

Abstract
Background:
In addition to cognitive deficits, Alzheimer’s disease (AD) is also associated with other neuropsychiatric symptoms, including severe depression. Indeed, depression often precedes cognitive deficits in patients with AD. Unfortunately, the field has seen only minimal therapeutic advances, underscoring the critical need for new treatments. P7C3 aminopropyl carbazoles promote neuronal survival by enhancing nicotinamide adenine dinucleotide flux in injured neurons. Neuroprotection with P7C3 compounds has been demonstrated in preclinical models of neurodegeneration by virtue of promoting neuronal survival independently of early disease-specific pathology, resulting in protection from cognitive deficits and depressive-like behavior. We hypothesize that P7C3 compounds might thus be uniquely applicable to patients with AD, given the co-morbid presentation of depression and cognitive deficits.
Methods:
Aging male and female wild-type and TgF344-AD rats, a well-characterized preclinical AD model, were administered daily (−)-P7C3-S243 for 9 and 18 months, beginning at 6 months of age. Behavioral phenotypes related to cognition and depression were assessed at 15 and 24 months, and brain pathology and biochemistry were assessed at 24 months.
Results:
(−)-P7C3-S243 safely protected aging male and female wild-type and TgF344-AD rats from cognitive deficits and depressive-like behavior. Depressive-like behavior occurred earlier than cognitive deficits in TgF344-AD rats, consistent with AD in many patients. Treatment with (−)-P7C3-S243 blocked neurodegeneration in TgF344-AD rats, without altering amyloid deposition or indicators of neuroinflammation.
Conclusions:
Neuronal cell death-specific treatment approaches, such as offered by P7C3 compounds, may represent a new treatment approach for patients suffering from the combination of cognitive deficits and depression associated with AD.
Keywords: Alzheimer’s disease, P7C3, neurodegeneration, neurogenesis, neuroprotection, TgF344-AD, depression
Alzheimer’s disease (AD), the most common form of dementia worldwide, progressively destroys cognition and other mental functions, including the ability to regulate mood and social behavior. AD was named after Dr. Alois Alzheimer, the psychiatrist and neuroanatomist who in 1906 described key changes in the brain of a woman who died from what was then described as an unusual form of mental illness, characterized by memory loss, language difficulty, and unpredictable behavior. Dr. Alzheimer discovered clumps and tangled bundles in her brain, which subsequently became identified as amyloid plaques and neurofibrillary tangles (NFTs), respectively (1, 2). Today, over a century later, amyloid plaque deposition, NFTs, and neuronal cell loss associated with vacuole formation and neuroinflammation are defining pathological hallmarks of AD (3–5).
Currently available treatment strategies for patients with AD include medications that augment synaptic signaling, or environmental management strategies that address the changing needs of patients (6, 7). While these interventions may temporarily improve symptoms or help patients accommodate to new challenges, they do not prevent the progressive neuronal cell death that drives deteriorating neuropsychiatric function in patients. The majority of efforts to develop new therapeutics have thus far focused predominantly on targeting early events of neuroinflammation or amyloid pathology that precede neuronal loss in AD (6). Unfortunately, this approach has thus far failed to help patients (8–10). As the AD crisis rapidly escalates, there is an ever-increasing need for disease-modifying treatments. Specifically targeting the late stage of neuronal cell death, independently of earlier pathological processes, is a therapeutic strategy that has not been previously explored. The novel P7C3-series of neuroprotective aminopropyl carbazoles provides a basis for specifically testing this approach.
P7C3 was discovered through a target-agnostic in vivo screen for drug-like molecules that could safely enhance the net magnitude of postnatal hippocampal neurogenesis, by increasing either proliferation or survival of newborn hippocampal neurons in the dentate gyrus of the hippocampus. In the screening platform, the third compound (C3) of the seventh pool (P7), thereby named P7C3, was discovered to augment postnatal hippocampal neurogenesis by protecting young hippocampal neurons from dying, without affecting their rate of proliferation, and was also shown to mitigate cognitive decline in terminally aging rats (11). Subsequent medicinal chemistry efforts led to more potent variations of the original P7C3 molecule (12–14) and also generated bioactive probes that facilitated the discovery that P7C3 molecules enhance flux of nicotinamide adenine dinucleotide (NAD) in mammalian cells (15). Treatment with P7C3 has recently been shown to indirectly mitigate other critical cell death signaling events as well (16). Thus, P7C3 treatment renders neurons able to survive under conditions of otherwise overwhelming energy crisis that normally lead to death. Broad therapeutic efficacy of P7C3 compounds has been demonstrated in multiple preclinical models of neurodegeneration, including amyotrophic lateral sclerosis (17), Parkinson’s disease (13, 16, 18, 19), traumatic brain injury (20–22), psychological stress-related hippocampal cell death (23, 24), peripheral nerve crush injury (25), and stroke (26). In these studies, behavioral protection was associated with direct neuroprotective efficacy on neurons, without modulation of preceding pathological hallmarks of the disease.
For this study, we utilized the TgF344-AD rat model of AD (27). These animals overexpress human mutant amyloid precursor protein gene (APPSW) and human Δ exon 9 mutant presenilin-1 gene (PS1ΔE9), two familial mutations known to cause AD and directly affect amyloid pathology. Although these two mutations have not been observed to occur simultaneously in patients, they nonetheless produce a preclinical model that develops the classic neuropathological features of AD. Specifically, TgF344-AD animals develop progressive amyloid plaque deposition, early onset of hyperphosphorylated tau, neuroinflammation, neuron cell loss, and learning and memory deficits—the essential hallmarks of the human disease (27). Furthermore, TgF344-AD rats also develop early ocular and neurovascular deficits, akin to what is observed in people, highlighting the translational value of this AD model (28, 29). Prior to this study, examination of AD-related depressive-like behavior in this model had not been conducted. Nonetheless, while other commonly used rodent models of AD faithfully produce some aspects of the disease, the TgF344 AD rat model is the only one that has been established as manifesting such an extensive spectrum of pathological and behavioral hallmarks of human AD. Rats are also 4-5 million years evolutionarily closer to humans than mice, and their larger size and more fully characterized physiology further increases their translational value over mouse models (30). Therefore, TgF344-AD rats provide a uniquely valuable preclinical platform for characterizing the efficacy of putative therapeutic treatments for patients with AD.
METHODS AND MATERIALS
Details regarding animals, behavioral tasks, reagents, tissue preparation, immunohistochemistry and microscopy, morphological analysis and whole brain neuronal estimates, biochemical analysis are provided in Supplement 1.
Animal behavior experimental design
Extensive animal behavior testing was conducted on two individual cohorts. To limit confounding factors of repeated animal behavior testing, the order of each behavior test conducted was kept consistent throughout the duration of the experiment (Supplemental Figure 1). Less aversive tasks were conducted first, with more stress inducing tasks conducted on subsequent days.
Statistical analysis
All data were normally distributed; therefore, in instances of multiple mean comparisons, analysis of variance (ANOVA) was used, followed by post-hoc comparison using Tukey’s method. In instances of direct comparison, a two-tailed Student’s t-test was performed. Alpha levels were set to 0.05, and analyses were conducted using GraphPad Prism (GraphPad Prism Software, Inc, CA.). Outliers were determined using ROUT method in Prism and were removed only with Q=0.1%. Significance is denoted as *p < 0.05; **p < 0.01; ***p<0.001; ****p<0.0001; n.s. = not significant. A blinded examiner conducted all analyses, and code was not broken until analyses were completed.
