

Abstract
Cardiac intracellular lipid accumulation (steatosis) is a pathophysiological phenomenon observed in starvation and diabetes mellitus. Perilipin 2 (PLIN2) is a lipid droplet (LD)-associated protein expressed in nonadipose tissues, including the heart. To explore the pathophysiological function of myocardial PLIN2, we generated transgenic (Tg) mice by cardiac-specific overexpression of PLIN2. Tg hearts showed accumulation of numerous small LDs associated with mitochondrial chains and high cardiac triacylglycerol (TAG) content [8-fold greater than wild-type (WT) mice]. Despite massive steatosis, cardiac uptake of glucose, fatty acids and VLDL, systolic function, and expression of metabolic genes were comparable in the two genotypes, and no morphological changes were observed by electron microscopy in the Tg hearts. Twenty-four hours of fasting markedly reduced steatosis in Tg hearts, whereas WT mice showed accumulation of LDs. Although activity of adipose triglyceride lipase in heart homogenate was comparable between WT and Tg mice, activity of hormone-sensitive lipase (HSL) was 40–50% less in Tg than WT mice under both feeding and fasting conditions, suggesting interference of PLIN2 with HSL. Mice generated through crossing of PLIN2-Tg mice and HSL-Tg mice showed cardiac-specific HSL overexpression and complete lack of steatosis. The results suggest that cardiac PLIN2 plays an important pathophysiological role in the development of dynamic steatosis and that the latter was prevented by upregulation of intracellular lipases, including HSL.
Keywords: energy metabolism, lipolysis, lipotoxicity, cardiomyopathy
cardiac intracellular lipid accumulation (steatosis) is a physiological phenomenon observed under specific conditions of unbalanced influx and utilization of fatty acids (FAs) (9). Starvation stimulates adipose lipolysis, and the subsequently released FAs are transferred to oxidative tissues, including heart, skeletal muscles, and liver. When FA influx exceeds its oxidation, the excess FAs are esterified to form triacylglycerol (TAG), which is subsequently stored in lipid droplets (LDs). Cardiac steatosis is also observed in morbid conditions such as obesity or diabetes (4, 36, 46). Under these conditions, plasma FA concentration is generally elevated by aberrant adipose lipolysis due to insulin resistance. Consequently, excess FAs are transferred into cardiomyocytes, esterified, and packaged in LDs. However, because cardiac intracellular lipolysis is also stimulated, excess FAs and their metabolites leak from LDs and stimulate the production of reactive oxygen species (ROS) and ceramide, which can cause apoptosis of cardiomyocytes (31, 48).
Recent studies have demonstrated that LDs and steatosis are not necessarily detrimental to the heart. It has been reported that LD accumulation by overexpression of diacylglycerol acyltransferase (DGAT) 1 ameliorates lipotoxic cardiomyopathy (27) and muscle insulin resistance (28). Others reported that lipolysis of TAG in LD provides FA ligands for the activation of peroxisome proliferator-activated receptor (PPAR) α, which is a master regulator of FA oxidation in the heart (15). Bailey et al. (2) have reported the antioxidant role of LDs, which limit ROS production from peroxidation of polyunsaturated FAs in neuroblasts of drosophila. Thus, LDs are thought to comprise a multifunctional organelle that controls energy metabolism, signaling molecules, and intracellular lipotoxicity.
LDs comprise a dynamic organelle whose metabolism is regulated precisely by cytosolic lipases and LD-associated molecules (22). In cardiomyocytes, breakdown of TAG in LDs is catalyzed by cytosolic lipases, e.g., adipose triglyceride lipase (ATGL), hormone-sensitive lipase (HSL), and, potentially, monoacylglycerol lipase (MGL). ATGL catalyzes the first step of TAG hydrolysis, which is activated by the coactivator comparative gene identification-58 (CGI-58), to generate diacylglycerol (DAG) and FA (25). HSL then hydrolyzes DAG to generate monoacylglycerol (MAG) and FA. Although HSL possesses significant activity against TAG, ATGL has been shown to be rate limiting for the first step of TAG hydrolysis (14), and HSL has been shown to play a pivotal role in DAG hydrolysis in adipose tissue and heart (16). The FAs released from lipolytic reactions are transferred to mitochondria, which are attached to LDs, and oxidized for energy production.
In nonadipose tissues, LDs are coated with specific members of the perilipin (PLIN) family (42). PLIN2 (ADRP, ADFP, or adipophilin) and PLIN3 (Tip47) are ubiquitously expressed in many tissues and cells, and PLIN5 (LSDP5, MLDP, or OXPAT) is expressed in cardiomyocytes, skeletal myocytes, and brown adipocytes (42, 53). PLIN5 is a dominant LD-associated protein in cardiomyocytes and has been shown to induce LD accumulation via the formation of a lipolytic barrier, which protects cardiomyocytes from FA-overload (24, 35). PLIN5 also plays a role in LD-mitochondria association (51) and CGI-58-ATGL interaction (13, 34). Thus PLIN5 seems to play a pivotal role in the control of TAG hydrolysis and FA oxidation in cardiomyocytes.
PLIN2 was discovered in LDs from preadipocytes and is replaced with PLIN1 upon differentiation of preadipocytes into mature adipocytes (3). Overexpression of PLIN2 stimulates LD accumulation in cultured fibroblasts (19) and human embryonic kidney cells by inhibiting the association of ATGL to LDs (26). In pancreatic β-cells, PLIN2 has been shown to play a role in LD formation and FA-induced insulin secretion (10). Studies using PLIN2-knockout (KO) mice have demonstrated that deletion of PLIN2 ameliorates hepatic steatosis (6, 30) and macrophage foam cell formation (33), whereas overproduction of PLIN2 induces severe hepatic steatosis (38). In mouse heart, we previously reported PLIN2 upregulation during fasting-induced steatosis (40, 41), whereas Trent et al. (43) reported a modest increase in PLIN2 protein expression with fasting. Thus PLIN2 appears to play an important role in LD formation in PLIN2-expressing tissues and cells, although the precise function of PLIN2, particularly in the heart, is not fully understood.
The present study was designed to determine the pathophysiological function of PLIN2 in the heart. For this purpose, we generated transgenic mice with cardiac-specific overexpression of PLIN2 and analyzed these mice under both feeding and fasting conditions. The results showed that cardiac PLIN2 plays an important role in the generation of dynamic steatosis and that the latter was readily resolved by HSL.
MATERIALS AND METHODS
Generation of heart-specific PLIN2-overexpressing mice.
The enhanced green fluorescent protein (EGFP) and mouse PLIN2 cDNA were inserted under the control of the myosin heavy chain (MHC)-α promoter kindly provided by Dr. J. Robbins (University of Cincinnati Medical Center, Cincinnati, OH) (Fig. 1A) (37). The transgene was injected into embryos derived from C57BL/6 × DBA2 background mice in the Laboratory Animal Resources at the University of Fukui. Two founders (AD17 and AD19) were identified by PCR screening, followed by Southern blot analysis. The founders were back-crossed to C57BL/6 mice for six to seven generations. The AD17 line was used primarily in these studies; the results were confirmed with the AD19 line. Both male and female mice were used unless otherwise noted. Cardiac-specific HSL-overexpressing (HSL-Tg) mice, which harbor rat HSL cDNA, were generated and used as reported previously (45). The homozygous ATGL-KO mice were generated from crossing heterozygous KO mice purchased from The Jackson Laboratory (14). All procedures were conducted in accordance with the Regulations for Animal Research at the University of Fukui and approved by the Animal Research Committee, the University of Fukui.
Fig. 1.
