol 1995; 90: 1401–1406. [PubMed] [Google Scholar]
- 22. Verdu EF, Armstrong D, Fraser R, et al. Effect of Helicobacter pylori status on intragastric pH during treatment with omeprazole. Gut 1995; 36: 539–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gillen D, Wirz AA, Neithercut WD, et al. Helicobacter pylori infection potentiates the inhibition of gastric acid secretion by omeprazole. Gut 1999; 44: 468–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kuipers EJ, Lundell L, Klinkenberg-Knol EC, et al. Atrophic gastritis and Helicobacter pylori infection in patients with reflux esophagitis treated with omeprazole or fundoplication. N Engl J Med 1996; 334: 1018–1022. [DOI] [PubMed] [Google Scholar]
- 25. Waldum HL, Sandvik AK, Brenna E, et al. Gastrin-histamine sequence in the regulation of gastric acid secretion. Gut 1991; 32: 698–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Betton GR, Dormer CS, Wells T, et al. Gastric ECL-cell hyperplasia and carcinoids in rodents following chronic administration of H2-antagonists SK&F 93479 and oxmetidine and omeprazole. Toxicol Pathol 1988; 16: 288–298. [DOI] [PubMed] [Google Scholar]
- 27. Berlin RG. Omeprazole. Gastrin and gastric endocrine cell data from clinical studies. Dig Dis Sci 1991; 36: 129–136. [DOI] [PubMed] [Google Scholar]
- 28. Waldum HL, Sandvik AK, Idle JR. Gastrin is the most important factor in ECL tumorigenesis. Gastroenterology 1998; 114: 1113–1115. [DOI] [PubMed] [Google Scholar]
- 29. Fiocca R, Mastracci L, Attwood SE, et al. Gastric exocrine and endocrine cell morphology under prolonged acid inhibition therapy: results of a 5-year follow-up in the LOTUS trial. Aliment Pharmacol Ther 2012; 36: 959–971. [DOI] [PubMed] [Google Scholar]
- 30. Attwood SE, Ell C, Galmiche JP, et al. Long-term safety of proton pump inhibitor therapy assessed under controlled, randomised clinical trial conditions: data from the SOPRAN and LOTUS studies. Aliment Pharmacol Ther 2015; 41: 1162–1174. [DOI] [PubMed] [Google Scholar]
- 31. Hayakawa Y, Chang W, Jin G, et al. Gastrin and upper GI cancers. Curr Opin Pharmacol 2016; 31: 31–37. [DOI] [PubMed] [Google Scholar]
- 32. Wang TC, Bonner-Weir S, Oates PS, et al. Pancreatic gastrin stimulates islet differentiation of transforming growth factor alpha-induced ductular precursor cells. J Clin Invest 1993; 92: 1349–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wang TC, Dangler CA, Chen D, et al. Synergistic interaction between hypergastrinemia and Helicobacter infection in a mouse model of gastric cancer. Gastroenterology 2000; 118: 36–47. [DOI] [PubMed] [Google Scholar]
- 34. Takaishi S, Cui G, Frederick DM, et al. Synergistic inhibitory effects of gastrin and histamine receptor antagonists on Helicobacter-induced gastric cancer. Gastroenterology 2005; 128: 1965–1983. [DOI] [PubMed] [Google Scholar]
- 35. Sekikawa A, Fukui H, Fujii S, et al. REG Ialpha protein may function as a trophic and/or anti-apoptotic factor in the development of gastric cancer. Gastroenterology 2005; 128: 642–653. [DOI] [PubMed] [Google Scholar]
- 36. Murphy G, Abnet CC, Choo-Wosoba H, et al. Serum gastrin and cholecystokinin are associated with subsequent development of gastric cancer in a prospective cohort of Finnish smokers. Int J Epidemiol 2017; 46: 914–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Borch K, Renvall H, Liedberg G. Gastric endocrine cell hyperplasia and carcinoid tumors in pernicious anemia. Gastroenterology 1985; 88: 638–648. [DOI] [PubMed] [Google Scholar]
- 38. Zamcheck N, Grable E, Ley A, et al. Occurrence of gastric cancer among patients with pernicious anemia at the Boston City Hospital. N Engl J Med 1955; 252: 1103–1110. [DOI] [PubMed] [Google Scholar]
- 39. Soga J, Koro T, Tazawa K, et al. Argyrophil cell microneoplasia in the Mastomys’ stomach–an observation on early carcinoid formation. J Natl Cancer Inst 1975; 55: 1001–1006. [DOI] [PubMed] [Google Scholar]
- 40. Azzopardi JG, Pollock DJ. Argentaffin and Argyrophil cells in gastric carcinoma. J Pathol Bacteriol 1963; 86: 443–451. [DOI] [PubMed] [Google Scholar]
- 41. Wilander E, El-Salhy M, Pitkanen P. Histopathology of gastric carcinoids: a survey of 42 cases. Histopathology 1984; 8: 183–193. [DOI] [PubMed] [Google Scholar]