RESULTS
We chose to evaluate efficacy of one of the most potent compounds in the P7C3 series, (−)-P7C3-S243, which lacks the potential liability of the aniline ring found in other P7C3 derivatives. (−)-P7C3-S243 readily crosses the blood brain barrier and is readily prepared as a single active enantiomer (13, 18, 22). Although the kinetics of (−)-P7C3-S243 have been previously established in mice (13), we wanted to ensure that we could achieve similar blood levels in rats before embarking on this study. Therefore, we administered 3 mg/kg/d of (−)-P7C3-S243 for two months and collected plasma samples at various time points for pharmacological analysis. As shown in Figure 1A, this dosing regimen achieved plasma (−)-P7C3-S243 concentrations previously reported to be sufficient for biological activity (13). Consistent with what has been seen in mice, (−)-P7C3-S243 showed a prolonged terminal elimination phase with good exposures maintained between the 24-hour dosing interval.
Figure 1.
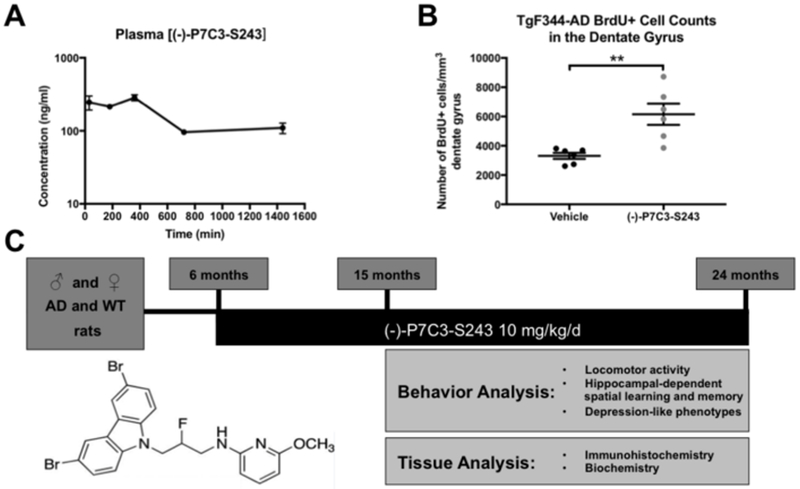
Experimental Design. (A) Peripherally-administered (−)-P7C3-S243 achieves blood levels in rats equivalent to that required for biological activity in mouse preclinical models, and (B) also enhances hippocampal neurogenesis in TgF344-AD rats. BrdU positive cell counts treatment effect was t=3.763, df=10, p=0.0037 using a two-tailed Student’s t-test. Once this effect was established, we proceeded to evaluate the efficacy of (−)-P7C3-S243 in TgF344-AD rats using the experimental design in panel (C). Beginning at 6 months of age, (−)-P7C3-S243 at 10 mg/kg was administered daily to male and female TgF344-AD rats and age/sex-matched WT littermates. Behavior was assessed at 15 and 24 months, following 9 and 18 months of (−)-P7C3-S243 or vehicle treatment, respectively, in the form of locomotor activity (open field), hippocampal-dependent spatial learning and memory (Barnes maze and Morris water maze), and depression-like behavior (Porsolt forced swim task). Pathological and biochemical analysis was subsequently conducted on tissue collected at both time points.
Although we have previously shown that P7C3 compounds are bioactive in aged Fischer 344 rats (11), and TgF344-AD rats are on Fisher 344 genetic background, we also wanted to ensure that (−)-P7C3-S243 was similarly bioactive in the transgenic animals before conducting the full study. To determine this, we turned to the classic postnatal hippocampal neurogenesis assay by which the original P7C3 compound was discovered. In this in vivo assay of biological activity, P7C3 compounds increase the net magnitude of postnatal hippocampal neurogenesis by blocking cell death of newborn hippocampal neurons in the dentate gyrus (11). We administered (−)-P7C3-S243 at 10 mg/kg/d for ten days to 3-month old male and female TgF344-AD rats, along with daily injections of bromodeoxyuridine (BrdU, 50 mg/kg/d). Twenty-four hours after administration of the final doses of BrdU and (−)-P7C3-S243, animals were transcardially perfused and brains were processed for immunohistochemical staining of BrdU to quantify hippocampal neurogenesis, per standard procedures (11). As shown in Figure 1B, treatment with (−)-P7C3-S243 significantly enhances neurogenesis in TgF344-AD rats by two fold, from a baseline of approximately 3000 BrdU+ cells / mm3 dentate gyrus in vehicle-treated TgF344-AD rats up to approximately 6000 BrdU+ cells / mm3 in (−)-P7C3-S243-treated TgF344-AD rats. Importantly, no difference in biological activity was observed between sexes.
Confident in the knowledge that (−)-P7C3-S243 achieved good exposure and was bioactive in this preclinical model of AD, we then chronically administered this agent daily to wild-type (WT) and TgF344-AD rats, starting at 6 months of age, before the onset of robust symptoms (27) (Figure 1C). Over the years, male animals have been disproportionately studied in basic science research, leading to a lack of awareness of sex-specific differences (31, 32). Therefore, we included equally robust numbers of both male and female animals in our study. Behavioral testing was conducted at 15 and 24 months of age, representing 9 and 18 months of daily treatment, respectively. Then, brain tissue was collected for histologic and biochemical analysis. Multiple behaviors were assessed within each animal cohort, with careful consideration taken to determine a behavioral testing sequence that reduced the confounding factors of stressful behavior testing on subsequent tests (Supplemental Figure 1). Animals were assigned an identification number to ensure that animal information remained coded for blinded data collection and analysis throughout the study. Furthermore, at 5 months of age, prior to initial treatment, all animals were subjected to Barnes maze testing to ensure that they were randomly and equally placed into vehicle or (−)-P7C3-S243 treatment groups. At this time point, no differences were observed in the learning or memory portion of the Barnes maze task, and thus no animals were removed from the study due to behavioral deficits.
15-month old TgF344-AD rats develop early depressive-like behavior that is blocked by (−)-P7C3-S243 treatment.
Numerous studies in humans have identified psychiatric disturbances as early indicators of AD, occurring before the classic memory deficits. For example, anxiety, depression, irritability, psychosis, and sleep disturbances are often observed early in AD patients (33), with depression frequently being the earliest manifestation of disease (34, 35). Notably, we observed the correlate of this pattern in our animal studies as well. At 15 months of age, neither male nor female TgF344-AD or WT littermate rats showed significant deficits in learning or memory in the Barnes maze (Figure 2A-F). However, both male and female 15-month old TgF344-AD rats did display significant depression-like behaviors in the Porsolt forced swim test (FST) (Figure 2G-I), with no effect on general locomotor activity (Figure 2J-L).
Figure 2.
