

Abstract
The Insulin-like growth factor type 1 receptor (IGF-1R) is part of the receptor tyrosine kinase (RTK) superfamily. Activation of IGF-1R regulates several key signaling pathways responsible for maintaining cellular homeostasis, including survival, growth and proliferation. In addition to mediating signal transduction at the plasma membrane, in serum-based models, IGF-1R undergoes SUMOylation by SUMO1 and translocates to the nucleus in response to IGF-1. In corneal epithelial cells grown in serum free culture however, IGF-1R has been shown to accumulate in the nucleus independent of IGF-1. In this study, we report that the insulin-like growth factor binding protein 3 (IGFBP-3) mediates nuclear translocation of IGF-1R in response to growth factor withdrawal. This occurs via SUMOylation by SUMO2/3. Further, IGF-1R and IGFBP-3 undergo reciprocal regulation independent of PI3k/Akt signaling. Thus, under healthy growth conditions, IGFBP-3 functions as a gatekeeper to arrest the cell cycle in G0/G1, but does not alter mitochondrial respiration in cultured cells. When stressed, IGFBP-3 functions as a caretaker to maintain levels of IGF-1R in the nucleus. These results demonstrate mutual regulation between IGF-1R and IGFBP-3 to maintain cell survival under stress. This is the first study to show a direct relationship between IGF-1R and IGFBP-3 in the maintenance of corneal epithelial homeostasis.
Keywords: IGF-1R, IGFBP-3, Insulin, SUMO, cornea, epithelium
Introduction
Insulin-like growth factor receptor (IGF-1R) is a hetero-tetrameric tyrosine kinase receptor that belongs to the receptor tyrosine kinase superfamily. IGF-1R shares a high homology with the closely related insulin receptor (INSR), with highest homology found within their tyrosine kinase domains. Due to the significant homology between these receptors, IGF-1R and INSR together can form heterodimeric receptors known as hybrids (Soos et al., 1990). The functional significance of hybrid receptors (Hybrid-R) in different cells and tissues is still largely undefined. In addition to Hybrid-Rs, the high structural similarity between IGF-1R and INSR also impacts ligand specificity and binding, resulting in crosstalk between the IGF-1 and insulin axes (Werner et al., 2008).
It is well established that the principal IGF-1R ligands, IGF-1 and IGF-2, bind IGF-1R, activating IGF-mediated signaling (Pandini et al., 2002). This leads to the induction of multiple cellular pathways including proliferation, differentiation, and survival (Laron, 2001). Over-expression of IGF-1R is frequently implicated in human malignancies. As such, the IGF system has been extensively characterized in cancer cells and has been shown to regulate tumor progression (Arteaga, 1992; Chen et al., 1998; Myal et al., 1984), angiogenesis and vascular endothelial repair (Jacobo and Kazlauskas, 2015), and metastasis (Cubbon et al., 2016; Imrie et al., 2012; Thum et al., 2007). The finding of a nuclear IGF-1R has complicated our previous understanding of the role of IGF-1R in tumor and epithelial cell biology. In their seminal study, Sehat et al demonstrated that IGF-1 binding of the IGF-1R triggers nuclear translocation of IGF-1R via SUMOylation by SUMO1 and that nuclear localized IGF-1R functions as a transcriptional modulator (Sehat et al., 2010). More recent work by this same group has since shown that nuclear localized IGF-1R functions as a transcriptional co-activator of the lymphoid enhancer factor/T cell factor (LEF1/TCF) to mediate proliferation and control of the cell cycle (Lin et al., 2017; Sehat et al., 2010).
Insulin-like growth factor binding protein-3 (IGFBP-3) is an N-linked glycosylated and phosphorylated secretory protein that belongs to the family of insulin-like growth factor binding proteins (IGFBPs) (Baxter et al., 1986). IGFBP-3 is the primary binding protein for IGF-1 in serum (Furstenberger and Senn, 2002). Binding of IGFPB-3 to IGF-1 functions to both block activation of IGF-1R and extend the half-life of IGF-1 in circulation. IGFBP-3 also has known IGF-independent functions (Kim et al., 2011). In a cell and tissue-specific manner, IGFBP-3 has been shown to regulate apoptosis (Butt et al., 2002), DNA repair, cell cycle, autophagy (Grkovic et al., 2013), angiogenesis, hypoxia and insulin resistance (Baxter, 2013). IGFBP-3 contains multiple p53 response elements and induces apoptosis in tumor cells in response to DNA damage or hypoxia (Grimberg et al., 2005; Grimberg et al., 2002). Moreover, in breast cancer cells, IGFBP-3 regulates autophagy through binding to the 78-kDa glucose-regulated protein, GRP78 (Grkovic et al., 2013). In contrast, exposure to hyperglycemia induces phosphorylation of IGFBP-3 by the DNA-dependent protein kinase, DNA-PK, to prevent apoptosis in retinal endothelial cells (Zhang and Steinle, 2013).
In the corneal epithelium, IGFBP-3 is lowly expressed in proliferating cells, but is upregulated upon reaching confluence (Robertson et al., 2007). IGFBP-3 is also upregulated in response to hypoxia (unpublished data) and hyperglycemia in vitro, suggesting that IGFBP-3 may function in corneal epithelial cells as an important stress response protein. In vivo, IGFBP-3 is present in the human tear film and is increased approximately 3 fold in diabetic tears (Wu et al., 2012a). Increased expression in diabetic tears is associated with damage to the corneal subbasal nerve plexus (Stuard et al., 2017). We have previously shown that IGF-1R is upregulated in response to stress induced by growth factor withdrawal (Titone et al., 2018). In the present study, we investigated the relationship between IGF-1R and IGFBP-3 in corneal epithelial cells to determine whether the stress induced increase in IGFBP-3 mediates IGF-1R. Importantly, we show for the first time that IGFBP-3, not IGF-1, induces translocation of IGF-1R into the nucleus of corneal epithelial cells via SUMOylation by SUMO2/3. This is the first report of an IGF-binding protein inducing the nuclear translocation of IGF-1R in any cell type.
Materials and Methods
Cell lines and primary cultures
Human telomerized corneal epithelial (hTCEpi) cells, previously developed and characterized by our laboratory, were used in this study (Robertson et al., 2005). hTCEpi cells were cultured in serum-free keratinocyte basal media containing 0.15 mM calcium with supplements at 37°C and 5% CO2 (KGM2, Lonza, Walkersville, MD). To induce growth factor deprivation, cells were cultured in keratinocyte basal media containing 0.15 mM calcium without supplements (KBM). For primary cultures of human corneal epithelial cells (HCECs), human eye bank corneas (Tissue Transplant Services, UT Southwestern Medical Center, Dallas, TX) were digested in dispase (Invitrogen, Carlsbad, CA) overnight at 4 °C. Intact epithelial cell sheets were carefully removed and subject to a second digestion in dispase for 2 hours at 37 °C. Individual cells were separated by gentle pipetting and seeded onto plastic tissue culture dishes coated with Type 4 collagen (Biocoat, BD Biosciences, San Jose, CA). Cells were cultured in CnT20 cell culture media enriched for progenitor cell culture for 10–15 days (Zen Bio, Research Triangle Park, NC). After their first passage, HCECs were then transitioned to serum-free keratinocyte basal media containing 0.15 mM calcium with supplements at 37°C and 5% CO2 (KGM2, Promocell, Germany). To induce growth factor deprivation, cells were cultured in keratinocyte basal media containing 0.15 mM calcium without supplements (KBM).
siRNA knockdown
For siRNA experiments, hTCEpi cells were seeded at 50–60% confluence in a 6-well tissue culture plate and grown overnight. Cells were transfected with double-stranded inhibitory RNA oligonucleotides (GeneSolution, IGF-1R #GS3480, INSR #GS3643, FlexiTube CAV1 SI00027720 and CLTC SI00299873, Qiagen, Germantown, MD) using Lipofectamine RNAiMAX (Invitrogen, Carlsbad, CA) in antibiotic-free basal media. Briefly, 12 pmol of siRNA oligonucleotides were added to 100 μl basal media and incubated for 5 minutes at room temperature. siRNAs were then mixed with 2 μl Lipofectamine and allowed to incubate for an additional 20 minutes, after which the transfection mixture was added directly to hTCEpi cells containing 1 ml of KBM and incubated for 24 hours. The media was removed and cells were then cultured in KGM (growth) or KBM (basal) for another 24 hours as indicated, with or without 10 μg/ml of human recombinant insulin (Sigma, St. Louis, MO) or 500 ng/ml of human recombinant IGFBP-3 (rhIGFBP-3, Sino Biological Inc., BDA, Beijing, China). AllStars negative control siRNA was used as a non-targeting control (#1027280, Qiagen, Germantown, MD) for all experiments.
Subcellular fractionation and immunoblotting
To examine the nuclear localization of IGF-1R, hTCEpi cells were subject to nuclear and cytoplasmic fractionation. Confluent hTCEpi cells were collected using trypsin-EDTA and washed with phosphate buffered saline (PBS). A NE-PER nuclear fractionation kit was used to separate soluble and insoluble nuclear and non-nuclear proteins (Thermo Fisher, Rockford, IL). Protein concentration for individual fractions was measured using a Qubit 3.0 Fluorometer (Thermo Fisher, Rockford, IL). Lysates were then electrophoresed through a 4–15% linear gradient precast polyacrylamide gel (BioRad, Hercules, CA) and immunoblotted as described below.