- 42. Waldum HL, Aase S, Kvetnoi I, et al. Neuroendocrine differentiation in human gastric carcinoma. Cancer 1998; 83: 435–444. [PubMed] [Google Scholar]
- 43. Bakkelund K, Fossmark R, Nordrum I, et al. Signet ring cells in gastric carcinomas are derived from neuroendocrine cells. J Histochem Cytochem 2006; 54: 615–621. [DOI] [PubMed] [Google Scholar]
- 44. Sordal O, Qvigstad G, Nordrum IS, et al. The PAS positive material in gastric cancer cells of signet ring type is not mucin. Exp Mol Pathol 2014; 96: 274–278. [DOI] [PubMed] [Google Scholar]
- 45. Qvigstad G, Qvigstad T, Westre B, et al. Neuroendocrine differentiation in gastric adenocarcinomas associated with severe hypergastrinemia and/or pernicious anemia. APMIS 2002; 110: 132–139. [DOI] [PubMed] [Google Scholar]
- 46. Sanduleanu S, Jonkers D, De Bruine A, et al. Double gastric infection with Helicobacter pylori and non-Helicobacter pylori bacteria during acid-suppressive therapy: increase of pro-inflammatory cytokines and development of atrophic gastritis. Aliment Pharmacol Ther 2001; 15: 1163–1175. [DOI] [PubMed] [Google Scholar]
- 47. Engstrand L, Lindberg M. Helicobacter pylori and the gastric microbiota. Best Pract Res Clin Gastroenterol 2013; 27: 39–45. [DOI] [PubMed] [Google Scholar]
- 48. Jakszyn P, Bingham S, Pera G, et al. Endogenous versus exogenous exposure to N-nitroso compounds and gastric cancer risk in the European Prospective Investigation into Cancer and Nutrition (EPIC-EURGAST) study. Carcinogenesis 2006; 27: 1497–1501. [DOI] [PubMed] [Google Scholar]
- 49. Parsons BN, Ijaz UZ. Comparison of the human gastric microbiota in hypochlorhydric states arising as a result of Helicobacter pylori-induced atrophic gastritis, autoimmune atrophic gastritis and proton pump inhibitor use. PLoS Pathog 2017; 13: e1006653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bray F, Jemal A, Grey N, et al. Global cancer transitions according to the Human Development Index (2008–2030): a population-based study. Lancet Oncol 2012; 13: 790–801. [DOI] [PubMed] [Google Scholar]
- 51. Waldum HL, Sordal O, Fossmark R. Proton pump inhibitors (PPIs) may cause gastric cancer - clinical consequences. Scand J Gastroenterol 2018; 53: 639–642. [DOI] [PubMed] [Google Scholar]
- 52. Eslami L, Nasseri-Moghaddam S. Meta-analyses: does long-term PPI use increase the risk of gastric premalignant lesions? Arch Iran Med 2013; 16: 449–458. [PubMed] [Google Scholar]
- 53. Song H, Zhu J, Lu D. Long-term proton pump inhibitor (PPI) use and the development of gastric pre-malignant lesions. Cochrane Database Syst Rev 2014: Cd010623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Tran-Duy A, Spaetgens B, Hoes AW, et al. Use of proton pump inhibitors and risks of fundic gland polyps and gastric cancer: systematic review and meta-analysis. Clin Gastroenterol Hepatol 2016; 14: 1706–1719.e5. [DOI] [PubMed] [Google Scholar]
- 55. Klinkenberg-Knol EC, Nelis F, Dent J, et al. Long-term omeprazole treatment in resistant gastroesophageal reflux disease: efficacy, safety, and influence on gastric mucosa. Gastroenterology 2000; 118: 661–669. [DOI] [PubMed] [Google Scholar]
- 56. Poulsen AH, Christensen S, McLaughlin JK, et al. Proton pump inhibitors and risk of gastric cancer: a population-based cohort study. Br J Cancer 2009; 100: 1503–1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Garcia Rodriguez LA, Lagergren J, Lindblad M. Gastric acid suppression and risk of oesophageal and gastric adenocarcinoma: a nested case control study in the UK. Gut 2006; 55: 1538–1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Singh PP, Singh S. Statins are associated with reduced risk of gastric cancer: a systematic review and meta-analysis. Ann Oncol 2013; 24: 1721–1730. [DOI] [PubMed] [Google Scholar]
- 59. Zhou XL, Xue WH, Ding XF, et al. Association between metformin and the risk of gastric cancer in patients with type 2 diabetes mellitus: a meta-analysis of cohort studies. Oncotarget 2017; 8: 55622–55631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Cheung KS, Chan EW, Wong AYS, et al. Aspirin and risk of gastric cancer after helicobacter pylori eradication: a territory-wide study. J Natl Cancer Inst 2018; 110: 743–749. [DOI] [PubMed] [Google Scholar]