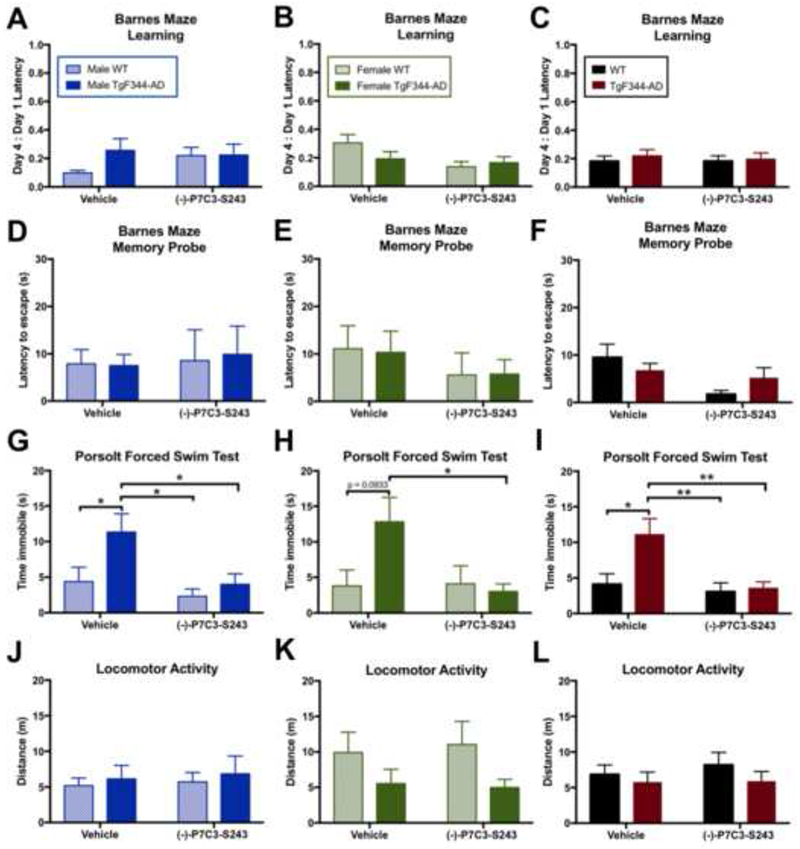
(−)-P7C3-S243 protects TgF344-AD rats from depression-like behavior at 15 months, before the onset of cognitive deficits. (A-F) No learning or memory deficits were observed at 15 months of age, regardless of sex, as assessed using Barnes maze. Barnes maze learning treatment effect: Male F(1, 32)=0.5419, p=0.4670; Female F(1,29)=3.861, p=0.0591; Combined F(1,65)=0.0734, p=0.7873. Barnes maze learning genotype effect: Male F(1, 32)=1.8020, p=0.1889; Female F(1,29)=0.7218, p=0.4025; Combined F(1,65)=0.3674, p=0.5465. Barnes maze memory probe treatment effect: Male F(1,30)=0.0979, p=0.7565; Female F(1,27)=1.2350, p=0.2763; Combined F(1,57)=4.7950, p=0.0327. Barnes maze memory probe genotype effect: Male F(1,30)=0.0092, p=0.9244; Female F(1,27)=0.0048, p=0.9454; Combined F(1,57)=0.0318, p=0.8591. (G-I) Significant depression like phenotypes in the Porsolt forced swim test (FST) were observed in TgF344-AD rats, also regardless of sex, and (−)-P7C3-S243 administration for 9 months protected against depression-like behavior. FST treatment effect: Male F(1,32)=6.456, p=0.0161; Female F(1,28)=3.133, p=0.0876; Combined F(1,65)=7.119, p=0.0096. FST genotype effect: Male F(1,32)=5.454, p=0.0260; Female F(1,28)=2.185, p=0.1505; Combined F(1,65)=5.253, p=0.0252. (J-L) Locomotor activity was comparable between all groups. Open field treatment effect: Male F(1,32)=0.1285, p=0.7226; Female F(1,28)=0.0150, p=0.9036; Combined F(1,60)=0.2470, p=0.6210. Open field genotype effect: Male F(1,32)=0.3605, p=0.5529; Female F(1,28)=4.5950, p=0.0412; Combined F(1,60)=1.296, p=0.2595. For each measurement, n=9-10/group for males, n=7-10/group for females, and n=16-19/group for combined sexes. Data are represented as mean ± SEM. Significance was determined using two-way ANOVA with Tukey’s post-hoc multiple comparison analysis. *p < 0.05; **p < 0.01.
FST is used to examine depression-like phenotypes by measuring the extent of effort exerted by an animal to escape a stressful situation, which in this case is a water-filled cylinder. In the FST, depression-like behavior is indicated by less time spent moving and more time spent immobile (36). (−)-P7C3-S243 proved protective against this depression-like phenotype in both male and female TgF344-AD rats, as the time spent immobile in the task was significantly reduced and comparable to that seen in WT animals (Figure 2G-I). The proneurogenic efficacy of P7C3 compounds has been previously reported to be responsible for their anti-depressant-like effects (23), which is consistent with the proneurogenic efficacy of (−)-P7C3-S243 that we observed in TgF344-AD animals (Figure 1B).
(−)-P7C3-S243 protects 24-month old TgF344-AD rats from developing cognitive deficits.
As there were no significant behavioral differences between sexes (Figure 2), we combined males and females for subsequent analysis. Extensive behavioral analysis was conducted on a second cohort of 24-month old animals, a time point by which impairments in cognition have been shown to manifest in TgF344-AD rats (27). Regardless of genotype or treatment group, survival at this time point was reduced roughly 50% relative to the 15 month time point. By this age, it is well known that almost all Fisher rats will have started to develop benign neoplasms that may reduce mobility and impede quality of life. The animal care staff discourages repeated removal of masses in aging animals, and thus animals were removed from the study if they required more than one surgical mass removal. All surviving animals at this time point, regardless of genotype or treatment group, performed similarly in the initial Morris water maze test of learning and memory (Figure 3A, B). However, more rigorous testing with the Morris water maze reversal task showed that hippocampal-dependent spatial memory was significantly impaired in TgF344-AD rats relative to WT littermate controls, while learning was only mildly affected (Figure 3C, D). Of note, this determination of the relearning task as mildly affected relates to the lack of statistical significance between TgF344-AD rats and WT littermates in the vehicle condition when compared via two-way ANOVA analysis. When these two groups are compared directly to one another by Student’s t-Test, however, a statistically significant difference is found for both Day3:Day1 Latency (t=2.067, df=23, p=0.05) and Day 3 Latency (t=2.077, df=29, p=0.0468). Thus, we propose that TgF344-AD rats did not relearn the task, while WT littermates did. Lack of relearning can be equated to behavioral inflexibility, which is often associated with deficits in prefrontal cortex function. In the reversal procedure, the hidden platform is moved to a different location in the water tank, and the animal’s ability to react to this change to learn and remember the new escape location is measured. This more challenging test of cognitive function in the MWM revealed that TgF344-AD rats required 2-3 times longer to travel the new location of escape platform than WT littermates in the memory probe phase (Figure 3D), which could indicate significant impairment in memory, as there were no differences in swim speed, as well as impaired learning (Figure 3E). Continuous daily treatment with (−)-P7C3-S243 for 18 months proved protective against this memory deficit, as evidenced by improving the TgF344-AD animal’s time to locate the target position in the reversal memory probe (Figure 3D). Importantly, no significant locomotor differences were observed across groups at 24 months of age (Figure 3E). With respect to depression-like behavior, 24-month old TgF344-AD animals continued to display deficits in the FST, which at this advanced stage of disease could no longer be corrected by treatment with (−)-P7C3-S243 (time spent immobile in seconds: TgF344-AD, Vehicle n=6: 31.3 ± 17.028; TgF344-AD, (−)-P7C3-S243 n=8: 32.2 ± 37.708). However, in contrast to the 15-month time point, 24-month old WT animals displayed depression-like behavior in the FST, which was indeed significantly ameliorated by treatment with (−)-P7C3-S243 (Figure 3F).
Figure 3.