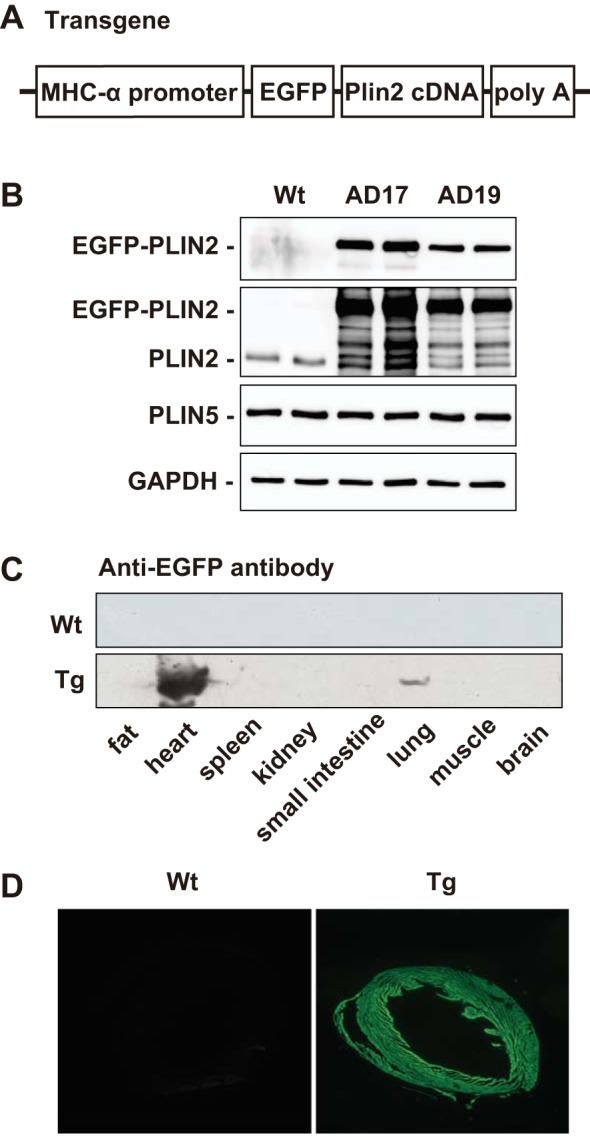
Generation and characterization of heart-specific perilipin 2 (PLIN2)-overexpressing mice. A: the transgene consists of the promoter of mouse myosin heavy chain (MHC)-α, cDNA of enhanced green fluorescent protein (EGFP), mouse PLIN2 cDNA, and a fragment containing human growth hormone poly(A). B: Western blot analysis for EGFP, PLIN2, PLIN5, and GAPDH. Tissue homogenates of ventricles of wild-type (WT), AD17-transgenic (Tg), and AD19-Tg mice were subjected to SDS-PAGE, blotted, and probed with anti-EGFP, -PLIN2, -PLIN5, and -GAPDH antibodies. C: tissue expression of EGFP-PLIN2 in WT and AD17-Tg mice assessed by Western blotting using anti-EGFP antibody. Tg mice show robust heart-specific PLIN2 expression. D: representative fluorescent micrographs showing EGFP-PLIN2 expression in cardiac ventricles of AD17-Tg mice. Magnification, ×40.
Animal studies.
Heterozygous EGFP-PLIN2-transgenic (PLIN2-Tg) mice and wild-type (WT) littermates aged 5–8 mo were used in all experiments. The mice were maintained on a chow diet (MF; Oriental Yeast) with a 12:12-h dark-light cycle and housed in individual cages 2 wk before the experiments. The mice were fed ad libitum or fasted for 24 h and used for the experiments. For some experiments, heterozygous PLIN2-Tg mice were crossed with heterozygous HSL-Tg mice to generate PLIN2-HSL double-Tg mice. The hearts of the latter group were analyzed and the results compared with those of their littermates.
Blood chemistry.
Plasma concentrations of FFAs, TAG, and total cholesterol (T-Chol) were measured by commercially available kits (WAKO). Plasma glucose concentration was measured using Freestyle (NIPRO).
Western blot analysis.
Immunoblot analysis was performed using specific antibodies against mouse PLIN2 (ab52356; Abcam), PLIN3 (3881; ProSci) (21), PLIN5 (PAB12542; Abnova) (35), EGFP (GF090R; Nacalai), HSL, phosphorylated HSLs, ATGL (4107, 4139, 4137, 4126, and 2439; Cell Signaling Technology), α-tubulin (ab18251; Abcam), β-actin (A5441; Sigma-Aldrich), and GAPDH (MAB374; Chemicon), as described previously (20), and analyzed by FluorChem IS-8000 (Alpha Innotech). Stable expression of GAPDH and α-tubulin upon fasting/feeding was confirmed by a total protein imaging system (Bio-Rad), and those proteins were used as a loading control. A specific antibody against rat HSL was used for the detection of transgenic rat HSL (23).
Tissue lipid content.
The heart was perfused with 3 ml of PBS from the left ventricle and excised. Thirty to 40 milligrams of the tissues were homogenized in 20 vol of PBS, and lipids were extracted with 20 vol of chloroform-methanol (2:1). After centrifugation, the chloroform phase was transferred into glass tubes, dried, and dissolved in isopropyl alcohol at 70°C for 1 h. TAG, T-Chol, and FFAs were measured using the respective assay kits (WAKO) with lipid standards, which were treated in parallel with the tissue samples from the extraction step.
Liquid chromatography-mass spectrometry.
Cardiac lipids were extracted from WT and Tg mice (n = 3), as described above. The extracted lipids were dissolved and subjected to liquid chromatography-mass spectrometry (LC-MS) analysis performed with an Agilent 1200 system (Agilent Technologies) coupled to a Finnigan LTQ Orbitrap XL (Thermo Fisher Scientific), as described previously (18). The resultant data were analyzed using LipidSearch (Mitsui Knowledge Industry) and SIEVE (Thermo Fisher Scientific) software. References for each annotated compound were searched for the KNApSAcK and KEGG database (1).
Microscopy.
Electron microscopy was performed as described previously using a transmission electron microscope (Hitachi H-7500) (45). For confocal microscopy, tissues were fixed with 4% paraformaldehyde/PBS for 1 h and embedded. The sections were stained with PLIN2-specific antibody, heat shock cognate protein of 70 kDa (hsc70; NB120-2788; Novus) and fluorescent secondary antibodies (ALEXA), or LipidTOX reagent (Thermo Fisher Scientific) for lipid staining. The sections were analyzed with a confocal microscope system (Leica TCS Sp2).
Echocardiography.
Cardiac function was studied by echocardiography in awake mice using ultrasonography equipped with a 13-MHz linear transducer (ALOKA), as previously described (41).
Gene expression analysis.
Total RNA was extracted from cardiac ventricles using TRIzol reagent (Invitrogen) and reverse-transcribed using a Quantitect reverse transcription kit (Qiagen). The target genes were amplified and analyzed in triplicate using TaqMan probes (Applied Biosystems), as described previously (45).
Cardiac uptake of energy sources.
Tissue glucose uptake was analyzed by injecting d-[14C]deoxyglucose (GE Healthcare) via the tail vein, as described previously (41). Briefly, the mouse was injected with 0.1 μCi/g body wt of d-[14C]deoxyglucose and euthanized 40 min later. Plasma glucose was measured, and cardiac ventricles and liver were excised. The tissues were then dissolved in Solvable (Perkin-Elmer), and radioactivity was measured using a liquid scintillation counter. Tissue glucose uptake was calculated from tissue radioactivity and plasma-specific activity of the tracer.
Tissue FA uptake was analyzed by injecting [125I]β-methyl iodophenyl pentadecanoic acid ([125I]BMIPP; Nihon Medi-Physics) (41). Briefly, 0.1 μCi/g body wt of [125I]BMIPP was injected via the tail vein, and mice were euthanzied 20 min later. Cardiac ventricles and liver were excised, tissue radioactivity and plasma FA concentration were measured, and tissue FA uptake was calculated using the plasma-specific activity of the tracer.
For tissue VLDL uptake, VLDL was obtained by ultracentrifugation from rabbits fed a high-cholesterol diet (0.5% wt/wt). The VLDL was radiolabeled with [125I] (GE Healthcare) using IODOBEASE (Pierce) and purified as described previously (41). The mice were injected with 120,000 counts·min−1·g body wt−1 [125I]VLDL via the tail vein and euthanized 20 min later. Cardiac ventricles and livers were collected, and VLDL uptake was calculated from tissue radioactivity and plasma-specific activity of the tracer.
Lipase assays.
Hearts were homogenized in 20 mM Tris and 1 mM EDTA, pH 7.4, containing 255 mM sucrose, 1 μM leupeptin, and 0.1 μM okadaic acid and centrifuged, and the supernatant was used for lipase assays, as described previously (41).
ATGL activity was measured using a specific ATGL inhibitor atglistatin (Cayman Chemical) (29). Atglistatin was dissolved in DMSO at a concentration of 10 mM, and 5 μl was added in the reaction mixture. Ninety-five microliters of the supernatant was incubated at 37°C for 30 min in 200 μl of a reaction mixture containing 105 μM [3H]trioleoylglycerol (99.4 μCi/µmol), 23.7 μM lecithin, and 5 mM sodium taurocholate in 100 mM potassium phosphate buffer (pH 7.4) in the absence (DMSO only) or presence of 250 μM atglistatin. ATGL activity was determined by subtracting the activity with atglistatin from that with DMSO only. Tissue homogenates of WT adipose tissue and hearts of homozygous ATGL-KO mice were included in the assay as positive and negative controls, respectively.
HSL activity was determined as neutral cholesteryl ester hydrolase (NCEH) activity using 100 μl of the heart samples and a micellar substrate, including cholesteryl-[14C]oleate, as described previously (23). Because hearts of HSL-KO mice have virtually no NCEH activity (41), cardiac NCEH activity represents the activity of HSL.
For lipoprotein lipase (LPL) assay, 60 μl of the supernatant was incubated at 37°C for 30 min in 200 μl of a reaction mixture containing 105 μM [3H]trioleoylglycerol (99.4 μCi/µmol), 23.7 μM lecithin, and 4.2% of heated rat serum in 100 mM potassium phosphate buffer (pH 8.0) in the absence or presence of 1 M NaCl. LPL activity was determined by subtracting the activity in 1 M NaCl from that in the absence of 1 M NaCl (5).