SDS PAGE and Immunoblotting
For experiments involving whole cell lysates, hTCEpi cells and HCECs were lysed directly in 6-well culture plates using radioimmunoprecipitation buffer (RIPA) containing a protease and phosphatase inhibitor cocktail (Thermo Fisher, Rockford, IL) on ice for 10 minutes, then centrifuged for 5 minutes at 12,000 rpm at 4°C in a microcentrifuge (BioRad, Hercules, CA). For experiments testing for SUMOylation, 10 mM N-Ethylmaleimide, an isopeptidase inhibitor, was added to the lysis buffer. Protein concentration was determined using a Qubit 3.0 Fluorometer (Thermo Fisher, Rockford, IL). Supernatants were removed and boiled for 5 minutes in 2× sample buffer, pH 6.8, containing 65.8 mM Tris-HCL, 26.3% (w/v) glycerol, 2.1% SDS, 5.0% β-mercaptoethanol and 0.01% bromophenol blue (Bio-rad, Hercules, CA). Samples were resolved on a 4–15% precast linear gradient polyacrylamide gel (Bio-rad, Hercules, CA) and transferred to a polyvinyl difluoride (PVDF) membrane (Millipore, Temecula, CA). Membranes were blocked in 5% non-fat milk (Bio-rad, Hercules, CA) for 1 hour at room temperature and incubated in primary antibody overnight at 4°C (IGF1-Rβ #3027 Caveolin-1 #3238, Histone 3 #9715, Sumo-1 #4930, Sumo-2/3 #4971, Cell Signaling, Danvers, MA; p-Akt # sc-7985-R, Akt1 #sc-5298, Insulin Rβ #sc-57342, GAPDH #sc-66163, Santa Cruz, CA; IGFBP-3 #MAB305, R&D Systems, Minneapolis, MN; and clathrin heavy chain #ab21679, SP1 #ab13370, Abcam, Cambridge, MA). Following a 1 hour incubation with an anti-mouse or anti-rabbit secondary antibody (Santa Cruz, CA), proteins were visualized using ECL Prime Detection Reagent (Amersham Biosciences, Piscataway, NJ) and imaged on an Amersham Imager 600 (Amersham Biosciences, Piscataway, NJ). β-actin was used as a loading control for whole cell lysates. Histone 3, SP1, and GAPDH were used as loading controls for the insoluble nuclear fraction, the soluble nuclear fraction, and the cytosolic fraction, respectively.
Enzyme-linked Immunoassay (ELISA)
Cell culture supernatants were concentrated using protein concentrators containing a polyether sulfone membrane (10K MWCO; Pierce, Rockford, IL). Cells were starved in basal media for 24 hours and then stimulated by 10 μg/ml of recombinant human insulin or 500 ng/ml of recombinant human IGFBP-3 for 24 hours. Whole cell lysates were harvested directly in the culture dish using RIPA buffer containing a protease and phosphatase inhibitor cocktail (Thermo Fisher, Rockford, IL) on ice for 10 minutes. Protein concentration was determined using a Qubit 3.0 Fluorometer (Thermo Fisher, Rockford, IL). IGFBP-3 levels were detected by a human IGFBP-3 Quantikine ELISA (R&DSystems, Minneapolis, MN). Media and whole cell lysate samples were assayed in triplicate. Each experiment was repeated a minimum of two additional times.
Cell Cycle Assay
Propidium Iodide (PI)/RNase staining solution (Cell Signaling, Danvers, MA) was used for cell cycle analysis. hTCEpi cells were seeded into 6-well plates and cultured overnight in KGM. Media was removed and replaced with either growth or basal media with or without rhIGFBP-3. At the end of the incubation period, cells were trypsinized and centrifuged at 1500 rpm. Cells were then washed with PBS and fixed with cold ethanol for 15 minutes. Following fixation, cells were stained with PI for 40 minutes at 37°C. The cell cycle was analyzed using a Cellometer K2 Fluorescent Viability Cell Counter (Nexcelom, Lawrence, MA). All assays were performed in triplicate and repeated a minimum of two additional times.
Quantitative ATP Analysis
hTCEpi cells were seeded into 6-well plates and cultured overnight in KGM. Media was removed and replaced with either growth or basal media with or without rhIGFBP-3 for 24 hours. At the end of the incubation period, cells were trypsinized and centrifuged at 1500 rpm. Cells were then washed with PBS and incubated at 37°C for 5 minutes in a low detergent lysis buffer containing 10 mM Tris pH 7.5, 100 μM NaCl, 1 mM EDTA, and 0.15% Triton X-100 (Thermo Fisher, Rockford, IL). ATP levels were measured using a bioluminescence assay with recombinant luciferase and its substrate D-luciferin that in the presence of ATP produces light (Thermo Fisher, Rockford, IL). Samples were incubated in a reaction solution at room temperature for 15 minutes and the measured ATP levels were compared against a standard curve of a series of known ATP concentrations. All samples were assayed in quadruplicate. Each experiment was repeated a minimum of three times.
Global Protein SUMOylation Assay
hTCEpi cells were cultured in KBM and transfected with siRNA oligonucleotides targeting IGFBP3 or a non-targeting control, as described under siRNA knockdown. Extracellular IGFBP-3 levels were rescued following knockdown by the addition of exogenous rhIGFBP-3. Nuclear fractions were isolated using an NE-PER nuclear fractionation kit as described above (Thermo Fisher, Rockford, IL). 10 mM N-Ethylmaleimide was added to the lysis buffer. To quantify SUMOylation of our target protein (IGF-1R), a global protein sumoylation colorimetric assay (Abcam, Cambridge, MA) was used. Two μg/ml of an immunoprecipitation-grade antibody against IGF-1R alpha (#MA5–13807, Thermo Fisher, Rockford, IL) or an irrevelant IgG negative control antibody (provided in the SUMOylation kit) were added to the wells and incubated overnight at 4 ͦ C. Nuclear fractions were then added to each pre-coated well containing the capture antibody. To detect SUMOylated IGF-1R, a pan-SUMO detection antibody was added to each well and incubated for 1 hour at room temperature using an orbital shaker (Talboys Advanced Microplate Shaker, Thermo Fisher, Rockford, IL) at 100 rpm. After addition of the developing solution, absorbance was read at 450 nm. Results are calculated in terms of ng of SUMOylated-IGF-1R normalized to mg of total protein. All samples were assayed in quadruplicate. Each experiment was repeated a minimum of three times.
Immunofluorescence
For immunofluorescence studies, hTCEpi cells and HCECs were seeded onto 35 mm glass bottom dishes (MatTek Corporation, Ashland, MA) and allowed to adhere overnight. Cells were transfected with double-stranded inhibitory RNA oligonucleotides (GeneSolution, IGFBP3 #GS3486, QIAGEN) with or without rhIGFBP-3. After treatment, cells were first rinsed with cold PBS and then fixed in 1% paraformaldehyde (Electron Microscopy Sciences, Fort Washington, PA) in PBS for 10 minutes. After 3 washes with PBS, cells were permeabilized in 0.1% Triton X-100 in PBS for 10 minutes and then blocked using 0.5% bovine serum albumin (Sigma, St. Louis, MO) in PBS for 30 minutes. Samples were then incubated in primary antibodies against IGF-1R and IGFBP-3 at 4°C overnight and subsequently washed in PBS and stained with Alexa Fluor 488 and 555 (Cell Signaling, Danvers, MA) secondary antibodies for 1 hour at room temperature. All samples were mounted on slides using Prolong gold antifade reagent with DAPI to label nuclei (Invitrogen, Carlsbad, CA). Cells were imaged on a Leica SP8 laser scanning confocal microscope (Leica Microsystems, Heidelberg, Germany) using a 63x oil objective. All images were sequentially scanned to avoid spectral crosstalk between channels.
In Situ Fluorescence Proximal Ligation Assay
A Proximal Ligation Assay (PLA) was used to determine the localization of SUMOylated IGF-1R (Sigma, St. Louis, MO). hTCEpi cells were seeded onto 35 mm glass bottom dishes (MatTek Corporation, Ashland, MA) and allowed to adhere overnight. Cells were then treated with KGM or KBM and fixed in 4% paraformaldehyde (Electron Microscopy Sciences, Fort Washington, PA) in PBS for 10 minutes. After 3 washes with PBS, cells were permeabilized in 0.2% Triton X-100 in PBS for 20 minutes and blocked using the Duolink blocking solution for 1 hour at room temperature. Samples were then incubated in primary antibodies recognizing IGF-1R β (#3027, Cell Signaling, Danvers, MA) and SUMO 2/3 (#FL-103, Santa Cruz, CA) overnight at 4°C. After washing, cells were incubated with secondary antibodies conjugated to oligonucleotides (PLA probe MINUS and PLA probe PLUS) for 1 hour at 37°C. Oligonucleotides were ligated using the ligation solution for 30 minutes at 37°C, followed by a 2 hour amplification step at 37°C. Amplified oligonucleotides were labeled using FITC and nuclei were counterstained using DAPI contained within the mounting media (Vectashield, Thermo Fisher, Rockfield, IL). PLA signals were detected by imaging with a Leica SP8 confocal microscope in sequential scan mode.
Statistical Analysis
All data are expressed as mean ± standard deviation. For comparison between three groups, a One-way or Two-way ANOVA was used with an appropriate post-hoc comparison. Statistical significance was set at P<0.05.
Results
IGFBP-3 levels are regulated by IGF-1R, not INSR, in human corneal epithelial cells
To measure the level of secreted IGFBP-3 in conditioned media from hTCEpi cells, conditioned media was collected and concentrated following transfection of non-targeting siRNA control oligonucleotides. In basal media, there was a significant increase in secreted IGFBP-3 compared to culture in growth media (Fig. 1A). The increase in IGFBP-3 was attenuated following the addition of insulin to culture media. To determine the effect of IGF-1R expression on secreted IGFBP-3, cells were transfected with siRNA oligonucleotides targeting IGF-1R in parallel to controls. Following knockdown, hTCEpi cells were cultured in growth (KGM) and basal (KBM) media for 24 hours and then treated with 10 μg/μl of human recombinant insulin. Concentration of secreted IGFBP-3 in the conditioned media was measured using ELISA (Fig. 1A). Reduced expression of IGF-1R following knockdown triggered a significantly attenuated IGFBP-3 secretion.