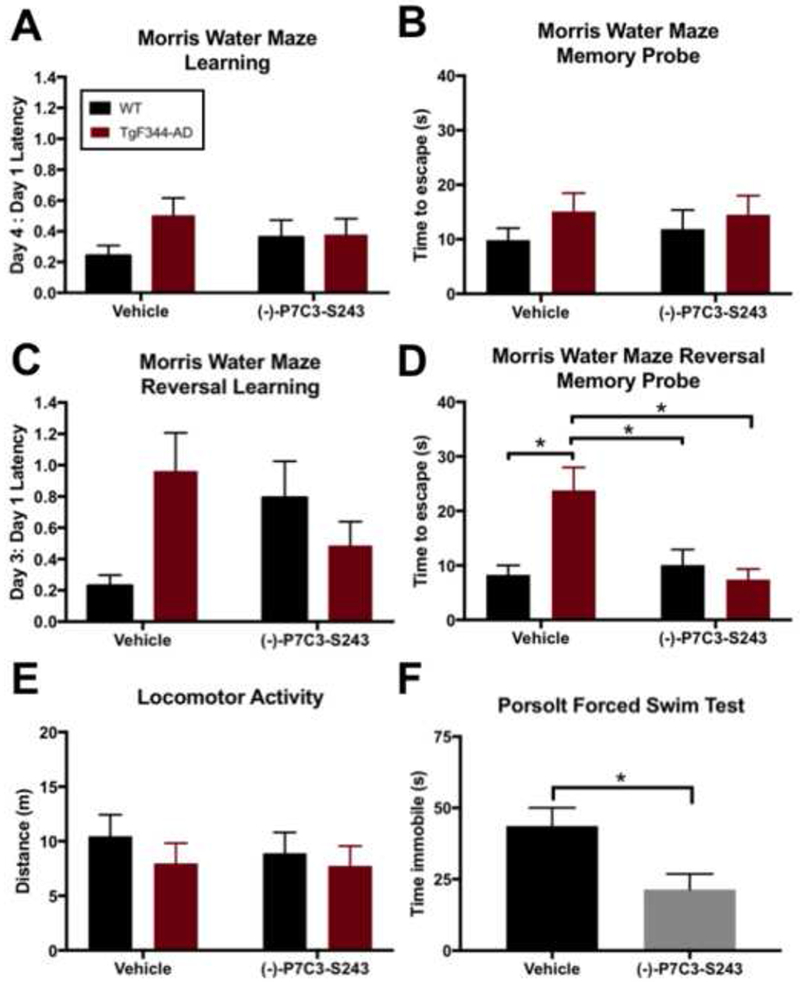
(−)-P7C3-S243 protects 24-month old TgF344-AD rats from memory deficits, and WT rats from depression-like behavior. (A, B) TgF344-AD and WT rats preformed similarly in initial Morris water maze learning and memory tasks. Morris water maze learning treatment effect was F(1,56)=0.0012, p=0.9730 and genotype effect was F(1,56)=1.627, p=0.2073. Morris water maze memory probe treatment effect was F(1,54)=0.0426, p=0.8373 and genotype effect was F(1,54)=1.296, p=0.2599. (C, D) However, in Morris water maze reversal testing, TgF344-AD rats performed significantly worse during the memory probe, and (−)-P7C3-S243 treatment was protective against this impairment. Morris water maze reversal learning treatment effect was F(1,55)=0.0347, p=0.8530 and genotype effect was F(1,55)=0.8550, p=0.3595. Morris water maze reversal memory probe treatment effect was F(1,55)=3.808, p=0.0561 and genotype effect was F(1,54)=2.953, p=0.0913. (E) Locomotor activity was equivalent in all groups. Open field treatment effect was F(1,56)=0.2017, p=0.6551 and genotype effect was F(1,56)=0.8566, p=0.3587. For each measurement, n=11-20/group. (F) WT rats exhibited significant depression-like behaviors in the FST and were protected from this deficit by treatment with (−)-P7C3-S243. FST treatment effect was t=2.613, df=18, p=0.0176. For FST measurements, n=9-12/group. Data are represented as mean ± SEM. Significance was determined using two-way ANOVA with Tukey’s post-hoc multiple comparison analysis or a two-tailed Student’s t-test. *p <0.05.
(−)-P7C3-S243 treatment does not alter amyloid plaque deposition, tau hyperphosphorylation, or glial reaction in TgF344-AD rats.
Brains from 24-month old animal were harvested to examine histopathology and classical markers of amyloid deposition, tau hyperphosphorylation, and neuroinflammation. Amyloid deposition is a well-characterized pathological hallmark of AD (4, 5). When normal processing of the amyloid precursor protein (APP) becomes dysregulated, a shift in Aβ1-40 and Aβ1-42 peptide fragment formation occurs, with Aβ1-42 fragments disproportionately formed and widely considered to be more toxic than Aβ1-40 peptides (37). Aβ1-42 fragments have strong binding affinity for one another and thus form insoluble Aβ plaques. Alternatively, some Aβ fragments also remain soluble in smaller fibril forms, which disrupt synaptic signaling (37).
In vehicle-treated 24-month old TgF344-AD animals, abundant amyloid plaque deposition was evident throughout the brain by both H&E and Congo Red staining (hippocampus, cerebral cortex, frontal cortex, and cerebellum), but neither the degree nor distribution of plaque deposition was significantly altered by treatment with (−)-P7C3-S243 (Figure 4A-E). To further examine whether (−)-P7C3-S243 could impact amyloid pathology in TgF344-AD rats, we next assayed amyloid peptide deposition in triton soluble (i.,e. soluble Aβ peptides) and 5 M guanidine HCl soluble (i.e. insoluble Aβ oligomers) brain extracts using Aβ1-40 and Aβ1-42 peptide ELISAs. Both soluble and insoluble Aβ1-40 and Aβ1-42 were significantly increased in male and female TgF344-AD rats relative to WT controls at 24 months (Figure 4F, G). However, no significant differences were observed in TgF344-AD rats treated with (−)-P7C3-S243, relative to vehicle-treated controls (Figure 4F, G). Therefore, (−)-P7C3-S243 exerts its protective efficacy independently of altering amyloid pathology in TgF344-AD rats.
Figure 4.
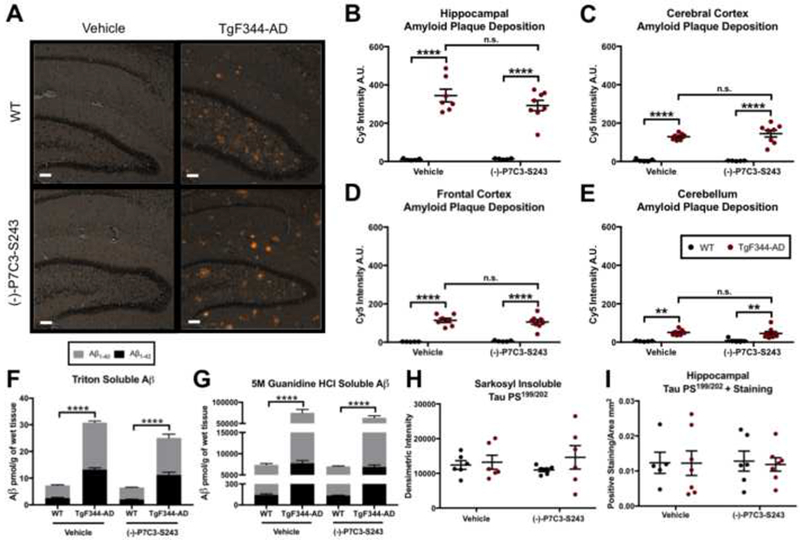
Amyloid plaque deposition and tau phosphorylation in TgF344-AD rats are unaffected by treatment with (−)-P7C3-S243. (A-E) Amyloid plaque deposition is significantly increased in TgF344-AD rats in the hippocampus, cerebral cortex, frontal cortex, and cerebellum, but is not affected by (−)-P7C3-S243 administration. (A) Representative images of Congo Red staining in the hippocampus are depicted and (B-E) quantified in the hippocampus, cerebral cortex, prefrontal cortex, and cerebellum using Cy5 intensity quantification. Hippocampus plaque treatment effect was F(1,21 )=0.8611, p=0.3640 and genotype effect was F(1,21 )=135.2, p<0.0001. Cerebral cortex plaque treatment effect was F(1,21 )=0.3161, p=0.5799 and genotype effect was F(1,21 )=115.3, p<0.0001. Prefrontal cortex plaque treatment effect was F(1,21 )=0.0571, p=0.8135 and genotype effect was F(1,21 )=104.3, p<0.0001. Cerebellum plaque treatment effect was F(1,21)=0.0203, p=0.8881 and genotype effect was F(1,21)=30.63, p<0.0001. The white bar represents 100 μm. (F, G) Soluble (triton) and insoluble (5 M guanidine HCl) Aβ 1-40 and Aβ 1- 42 peptides are elevated in TgF344-AD rats relative to WT animals at 15 months, regardless of sex or treatment. (H) Pathological tau in the form of insoluble (sarkosyl insoluble) hyperphosphorylated tau is elevated in all groups. (I) Flyperphosphorylated tau (anti-Tau PS199/202) immunostaining in the hippocampus is also similar between all experimental groups. Hippocampal tau ps199/202 treatment effect was F(1,21)=0.0005, p=0.9832 and genotype effect was F(1,21)=0.0324, p=0.8589. Whole brain tau ps199/202 treatment effect was F(1,20)<0.0001, p=0.9973 and genotype effect was F(1,20)=1.126, p=0.3012. For each measurement, n=6-7/group. Data are represented as mean ± SEM. Significance was determined using two-way ANOVA with Tukey’s post-hoc multiple comparison analysis. *p < 0.05; **p < 0.01; ***p<0.001; ****p<0.0001.