Transfection experiments.
Chinese hamster ovary (CHO)-K1 cells were transfected with the expression plasmids pDsRed, pDsRed-HSL, pEGFPC1, and pEGFPC1-PLIN2, as described previously (44). The cells were harvested 60 h after the removal of the transfection mixture, sonicated, and centrifuged, and HSL activity of the supernatant was measured as NCEH activity, as described above. The expression of each protein was analyzed by Western blotting.
Statistical analysis.
All values are expressed as means ± SE. Significance was determined by ANOVA, followed by Fisher’s protected least significant difference. P < 0.05 was considered significant, and the differences between genotypes or conditions are indicated in the figures unless otherwise noted.
RESULTS
Characterization of heart-specific PLIN2-overexpresssing mice.
Both of the established Tg lines (AD17 and AD19) showed robust expression of EGFP-PLIN2 fusion protein in a heart-specific manner (Fig. 1, B and C), and expression of the PLIN2 fusion protein was detected throughout the heart (Fig. 1D). EGFP-PLIN2 was also detected in the main trunk of the pulmonary arteries, which showed a faint band in lung tissues on Western blotting (Fig. 1C). PLIN2 overexpression did not affect the expression of PLIN5 in the hearts of Tg mice (Fig. 1B). The Tg heart appeared normal macroscopically, and heart weight was comparable between WT and Tg mice. Light microscopic examination of Oil Red O-stained Tg myocardium showed marked lipid accumulation (Fig. 2A). Electron microscopy showed accumulation of numerous small LDs in Tg hearts compared with virtually no LDs in WT hearts. It was noteworthy that LDs in Tg hearts were similar in size to mitochondria, smooth and round-shaped, and were localized within the “mitochondrial chain” (Fig. 2A). Cardiac TAG content was eight- to ninefold higher in both Tg lines than in WT hearts (Fig. 2B). The LC-MS analysis detected ∼3,000 compounds from WT or Tg hearts. A comparison analysis using LipidSeach software showed exclusive increases in TAG species (21 species, 3.5- to 10-fold, P < 0.05) in the hearts of Tg mice compared with WT mice. In addition, SIEVE software identified DAG species, which were significantly increased in Tg hearts compared with WT hearts (2 species, 6.3- and 6.8-fold, P < 0.01), whereas ceramide species were comparable in both genotypes. Despite the massive steatosis, electron microscopy showed no abnormal findings in the mitochondria.
Fig. 2.

Cardiac steatosis in PLIN2-Tg mice. A: representative light micrographs with Oil Red O staining (left) and electron micrographs (middle and right) of the left ventricles of WT and AD17-Tg mice. Arrowheads, lipid droplets (LDs). Scale bars, 46 (left), 6.7 (middle), or 1 μm (right). B: cardiac triacylglycerol (TAG) content in WT, AD17-Tg, and AD19-Tg mice. The hearts were perfused with 3 ml of PBS from the left ventricle and excised. Tissues were homogenized in PBS, and lipids were extracted with chloroform-methanol (2:1), dried, and resolved with isopropyl alcohol and measured with a TAG assay kit. Values are means ± SE of 4–5 mice in each group. **P < 0.01 vs. WT mice.
Effects of fasting on PLIN2-induced steatosis.
To test whether fasting had an impact on PLIN2-induced steatosis, mice were fasted for 24 h, and hearts were analyzed. As shown in Table 1, plasma glucose concentration was markedly lower after 24 h of fasting, whereas FFA concentration was ≥1.4 mM/l in both genotypes. Plasma TAG and T-Chol concentration were not altered significantly. Under these conditions (24 h of fasting), electron microscopy showed accumulation of small LDs in WT hearts, which were smaller in size than mitochondria and associated with the mitochondrial chain (Fig. 3A). In contrast, the fasted Tg hearts displayed smaller and “shrunken” LDs compared with the fed Tg hearts. Consistent with the microscopic findings, fasting tended to increase cardiac TAG content in WT hearts, whereas it decreased the contents to 45% in Tg hearts (Fig. 3B). Cardiac FFA content was slightly higher in Tg hearts compared with WT hearts in the fed state, and T-Chol content was comparable in both genotypes; neither was affected by fasting (Fig. 3B). Despite the lower LD and TAG contents in the hearts of fasted Tg mice, EGFP-PLIN2 expression was comparable in fed and fasted animals by Western blot analysis (Fig. 3C). The cardiac expression of endogenous PLIN2 was also comparable in the fed and fasted WT mice. In addition, the expression levels of PLIN3 and PLIN5 were not altered with fasting or with EGFP-PLIN2 overexpression (Fig. 3C). Confocal microscopy showed spotty expression of PLIN2 in cardiomyocytes of fed WT mice, where it showed ring-shaped staining in those of fasted WT mice (Fig. 3D). In cardiomyocytes of fed Tg mice, PLIN2 was detected as much larger ring-shaped staining, whereas they became smaller rings in those of fasted Tg mice. Recently, PLIN2 and -3 have been shown to be substrates of chaperon-mediated autophagy (CMA), which regulates LD metabolism in hepatocytes (21). To test the involvement of CMA in the decrease in LDs upon fasting in Tg hearts, colocalization of a chaperone protein, hsc70, with PLIN2 was investigated using specific antibodies and confocal microscopy. However, hsc70 was not localized around LDs in WT or Tg hearts in either feeding or fasting conditions (data not shown).
Table 1.
Body weight and plasma parameters of WT and Tg mice in feeding or 24-h-fasting condition
| Feeding |
Fasting |
|||
|---|---|---|---|---|
| WT | Tg | WT | Tg | |
| n (Males/females) | 5/5 | 5/5 | 5/5 | 5/5 |
| Body weight | ||||
| Males | 34.8 ± 1.6 | 31.2 ± 1.2 | 33.5 ± 2.1 | 31.3 ± 0.9 |
| Females | 23.8 ± 0.6 | 23.8 ± 0.5 | 21.5 ± 0.6 | 21.9 ± 0.8 |
| Glucose, mg/dl | 171 ± 10 | 179 ± 8 | 87 ± 4† | 90 ± 4† |
| TAG, mg/dl | 59 ± 6 | 54 ± 3 | 67 ± 4 | 71 ± 6 |
| T-Chol, mg/dl | 72 ± 5 | 70 ± 4 | 60 ± 7 | 66 ± 6 |
| FFA, mmol/l | 0.46 ± 0.04 | 0.58 ± 0.06 | 1.48 ± 0.11† | 1.43 ± 0.08† |
Values are means ± SE. WT, wild-type; Tg, transgenic; TAG, triacylglycerol; T-Chol, total cholesterol; FFA, free fatty acids.
P < 0.01 between feeding conditions.
Fig. 3.

Effect of fasting on PLIN2-induced cardiac steatosis. A: representative electron micrographs showing LD accumulation in the left ventricles of WT and Tg mice under feeding and 24-h-fasting conditions. Note the accumulation of small LDs in WT mice (arrowheads) after 24 h of fasting (top right) compared with smaller and “shrunken” LDs after 24 h of fasting in Tg hearts (bottom). Scale bars, 2 μm. B: cardiac contents of TAG, free fatty acid (FFA), and total cholesterol (T-Chol) in WT and Tg mice under feeding and 24-h-fasting conditions. Values are means ± SE of 4–6 mice in each group. *P < 0.05 and **P < 0.01 vs. WT mice; †P < 0.05 vs. feeding condition. C: Western blot analysis of PLIN2, PLIN3, and PLIN5 in ventricles of WT and Tg mice under feeding and 24-h-fasting conditions. Anti-PLIN2, -PLIN3, -PLIN5, and -α-tubulin antibodies were used in these experiments. Six mice per group were analyzed, and representative blots are presented. Graphs: densitometric analysis of the protein expression normalized by α-tubulin. Values are means ± SE of 4 mice/group. D: PLIN2 expression in cardiomyocytes of WT and Tg mice under feeding and 24-h-fasting conditions. The tissue sections were stained with anti-PLIN2 antibody and analyzed by confocal microscopy. Four mice in each group were studied, and representative images are shown. Scale bars, 10 μm.
Thus the results indicate that PLIN2-induced LDs were sensitive to lipolytic stimuli upon fasting, which resulted in the reduction of LD accumulation despite the high concentrations of circulating plasma FFAs.
We also analyzed cardiac function with echocardiography. As shown in Table 2, Tg hearts displayed comparable systolic function indicated by fractional shortening, compared with WT hearts, under both feeding and fasting conditions. Other cardiac parameters were comparable in the two genotypes despite the massive steatosis in Tg hearts.
Table 2.
