

Abstract
A series of 13 acyclic nucleoside phosphonates (ANPs) as bisamidate prodrugs was prepared. Five compounds were found to be non-cytotoxic and selective inhibitors of Bordetella pertussis adenylate cyclase toxin (ACT) in J774A.1 macrophage cell-based assays. The 8-aza-7-deazapurine derivative of adefovir (PMEA) was the most potent ACT inhibitor in the series (IC50 = 16 nM) with substantial selectivity over mammalian adenylate cyclases (mACs). AC inhibitory properties of the most potent analogues were confirmed by direct evaluation of the corresponding phosphonodiphosphates in cell-free assays and were found to be potent inhibitors of both ACT and edema factor (EF) from Bacillus anthracis (IC50 values ranging from 0.5 to 21 nM). Moreover, 7-halo-7-deazapurine analogues of PMEA were discovered to be potent and selective mammalian AC1 inhibitors (no inhibition of AC2 and AC5) with IC50 values ranging from 4.1–5.6 µM in HEK293 cell-based assays.
Keywords: adenylate cyclase, inhibitor, Bordetella pertussis, Bacillus anthracis, adefovir
Graphical Abstract
A novel series of acyclic nucleoside phosphonates derived from adefovir (PMEA) with modified purine nucleobases was prepared where some compounds (e.g. 8-aza-7-deazapurine analogue) are potent and selective inhibitors of bacterial adenylate cyclases (adenylate cyclase toxin from B. pertussis and edema factor from B. anthracis) and some analogues (7-halo-7-deazapurine analogues) are selective inhibitors of mammalian AC1 over AC2 and AC5.
Introduction
Bordetella pertussis, a strictly human pathogen, causes acute or chronic respiratory infections in the tracheobronchial tree, called whooping cough.1 B. pertussis is easily transmitted from infected to susceptible people through droplets. It is estimated that there were 24,1 million pertussis cases and 160 700 deaths from pertussis in children younger than 5 years in 2014,2 whereas the most endangered group are children aged less than 12 months.3,4
B. pertussis is usually treated with antibiotics.5,6 Despite the fact that B. pertussis resistance to antibiotics has been reported sporadically,7 the level of bacterial resistance keeps rising.8 Bordetella pertussis produces several virulent factors,9–11 which help it to evade the host organism during the infection.10 The calmodulin-dependent adenylate cyclase toxin (ACT) is considered to be an essential virulence factor,11 since it has been demonstrated that the virulence of B. pertussis ACT-deficient mutant was significantly reduced.12–14 Thus, the development of potential antitoxin therapies represents a viable approach for whooping cough treatment.
ACT binds to the surface receptors (integrin CD11b/CD18) on myeloid phagocytic cells and transports its AC domain into the cytosol in a two-step process.15 The pore-forming activity16,17 leads to an increased intracellular calcium concentration and calmodulin-mediated activation of ACT which is responsible for the conversion of intracellular ATP into the second messenger cAMP.10,13,18–20 This massive and non-physiological increases of intracellular cAMP levels completely disrupt cellular signalling pathways. ACT has been shown to impair a number of key metabolic functions of human immune effector cells,21–26 facilitating an effective invasion and colonization of the toxin-producing bacteria in the host organism. Moreover, the suppression of the immune response can make the host organism more vulnerable to additional infections.
Acyclic nucleoside phosphonates (ANPs)27 possess a broad spectrum of biological activities. The most pronounced is their antiviral effect,28 however ANPs exhibit also cytostatic,29,30, antiparasitic,31–35 antibacterial,36 and immuno-modulatory37–40 properties. It was also found that the approved anti-HBV drug adefovir dipivoxil (bis(POM)PMEA, I, Fig. 1), upon its intracellular conversion into the active metabolite adefovir diphosphate (PMEApp, II, Fig. 1), inhibited Bordetella pertussis ACT.41 Bis(POM)PMEA (I) was also active against adenylate cyclase-based edema factor (EF) from Bacillus anthracis in CHO and BMMΦ cells.42 Recently, various bisamidate prodrugs of PMEA, e.g. bis(L-phenylalanine isopropyl ester) PMEA (III, Fig. 1), with enhanced plasma stability profile and low toxicity were evaluated as potentially more suitable drug candidates for antitoxin therapy despite their somewhat lower efficacy to inhibit ACT compared with bis(POM)PMEA (I).43 Based on these results,43 and taking into account various aspects of the prodrug strategy (stability, cytotoxicity, cell membrane permeability, ease of synthesis and handling), L-phenylalanine isopropyl ester moiety (as in compound III, Fig. 1) has been selected as a representative type of bisamidate prodrug for the evaluation of any other novel nucleotide analogues.
Figure 1.