Fig. 1:
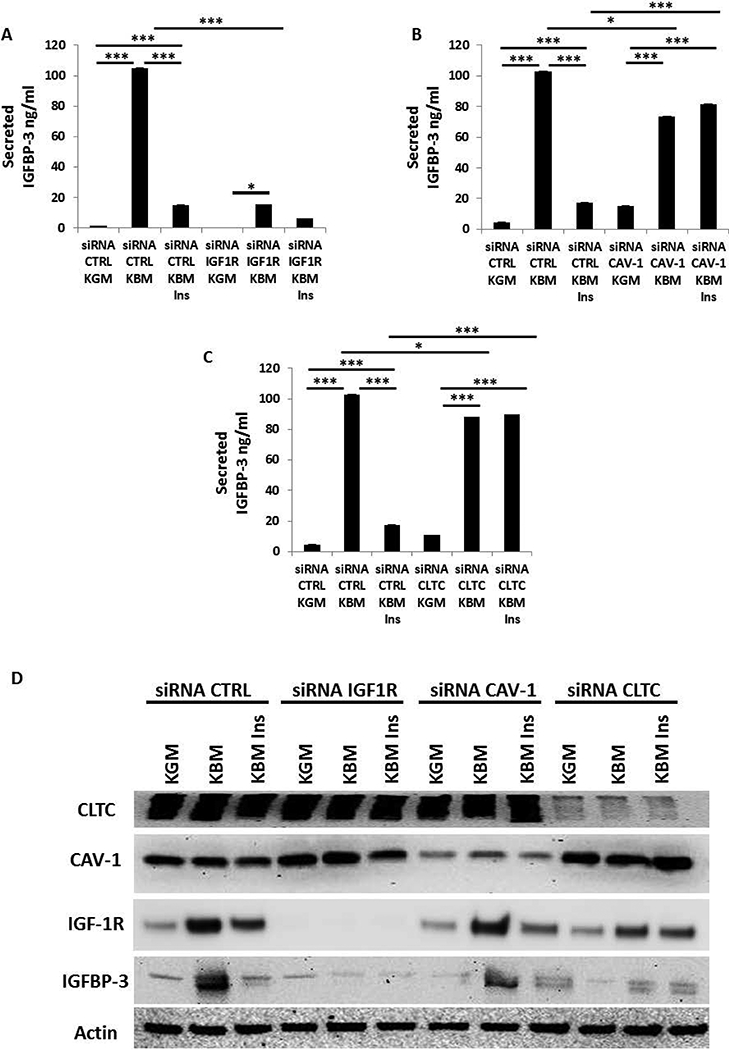
IGF-1R, CAV-1, and CLTC regulate IGFBP-3 secretion in conditioned media. An IGFBP-3 ELISA was used to analyze the concentration of secreted IGFBP-3. (A) hTCEpi cells treated with siRNA oligonucleotides targeting IGF-1R significantly decreased IGFBP-3 secretion into culture media compared to the non-targeting control (**P<0.001, One-way ANOVA, Holm-Sidak post hoc multiple comparison test). (B, C) hTCEpi cells were treated with siRNA oligonucleotides targeting CAV-1 and CLTC. Knockdown of each protein increased secretion of IGFBP-3 in basal media. Unlike the non-targeting control, secreted levels of IGFBP-3 were unchanged by the addition of 10 μg/ml of human recombinant insulin in basal media (***P<0.001, One-way ANOVA, Holm-Sidak post hoc multiple comparison test). (D) Immunoblotting for IGF-1R, Cav-1 and CLTC confirmed knockdown in whole cell lysates. IGFBP-3 immunoblotting of whole cell lysates paralleled secreted levels of IGFBP-3. β-actin was used as a loading control. KGM: keratinocyte growth media; KBM: keratinocyte basal media; IGF-1R: insulin-like growth factor type 1 receptor; IGFBP-3: insulin-like growth factor binding protein-3; Cav-1: caveolin-1; CLTC: clathrin; CTRL: control; ins: insulin. Data representative of 6 independent experiments performed in triplicate. ELISA data presented as mean ± standard deviation.
To investigate the mechanism by which insulin mediates the downregulation of IGFBP-3, caveolin 1 (Cav-1) and clathrin (CLTC), two main proteins involved in cellular trafficking and surface membrane receptor uptake, were knocked down using siRNA. As shown in Figures 1B and 1C, cells transfected with the negative control siRNA showed an increase in secretion of IGFBP-3 in conditioned media under basal conditions, which was reduced by the addition of insulin (Fig. 1B and 1C). Knockdown of either CAV-1 or CLTC however, blocked insulin uptake. This blunted the effect of insulin on IGFBP-3.
Upregulation of IGFBP-3 in basal media was further confirmed in whole cell lysates by immunoblotting (Fig. 1D). Similar to expression in conditioned media, IGFBP-3 was increased in whole cell lysates from basal media compared to growth media. Secretion of IGFBP-3 was similarly reduced in the presence of insulin. The efficiency of siRNA knockdown of IGF-1R, CAV-1, and CLTC was confirmed by immunoblotting (Fig. 1D). Compared to the non-targeting control, blocking CLTC blunted the increase in IGF-1R in basal media. This paralleled a decrease in IGFBP-3. These observations suggest that IGF-1R regulates expression of IGFBP-3 and that CLTC, and to a lesser extent, Cav-1, is essential for insulin uptake by the receptors and their consequent activation.
We next investigated whether IGF1-R and INSR both played a role in regulating IGFBP-3 secretion. To determine if INSR was involved in IGFBP-3 regulation, INSR and IGF-1R were each knocked down using siRNA and secreted IGFBP-3 was again measured using ELISA. As expected, there was a significant increase in secreted IGFBP-3 in basal media that was abrogated following IGF-1R knockdown (Fig. 2A). In cells transfected with siRNA targeting INSR, IGFBP-3 secretion in basal media was slightly increased compared to controls, indicating that IGF-1R and not INSR, mediates IGFBP-3 expression. Similar to the negative controls, secretion of IGFBP-3 after INSR knockdown was decreased after treatment with insulin. The efficiency of each knockdown was evaluated by immunoblotting (Fig. 2B). Knockdown of INSR in basal media produced a corresponding increase in IGF-1R. The increase in IGF-1R paralleled the increase in secreted IGFBP-3. Taken together, these data suggest that the presence of insulin regulates IGFBP-3 expression independent of INSR. To confirm that there is a decrease in IGFBP-3 secretion following depletion of IGF-1R was not a cell line artifact, we transfected HCECs with siRNA targeting IGF-1R. Transfected HCECs were cultured in growth media, basal media, and basal media supplemented with insulin. Similar to hTCEpi cells, knockdown of IGF-1R blunted secretion of IGFBP-3 in basal media (Fig. 3A, B).
Fig. 2:
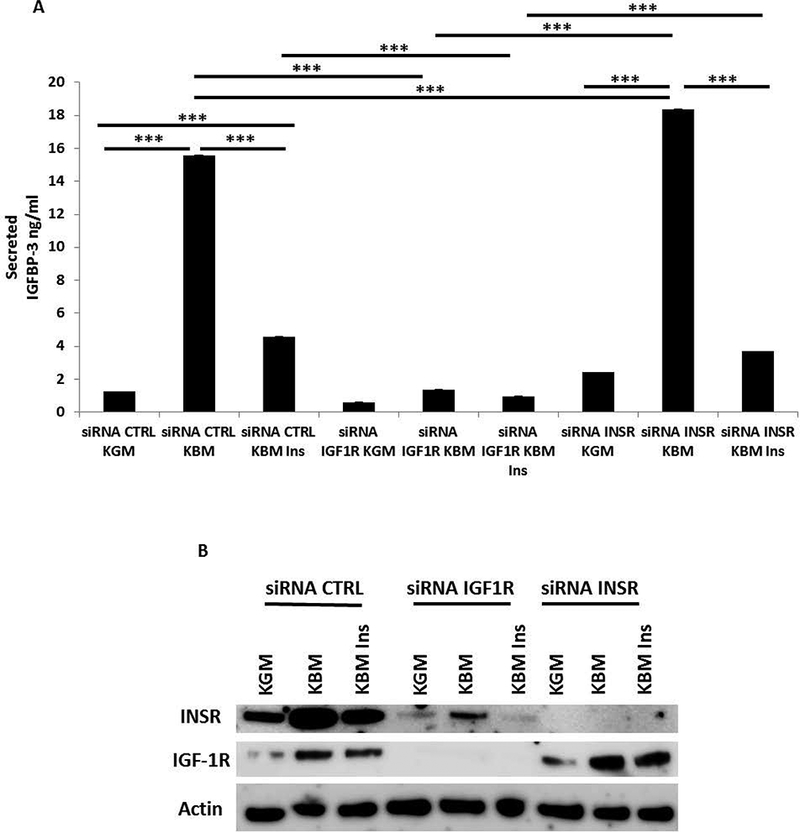
Depletion of INSR does not inhibit IGFBP-3 secretion in hTCEpi cells. Secreted IGFBP-3 in conditioned media was measured using an IGFBP-3 ELISA. (A) hTCEpi cells were treated with siRNA oligonucleotides targeting IGF-1R or INSR. In basal media, knockdown of IGF-1R blunted secretion of IGFBP-3 compared to the non-targeting control (***P<0.001, One-way ANOVA, Holm-Sidak post hoc multiple comparison test). In contrast, knockdown of INSR in basal media resulted in a small but significant increase in secreted IGFBP-3 compared to the non-targeting control (***P<0.001, Two-way ANOVA, Holm-Sidak post hoc multiple comparison test). (B) Immunoblotting for IGF-1R and INSR confirmed knockdown in whole cell lysates. As expected, in basal media, knockdown of INSR upregulated expression of IGF-1R I whole cell lysates. β-actin was used as a loading control. KGM: keratinocyte growth media; KBM: keratinocyte basal media; CTRL: control; ins: insulin. Data representative of 4 independent experiments performed in triplicate. ELISA data presented as mean ± standard deviation.