Next, we assessed tauopathy in the brain at 24 months. Tau proteins function primarily in the axons of neurons to stabilize microtubules by interacting with tubulin, and disruption of this function by mutations or excessive phosphorylation of tau is associated with neurodegeneration (38). Tau also becomes hyperphosphorylated in normal aging (39). When tau becomes hyperphosphorylated, it forms intracellular NFTs predominantly in large pyramidal neurons of the entorhinal cortex, hippocampus, association cortex, and other cortical regions as the disease advances (4, 5). Although it is unclear whether the NFTs themselves, tau oligomers released prior to NFT formation, or perhaps both, lead to cell death (40), the endpoint of NFT accumulation is a classic hallmark seen on post-mortem examination of the brains of patients with AD. NFTs can be measured using routine detergent (sarkosyl) extractions, and sarkosyl-insoluble tau correlates with the pathological features of this altered protein that accumulates in vivo. We applied this method to measure the immunoreactivity of a commonly phosphorylated phosphoserine site on tau using an antibody that recognizes Tau PS199/202, and found no significant increases in TgF344-AD rats relative to WT controls (Figure 4H, I). This is consistent with what has been previously reported in older TgF344-AD rats (27). Tau PS199/202 was also not impacted by (−)-P7C3-S243 treatment in aged WT or TgF344-AD animals (Figure 4H, I).
Finally, we investigated the effects of (−)-P7C3-S243 on indicators of neuroinflammation. At 24 months of age, astrogliosis was not apparent in TgF344-AD rats relative to control groups, and astrocyte activation was also not affected by treatment with (−)-P7C3-S243 (Figure 5A, B). Microgliosis, both independently located and in close proximity to amyloid deposition, was evident in TgF344-AD rats, as previously observed by others (27), but also was not affected by (−)-P7C3-S243 treatment (Figure 5C-E).
Figure 5.
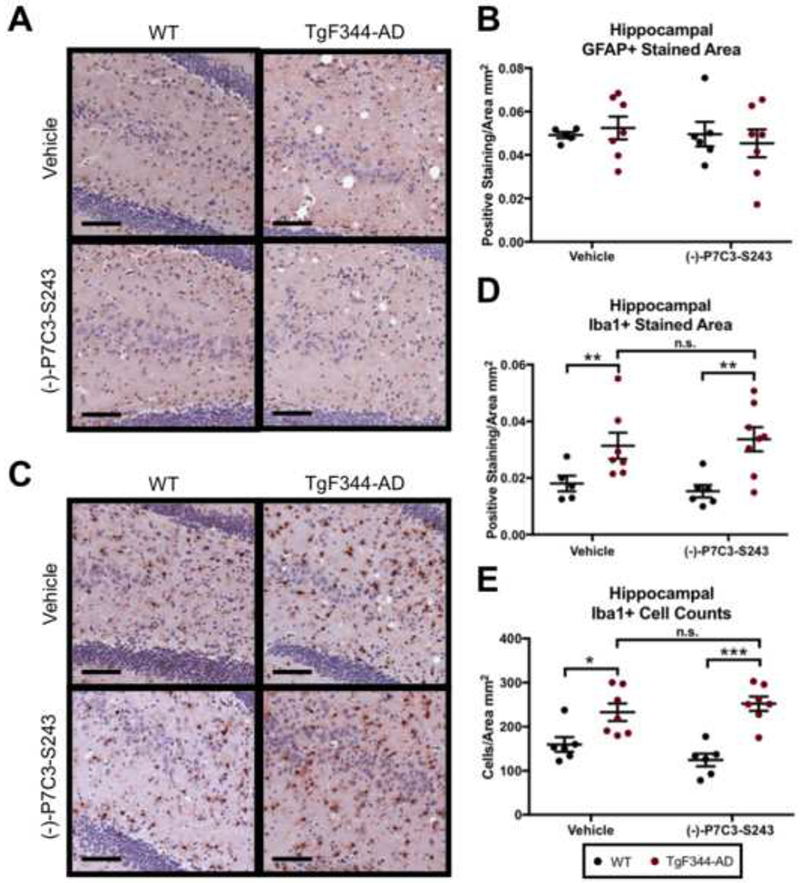
Neuroinflammation in TgF344-AD rats is unaffected by treatment with (−)-P7C3-S243. (A, B) GFAP staining is similar between all experimental groups, indicating that at 24 months of age astrocyte activation is not increased in TgF344-AD rats or altered by (−)-P7C3-S243 treatment. (A) Representative images of GFAP staining in the hippocampus are presented and (B) positive staining is quantified. GFAP positive staining treatment effect was F(1,21 )=0.3610, p=0.5544 and genotype effect was F(1,21)=0.0079, p=0.9302. (C-E) Microglia cell populations are increased in TgF344-AD rats at 24 months and unaltered by treatment with (−)-P7C3-S243. (C) Representative images of Iba1 staining in the hippocampus are presented and (D) positive staining and (E) cell counts are quantified. Iba1 positive staining treatment effect was F(1,22)=0.0030, p=0.9569 and genotype effect was F(1,22)=15.83, p=0.0001. Iba1 cell counts treatment effect was F(1,22)=0.2125, p=0.6494 and genotype effect was F(1,22)=34.18, p<0.0001. The black bars represent 100 μm. For each measurement, n=6-7/group. Data are represented as mean ± SEM. Significance was determined using two-way ANOVA with Tukey’s post-hoc multiple comparison analysis. *p < 0.05; **p < 0.01.
(−)-P7C3-S243 treatment reduces neurodegeneration in TgF344-AD rats.
Neuronal loss leads to vacuole formation following a sequence of cellular swelling, death, and ultimately microglia-mediated clearage of cellular debris. Thus, this pathological hallmark is indicative of a later stage of neuronal damage in AD, distal to plaque and NFT formation. Indeed, we often noted degenerating neuronal cell bodies within regions dense with amyloid plaques, typically in combination with glial and microglial responses (Figure 6A), and microglial engulfment of neuronal debris was also visualized in the original report characterizing TgF344-AD rats (27). Due to the general neuroprotective activity of P7C3 compounds in other models, we wondered whether the protective effects of prolonged (−)-P7C3-S243 administration seen here might be associated with reduced vacuole formation. Indeed, blinded manual counting of vacuole formation in the hippocampus and cortex of male and female TgF344-AD rats and WT littermates demonstrated that (−)-P7C3-S243 treated groups had significantly reduced numbers of vacuoles in both regions (Figure 6B-E).
Figure 6.