- 61. Cheung KS, Chan EW, Wong AYS, et al. Long-term proton pump inhibitors and risk of gastric cancer development after treatment for Helicobacter pylori: a population-based study. Gut 2018; 67: 28–35. [DOI] [PubMed] [Google Scholar]
- 62. Rugge M, Meggio A, Pennelli G, et al. Gastritis staging in clinical practice: the OLGA staging system. Gut 2007; 56: 631–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Brusselaers N, Wahlin K, Engstrand L, et al. Maintenance therapy with proton pump inhibitors and risk of gastric cancer: a nationwide population-based cohort study in Sweden. BMJ Open 2017; 7: e017739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Peng YC, Huang LR, Lin CL, et al. Association between proton pump inhibitors use and risk of gastric cancer in patients with GERD. Gut 2018; 68: 374–376. [DOI] [PubMed] [Google Scholar]
- 65. Lai SW, Lai HC, Lin CL, et al. Proton pump inhibitors and risk of gastric cancer in a case-control study. Gut. Epub ahead of print 16 April 2018. DOI: 10.1136/gutjnl-2018-316371. [DOI] [PubMed] [Google Scholar]
- 66. Niikura R, Hayakawa Y, Hirata Y, et al. Long-term proton pump inhibitor use is a risk factor of gastric cancer after treatment for Helicobacter pylori: a retrospective cohort analysis. Gut 2018; 67: 1908–1910. [DOI] [PubMed] [Google Scholar]
- 67. Cheung KS, Leung WK. Response to letter to the editor by Moayyedi et al. Gut. Epub ahead of print 18 August 2018. DOI: 10.1136/gutjnl-2018-317127. [DOI] [PubMed] [Google Scholar]
- 68. Wan QY, Wu XT, Li N, et al. Long-term proton pump inhibitors use and risk of gastric cancer: a meta-analysis of 926 386 participants. Gut. Epub ahead of print 3 April 2018. DOI: 10.1136/gutjnl-2018-316416. [DOI] [PubMed] [Google Scholar]
- 69. Song H, Held M, Sandin S, et al. Increase in the prevalence of atrophic gastritis among adults age 35 to 44 years old in northern sweden between 1990 and 2009. Clin Gastroenterol Hepatol 2015; 13: 1592–1600.e1. [DOI] [PubMed] [Google Scholar]
- 70. Tavares A, Gandra A, Viveiros F, et al. Analysis of clinicopathologic characteristics and prognosis of gastric cancer in young and older patients. Pathol Oncol Res 2013; 19: 111–117. [DOI] [PubMed] [Google Scholar]
- 71. Freston JW. Omeprazole, hypergastrinemia, and gastric carcinoid tumors. Ann Intern Med 1994; 121: 232–233. [DOI] [PubMed] [Google Scholar]
- 72. Graham DY, Tansel A. Interchangeable use of proton pump inhibitors based on relative potency. Clin Gastroenterol Hepatol 2018; 16: 800–808.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Wang WH, Huang JQ, Zheng GF, et al. Non-steroidal anti-inflammatory drug use and the risk of gastric cancer: a systematic review and meta-analysis. J Natl Cancer Inst 2003; 95: 1784–1791. [DOI] [PubMed] [Google Scholar]
- 74. Algra AM, Rothwell PM. Effects of regular aspirin on long-term cancer incidence and metastasis: a systematic comparison of evidence from observational studies versus randomised trials. Lancet Oncol 2012; 13: 518–527. [DOI] [PubMed] [Google Scholar]
- 75. Cheung KS, Leung WK. Modification of gastric cancer risk associated with proton pump inhibitors by aspirin after Helicobacter pylori eradication. Oncotarget 2018; 9: 36891–36893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Cheung KS, Chan EW, Wong AYS, et al. Metformin use and gastric cancer risk in diabetic patients after Helicobacter pylori eradication. J Natl Cancer Inst. Epub ahead of print 16 October 2018. DOI: 10.1093/jnci/djy144. [DOI] [PubMed] [Google Scholar]
- 77. Lanza FL, Chan FK, Quigley EM. Guidelines for prevention of NSAID-related ulcer complications. Am J Gastroenterol 2009; 104: 728–738. [DOI] [PubMed] [Google Scholar]
- 78. Shaheen NJ, Falk GW, Iyer PG, et al. ACG clinical guideline: diagnosis and management of barrett’s esophagus. Am J Gastroenterol 2016; 111: 30–50; quiz 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Malfertheiner P, Megraud F, O’Morain CA, et al. Management of Helicobacter pylori infection-the Maastricht V/Florence consensus report. Gut 2017; 66: 6–30. [DOI] [PubMed] [Google Scholar]
- 80. Tamim H, Duranceau A, Chen LQ, et al. Association between use of acid-suppressive drugs and risk of gastric cancer. A nested case-control study. Drug Saf 2008; 31: 675–684. [DOI] [PubMed] [Google Scholar]