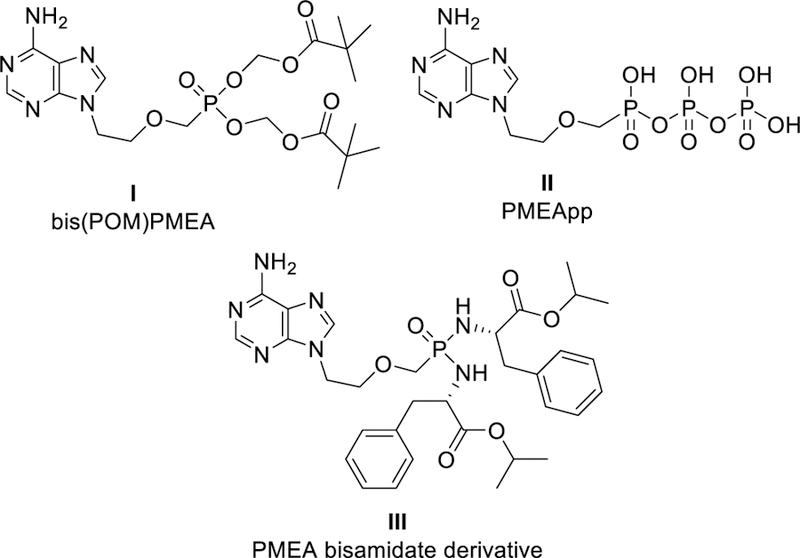
PMEA prodrugs I and III and metabolically active PMEA diphosphate II.
The present work is a part of a broad structure-activity relationship (SAR) study mapping the influence of each structural part of the acyclic nucleotide analogues, i.e. an aliphatic moiety and a nucleobase, on the inhibition of bacterial adenylate cyclases, namely ACT and EF (Fig. 2). In recent years, the molecule of adefovir (PMEA) was structurally modified and evaluated in this regard, but no significant improvement of the ACT inhibition was observed with ANPs bearing variously modified aliphatic chains (chain length, oxygen position and/or branching, unpublished results). Thus, the presence of the unmodified 2-(phosphonomethoxy)ethyl (PME) moiety turned out to be crucial for the preservation of the ACT inhibition.
Figure 2.
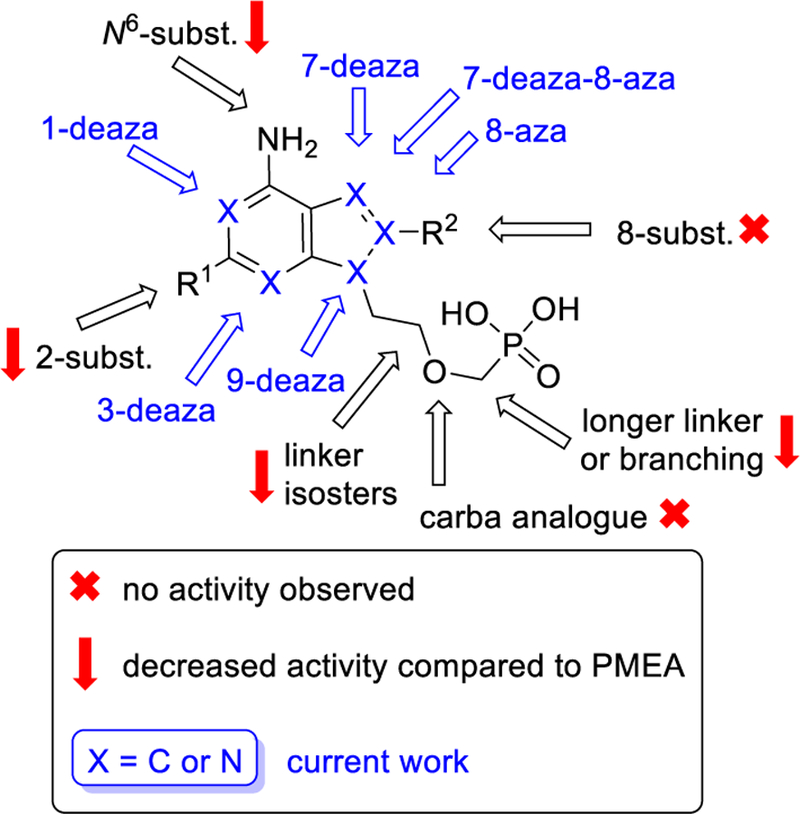
Overview of the structure activity relationship studies of adefovir (PMEA) analogues as ACT inhibitors. Blue colour depicts the positions on the purine scaffold modified in this work, namely positions C-1, C-3, C-7, C-8, and C-9.
Next, we tried to modify the adenine moiety by means of various substitutions. A modification of the 6-amino group by its alkylation led to a decrease of activity, while a substitution of the C-8 position (e.g. amino, oxo, thio, methylthio derivatives, Fig. 2) resulted in a complete loss of the antitoxin activity (unpublished results). On the other hand, PMEA bisamidates substituted with various functional groups in the C-2 position of the purine moiety showed activity against ACT in a cell-based assay, however, they were weaker inhibitors than the corresponding base-unsubstituted PMEA derivative III (Fig. 1).44
Further structural changes of the heterocyclic scaffold, namely aza- and deaza-modifications, supported by the docking into the crystal structure of the ACT – PMEApp complex,41,45 represent the next logical step in the design of future ACT inhibitors. Herein, we prepared and evaluated a series of ANPs derived from adefovir (PMEA) derivative III with modified nucleobases that are more or less able to mimic the adenine scaffold (Fig. 2). The common feature of such nucleobases is the bicyclic heterocycle (usually 6 + 5 atoms) and the presence of an unmodified “6-aminopurine” group for potential hydrogen bond interactions with the target enzymes (ACT and EF).