Echocardiographic parameters of WT and Tg mice in feeding or 24-h-fasting condition
| Feeding |
Fasting |
|||
|---|---|---|---|---|
| WT | Tg | WT | Tg | |
| HR, beats/min | 659 ± 9 | 660 ± 10 | 603 ± 11† | 608 ± 12† |
| FS, % | 47.0 ± 1.4 | 46.4 ± 1.4 | 45.4 ± 1.7 | 46.7 ± 1.8 |
| LVIDd, mm | 2.94 ± 0.12 | 3.03 ± 0.12 | 2.94 ± 0.13 | 3.14 ± 0.12 |
| LVIDs, mm | 1.56 ± 0.09 | 1.59 ± 0.05 | 1.63 ± 0.10 | 1.70 ± 0.09 |
| IVS, mm | 0.78 ± 0.03 | 0.81 ± 0.03 | 0.76 ± 0.02 | 0.74 ± 0.02 |
| LVPW, mm | 0.71 ± 0.04 | 0.73 ± 0.04 | 0.69 ± 0.04 | 0.67 ± 0.02 |
Values are means ± SE of 9 male mice. HR, heart rate; FS, fractional shortening; LVIDd, left ventricular internal dimension in diastole; LVIDs, left ventricular internal dimension in systole; IVS, interventricular septum; LVPW, left ventricular posterior wall.
P < 0.01 between feeding conditions.
Cardiac gene expression.
Cardiac gene expression was analyzed to explore the impact of PLIN2 overexpression on the expression of genes related to energy metabolism and cardiac stress. As shown in Fig. 4, the hearts of Tg mice showed robust expression of PLIN2 mRNA compared with WT mice under the feeding condition. However, it was markedly downregulated by fasting, whereas PLIN2 was upregulated with fasting in the hearts of WT mice. Fasting also increased PLIN5 mRNA expression in both genotypes, although the change did not reach statistical significance in Tg mice. The mRNA expression levels of HSL, ATGL, and DGAT 2 (which catalyzes the final step of TAG synthesis) were increased with fasting in the hearts of both genotypes. Fasting also upregulated the expression of genes relevant to FA metabolism, including CD36, HMG-CoA synthase 2, and uncoupling protein 2 in both genotypes. The expression of genes related to cardiac stress, such as natriuretic peptide type A and B, tumor necrosis factor-α, and myosin heavy chain-β, were not altered in cardiac tissues of Tg mice compared with WT mice in both feeding and fasting conditions. Thus the gene expression profile indicated that PLIN2-induced steatosis does not seem to provoke intracellular lipotoxicity.
Fig. 4.

Cardiac expression of the genes related to LD metabolism (top), energy metabolism (middle), and cardiac stress (bottom). Total RNA was extracted from ventricles of WT and Tg mice under feeding and 24-h-fasting conditions. The mRNA expression levels were determined by RT-quantitative PCR and normalized to GAPDH. Values are means ± SE of 5 mice/group. *P < 0.05 and **P < 0.01 between genotypes; †P < 0.05 and ††P < 0.01 between conditions. Lipe, hormone-sensitive lipase; Pnpla2, adipose triglyceride lipase; Slc2a4, glucose transporter 4; Ppp1r3c, protein phosphatase 1; Nppa, natriuretic peptide type A; Nppb, natriuretic peptide type B; Tnf, tumor necrosis factor-α; Myh6, myosin heavy chain-α; Myh7, myosin heavy chain-β.
Cardiac uptake of energy sources.
To explore how PLIN2 overexpression affects the uptake of major energy sources for the heart, radioactive tracers of glucose, FAs, and VLDL were injected into the mice, and the tissue uptake of each energy source was analyzed. As shown in Fig. 5A, cardiac glucose uptake was comparable in WT and Tg mice under both feeding and fasting conditions, whereas it was decreased ∼95% with fasting in both genotypes. Hepatic glucose uptake was relatively stable under fasting, reflecting the differential expression and regulation of glucose transporters (GLUT) in the heart (GLUT4) and liver (GLUT2). In contrast to glucose uptake, cardiac FA uptake was approximately twofold higher under fasting in both genotypes, although the change did not reach statistical significance in Tg mice, and it was comparable in the two genotypes under both feeding and fasting conditions (Fig. 5B). Cardiac VLDL uptake was also comparable between the two genotypes under both feeding and fasting conditions (Fig. 5C). These results indicate that overexpression of PLIN2 in the myocardium induces massive LD accumulation without affecting the uptake of major energy sources.
Fig. 5.
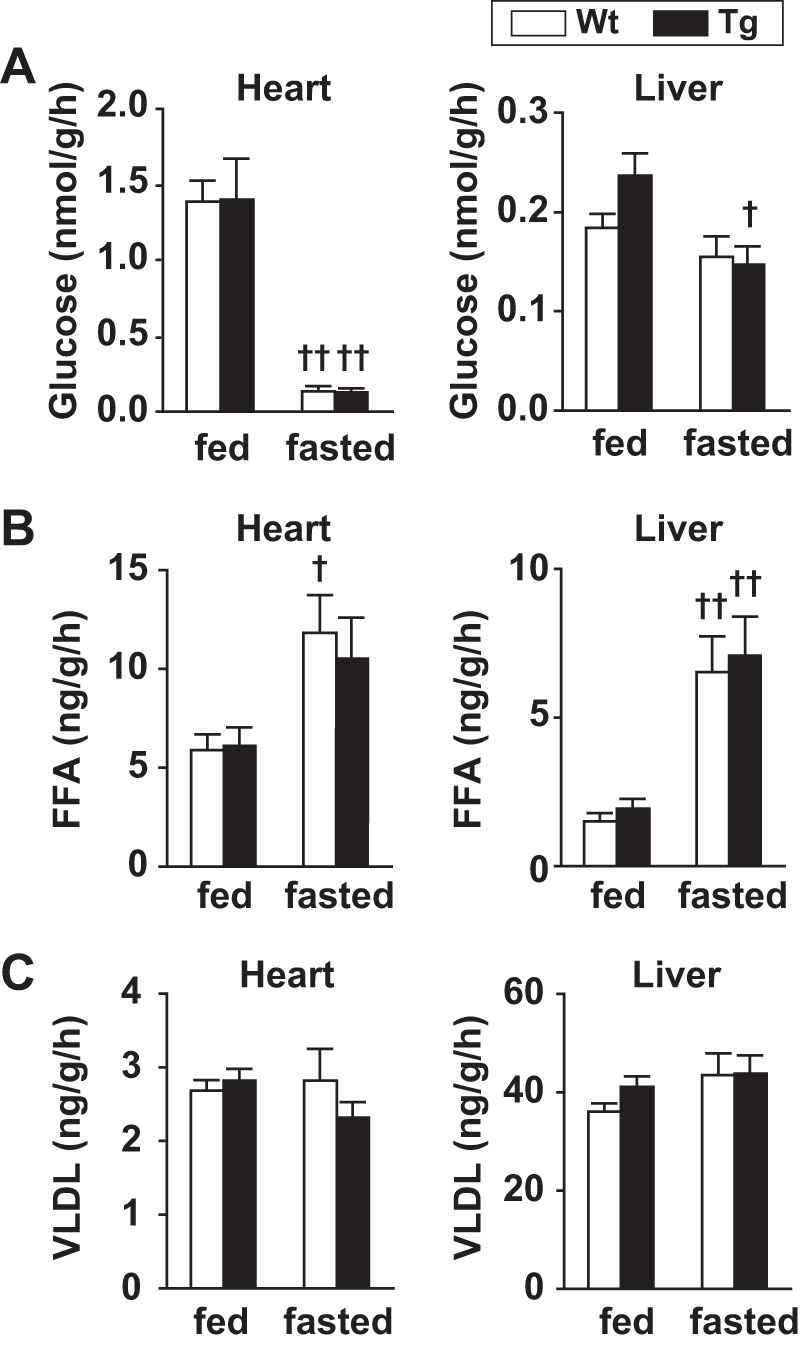
Cardiac uptake of energy sources. A: tissue glucose uptake. Mice were injected with d-[14C]deoxyglucose, and tissue glucose uptake was calculated from tissue radioactivity and plasma-specific activity of the tracer. Values are means ± SE of 5–6 mice/group. †P < 0.05 and ††P < 0.01 between conditions. B: tissue FFA uptake. [125I]β-methyl iodophenyl pentadecanoic acid ([125I]BMIPP) was injected, and tissue FFA uptake was calculated from tissue radioactivity and plasma-specific activity of the tracer. Values are means ± SE of 5–6 mice/group. †P < 0.05 and ††P < 0.01 between conditions. C: tissue VLDL uptake. VLDL obtained from rabbits fed high-cholesterol diet was labeled with [125I] and purified. Mice were injected with [125I]VLDL, and tissue VLDL uptake was calculated from tissue radioactivity and plasma-specific activity of the tracer. Values are means ± SE of 5–8 mice/group.
Cardiac lipase activities.