Fig. 3:
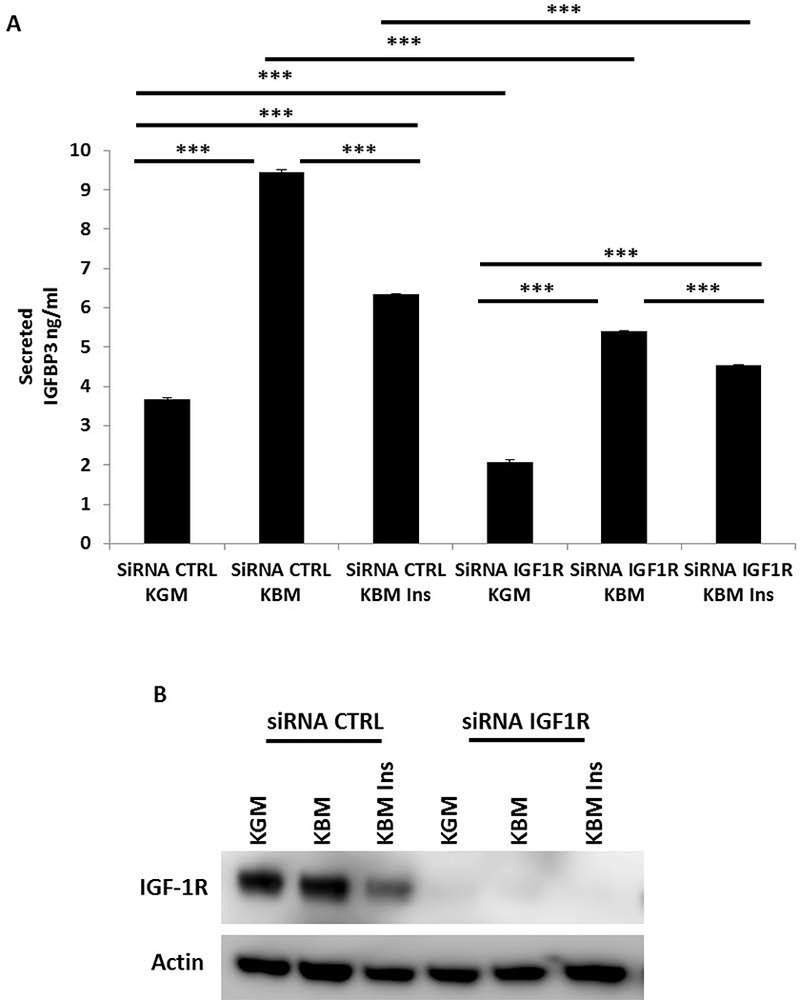
Knockdown of IGF-1R attenuates IGFBP-3 secretion in HCECs. Secreted IGFBP-3 in conditioned media was measured using an IGFBP-3 ELISA. (A) HCECs were treated with siRNA oligonucleotides targeting IGF-1R. In basal media, IGF-1R knockdown resulted in a large decrease in secreted IGFBP-3 compared to the non-targeting control (***P<0.001, Two-way ANOVA, Holm-Sidak post hoc multiple comparison test). (B) Immunoblotting for IGF-1R confirmed knockdown in whole cell lysates. β-actin was used as a loading control. KGM: keratinocyte growth media; KBM: keratinocyte basal media; CTRL: control; ins: insulin. Data representative of 3 independent experiments performed in triplicate. All independent experiments used HCECs derived from different donors. ELISA data presented as mean ± standard deviation.
Our prior studies have shown that phosphorylation of IGF-1R in hTCEpi cells activates the PI3k/Akt signaling pathway. To test whether IGF-1R regulates IGFBP-3 secretion via PI3k/Akt signaling, we measured IGFBP-3 secretion in hTCEpi cells treated with 50 μM of PI3 kinase inhibitor LY 294002 in basal and growth media, with or without insulin. IGFBP-3 secretion in conditioned media was measured using ELISA (Supplementary data 1. A). Neither treatment with the inhibitor nor the DMSO control altered secretion of IGFBP-3 from hTCEpi cells. Similarly, the addition of insulin induced the expected decrease in secretion of IGFBP-3. The inhibition of Akt phosphorylation by the PI3 kinase inhibitor was assessed by immunoblotting (Supplementary data 1. B). In control cells treated without the inhibitor, growth media and basal media supplemented with insulin induced phosphorylation of Akt. In contrast, phosphorylation of Akt was completely abrogated following treatment with the inhibitor LY 294002. These observations indicate that regulation of IGFBP-3 by IGF1-R is not through activation of the canonical PI3k/Akt pathway.
In growth condition, exogenous IGFBP-3 arrests the cell cycle, but does not alter cellular respiration
Our data demonstrate a role for IGF-1R in the regulation of IGFBP-3. To determine if there is reciprocal regulation between IGF-1R and IGFBP-3, we used siRNA oligonucleotides to knock down IGFBP-3 in hTCEpi cells in basal media. We then used ELISA (Fig. 4A, B) and immunoblotting (Fig. 4C) to verify the transfection efficiency in whole cell lysates (Fig. 4A) and in conditioned media (Fig. 4B). In both whole cell lysates and conditioned media, knockdown of IGFBP-3 decreased IGFBP-3 to undetectable levels. Knockdown of IGFBP-3 in basal media triggered an increase in IGF-1R (Fig. 4C). The subsequent addition of exogenous rhIGFBP-3 to cells deficient in IGFBP-3 appeared to further increase IGF-1R (Fig. 4C). We then tested whether rhIGFBP-3 was internalized in epithelial cells cultured in growth media. Immuoblotting for IGFBP-3 showed robust uptake of the exogenous protein (Fig. 4D). IGF-1R, which was decreased in growth media compared to basal, was unaffected by the addition of rhIGFBP-3 under growth conditions. These data demonstrate that IGF-1R expression is stimulated by the addition of IGFBP-3 in basal media, but is unaltered by the addition of exogenous IGFBP-3 in growth media. To test whether there is reciprocal regulation between IGF-1R and IGFBP-3 in HCECs, we knocked down IGFBP-3 in HCECs cultured in basal media. We used ELISA (Fig. 5A, B) and immunoblotting (Fig. 5C) to confirm the transfection efficiency in the whole cell lysates and in conditioned media. Our results confirmed our cell line findings and showed the same increase of IGF-1R expression after IGFBP-3 knock down and a further increase by the addition of rhIGFBP-3.
Fig. 4:
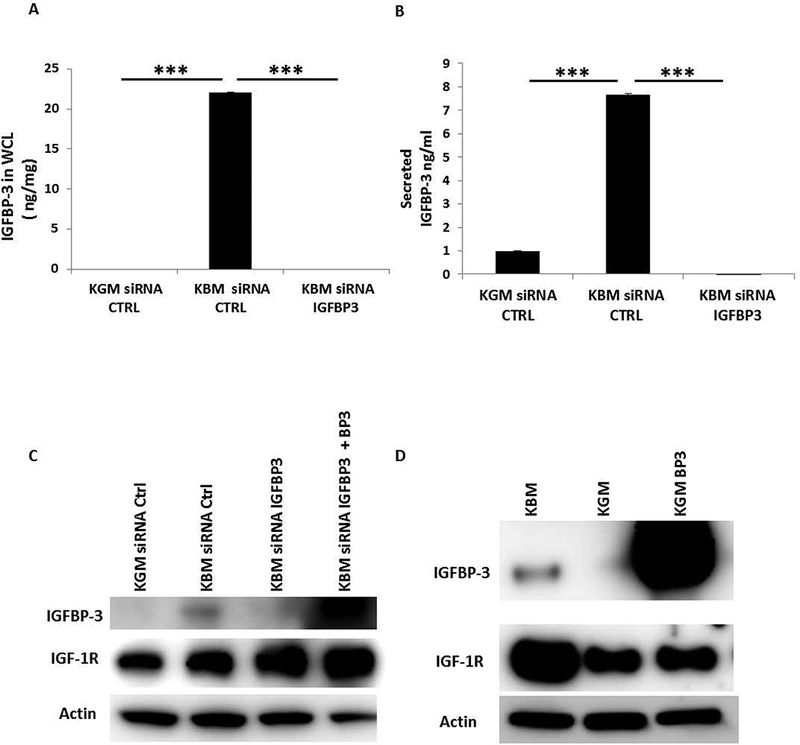
Knockdown of IGFBP-3 increases expression of IGF-1R in whole cell lysates. hTCEpi cells were treated with siRNA oligonucleotides targeting IGFBP-3 in basal media. (A, B) An IGFBP-3 ELISA was used to confirm knockdown of IGFBP-3 in whole cell lysates (A, ***P<0.001, One-way ANOVA, post hoc multiple comparison test) and secretion of IGFBP-3 in conditioned media (B, ***P<0.001, One-way ANOVA, post hoc multiple comparison test). (C) Immunoblotting for IGF-1R in whole cell lysates demonstrated an increase in receptor expression after siRNA knockdown of IGFBP-3 in basal media compared to the non-targeting control. Rescue with 100 ng/ml exogenous rhIGFBP-3 further increased IGF-1R expression. (D) Immunoblotting for IGFBP-3 was used to confirm knockdown of IGFBP-3. β-actin was used as a loading control. hTCEpi cells were cultured in basal or growth media with or without 500 ng/ml of rhIGFBP-3 for 24 hours. Addition of rhIGFBP-3 in growth media did not alter IGF-1R expression compared to cells cultured in growth media alone. β-actin was used as a loading control. KGM: keratinocyte growth media; KBM: keratinocyte basal media; BP3: IGFBP-3; WCL: whole cell lysates; CTRL: control. Data representative of 3 independent experiments performed in triplicate. ELISA data presented as mean ± standard deviation.