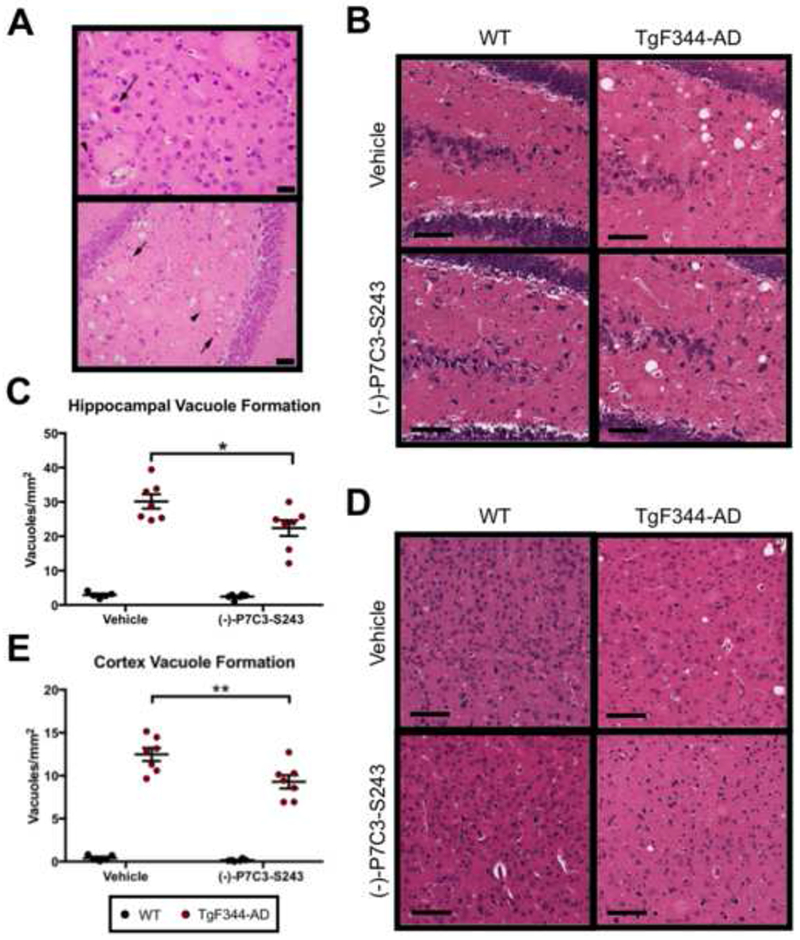
Vacuole formation in the brains of TgF344-AD rats 455 is significantly reduced by treatment with (−)-P7C3-S243. (A) H&E images of cortex (top) and hippocampus (bottom) of TgF344-AD rats at 24 months show large, pale, eosinophilic amyloid plaques surrounded by reactive microglia (arrowheads). In close proximity are neurons in variable stages of degeneration (arrows). The black bars represent 50 and 100 μm, respectively. (B-E) Vacuole formation, a common marker of neurodegeneration, was readily apparent in TgF344-AD rats. Representative H&E images of vacuoles in (B) the hippocampus and (D) cortex are depicted. Vacuoles were manually quantified in (C) hippocampus and (E) cerebral cortex in a blinded manner and found to be significantly decreased by (−)-P7C3-S243 treatment. Hippocampal vacuole formation treatment effect was F(1,21 )=5.285, p=0.0319 and genotype effect was F(1,21 )=174.3, p<0.0001. Cortex vacuole formation treatment effect was F(1,21 )=7.528, p=0.0122 and genotype effect was F(1,21 )=285.9, p<0.0001. For each measurement, n=6–7/group. Data are represented as mean ± SEM. Significance was determined using two-way ANOVA with Tukey’s post-hoc multiple comparison analysis. *p < 0.05; **p < 0.01.
Next, we quantified neuronal cell counts in the hippocampus and cortex of male and female TgF344-AD rats and WT littermates. We initially quantified neurons using the neuron specific cell marker NeuN. However, we also observed NeuN+ microglial cells throughout the brain, as microglia routinely phagocytize debris from dying neurons and thereby physically incorporate markers for neurons. This complicated our automated cell counting analysis, and artificially increased neuronal cell counts. We thus turned to cresyl violet, another neuronal marker, that in cases of broad microglial activation serves as a more selective stain for neurons by virtue of staining fewer microglia (Supplemental Figure 2). Neuronal cell counts quantified using cresyl violet staining were significantly reduced in the hippocampus and trending down in the cortex in TgF344-AD rats relative to WT controls at 24 months of age, and treatment with (−)-P7C3-S243 protected TgF344-AD rats from neuronal cell loss in both regions (Figure 7A, B). Microglia were rarely identified in the granule cell layer of the dentate gyrus, and thus we conducted neuron cell counts using the NeuN marker in this densely packed region. Again, we observed significantly increased cell counts in the dentate gyrus of the hippocampus in TgF344-AD rats treated with (−)-P7C3-S243 relative to those treated with vehicle (Figure 7C).
Figure 7.
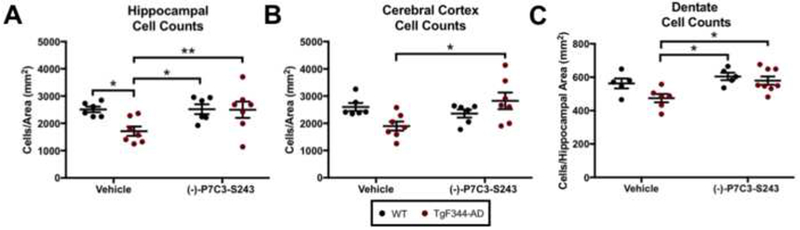
(−)-P7C3-S243 administration protects against neuronal cell loss in TgF344-AD rats. (A, B) (−)-P7C3-S243 treatment for 15 months (24 months of age), protected TgF344-AD rats from neuronal cell loss in the (A) hippocampus and (B) cortex. Hippocampal cell counts treatment effect was F(1,22)=3.841, p=0.0128 and genotype effect was F(1,22)=3.829, p=0.0632. Cortex cell counts treatment effect was F(1,22)=2.630, p=0.1191 and genotype effect was F(1,22)=0.3121, p=0.5821. Dentate cell counts treatment effect was F(1,20)=7.796, p=0.0113 and genotype effect was F(1,20)=4.631, p=0.0438. (C) Cell counts were also significantly increased in the granule cell layer of the dentate gyrus of the hippocampus.
Discussion
Identifying effective treatment strategies for AD, the most abundant neurodegenerative disease worldwide, is crucial. Unfortunately, despite tremendous efforts in academic medicine and the pharmaceutical industry, experimental therapies have thus far failed to translate to effective treatments for patients. Here, we have investigated whether neuronal protection at a more distal time point, at the level of actually blocking neuronal cell death, irrespective of upstream events, might have protective efficacy in a preclinical model of AD. P7C3 compounds have previously displayed protective efficacy in an array of preclinical models of central and peripheral neurodegeneration, with the common feature that regardless of earlier disease-specific pathological events, neurons across systems are critically dependent on NAD salvage, a process augmented by P7C3 compounds. Here, we now report efficacy of (−)-P7C3-S243 against acquisition of early depressive-like behavior, later cognitive impairment, and ultimately neuronal cell loss in the TgF344-AD rat model of AD. Notably, (−)-P7C3-S243 was observed to be equally efficacious in both males and females.
Early in the disease process, before the onset of cognitive deficits, (−)-P7C3-S243 was protective against depression-like phenotypes, which is often the earliest neuropsychiatric symptom of AD in patients. Later in the disease process, when hippocampal-dependent spatial memory became impaired at 24 months, treatment with (−)-P7C3-S243 also protected from this deficit as well. Also at this later stage of 24 months of age, WT animals additionally acquired depressive-like behavior, which was also blocked by treatment with (−)-P7C3-S243. This antidepressant effect of (−)-P7C3-S243 is likely due to the proneurogenic efficacy achieved by P7C3 compounds by virtue of protecting young newborn hippocampal neurons from cell death, while the cognitive protection afforded by (−)-P7C3-S243 is likely due to the combined effect of protecting both newborn hippocampal neurons and mature neurons in the cortex and hippocampus. Strikingly, examination of histological and biochemical markers in the brains of TgF344-AD rats revealed that (−)-P7C3-S243 protects against neuronal damage without altering the traditional markers of AD pathology: amyloid deposition, tau phosphorylation, and reactive glia. TgF344-AD rats treated with (−)-P7C3-S243 did, however, display reduced neurodegeneration in the hippocampus and cortex, two brain regions critical for learning and memory, as measured by vacuole formation and neuron cell counts.