We then analyzed cardiac hydrolytic activities of TAG and cholesteryl ester (CE) to assess the function of lipases and explain the mechanism of LD accumulation. As shown in Fig. 6A, the activity of ATGL was markedly increased with fasting, although it was comparable between WT and Tg mice in both feeding and fasting conditions. Cardiac HSL activity was 50 (feeding condition) and 40% (fasting condition) lower in Tg compared with WT hearts. Cardiac LPL activity was not significantly altered in Tg mice under either condition. The decrease in HSL activity in Tg hearts suggests an inhibitory effect for PLIN2 on HSL function.
Fig. 6.
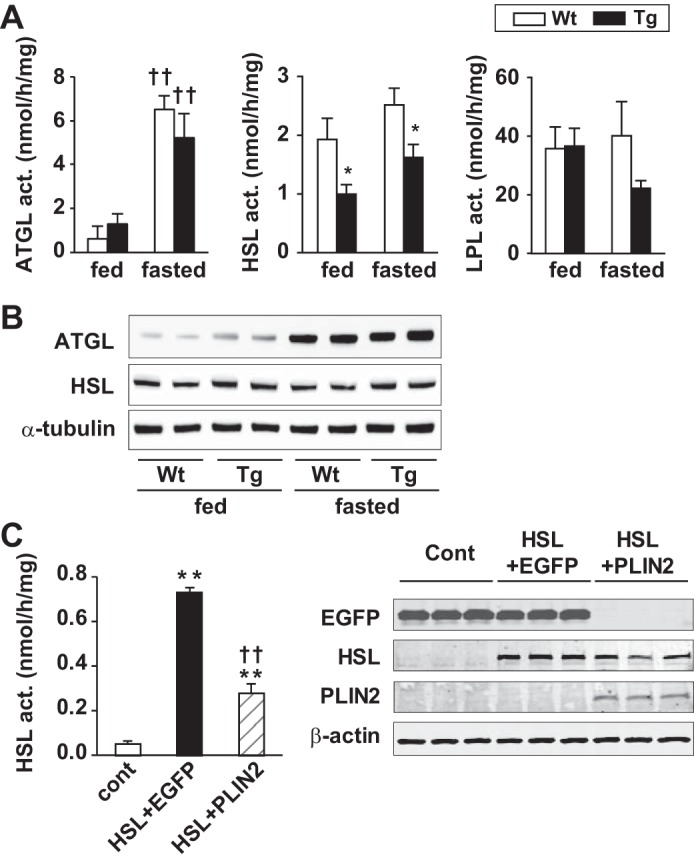
A: cardiac activities of adipose triglyceride lipase (ATGL), hormone-sensitive lipase (HSL), and lipoprotein lipase (LPL). Tissue extracts of ventricles of fed or 24-h-fasted mice were subjected to the relevant lipase assay using radiolabeled micellar substrates. Values are means ± SE of 4–6 mice/group, and the graphs represent 2–3 independent experiments. *P < 0.05 between genotypes; ††P < 0.01 vs. fed group. B: Western blot analysis of ATGL, HSL, and α-tubulin in mouse hearts. Tissue homogenates of ventricles of WT and PLIN2-Tg mice under feeding and 24-h-fasting conditions were subjected to SDS-PAGE, blotted, and probed with anti-ATGL, -HSL, and -α-tubulin antibodies. C: effects of PLIN2 overexpression on HSL activity in CHO-K1 cells (left). The cells were transfected in triplicate with pDsRed and pEGFPC1 [control (cont)], pDsRed-HSL and pEGFPC1 (HSL + EGFP), or pDsRed-HSL and pEGFPC1-PLIN2 (HSL + PLIN2). Cell extracts were subjected to HSL activity assay using radiolabeled micellar substrates. Graph represents 3 independent transfection experiments. Values are means ± SE. **P < 0.01 vs. cont; ††P < 0.01 vs. HSL + EGFP. Right: Western blot analysis of EGFP, HSL, PLIN2, and β-actin in the transfected cells. Cell extracts were subjected to SDS-PAGE, blotted, and probed with antibodies against each protein.
Next, we examined the potential mechanism of decreased lipase activity. We analyzed the protein expression of ATGL and HSL by Western blotting. As shown in Fig. 6B, cardiac ATGL protein expression tended to increase in Tg mice compared with WT mice under feeding condition and markedly increased with fasting in both genotypes. HSL protein expression was not altered in Tg hearts compared with WT hearts in both feeding and fasting conditions. The specific antibodies against phosphorylated HSL at 423, 563, and 565 showed similar expression of HSL in both genotypes in fed and fasted animals (data not shown). To confirm the inhibitory effect of PLIN2 on HSL activity in vitro, CHO-K1 cells were transfected with expression vectors with DsRed-HSL and/or EGFP-PLIN2, and cellular HSL activity was measured. As shown in Fig. 6C, PLIN2 expression significantly (∼60%) decreased HSL activity in CHO-K1 cells, suggesting that PLIN2 can potentially interfere with HSL, which hydrolyzes LD-like micellar substrate.
Effect of HSL overexpression on PLIN2-induced cardiac steatosis.
Based on the above results, we hypothesized that an increase in HSL activity can normalize PLIN2-induced steatosis in the Tg mice. To address this, we crossed PLIN2-Tg mice with HSL-Tg mice (which overexpress rat HSL in a cardiac-specific manner; see Ref. 45). As shown in Fig. 7A, the double-Tg mice displayed robust cardiac protein expression of both EGFP-PLIN2 and rat HSL. Electron microscopy of hearts of double-Tg mice demonstrated that overexpression of HSL completely eliminated LDs, which were seen in PLIN2-Tg mice (Fig. 7B). Compatible with the electron microscopic findings, cardiac TAG content in the double-Tg hearts was as low as that of WT hearts (Fig. 7C). Interestingly, cardiac EGFP-PLIN2 expression remained high in double-Tg mice compared with WT mice (Fig. 7A). Confocal microscopy revealed dense deposition of PLIN2 in cardiomyocytes of double-Tg mice despite the disappearance of the lipid core (Fig. 7D).
Fig. 7.

Effects of HSL overexpression on PLIN2-induced cardiac steatosis in HSL-PLIN2 double-Tg mice. A: Western blot analysis of EGFP-PLIN2, rat HSL, and GAPDH using specific antibodies against each protein. Representative data of 4–5 mice/group. PLIN2, PLIN2-Tg mice; HSL, HSL-Tg mice; PLIN2 + HSL, PLIN2-HSL double-Tg mice. B: electron micrographs of the left ventricles. Arrowheads, LDs. Scale bars, 6.7 (top) or 1 μm (bottom). C: cardiac TAG content in WT and Tg mice. Tissue lipids were extracted with chloroform-methanol, and TAG contents were measured using a TAG assay kit. Values are means ± SE of 4–5 mice/group. **P < 0.01 vs. WT mice. D: PLIN2 expression and LD accumulation in left ventricles from WT, PLIN2-Tg, and PLIN2-HSL double-Tg mice. The tissue sections were stained with anti-PLIN2 antibody (top) or LipidTOX (bottom) and analyzed by confocal microscopy. At the bottom are merged images of those focused on LDs (red) and EGFP (green). Four mice in each group were studied, and representative micrographs are presented. Scale bars, 5 μm.
These results suggest that overexpression of HSL overcomes the steatotic propensity of PLIN2 and support the hypothesis that PLIN2 induces dynamic steatosis in which LDs are readily hydrolyzed by HSL in response to lipolytic stimuli, e.g., fasting.
DISCUSSION
In the present study, we demonstrate that cardiac overexpression of PLIN2 provoked severe and dynamic steatosis, which was reduced by fasting and completely prevented by overexpression of HSL. Based on the massive accumulation and presence of LD throughout the entire heart and the superphysiological cardiac TAG content (∼9-fold higher than WT), we also examined whether steatosis had a negative effect on the heart. Surprisingly, PLIN2-Tg mice showed normal cardiac function, gene expression, and unaltered uptake of energy sources under both feeding and fasting conditions. These features led us to interpret PLIN2-induced steatosis to mimic “physiological” steatosis, as observed under fasting conditions in WT mice, despite the extreme level of TAG content (32).
To date, many mouse models of lipotoxic cardiomyopathy have been reported. For instance, streptozotocin-induced diabetic mice or Zucker diabetic fatty rats (45, 54), cardiac-specific PPARα-overexpressing mice (11), or long-chain acyl-CoA synthase-Tg mice (8) developed cardiac dysfunction with steatosis, and Tg mice with overexpression of fatty acid transport protein 1 developed diastolic dysfunction with increased cardiac uptake of FAs (7). These mouse models share features of cardiac stress, including increased FA oxidation, mitochondrial overload, and ceramide accumulation in cardiomyocytes. In contrast to these models, the heart of PLIN2-Tg mice displays none of these cardiotoxic features, supporting the reports showing that PLIN2 functions in partitioning toxic lipids and their metabolites in LDs (38, 47).