Fig. 5:
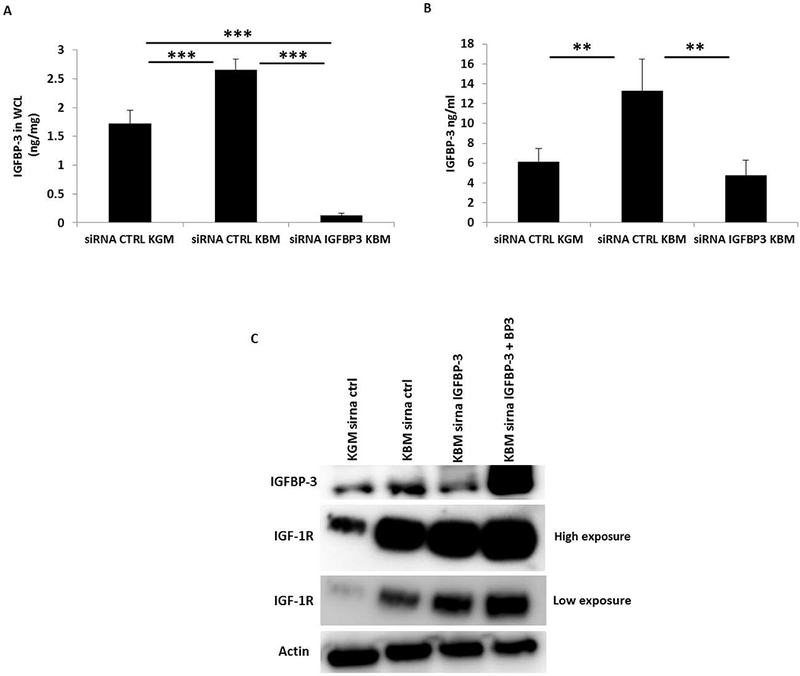
IGFBP-3 regulates nuclear accumulation of IGF-1R. HCECs were treated with siRNA oligonucleotides targeting IGFBP-3 in basal media. (A, B) An IGFBP-3 ELISA was used to confirm knockdown of IGFBP-3 in whole cell lysates (A, ***P<0.001, One-way ANOVA, post hoc multiple comparison test) and in conditioned media (B, **P<0.01, One-way ANOVA, post hoc multiple comparison test). (C) Immunoblotting for IGF-1R in basal media demonstrated an increase in receptor expression following siRNA knockdown of IGFBP-3 compared to the non-targeting control. Rescue with 100 ng/ml exogenous rhIGFBP-3 further increased IGF-1R expression. Immunoblot for IGF-1R is shown at both high and low exposures during imaging. Immunoblotting for IGFBP-3 confirmed knockdown of IGFBP-3 in basal media. β-actin was used as a loading control. KGM: keratinocyte growth media; KBM: keratinocyte basal media; BP3: IGFBP-3; WCL: whole cell lysates; CTRL: control. Data representative of 3 independent experiments performed in triplicate. All independent experiments used HCECs derived from different donors. ELISA data presented as mean ± standard deviation.
Previous studies in our laboratory have shown that IGFBP-3 is expressed at very low levels during normal growth conditions and is upregulated upon reaching confluence (Robertson et al., 2007). To examine the effects of supplementation of exogenous rhIGFBP-3 on the cell cycle during growth, epithelial cells were stained with PI and the cell cycle was analyzed using a Cellometer. As expected, due to the lack of growth factors in basal media, there was a significant increase in cells arrested in G0/G1 compared to cells cultured in growth media. This was associated with a significant reduction in cells in S phase and in G2/M (Fig. 6A-F). When exogenous rhIGFBP-3 was added to epithelial cells cultured in growth media for 24 hours, there was an increase in the number of cells arrested in G0/G1 compared to growth media alone. There was a similar reduction in the number of cells in S phase and in G2/M after treatment with IGFBP-3 in growth media (Fig. 6A-F). Together, these data confirm that IGFBP-3 functions as an inhibitor of cell growth in proliferating cells. This is in line with our previously reported data showing that IGFBP-3 can inhibit cell proliferation in an IGF-1 dependent manner (Wu et al., 2012b).
Fig. 6:
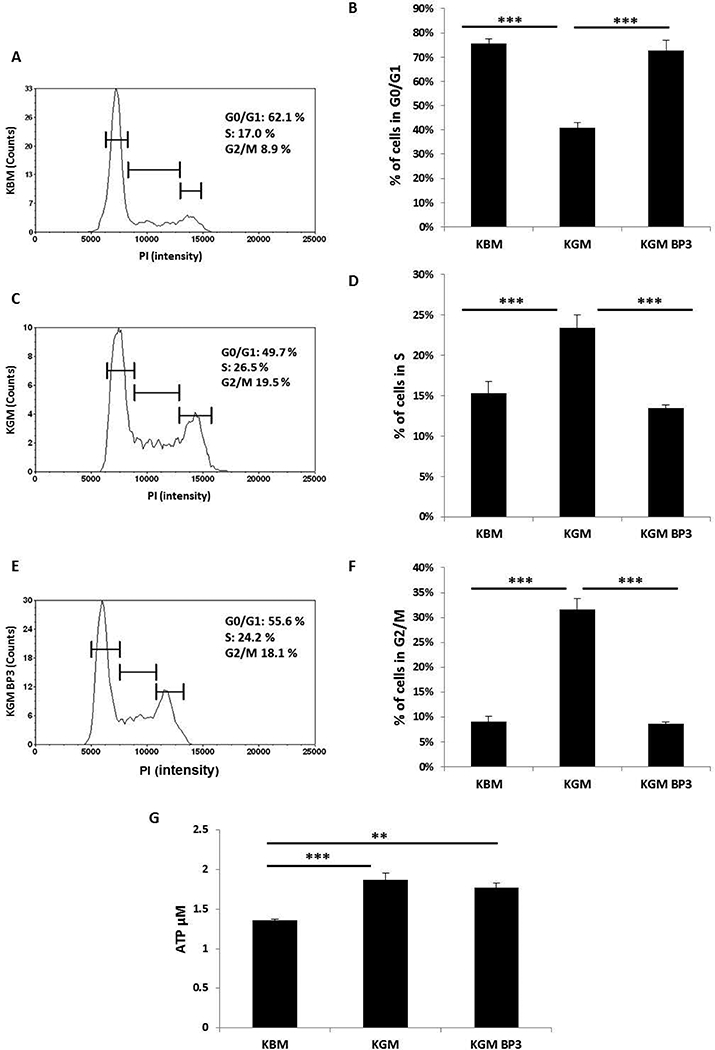
IGFBP-3 induces cell cycle arrest but does not alter cellular respiration. (A, C and E) Representative graphs of cell count as a function of PI intensity in (A) basal media; (C) growth media; (E) growth media supplemented with 100 ng/ml rhIGFBP-3. (B) hTCEpi cells cultured in basal media showed an increase in the proportion of cells in G0/G1 compared to growth conditions (***P<0.001, One-way ANOVA, Holm-Sidak multiple comparison test). Treatment with rhIGFBP-3 also produced a corresponding arrest in the cell cycle (***P<0.001, One-way ANOVA, Holm-Sidak multiple comparison test). (D) hTCEpi cells cultured in growth media showed an increase in the number of cells in S phase compared to cells cultured in basal media and growth media supplemented with rhIGFBP-3 (***P<0.001, One-way ANOVA, Holm-Sidak multiple comparison test). (F) Cells cultured in basal media showed a corresponding decrease in the number of cells in the G2/M phase of the cell cycle compared to growth media and growth media supplemented with rhIGFBP-3 (***P<0.001, One-way ANOVA, Holm-Sidak multiple comparison test). Data shown as a composite of 3 independent experiments performed in triplicate. (G) Quantitative determination of ATP levels (μM). hTCEpi cells were cultured in basal or growth media with or without 100 ng/ml rhIGFBP-3 for 24 hours. ATP levels were significantly higher in growth media with or without IGFBP-3 compared to basal media (***P<0.001; **P<0.01, One-way ANOVA, Holm-Sidak multiple comparison test). KGM: keratinocyte growth media; KBM: keratinocyte basal media; BP3: IGFBP-3. Data representative of 5 independent experiments performed in triplicate. All data represented as mean ± standard deviation.
In order to investigate the effects of IGFBP-3 on cellular respiration, we measured the level of ATP in epithelial cells incubated in basal media and after supplementation with rhIGFBP-3 in growth media for 24 hours. As expected, the level of ATP decreased significantly after culture in basal media (Fig. 6G). However, there was no significant difference in ATP levels in hTCEpi cells cultured in growth media with or without rhIGFBP-3. These data show that IGFBP-3 functions to regulate the cell cycle during proliferative growth conditions, but does not play a role in mediating cellular respiration.
IGF-1R nuclear translocation is regulated by IGFBP-3 and SUMOylation of IGF-1R
As described above, we observed an increase in IGF-1R following siRNA knockdown of IGFBP-3 in our cells. To determine whether IGFBP-3 impacts nuclear translocation and accumulation of IGF-1R after treatment with siRNAs targeting IGFBP-3 in basal media, we performed a nuclear fractionation (Fig. 7A). Immunoblotting for IGFBP-3 confirmed the efficiency of the knockdown. The addition of rhIGFBP-3 following knockdown restored IGFBP-3 levels in the cytosol, confirming cellular uptake. There was also an observed increase in IGFBP-3 in the insoluble nucleus. No IGFBP-3 was detected in the soluble nucleus, suggesting that nuclear IGFBP-3 may be complexed with DNA in the epithelial cell nucleus.