P7C3 compounds accelerate flux of NAD salvage in neurons through enhancing activity of nicotinamide phosphoribosyltransferase (NAMPT), the rate-limiting enzyme in this process. NAMPT synthesizes nicotinamide mononucleotide (NMN) from nicotinamide, and normal aging is associated with considerable reduction in NAMPT and NAD levels in the hippocampus, along with reduced proliferation of hippocampal neural precursor cells. Thus, NAMPT activation or other strategies aimed at enhancing hippocampal neurogenesis could be a useful therapeutic intervention to increase survival of functioning newborn neurons in the hippocampus (41, 42). Of note, NAMPT has also been proposed as a candidate susceptibility gene for AD, by virtue of a rare copy number variation in NAMPT that was identified in a patient with late-onset AD (43). In addition, other investigators have recently shown some measure of protective efficacy with administration of NMN, the product of NAMPT, in other rodent models of AD (44–46).
Although our results do not rule out that traditionally-considered pathological events in AD may contribute to impaired functioning in patients, we clearly show that preserving neuron survival without attempting to intervene in other pathological aspects of the disease results in significant protective efficacy in measures of depression-like behavior, cognitive function, and neuronal cell death associated with AD. Thus, P7C3 compounds may form the basis for a new class of neuroprotective drugs for mitigating the symptoms in patients with AD by preserving neuronal cell survival, irrespective of other pathological events. P7C3 compounds represent a novel route to treating depression, and new onset depression in elderly patients may herald the development of Alzheimer’s disease with later cognitive impairments to follow. Thus, it is reasonable to propose that a drug from the P7C3 class of neuroprotective chemicals might one day offer a novel first-line treatment for new onset depression in the elderly, by virtue of its combined antidepressant and neuroprotective properties that would simultaneously help normalize mood and protect from progression to other cognitive symptoms of Alzheimer’s disease.
Supplementary Material
IN THIS ISSUE Statement.
Voorhees et al. demonstrate protective efficacy of P7C3 neuroprotective compounds in a rat model of Alzheimer’s disease (AD), with respect to early depressive-like symptoms and later problems with learning and memory. The protective effect correlated with direct protection from neuronal cell death, without altering earlier classical hallmarks of AD, such as amyloid plaque formation. Therapeutic strategies targeting cell survival irrespective of earlier pathology may represent a viable path forward in treating patients with AD.
Acknowledgements:
We thank Dr. Robert Cohen for generously providing a male TgF344-AD hemizygous breeder to our laboratory for establishment of the animal colony. This work was supported by the National Institute for Environmental Health Sciences through the University of Iowa Environmental Health Sciences Research Center (NIEHS/NIH P30 ES005605) to JRV and AAP, funds to AAP from an anonymous donor to the Mary Alice Smith Fund for Neuropsychiatry Research, funds to AAP from the Titan Neurology Research Fund, funds to AAP from the Brockman Medical Research Foundation, by Department of Veterans Affairs Merit Review 1IO1BX002444 to AAP, and by CDA number IK2 RX002003 from US Dept, of Veterans Affairs RR&D to LMD. The contents of this manuscript do not represent the views of the U.S. Department of Veterans Affairs or the U.S. Government.
Footnotes
Financial Disclosures
Andrew Pieper is co-founder and consultant of Neuroprotective Therapeutics, and holds patents related to the P7C3 series of neuroprotective compounds. JR Voorhees, MT Remy, CJ Cintrón-Pérez, E El Rassi, MZ Kahn, LM Dutca, TC Yin, LM McDaniel, NS Williams, and DJ Brat have no biomedical financial interests or potential conflicts of interest.
References
- 1.Alzheimer A Fau - Stelzmann RA, Stelzmann Ra Fau - Schnitzlein HN, Schnitzlein Hn Fau - Murtagh FR, Murtagh FR An English translation of Alzheimer’s 1907 paper, “Uber eine eigenartige Erkankung der Hirnrinde”. [DOI] [PubMed]
- 2.Alzheimer AF (1907): Uber eine eigenartige Erkrankung der Hirnrinde. Allgemeine Zeitschrift fur Psychiatrie und phy- chish-Gerichtliche Medizin (Berlin). 64:146–148. [Google Scholar]
- 3.Selkoe DJ (1994): Cell biology of the amyloid beta-protein precursor and the mechanism of Alzheimer’s disease. Annual review of cell biology. 10:373–403. [DOI] [PubMed] [Google Scholar]
- 4.Montine TJ, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Dickson DW, et al. (2012): National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: a practical approach. Acta neuropathologica. 123:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hyman BT, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Carrillo MC, et al. (2012): National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimer’s & dementia : the journal of the Alzheimer’s Association. 8:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cummings JL, Morstorf T, Zhong K (2014): Alzheimer’s disease drug-development pipeline: few candidates, frequent failures. Alzheimer’s research & therapy. 6:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kumar A, Singh A, Ekavali (2015): A review on Alzheimer’s disease pathophysiology and its management: an update. Pharmacological reports : PR. 67:195–203. [DOI] [PubMed] [Google Scholar]
- 8.Kumar NS, Nisha N (2014): Phytomedicines as potential inhibitors of beta amyloid aggregation: significance to Alzheimer’s disease. Chinese journal of natural medicines. 12:801–818. [DOI] [PubMed] [Google Scholar]
- 9.Kumar J, Sim V (2014): D-amino acid-based peptide inhibitors as early or preventative therapy in Alzheimer disease. Prion. 8:119–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Anand R, Gill KD, Mahdi AA (2014): Therapeutics of Alzheimer’s disease: Past, present and future. Neuropharmacology. 76 Pt A:27–50. [DOI] [PubMed] [Google Scholar]
- 11.Pieper AA, Xie S, Capota E, Estill SJ, Zhong J, Long JM, et al. (2010): Discovery of a proneurogenic, neuroprotective chemical. Cell. 142:39–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.MacMillan KS, Naidoo J, Liang J, Melito L, Williams NS, Morlock L, et al. (2011): Development of proneurogenic, neuroprotective small molecules. Journal of the American Chemical Society. 133:1428–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Naidoo J, De Jesus-Cortes H, Huntington P, Estill S, Morlock LK, Starwalt R, et al. (2014): Discovery of a neuroprotective chemical, (S)-N-(3-(3,6-dibromo-9H-carbazol-9-yl)-2-fluoropropyl)-6-methoxypyridin-2-amine [(−)-P7C3-S243], with improved drug-like properties. Journal of medicinal chemistry. 57:3746–3754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pieper AA, McKnight SL, Ready JM (2014): P7C3 and an unbiased approach to drug discovery for neurodegenerative diseases. Chemical Society reviews. 43:6716–6726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang T, Zhao L, Liu M, Xie F, Ma X, Zhao P, et al. (2014): Oral intake of hydrogen-rich water ameliorated chlorpyrifos-induced neurotoxicity in rats. Toxicology and applied pharmacology. 280:169–176. [DOI] [PubMed] [Google Scholar]