Although there are limitations to comparing PLIN2-Tg and PLIN5-Tg mice based on published literature rather than on direct experimental comparisons, PLIN2-induced steatosis appears to be less harmful compared with that induced by PLIN5. PLIN5-Tg mice display decreased FA oxidation, downregulation of PPARα-target genes, increased cardiac glucose uptake, and enhanced glucose clearance (34, 35, 50). In contrast, PLIN2-Tg mice showed normal uptake of energy sources and gene expression profile under both feeding and fasting states compared with WT mice. In addition, the mitochondria appeared normal in PLIN2-Tg mice in contrast to PLIN5-Tg mice (50). These differences between the two Tg models might be related to the composition of LD-associated molecules; LDs in PLIN2-Tg hearts are exclusively rich in PLIN2, whereas LDs in PLIN5-Tg hearts are rich in many LD-associated proteins, such as PLIN-2, -4, and -5, ATGL, and CGI-58 compared with WT hearts (34, 35, 50). In addition, Wang et al. (49) reported that HSL is localized within the cytosol in PLIN2-overexpressing CHO-K1 cells, whereas HSL localizes in LDs in the presence of PLIN5 overexpression. Thus the distinct function of each PLIN might contribute to the differential phenotype of each PLIN-Tg mouse.
One unexpected finding was the shrinkage of LDs in the hearts of fasting PLIN2-Tg mice, in contrast to WT mice, which showed accumulation of LDs during fasting. Fasting has double-edged effects on LD accumulation; high plasma levels of FFAs and increased FA influx can stimulate LD formation, whereas activated cytosolic lipases can hydrolyze TAG in LDs. Our results suggest that under fasting, increased lipolysis with ATGL induction surpasses LD formation in PLIN2-enriched LDs in the Tg hearts, in contrast to the WT hearts in which LDs are relatively rich in PLIN5 and accumulate upon fasting. The results are consistent with the previous findings that PLIN2-enriched LDs are more susceptible to lipolysis by endogenous lipases compared with PLIN5-enriched LDs in CHO-K1 cells (49). Pollak et al. (34) have also reported recently that PLIN2-enriched LDs can be hydrolyzed more effectively by ATGL/CGI-58 or HSL compared with PLIN5-enriched LD. Based on these reports, the PLIN2-enriched “barrier” seems to be more permissive to lipase access upon fasting, leading to lipolysis and shrinkage of LDs in PLIN2-Tg hearts. Thus the nature of PLIN2 seems to play a role in dynamic steatosis in the PLIN2-Tg heart.
Lipase assay experiments using micellar substrates demonstrated lower HSL activity in tissue homogenates of PLIN2-Tg hearts compared with those of WT hearts. In contrast, ATGL activity was markedly increased with fasting in both genotypes, whereas it was comparable in WT and PLIN2-Tg hearts (Fig. 6A). In support of the results of lipase assays using heart homogenate, in vitro transfection experiments using CHO-K1 cells also showed that PLIN2 overexpression significantly reduced HSL activity (Fig. 6C). Although the results of in vitro lipase assays do not necessarily reflect the phenomenon in vivo, the results indicate the potential capacity of PLIN2 to interfere with the function of cardiac lipases, presumably by covering the surface of micellar substrates, which have phospholipid monolayer surface and lipid core, similar to LDs in living cardiomyocytes (17).
Although our lipase assay using heart homogenates did not show an inhibitory effect of PLIN2 on ATGL, PLIN2 has been reported to inhibit ATGL binding to LDs in cultured HEK-293 cells (26). The discrepancy between PLIN2 effects on ATGL and HSL in our activity assay might indicate the differential function of PLIN2 toward ATGL and HSL, or it might be due to substrate preference of PLIN2 toward TAG or CE micelles in the in vitro assay conditions. The mechanism is not yet known; nonetheless, the present research and the results of previous studies suggest that regulation of lipase access to LDs might be at least one of the mechanisms through which PLIN2 induces LD accumulation in PLIN2-Tg hearts. Other alternative mechanisms include increased long-chain FA uptake by PLIN2 (12), CMA, and yet-unknown functions of PLIN2 that interfere with HSL or the involvement of unexpected effects of exogenous protein overexpression. Further studies are required to clarify the precise mechanisms by which PLIN2 provokes cardiac steatosis.
Because cardiac HSL activity was decreased with PLIN2 overexpression, it was of interest to examine whether HSL overexpression can reverse steatosis in PLIN2-Tg hearts (39). Crossing PLIN2-Tg mice with HSL-overexpressing mice showed that HSL overexpression completely prevented the steatosis (Fig. 7), indicating that the hydrolytic effect of HSL can overcome the steatotic effect of PLIN2. This result is consistent with the diminished steatosis in fasting mice when HSL is activated (Fig. 3) (41) and indicates again that cardiac PLIN2 is permissive to HSL upon lipolytic stimuli, as reported in vitro (49). Interestingly, EGFP-PLIN2 protein expression in the hearts of PLIN2-HSL double-Tg mice was comparable to that in PLIN2-Tg mice despite the disappearance of LDs, and confocal microscopy demonstrated dense and spotty expression of PLIN2 in cardiomyocytes of double-Tg mice (Fig. 7, A and D). The hearts of WT mice under feeding conditions also showed comparable PLIN2 protein expression despite scarce LDs compared with fasting WT mice (Fig. 3, C and D). These data indicate that although increased PLIN2 expression induces lipid accumulation, disappearance of the lipid core does not necessarily lead to a decrease in PLIN2 expression. Thus PLIN2 protein expression seems to be regulated by mechanisms that are independent of lipid levels in LDs (52). Further investigation will be required to clarify the precise mechanism.
Based on our results and those reported in previous studies, a potential scenario for the findings in the hearts of PLIN2-Tg mice could be that PLIN2 generates LDs by preventing HSL (and ATGL) from functionally accessing LDs and by producing a unique barrier that is permissive to the access of lipases upon lipolytic stimulation. Once stimulation occurs, e.g., by fasting, ATGL is upregulated, phosphorylated PLIN5 promotes ATGL-CGI-58 interaction, and phosphorylated HSL translocates to LDs, initiating the lipolytic reaction. Thus unraveling the pathophysiological functions of PLINs in cardiomyocytes could be beneficial in the management of an increasingly prevalent human disease: lipotoxic cardiomyopathy.
GRANTS
This work is supported by a research grant from the Ministry of Education, Culture, Sports, Science, and Technology in Japan (to J. Suzuki).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
M.U., J.S., M.H., S.S., S.I., and T. Koizumi performed experiments; M.U., J.S., and M.H. analyzed data; M.U., J.S., M.H., S.S., M.I., Y.Z., S.T., and F.B.K. interpreted results of experiments; M.U. and J.S. prepared figures; M.U., J.S., and F.B.K. drafted manuscript; M.U., J.S., M.H., S.T., S.I., T. Konoshita, F.B.K., and T.I. approved final version of manuscript; J.S. and T. Koizumi conceived and designed research; J.S. and F.B.K. edited and revised manuscript.
ACKNOWLEDGMENTS
Special thanks to F. Kitaguchi, M. Uno, and S. Patel for skillful technical assistance. We thank J. Yamamoto (Life Science Research Laboratory at the University of Fukui) for performing confocal microscopy.