Fig. 7:
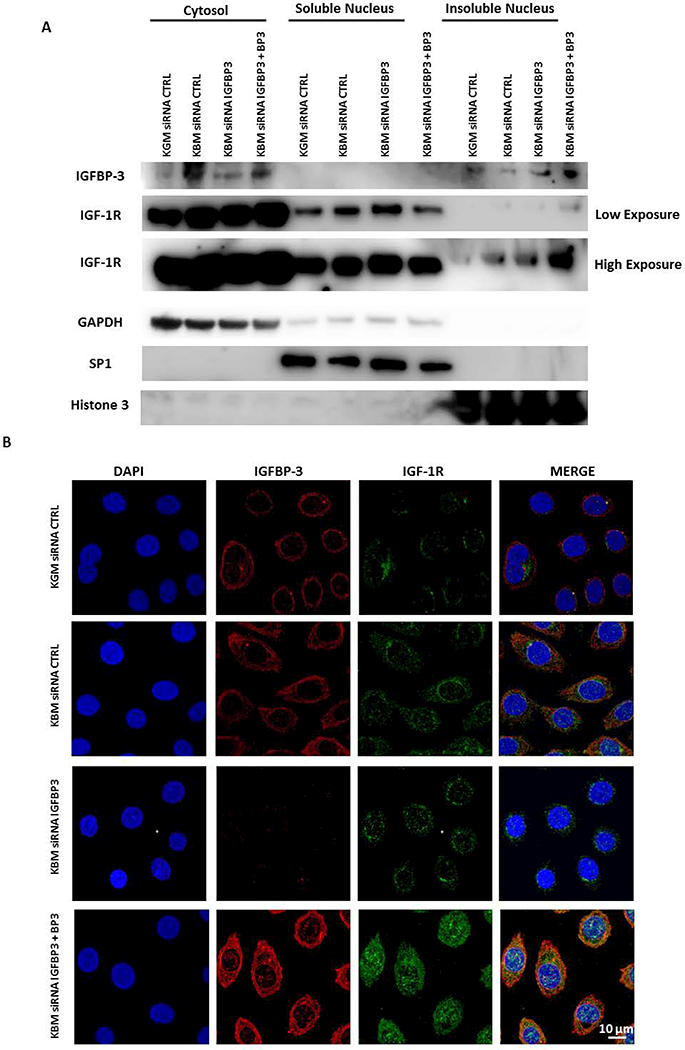
IGFBP-3 regulates IGF-1R nuclear translocation. hTCEpi cells were cultured in basal media and transfected with siRNAs for IGFBP-3 with or without 100 ng/ml rhIGFBP-3. (A) Cells were lysed and fractionated into cytosolic, soluble and insoluble nuclear fractions. Immunoblotting for IGF-1R showed an increase in IGF-1R expression in basal media compared to growth media in all fractions. The increase in IGF-1R was higher in the nuclear fraction after IGFBP-3 knockdown and further increased in the insoluble fraction after addition of exogenous rhIGFBP-3. Immunoblotting for IGFBP-3 confirmed knockdown and uptake of rhIGFBP-3. Immunoblotting for GAPDH, SP1 and Histone H3 were used for cytosolic, soluble nucleus, and insoluble nucleus fractionation controls, respectively. Data representative of 3 independent experiments. Low and high exposures shown for IGF-1R immunoblot. (B) Immunofluorescence for IGF-1R (green) and IGFBP-3 (red). Nuclei were counterstained with DAPI (blue). There was an increase in IGF-1R expression and nuclear accumulation in cells cultured in basal media compared to growth media. IGF-1R remained in the nucleus following IGFBP-3 knockdown. There was a robust increase in nuclear IGF-1R in the nucleus after rescue with exogenous rhIGFBP-3. Scale bar: 10 μm. KGM: keratinocyte growth media; KBM: keratinocyte basal media; BP3: IGFBP-3; CTRL: control. Data representative of 3 independent experiments.
Immunoblotting for IGF-1R under these same conditions demonstrated an increase in IGF-1R in basal media compared with cells cultured in growth media in all subcellular compartments tested (Fig. 7A). There was a small increase in IGF-1R in the soluble and insoluble nuclear fractions after siRNA knockdown of IGFBP-3. Subsequent rescue with rhIGFBP-3 further increased expression of IGF-1R in the insoluble nucleus. This coincided with a reduction in IGF-1R in the soluble nucleus. Taken together, the fractionation results confirm that IGF-1R is present in the soluble and insoluble nuclear fraction and that IGFBP-3 mediates nuclear translocation of IGF-1R. GAPDH, SP1, and Histone H3 for cytosolic, soluble nucleus, and insoluble nucleus loading controls, respectively, confirmed successful fractionation (Fig. 7A).
To further assess the effects of IGFBP-3 on nuclear accumulation of IGF-1R, we performed immunofluorescence staining for IGF-1R (Fig 7B). We found that there was low expression of IGF-1R in the nucleus of corneal epithelial cells cultured in growth media. Consistent with our immunoblot analysis, culture in basal media increased expression of IGF-1R in the nucleus and appeared to further increased, albeit slightly, following knockdown of IGFBP-3. Consistent with our immunoblotting data, we also found that rescue with rhIGFBP-3 induced robust nuclear accumulation (Fig. 7B). These data indicate a novel role for IGFBP-3 in the regulation of IGF-1R nuclear translocation in human corneal epithelial cells. We then used immunofluorescence to verify nuclear accumulation of IGF-1R in HCECs (Supplementary data Fig. 2). As we showed in hTCEpi cells, the addition of rhIGFBP-3 triggered an increase in nuclear localization of IGF-1R and IGFBP-3 (Supplementary data Fig. 2).
To determine whether SUMOylation mediated nuclear translocation of IGF-1R in corneal epithelial cells, we first analyzed global SUMOylation for SUMO-modifiers SUMO1 and SUMO2/3 in growth and basal conditions by immunoblotting (Fig. 8A). Our results indicate that there is greater expression of SUMO1 in cells cultured in growth media compared to basal media. In contrast to this, SUMO1 was decreased in basal media. This decrease coincided with an increase in SUMO-2/3.
Fig. 8:
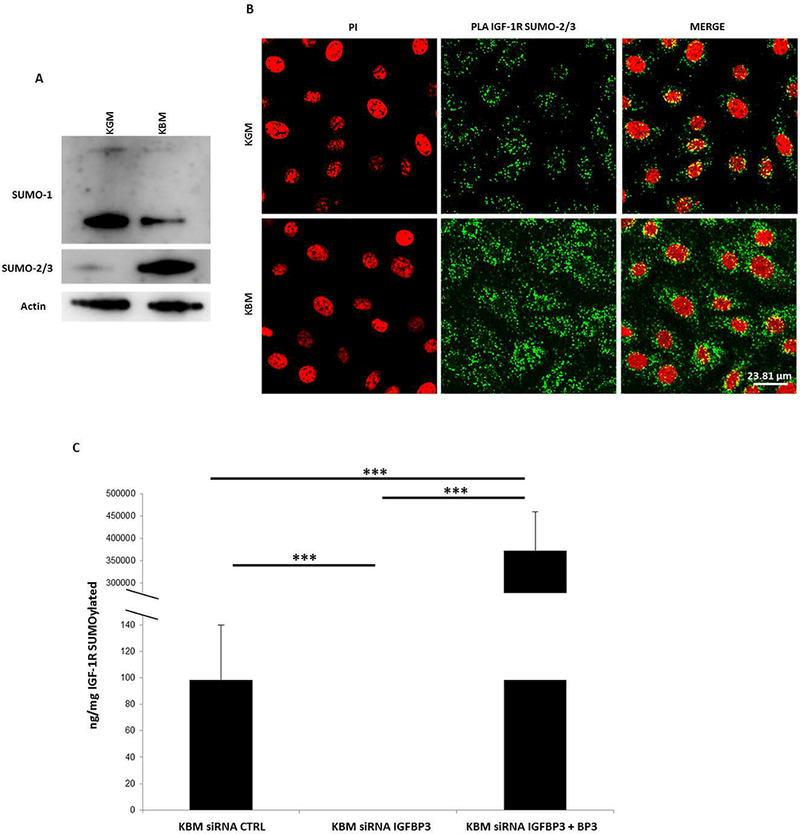
SUMO-2/3 but not SUMO-1 triggers IGF-1R nuclear translocation and DNA binding in corneal epithelial cells. (A) Immunoblotting for SUMO-1 and SUMO-2/3 showed a decrease in SUMO-1 in basal media compared with growth. Instead in basal media, there was an observable increase in SUMO-2/3. Data representative of 3 independent experiments. (B) Proximal Ligation Assay (PLA) shows an increase in IGF-1R SUMOylation by SUMO-2/3 (green) in the cytoplasm and nucleus in basal media compared with growth media. Scale bar: 23 μm. Data representative of 3 independent experiments. (C) The global SUMOylation assay confirmed a significant decrease in SUMOylated IGF-1R in nuclear extracts after knock down of IGFBP-3 in basal media (siRNA control versus siRNA IGFBP3 *P<0.05, siRNA IGFBP3 plus BP3 versus siRNA control and siRNA IGFBP3 in basal ***P<0.001, One-way ANOVA, Holm-Sidak multiple comparison test). KGM: keratinocyte growth media; KBM: keratinocyte basal media; BP3: IGFBP-3; CTRL: control; PI: propidium iodide. Data representative of 4 independent experiments performed in triplicate. Data shown as mean ± standard deviation.