- 16.Gu C, Zhang Y, Hu Q, Wu J, Ren H, Liu CF, et al. (2017): P7C3 inhibits GSK3beta activation to protect dopaminergic neurons against neurotoxin-induced cell death in vitro and in vivo. Cell death & disease. 8:e2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tesla R, Wolf HP, Xu P, Drawbridge J, Estill SJ, Huntington P, et al. (2012): Neuroprotective efficacy of aminopropyl carbazoles in a mouse model of amyotrophic lateral sclerosis. Proceedings of the National Academy of Sciences of the United States of America. 109:17016–17021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.De Jesus-Cortes H, Miller AD, Britt JK, DeMarco AJ, De Jesus-Cortes M, Stuebing E, et al. (2015): Protective efficacy of P7C3-S243 in the 6-hydroxydopamine model of Parkinson’s disease. NPJ Parkinson’s disease. 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.De Jesus-Cortes H, Xu P, Drawbridge J, Estill SJ, Huntington P, Tran S, et al. (2012): Neuroprotective efficacy of aminopropyl carbazoles in a mouse model of Parkinson disease. Proceedings of the National Academy of Sciences of the United States of America. 109:17010–17015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Blaya MO, Bramlett HM, Naidoo J, Pieper AA, Dietrich WD (2014): Neuroprotective efficacy of a proneurogenic compound after traumatic brain injury. Journal of neurotrauma. 31:476–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dutca LM, Stasheff SF, Hedberg-Buenz A, Rudd DS, Batra N, Blodi FR, et al. (2014): Early detection of subclinical visual damage after blast-mediated TBI enables prevention of chronic visual deficit by treatment with P7C3-S243. Investigative ophthalmology & visual science. 55:8330–8341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yin TC, Britt JK, De Jesus-Cortes H, Lu Y, Genova RM, Khan MZ, et al. (2014): P7C3 neuroprotective chemicals block axonal degeneration and preserve function after traumatic brain injury. Cell reports. 8:1731–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Walker AK, Rivera PD, Wang Q, Chuang JC, Tran S, Osborne-Lawrence S, et al. (2015): The P7C3 class of neuroprotective compounds exerts antidepressant efficacy in mice by increasing hippocampal neurogenesis. Molecular psychiatry. 20:500–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee AS, De Jesus-Cortes H, Kabir ZD, Knobbe W, Orr M, Burgdorf C, et al. (2016): The Neuropsychiatric Disease-Associated Gene cacna1c Mediates Survival of Young Hippocampal Neurons. eNeuro. 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kemp SW, Szynkaruk M, Stanoulis KN, Wood MD, Liu EH, Willand MP, et al. (2015): Pharmacologic rescue of motor and sensory function by the neuroprotective compound P7C3 following neonatal nerve injury. Neuroscience. 284:202–216. [DOI] [PubMed] [Google Scholar]
- 26.Loris ZB, Pieper AA, Dalton Dietrich W (2017): The neuroprotective compound P7C3-A20 promotes neurogenesis and improves cognitive function after ischemic stroke. Experimental neurology. 290:63–73. [DOI] [PubMed] [Google Scholar]
- 27.Cohen RM, Rezai-Zadeh K, Weitz TM, Rentsendorj A, Gate D, Spivak I, et al. (2013): A transgenic Alzheimer rat with plaques, tau pathology, behavioral impairment, oligomeric abeta, and frank neuronal loss. The Journal of neuroscience : the official journal of the Society for Neuroscience. 33:6245–6256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tsai Y, Lu B, Ljubimov AV, Girman S, Ross-Cisneros FN, Sadun AA, et al. (2014): Ocular changes in TgF344-AD rat model of Alzheimer’s disease. Investigative ophthalmology & visual science. 55:523–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Joo IL, Lai AY, Bazzigaluppi P, Koletar MM, Dorr A, Brown ME, et al. (2017): Early neurovascular dysfunction in a transgenic rat model of Alzheimer’s disease. Scientific reports. 7:46427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rats! Nat Methods, 7 (2010), p. 413. [DOI] [PubMed] [Google Scholar]
- 31.Miller LR, Marks C, Becker JB, Hurn PD, Chen WJ, Woodruff T, et al. (2017): Considering sex as a biological variable in preclinical research. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 31:29–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Miller VM, Rocca WA, Faubion SS (2015): Sex Differences Research, Precision Medicine, and the Future of Women’s Health. Journal of women’s health. 24:969–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Muller-Spahn F (2003): Behavioral disturbances in dementia. Dialogues in clinical neuroscience. 5:49–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lyketsos CG, Olin J (2002): Depression in Alzheimer’s disease: overview and treatment. Biological psychiatry. 52:243–252. [DOI] [PubMed] [Google Scholar]
- 35.Lyketsos CG, Lopez O, Jones B, Fitzpatrick AL, Breitner J, DeKosky S (2002): Prevalence of neuropsychiatric symptoms in dementia and mild cognitive impairment: results from the cardiovascular health study. Jama. 288:1475–1483. [DOI] [PubMed] [Google Scholar]
- 36.Yan HC, Cao X, Das M, Zhu XH, Gao TM (2010): Behavioral animal models of depression. Neuroscience bulletin. 26:327–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Iwatsubo T, Odaka A, Suzuki N, Mizusawa H, Nukina N, Ihara Y (1994): Visualization of A beta 42(43) and A beta 40 in senile plaques with end-specific A beta monoclonals: evidence that an initially deposited species is A beta 42(43). Neuron. 13:45–53. [DOI] [PubMed] [Google Scholar]
- 38.Iqbal K, Alonso Adel C, Chen S, Chohan MO, El-Akkad E, Gong CX, et al. (2005): Tau pathology in Alzheimer disease and other tauopathies. Biochimica et biophysica acta. 1739:198–210. [DOI] [PubMed] [Google Scholar]
- 39.Hartig W, Klein C, Brauer K, Schuppel KF, Arendt T, Bruckner G, et al. (2000): Abnormally phosphorylated protein tau in the cortex of aged individuals of various mammalian orders. Acta neuropathologica. 100:305–312. [DOI] [PubMed] [Google Scholar]
- 40.Shafiei SS, Guerrero-Munoz MJ, Castillo-Carranza DL (2017): Tau Oligomers: Cytotoxicity, Propagation, and Mitochondrial Damage. Frontiers in aging neuroscience. 9:83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McAvoy KM, Sahay A (2017): Targeting Adult Neurogenesis to Optimize Hippocampal Circuits in Aging. Neurotherapeutics : the journal of the American Society for Experimental NeuroTherapeutics. 14:630–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stein LR, Imai S (2014): Specific ablation of Nampt in adult neural stem cells recapitulates their functional defects during aging. The EMBO journal. 33:1321–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Villela D, Schlesinger D, Suemoto CK, Grinberg LT, Rosenberg C (2014): A microdeletion in Alzheimer’s disease disrupts NAMPT gene. Journal of genetics. 93:535–537. [DOI] [PubMed] [Google Scholar]
- 44.Long AN, Owens K, Schlappal AE, Kristian T, Fishman PS, Schuh RA (2015): Effect of nicotinamide mononucleotide on brain mitochondrial respiratory deficits in an Alzheimer’s disease-relevant murine model. BMC neurology. 15:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang X, Hu X, Yang Y, Takata T, Sakurai T (2016): Nicotinamide mononucleotide protects against beta-amyloid oligomer-induced cognitive impairment and neuronal death. Brain research. 1643:1–9. [DOI] [PubMed] [Google Scholar]
- 46.Yao Z, Yang W, Gao Z, Jia P (2017): Nicotinamide mononucleotide inhibits JNK activation to reverse Alzheimer disease. Neuroscience letters. 647:133–140. [DOI] [PubMed] [Google Scholar]
- 47.Kempermann G, Kuhn HG, Gage FH (1997): Genetic influence on neurogenesis in the dentate gyrus of adult mice. Proceedings of the National Academy of Sciences of the United States of America. 94:10409–10414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nilsson M, Perfilieva E, Johansson U, Orwar O, Eriksson PS (1999): Enriched environment increases neurogenesis in the adult rat dentate gyrus and improves spatial memory. Journal of neurobiology. 39:569–578. [DOI] [PubMed] [Google Scholar]
- 49.Johnson-Wood K, Lee M, Motter R, Hu K, Gordon G, Barbour R, et al. (1997): Amyloid precursor protein processing and A beta42 deposition in a transgenic mouse model of Alzheimer disease. Proceedings of the National Academy of Sciences of the United States of America. 94:1550–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Greenberg SG, Davies P (1990): A preparation of Alzheimer paired helical filaments that displays distinct tau proteins by polyacrylamide gel electrophoresis. Proceedings of the National Academy of Sciences of the United States of America. 87:5827–5831. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.