REFERENCES
- 1.Afendi FM, Okada T, Yamazaki M, Hirai-Morita A, Nakamura Y, Nakamura K, Ikeda S, Takahashi H, Altaf-Ul-Amin M, Darusman LK, Saito K, Kanaya S. KNApSAcK family databases: integrated metabolite-plant species databases for multifaceted plant research. Plant Cell Physiol 53: e1, 2012. doi: 10.1093/pcp/pcr165. [DOI] [PubMed] [Google Scholar]
- 2.Bailey AP, Koster G, Guillermier C, Hirst EM, MacRae JI, Lechene CP, Postle AD, Gould AP. Antioxidant role for lipid droplets in a stem cell niche of drosophila. Cell 163: 340–353, 2015. doi: 10.1016/j.cell.2015.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brasaemle DL, Barber T, Wolins NE, Serrero G, Blanchette-Mackie EJ, Londos C. Adipose differentiation-related protein is an ubiquitously expressed lipid storage droplet-associated protein. J Lipid Res 38: 2249–2263, 1997. [PubMed] [Google Scholar]
- 4.Brindley DN, Kok BP, Kienesberger PC, Lehner R, Dyck JR. Shedding light on the enigma of myocardial lipotoxicity: the involvement of known and putative regulators of fatty acid storage and mobilization. Am J Physiol Endocrinol Metab 298: E897–E908, 2010. doi: 10.1152/ajpendo.00509.2009. [DOI] [PubMed] [Google Scholar]
- 5.Briquet-Laugier V, Ben-Zeev O, Doolittle MH. Determining lipoprotein lipase and hepatic lipase activity using radiolabeled substrates. Methods Mol Biol 109: 81–94, 1999. [DOI] [PubMed] [Google Scholar]
- 6.Chang BH, Li L, Paul A, Taniguchi S, Nannegari V, Heird WC, Chan L. Protection against fatty liver but normal adipogenesis in mice lacking adipose differentiation-related protein. Mol Cell Biol 26: 1063–1076, 2006. doi: 10.1128/MCB.26.3.1063-1076.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chiu HC, Kovacs A, Blanton RM, Han X, Courtois M, Weinheimer CJ, Yamada KA, Brunet S, Xu H, Nerbonne JM, Welch MJ, Fettig NM, Sharp TL, Sambandam N, Olson KM, Ory DS, Schaffer JE. Transgenic expression of fatty acid transport protein 1 in the heart causes lipotoxic cardiomyopathy. Circ Res 96: 225–233, 2005. doi: 10.1161/01.RES.0000154079.20681.B9. [DOI] [PubMed] [Google Scholar]
- 8.Chiu HC, Kovacs A, Ford DA, Hsu FF, Garcia R, Herrero P, Saffitz JE, Schaffer JE. A novel mouse model of lipotoxic cardiomyopathy. J Clin Invest 107: 813–822, 2001. doi: 10.1172/JCI10947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Evans RD, Hauton D. The role of triacylglycerol in cardiac energy provision. Biochim Biophys Acta 1861: 1481–1491, 2016. doi: 10.1016/j.bbalip.2016.03.010. [DOI] [PubMed] [Google Scholar]
- 10.Faleck DM, Ali K, Roat R, Graham MJ, Crooke RM, Battisti R, Garcia E, Ahima RS, Imai Y. Adipose differentiation-related protein regulates lipids and insulin in pancreatic islets. Am J Physiol Endocrinol Metab 299: E249–E257, 2010. doi: 10.1152/ajpendo.00646.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Finck BN, Lehman JJ, Leone TC, Welch MJ, Bennett MJ, Kovacs A, Han X, Gross RW, Kozak R, Lopaschuk GD, Kelly DP. The cardiac phenotype induced by PPARalpha overexpression mimics that caused by diabetes mellitus. J Clin Invest 109: 121–130, 2002. doi: 10.1172/JCI0214080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gao J, Serrero G. Adipose differentiation related protein (ADRP) expressed in transfected COS-7 cells selectively stimulates long chain fatty acid uptake. J Biol Chem 274: 16825–16830, 1999. doi: 10.1074/jbc.274.24.16825. [DOI] [PubMed] [Google Scholar]
- 13.Granneman JG, Moore HP, Mottillo EP, Zhu Z, Zhou L. Interactions of perilipin-5 (Plin5) with adipose triglyceride lipase. J Biol Chem 286: 5126–5135, 2011. doi: 10.1074/jbc.M110.180711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Haemmerle G, Lass A, Zimmermann R, Gorkiewicz G, Meyer C, Rozman J, Heldmaier G, Maier R, Theussl C, Eder S, Kratky D, Wagner EF, Klingenspor M, Hoefler G, Zechner R. Defective lipolysis and altered energy metabolism in mice lacking adipose triglyceride lipase. Science 312: 734–737, 2006. doi: 10.1126/science.1123965. [DOI] [PubMed] [Google Scholar]
- 15.Haemmerle G, Moustafa T, Woelkart G, Büttner S, Schmidt A, van de Weijer T, Hesselink M, Jaeger D, Kienesberger PC, Zierler K, Schreiber R, Eichmann T, Kolb D, Kotzbeck P, Schweiger M, Kumari M, Eder S, Schoiswohl G, Wongsiriroj N, Pollak NM, Radner FP, Preiss-Landl K, Kolbe T, Rülicke T, Pieske B, Trauner M, Lass A, Zimmermann R, Hoefler G, Cinti S, Kershaw EE, Schrauwen P, Madeo F, Mayer B, Zechner R. ATGL-mediated fat catabolism regulates cardiac mitochondrial function via PPAR-α and PGC-1. Nat Med 17: 1076–1085, 2011. doi: 10.1038/nm.2439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Haemmerle G, Zimmermann R, Hayn M, Theussl C, Waeg G, Wagner E, Sattler W, Magin TM, Wagner EF, Zechner R. Hormone-sensitive lipase deficiency in mice causes diglyceride accumulation in adipose tissue, muscle, and testis. J Biol Chem 277: 4806–4815, 2002. doi: 10.1074/jbc.M110355200. [DOI] [PubMed] [Google Scholar]
- 17.Hapala I, Marza E, Ferreira T. Is fat so bad? Modulation of endoplasmic reticulum stress by lipid droplet formation. Biol Cell 103: 271–285, 2011. doi: 10.1042/BC20100144. [DOI] [PubMed] [Google Scholar]
- 18.Hashioka S, Suzuki H, Nakajima D, Miyaoka T, Wake R, Hayashiba M, Horiguchi J, Klegeris A. Metabolomics analysis implies noninvolvement of the kynurenine pathway neurotoxins in the interferon-g-induced eurotoxicity of adult human astrocytes. Neuropsychiatry (London) 7: 610–617, 2016. [Google Scholar]
- 19.Imamura M, Inoguchi T, Ikuyama S, Taniguchi S, Kobayashi K, Nakashima N, Nawata H. ADRP stimulates lipid accumulation and lipid droplet formation in murine fibroblasts. Am J Physiol Endocrinol Metab 283: E775–E783, 2002. doi: 10.1152/ajpendo.00040.2002. [DOI] [PubMed] [Google Scholar]
- 20.Kanehara H, Suzuki J, Zenimaru Y, Takahashi S, Oida K, Shen WJ, Kraemer FB, Miyamori I. Function of hormone-sensitive lipase in diacylglycerol-protein kinase C pathway. Diabetes Res Clin Pract 65: 209–215, 2004. doi: 10.1016/j.diabres.2004.02.006. [DOI] [PubMed] [Google Scholar]
- 21.Kaushik S, Cuervo AM. Degradation of lipid droplet-associated proteins by chaperone-mediated autophagy facilitates lipolysis. Nat Cell Biol 17: 759–770, 2015. doi: 10.1038/ncb3166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Khor VK, Shen WJ, Kraemer FB. Lipid droplet metabolism. Curr Opin Clin Nutr Metab Care 16: 632–637, 2013. doi: 10.1097/MCO.0b013e3283651106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kraemer FB, Patel S, Saedi MS, Sztalryd C. Detection of hormone-sensitive lipase in various tissues. I. Expression of an HSL/bacterial fusion protein and generation of anti-HSL antibodies. J Lipid Res 34: 663–671, 1993. [PubMed] [Google Scholar]
- 24.Kuramoto K, Okamura T, Yamaguchi T, Nakamura TY, Wakabayashi S, Morinaga H, Nomura M, Yanase T, Otsu K, Usuda N, Matsumura S, Inoue K, Fushiki T, Kojima Y, Hashimoto T, Sakai F, Hirose F, Osumi T. Perilipin 5, a lipid droplet-binding protein, protects heart from oxidative burden by sequestering fatty acid from excessive oxidation. J Biol Chem 287: 23852–23863, 2012. doi: 10.1074/jbc.M111.328708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lass A, Zimmermann R, Haemmerle G, Riederer M, Schoiswohl G, Schweiger M, Kienesberger P, Strauss JG, Gorkiewicz G, Zechner R. Adipose triglyceride lipase-mediated lipolysis of cellular fat stores is activated by CGI-58 and defective in Chanarin-Dorfman Syndrome. Cell Metab 3: 309–319, 2006. doi: 10.1016/j.cmet.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 26.Listenberger LL, Ostermeyer-Fay AG, Goldberg EB, Brown WJ, Brown DA. Adipocyte differentiation-related protein reduces the lipid droplet association of adipose triglyceride lipase and slows triacylglycerol turnover. J Lipid Res 48: 2751–2761, 2007. doi: 10.1194/jlr.M700359-JLR200. [DOI] [PubMed] [Google Scholar]