To test whether IGF-1R is SUMOylated by SUMO2/3 in basal conditions, we performed a proximal ligation assay. Nuclei were counterstained with propidium iodide. We detected IGF-1R-SUMO2/3 in both growth and basal conditions. However, there was a clear increase in IGF-1R-SUMO-2/3 in basal conditions in both the cytosol and the nucleus (Fig. 8B). To further confirm that IGF-1R is SUMOylated in basal conditions, we performed a global SUMOylation colorimetric assay using a detection antibody specific for the alpha subunit of IGF-1R and a pan-SUMO detection antibody that recognized both SUMO1 and SUMO2/3 (Fig 8B). Epithelial cells were fractionated into nuclear extracts. SUMOylated IGF-1R was detectable in nuclear fractions in basal conditions. This was completely inhibited following knockdown of IGFBP-3. Rescue with rhIGFBP-3 not only restored but significantly increased SUMOylation of IGF-1R over basal levels. Given our finding of the shift from the soluble to insoluble nuclear fraction following rescue with rhIGFBP3, together, this data suggests that SUMOylation of IGF-1R may regulate IGF-1R/DNA interactions.
Discussion
The key finding in this manuscript is the novel mechanism in which IGFBP-3 triggers nuclear translocation of IGF-1R. Nuclear localization of IGF-1R has been shown in several other cell lines. In these prior studies, IGF-1 triggers the translocation of IGF-1R from the plasma membrane to the nucleus in serum-starved cells (Sehat et al., 2010). It has further been shown that nuclear translocation of IGF-1R is mediated by SUMO1 and the nuclear transport protein importin-β. In corneal epithelial cells however, which are cultured in serum-free conditions, there is an upregulation and nuclear accumulation in response to stress-induced by growth factor withdrawal. This is further enhanced following knockdown of the putative stress-response protein, IGFBP-3. More interestingly however, is the finding that knockdown of IGFBP-3 induces a compensatory increase in IGF-1R in the soluble nucleus, whereas rescue with exogenous rhIGFBP-3 triggers a shift in nuclear localized IGF-1R from the soluble to the insoluble nuclear fraction. This suggests that there is a potential increase in DNA binding. This finding is in agreement with studies by our laboratory and others that indicate IGF-1R binds DNA and functions as a transcriptional modulator (Sehat et al., 2010; Wu et al., 2012b).
One potential difference that may account for our findings is the presence of a nuclear IGF-1R/INSR hybrid. We previously reported that IGF-1R present in the corneal epithelial nucleus is a Hybrid receptor (Wu et al., 2012b). In contrast to this, tumor cells or cell lines that over-express IGF-1R may function in a divergent manner. IGF-1R has been shown to have oncogenic effects in cancer, and has been implicated in tumor growth, migration and angiogenesis (Heidegger et al., 2014). Likewise, the role of IGF-1R in the regulation of the cell cycle has already beem well documented in cancer cells (Warsito et al., 2012). Other studies have shown that knockdown of IGF-1R diminishes tumor cell proliferation and increases susceptibility of cancer cells to apoptosis (Ofer et al., 2015). In addition to the presence of a nuclear localized hybrid receptor (Hybrid-R), in the current study, we have further shown that under basal conditions that upregulate IGF-1R and INSR, IGF-1R becomes SUMOylated by SUMO 2/3. The functional differences between SUMO1 and SUMO 2/3 are not well described. There is speculation that there is functional redundancy between the two modifiers (Yuan et al., 2010). Others posit that SUMO 2/3 may be implicated in stress response pathways (Guo and Henley, 2014). The addition of SUMO modifiers have been shown to mediate and stabilize DNA binding (Wei et al., 2007). This potential role would be consistent with our finding of the shift in IGF-1R from the soluble to insoluble fraction.
The role of IGFBP-3 in corneal epithelial cells is not well defined. Previous work in our laboratory has shown that IGFBP-3 secretion by corneal epithelial cells is upregulated in response to hypoxia (data not shown) and hyperglycemia (Wu et al., 2012a). In human studies in vivo, we have further shown that IGFBP-3 is increased in human diabetic tears and is associated with damage to the corneal subbasal nerve plexus. Collectively, these findings suggest that IGFBP-3 functions as a stress-response protein in the corneal epithelium. Unlike IGF-1R, IGFBP-3 does contain a nuclear localization site. IGFBP-3 is also present within the insoluble nuclear fraction. Future studies are needed to determine whether IGFBP-3 is directly or indirectly involved in triggering the shift in localization of IGF-1R from the soluble to insoluble nucleus.
The second key finding in this paper is the mutual relationship between IGFBP-3 and IGF-1R. Under basal conditions, IGF-1R and INSR are transcriptionally upregulated (Titone et al., 2018). However, knockdown of IGF-1R and not INSR blocks the stress-induced upregulation of IGFBP-3. Inclusion of the Akt inhibitor confirms that this is not due to signaling from the Akt/PI3K pathway. Conversely, knockdown of IGFBP-3 in basal stress conditions increases expression levels of IGF-1R, whereas during growth, IGF-1R expression is unaltered by exogenous rhIGFBP-3. This suggests that the interplay between IGF-1R and IGFBP-3 is critical in modulating the epithelial stress response.
Given the pleiotropic roles for IGFBP-3, many of its reported functions are cell and tissue specific. It has been reported that IGFBP-3 may function as a gatekeeper or caretaker in different cells (Baxter, 2013). Here our data suggests that IGFBP-3 has dual roles in the corneal epithelium (Fig. 9). During growth, IGFBP-3 binds IGF-1 to prevent IGF-1 from activating IGF-1R or Hybrid-R, but does not alter cellular respiration, as shown by measured ATP levels. The ability of IGFBP-3 to bind IGF-1 and induce cell cycle arrest is consistent with a gatekeeper role. In contrast to this, the addition of rhIGFBP-3 to stressed epithelial cells with already reduced levels of IGFBP-3, selectively targets IGF-1R to the insoluble nuclear fraction where it can bind DNA. This finding indicates a caretaker role for IGFBP-3. Studies to identify the nuclear targets of IGF-1R are on-going.
Fig. 9:
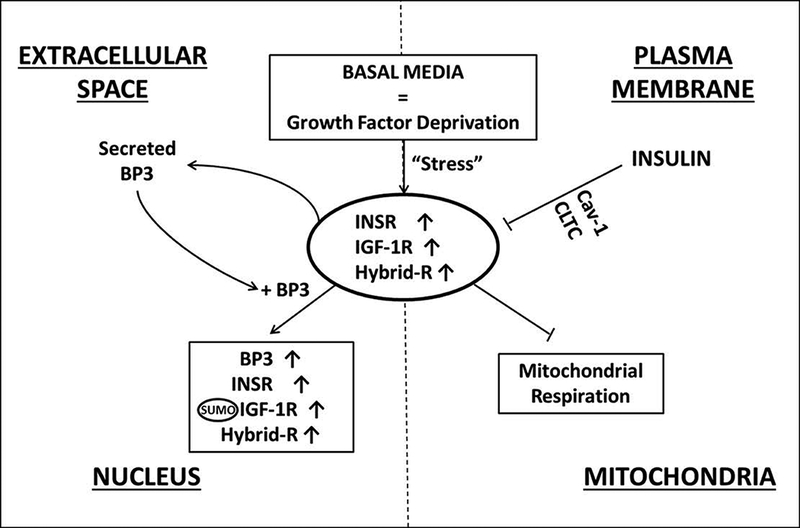
Schematic summary of IGF-1R and IGFBP-3 in corneal epithelial cells in response to stress induced by growth factor withdrawal. INSR, IGF-1R, and Hybrid-R are increased when cultured in basal media devoid of growth factors. This effect is mediated by the action of insulin at the plasma membrane. The induction of cellular stress triggers an increase in secreted IGFBP-3 into the extracellular space. During the growth phase, IGFBP-3 mediates cell cycle arrest. During stress however, IGFBP-3 mediates translocation of IGF-1R to the insoluble nucleus and potentially increases DNA binding and transcriptional modulation. In contrast to other studies, IGF-1R is SUMOylated by SUMO2/3, which may function to drive the translocation of IGF-1R from the soluble to insoluble nuclear fraction. BP3: IGFBP-3.
The membrane proteins Cav-1 and CLTC have been shown to mediate the internalization of IGF-1R and INSR (Huo et al., 2003; Salani et al., 2010). Recent studies show that Cav-1 plays a role in insulin uptake and in insulin resistance in diabetes (Stralfors, 2012). Here we show that in corneal epithelial cells, at the plasma membrane insulin uptake is mediated by Cav-1 and CLTC. Our data further shows that knock down of CAV-1 and CLTC does not alter secretion of IGFBP-3. Knockdown of CAV-1 and CLTC does however, blunt the ability of insulin to restore IGFBP-3 secretion to normal growth levels. We speculate that insulin regulates IGFBP-3 through interactions with the IGF-1R or Hybrid-R at the cell surface, since knockdown of INSR failed to alter IGFBP-3 secretion.
In summary, our data shows, for the first time, the reciprocal regulation between IGF-1R and IGFBP-3. These data also suggest that IGFBP-3 may play a critical role in mediating corneal epithelial stress responses by triggering nuclear translocation of IGF-1R independent of IGF-1. The corneal epithelium is continuously subjected to high amounts of stress, including hyperosmolarity, hyperglycemia, oxidative stress, and sheer stress from blinking. Further studies are necessary to identify key regulatory targets for IGF-1R, including a potential role for IGF-1R in gene modulation in the cornea.
Supplementary Material
Acknowledgements:
D.M.R. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis. Author contributions: RT designed and performed the experiments, analyzed data and wrote the manuscript. MZ designed and performed experiments. DMR designed experiments, analyzed data and wrote the manuscript.
Funding information: This study was funded by NIH/NEI grants EY02443 (DMR), EY024546 (DMR), NIH Core Grant EY020799, and an unrestricted grant from Research to Prevent Blindness, New York, NY.
Footnotes
None of the authors have any conflicts of interest to disclose.