- 27.Liu L, Shi X, Bharadwaj KG, Ikeda S, Yamashita H, Yagyu H, Schaffer JE, Yu YH, Goldberg IJ. DGAT1 expression increases heart triglyceride content but ameliorates lipotoxicity. J Biol Chem 284: 36312–36323, 2009. doi: 10.1074/jbc.M109.049817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu L, Zhang Y, Chen N, Shi X, Tsang B, Yu YH. Upregulation of myocellular DGAT1 augments triglyceride synthesis in skeletal muscle and protects against fat-induced insulin resistance. J Clin Invest 117: 1679–1689, 2007. doi: 10.1172/JCI30565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mayer N, Schweiger M, Romauch M, Grabner GF, Eichmann TO, Fuchs E, Ivkovic J, Heier C, Mrak I, Lass A, Höfler G, Fledelius C, Zechner R, Zimmermann R, Breinbauer R. Development of small-molecule inhibitors targeting adipose triglyceride lipase. Nat Chem Biol 9: 785–787, 2013. doi: 10.1038/nchembio.1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McManaman JL, Bales ES, Orlicky DJ, Jackman M, MacLean PS, Cain S, Crunk AE, Mansur A, Graham CE, Bowman TA, Greenberg AS. Perilipin-2-null mice are protected against diet-induced obesity, adipose inflammation, and fatty liver disease. J Lipid Res 54: 1346–1359, 2013. doi: 10.1194/jlr.M035063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Park TS, Goldberg IJ. Sphingolipids, lipotoxic cardiomyopathy, and cardiac failure. Heart Fail Clin 8: 633–641, 2012. doi: 10.1016/j.hfc.2012.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Park TS, Yamashita H, Blaner WS, Goldberg IJ. Lipids in the heart: a source of fuel and a source of toxins. Curr Opin Lipidol 18: 277–282, 2007. doi: 10.1097/MOL.0b013e32814a57db. [DOI] [PubMed] [Google Scholar]
- 33.Paul A, Chang BH, Li L, Yechoor VK, Chan L. Deficiency of adipose differentiation-related protein impairs foam cell formation and protects against atherosclerosis. Circ Res 102: 1492–1501, 2008. doi: 10.1161/CIRCRESAHA.107.168070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pollak NM, Jaeger D, Kolleritsch S, Zimmermann R, Zechner R, Lass A, Haemmerle G. The interplay of protein kinase A and perilipin 5 regulates cardiac lipolysis. J Biol Chem 290: 1295–1306, 2015. doi: 10.1074/jbc.M114.604744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pollak NM, Schweiger M, Jaeger D, Kolb D, Kumari M, Schreiber R, Kolleritsch S, Markolin P, Grabner GF, Heier C, Zierler KA, Rülicke T, Zimmermann R, Lass A, Zechner R, Haemmerle G. Cardiac-specific overexpression of perilipin 5 provokes severe cardiac steatosis via the formation of a lipolytic barrier. J Lipid Res 54: 1092–1102, 2013. doi: 10.1194/jlr.M034710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schulze PC, Drosatos K, Goldberg IJ. Lipid Use and Misuse by the Heart. Circ Res 118: 1736–1751, 2016. doi: 10.1161/CIRCRESAHA.116.306842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Subramaniam A, Jones WK, Gulick J, Wert S, Neumann J, Robbins J. Tissue-specific regulation of the alpha-myosin heavy chain gene promoter in transgenic mice. J Biol Chem 266: 24613–24620, 1991. [PubMed] [Google Scholar]
- 38.Sun Z, Miller RA, Patel RT, Chen J, Dhir R, Wang H, Zhang D, Graham MJ, Unterman TG, Shulman GI, Sztalryd C, Bennett MJ, Ahima RS, Birnbaum MJ, Lazar MA. Hepatic Hdac3 promotes gluconeogenesis by repressing lipid synthesis and sequestration. Nat Med 18: 934–942, 2012. doi: 10.1038/nm.2744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Suzuki J, Shen WJ, Nelson BD, Patel S, Veerkamp JH, Selwood SP, Murphy GM Jr, Reaven E, Kraemer FB. Absence of cardiac lipid accumulation in transgenic mice with heart-specific HSL overexpression. Am J Physiol Endocrinol Metab 281: E857–E866, 2001. [DOI] [PubMed] [Google Scholar]
- 40.Suzuki J, Shen WJ, Nelson BD, Selwood SP, Murphy GM Jr, Kanefara H, Takahashi S, Oida K, Miyamori I, Kraemer FB. Cardiac gene expression profile and lipid accumulation in response to starvation. Am J Physiol Endocrinol Metab 283: E94–E102, 2002. doi: 10.1152/ajpendo.00017.2002. [DOI] [PubMed] [Google Scholar]
- 41.Suzuki J, Ueno M, Uno M, Hirose Y, Zenimaru Y, Takahashi S, Osuga J, Ishibashi S, Takahashi M, Hirose M, Yamada M, Kraemer FB, Miyamori I. Effects of hormone-sensitive lipase disruption on cardiac energy metabolism in response to fasting and refeeding. Am J Physiol Endocrinol Metab 297: E1115–E1124, 2009. doi: 10.1152/ajpendo.91031.2008. [DOI] [PubMed] [Google Scholar]
- 42.Sztalryd C, Kimmel AR. Perilipins: lipid droplet coat proteins adapted for tissue-specific energy storage and utilization, and lipid cytoprotection. Biochimie 96: 96–101, 2014. doi: 10.1016/j.biochi.2013.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Trent CM, Yu S, Hu Y, Skoller N, Huggins LA, Homma S, Goldberg IJ. Lipoprotein lipase activity is required for cardiac lipid droplet production. J Lipid Res 55: 645–658, 2014. doi: 10.1194/jlr.M043471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ueno M, Shen WJ, Patel S, Greenberg AS, Azhar S, Kraemer FB. Fat-specific protein 27 modulates nuclear factor of activated T cells 5 and the cellular response to stress. J Lipid Res 54: 734–743, 2013. doi: 10.1194/jlr.M033365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ueno M, Suzuki J, Zenimaru Y, Takahashi S, Koizumi T, Noriki S, Yamaguchi O, Otsu K, Shen WJ, Kraemer FB, Miyamori I. Cardiac overexpression of hormone-sensitive lipase inhibits myocardial steatosis and fibrosis in streptozotocin diabetic mice. Am J Physiol Endocrinol Metab 294: E1109–E1118, 2008. doi: 10.1152/ajpendo.00016.2008. [DOI] [PubMed] [Google Scholar]
- 46.Unger RH, Clark GO, Scherer PE, Orci L. Lipid homeostasis, lipotoxicity and the metabolic syndrome. Biochim Biophys Acta 1801: 209–214, 2010. doi: 10.1016/j.bbalip.2009.10.006. [DOI] [PubMed] [Google Scholar]
- 47.Urahama Y, Ohsaki Y, Fujita Y, Maruyama S, Yuzawa Y, Matsuo S, Fujimoto T. Lipid droplet-associated proteins protect renal tubular cells from fatty acid-induced apoptosis. Am J Pathol 173: 1286–1294, 2008. doi: 10.2353/ajpath.2008.080137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.van de Weijer T, Schrauwen-Hinderling VB, Schrauwen P. Lipotoxicity in type 2 diabetic cardiomyopathy. Cardiovasc Res 92: 10–18, 2011. doi: 10.1093/cvr/cvr212. [DOI] [PubMed] [Google Scholar]
- 49.Wang H, Hu L, Dalen K, Dorward H, Marcinkiewicz A, Russell D, Gong D, Londos C, Yamaguchi T, Holm C, Rizzo MA, Brasaemle D, Sztalryd C. Activation of hormone-sensitive lipase requires two steps, protein phosphorylation and binding to the PAT-1 domain of lipid droplet coat proteins. J Biol Chem 284: 32116–32125, 2009. doi: 10.1074/jbc.M109.006726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang H, Sreenivasan U, Gong DW, O’Connell KA, Dabkowski ER, Hecker PA, Ionica N, Konig M, Mahurkar A, Sun Y, Stanley WC, Sztalryd C. Cardiomyocyte-specific perilipin 5 overexpression leads to myocardial steatosis and modest cardiac dysfunction. J Lipid Res 54: 953–965, 2013. doi: 10.1194/jlr.M032466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang H, Sreenivasan U, Hu H, Saladino A, Polster BM, Lund LM, Gong DW, Stanley WC, Sztalryd C. Perilipin 5, a lipid droplet-associated protein, provides physical and metabolic linkage to mitochondria. J Lipid Res 52: 2159–2168, 2011. doi: 10.1194/jlr.M017939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xu G, Sztalryd C, Lu X, Tansey JT, Gan J, Dorward H, Kimmel AR, Londos C. Post-translational regulation of adipose differentiation-related protein by the ubiquitin/proteasome pathway. J Biol Chem 280: 42841–42847, 2005. doi: 10.1074/jbc.M506569200. [DOI] [PubMed] [Google Scholar]
- 53.Yamaguchi T, Matsushita S, Motojima K, Hirose F, Osumi T. MLDP, a novel PAT family protein localized to lipid droplets and enriched in the heart, is regulated by peroxisome proliferator-activated receptor alpha. J Biol Chem 281: 14232–14240, 2006. doi: 10.1074/jbc.M601682200. [DOI] [PubMed] [Google Scholar]
- 54.Zhou YT, Grayburn P, Karim A, Shimabukuro M, Higa M, Baetens D, Orci L, Unger RH. Lipotoxic heart disease in obese rats: implications for human obesity. Proc Natl Acad Sci USA 97: 1784–1789, 2000. doi: 10.1073/pnas.97.4.1784. [DOI] [PMC free article] [PubMed] [Google Scholar]