References
- Arteaga CL. 1992. Interference of the IGF system as a strategy to inhibit breast cancer growth. Breast cancer research and treatment 22(1):101–106. [DOI] [PubMed] [Google Scholar]
- Baxter RC. 2013. Insulin-like growth factor binding protein-3 (IGFBP-3): Novel ligands mediate unexpected functions. Journal of cell communication and signaling 7(3):179–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baxter RC, Martin JL, Tyler MI, Howden ME. 1986. Growth hormone-dependent insulin-like growth factor (IGF) binding protein from human plasma differs from other human IGF binding proteins. Biochemical and biophysical research communications 139(3):1256–1261. [DOI] [PubMed] [Google Scholar]
- Butt AJ, Fraley KA, Firth SM, Baxter RC. 2002. IGF-binding protein-3-induced growth inhibition and apoptosis do not require cell surface binding and nuclear translocation in human breast cancer cells. Endocrinology 143(7):2693–2699. [DOI] [PubMed] [Google Scholar]
- Chen H, Yan GC, Gishizky ML. 1998. Identification of structural characteristics that contribute to a difference in antiapoptotic function between human insulin and insulin-like growth factor 1 receptors. Cell growth & differentiation : the molecular biology journal of the American Association for Cancer Research 9(11):939–947. [PubMed] [Google Scholar]
- Cubbon RM, Kearney MT, Wheatcroft SB. 2016. Endothelial IGF-1 Receptor Signalling in Diabetes and Insulin Resistance. Trends in endocrinology and metabolism: TEM 27(2):96–104. [DOI] [PubMed] [Google Scholar]
- Furstenberger G, Senn HJ. 2002. Insulin-like growth factors and cancer. The Lancet Oncology 3(5):298–302. [DOI] [PubMed] [Google Scholar]
- Grimberg A, Coleman CM, Burns TF, Himelstein BP, Koch CJ, Cohen P, El-Deiry WS. 2005. p53-Dependent and p53-independent induction of insulin-like growth factor binding protein-3 by deoxyribonucleic acid damage and hypoxia. The Journal of clinical endocrinology and metabolism 90(6):3568–3574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimberg A, Liu B, Bannerman P, El-Deiry WS, Cohen P. 2002. IGFBP-3 mediates p53-induced apoptosis during serum starvation. International journal of oncology 21(2):327–335. [PMC free article] [PubMed] [Google Scholar]
- Grkovic S, O’Reilly VC, Han S, Hong M, Baxter RC, Firth SM. 2013. IGFBP-3 binds GRP78, stimulates autophagy and promotes the survival of breast cancer cells exposed to adverse microenvironments. Oncogene 32(19):2412–2420. [DOI] [PubMed] [Google Scholar]
- Guo C, Henley JM. 2014. Wrestling with stress: roles of protein SUMOylation and deSUMOylation in cell stress response. IUBMB life 66(2):71–77. [DOI] [PubMed] [Google Scholar]
- Heidegger I, Kern J, Ofer P, Klocker H, Massoner P. 2014. Oncogenic functions of IGF1R and INSR in prostate cancer include enhanced tumor growth, cell migration and angiogenesis. Oncotarget 5(9):2723–2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huo H, Guo X, Hong S, Jiang M, Liu X, Liao K. 2003. Lipid rafts/caveolae are essential for insulin-like growth factor-1 receptor signaling during 3T3-L1 preadipocyte differentiation induction. The Journal of biological chemistry 278(13):11561–11569. [DOI] [PubMed] [Google Scholar]
- Imrie H, Viswambharan H, Sukumar P, Abbas A, Cubbon RM, Yuldasheva N, Gage M, Smith J, Galloway S, Skromna A, Rashid ST, Futers TS, Xuan S, Gatenby VK, Grant PJ, Channon KM, Beech DJ, Wheatcroft SB, Kearney MT. 2012. Novel role of the IGF-1 receptor in endothelial function and repair: studies in endothelium-targeted IGF-1 receptor transgenic mice. Diabetes 61(9):2359–2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobo SM, Kazlauskas A. 2015. Insulin-like growth factor 1 (IGF-1) stabilizes nascent blood vessels. The Journal of biological chemistry 290(10):6349–6360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, Choi DS, Lee OH, Oh SH, Lippman SM, Lee HY. 2011. Antiangiogenic antitumor activities of IGFBP-3 are mediated by IGF-independent suppression of Erk1/2 activation and Egr-1-mediated transcriptional events. Blood 118(9):2622–2631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laron Z 2001. Insulin-like growth factor 1 (IGF-1): a growth hormone. Molecular pathology : MP 54(5):311–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y, Liu H, Waraky A, Haglund F, Agarwal P, Jernberg-Wiklund H, Warsito D, Larsson O. 2017. SUMO-modified insulin-like growth factor 1 receptor (IGF-1R) increases cell cycle progression and cell proliferation. Journal of cellular physiology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myal Y, Shiu RP, Bhaumick B, Bala M. 1984. Receptor binding and growth-promoting activity of insulin-like growth factors in human breast cancer cells (T-47D) in culture. Cancer research 44(12 Pt 1):5486–5490. [PubMed] [Google Scholar]
- Ofer P, Heidegger I, Eder IE, Schopf B, Neuwirt H, Geley S, Klocker H, Massoner P. 2015. Both IGF1R and INSR Knockdown Exert Antitumorigenic Effects in Prostate Cancer In Vitro and In Vivo. Molecular endocrinology (Baltimore, Md) 29(12):1694–1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandini G, Frasca F, Mineo R, Sciacca L, Vigneri R, Belfiore A. 2002. Insulin/insulin-like growth factor I hybrid receptors have different biological characteristics depending on the insulin receptor isoform involved. The Journal of biological chemistry 277(42):39684–39695. [DOI] [PubMed] [Google Scholar]
- Robertson DM, Ho SI, Hansen BS, Petroll WM, Cavanagh HD. 2007. Insulin-like Growth Factor Binding Protein-3 expression in the human corneal epithelium. Experimental eye research 85(4):492–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson DM, Li L, Fisher S, Pearce VP, Shay JW, Wright WE, Cavanagh HD, Jester JV. 2005. Characterization of growth and differentiation in a telomerase-immortalized human corneal epithelial cell line. Investigative ophthalmology & visual science 46(2):470–478. [DOI] [PubMed] [Google Scholar]
- Salani B, Passalacqua M, Maffioli S, Briatore L, Hamoudane M, Contini P, Cordera R, Maggi D. 2010. IGF-IR internalizes with Caveolin-1 and PTRF/Cavin in HaCat cells. PLoS One 5(11):e14157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sehat B, Tofigh A, Lin Y, Trocme E, Liljedahl U, Lagergren J, Larsson O. 2010. SUMOylation mediates the nuclear translocation and signaling of the IGF-1 receptor. Science signaling 3(108):ra10. [DOI] [PubMed] [Google Scholar]
- Soos MA, Whittaker J, Lammers R, Ullrich A, Siddle K. 1990. Receptors for insulin and insulin-like growth factor-I can form hybrid dimers. Characterisation of hybrid receptors in transfected cells. The Biochemical journal 270(2):383–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stralfors P 2012. Caveolins and caveolae, roles in insulin signalling and diabetes. Advances in experimental medicine and biology 729:111–126. [DOI] [PubMed] [Google Scholar]
- Stuard WL, Titone R, Robertson DM. 2017. Tear Levels of Insulin-Like Growth Factor Binding Protein 3 Correlate With Subbasal Nerve Plexus Changes in Patients With Type 2 Diabetes Mellitus. Investigative ophthalmology & visual science 58(14):6105–6112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thum T, Fleissner F, Klink I, Tsikas D, Jakob M, Bauersachs J, Stichtenoth DO. 2007. Growth hormone treatment improves markers of systemic nitric oxide bioavailability via insulin-like growth factor-I. The Journal of clinical endocrinology and metabolism 92(11):4172–4179. [DOI] [PubMed] [Google Scholar]
- Titone R, Zhu M, Robertson DM. 2018. Insulin mediates de novo nuclear accumulation of the IGF-1/insulin Hybrid Receptor in corneal epithelial cells. Scientific reports 8(1):4378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warsito D, Sjostrom S, Andersson S, Larsson O, Sehat B. 2012. Nuclear IGF1R is a transcriptional co-activator of LEF1/TCF. EMBO reports 13(3):244–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei F, Scholer HR, Atchison ML. 2007. Sumoylation of Oct4 enhances its stability, DNA binding, and transactivation. The Journal of biological chemistry 282(29):21551–21560. [DOI] [PubMed] [Google Scholar]
- Werner H, Weinstein D, Bentov I. 2008. Similarities and differences between insulin and IGF-I: structures, receptors, and signalling pathways. Archives of physiology and biochemistry 114(1):17–22. [DOI] [PubMed] [Google Scholar]
- Wu YC, Buckner BR, Zhu M, Cavanagh HD, Robertson DM. 2012a. Elevated IGFBP3 levels in diabetic tears: a negative regulator of IGF-1 signaling in the corneal epithelium. The ocular surface 10(2):100–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu YC, Zhu M, Robertson DM. 2012b. Novel nuclear localization and potential function of insulin-like growth factor-1 receptor/insulin receptor hybrid in corneal epithelial cells. PLoS One 7(8):e42483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan H, Zhou J, Deng M, Liu X, Le Bras M, de The H, Chen SJ, Chen Z, Liu TX, Zhu J. 2010. Small ubiquitin-related modifier paralogs are indispensable but functionally redundant during early development of zebrafish. Cell research 20(2):185–196. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Steinle JJ. 2013. DNA-PK phosphorylation of IGFBP-3 is required to prevent apoptosis in retinal endothelial cells cultured in high glucose. Investigative ophthalmology & visual science 54(4):3052–3057. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.