Results and Discussion
Synthesis.
First, we aimed to prepare all four possible mono-deazaadenine analogues of PMEA (adefovir), namely 1-deaza-PMEA, 3-deaza-PMEA, 7-deaza-PMEA, and 9-deaza-PMEA, in the form of their isopropyl ester bis(L-phenylalanine) bisamidate prodrugs. Phosphonates 1a and 1b (Scheme 1), prepared according to previously reported procedures,46, 47 served as the starting material for the synthesis of 1-deaza- and 3-deazaadenine analogues, compounds 2a and 2b, respectively. Silyl esters, preformed from isopropyl esters 1a and 1b (using TMSBr in pyridine at room temperature) were treated with L-phenylalanine isopropyl ester under standard reaction conditions (Aldrithiol-2, pyridine, Et3N, 70 °C), developed previously in our laboratory,48 to give desired bisamidates 2a and 2b in 60% and 16% yields, respectively.
Scheme 1.
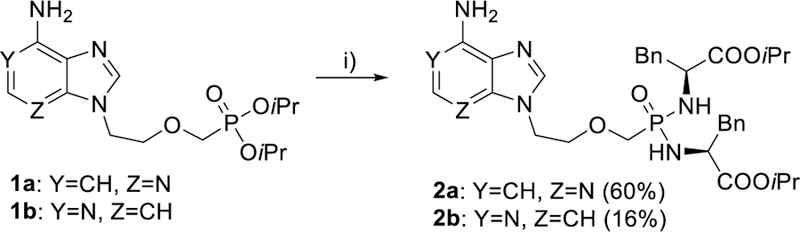
Synthesis of 1-deaza- and 3-deazaadenine derivatives 2a and 2b. Reagents and conditions: i) TMSBr, pyridine, rt; then (L)-NH2CH(Bn)COOiPr·HCl, PPh3, Aldrithiol-2, pyridine, Et3N, 70 °C.
The synthesis of 7-deazaadenine analogue 6a and its 7-halogenated versions 6b–6e (Scheme 2) started from commercially available 7-deazapurine (7H-pyrrolo[2,3-d]pyrimidine) derivatives 3a–3e. Although phosphonate diester 5a (Scheme 2) has been reported by Holý et al.,49 here we developed a more efficient method of its synthesis. Alkylation of 6-chloro-7-deazapurine (3a) with diisopropyl [(2-chloroethoxy)-methyl]phosphonate49 in the presence of Cs2CO3 in DMSO at 80 °C, followed by ammonolysis with ethanolic ammonia at 100 °C afforded compound 5a in a 39% overall yield (compared to an overall 23% yield of 5a,49 when starting from 2-methylsulfanyl-7-deazaadenine). Compounds 3b–3e were analogously converted into 7-halo-7-deazaadenine derivatives 5b–5e (Scheme 2) in two steps and with good yields. Corresponding bisamidates 6a–6e (Scheme 2) were then prepared from phosphonate diesters 5a–5e by the standard procedure.48
Scheme 2.
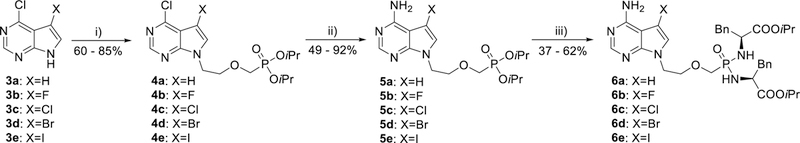
Synthesis of 7-deazadenine derivatives 6a–6e. Reagents and conditions: i) Cl(CH2)2OCH2P(O)(OiPr)2, Cs2CO3, DMSO, 80 °C; ii) EtOH/NH3, 100 °C; iii) TMSBr, pyridine, rt; then (L)-NH2CH(Bn)COOiPr·HCl, PPh3, Aldrithiol-2, pyridine, Et3N, 70 °C.
To the best of our knowledge, synthesis of 9-deaza-PMEA (14, Scheme 3) has not been reported so far. This might have been due to its demanding synthesis and/or inaccessibility of suitable C-C cross-coupling methods at times, when the other aza/deaza analogues were prepared.46,47,49 In order to synthesize 9-deaza-PMEA and its derivatives for biological evaluation, commercially available 6-chloro-9-deazapurine (4-chloro-5H-pyrrolo[3,2-d]pyrimidine, 7, Scheme 3) was first subjected to iodination with N-iodosuccinimide in THF to give 6-chloro-9-iodo-9-deazapurine (8) in a 85% yield.50 After the introduction of SEM protecting group using (2-chloromethoxyethyl)trimethylsilane (SEMCl) and NaH in DMF, SEM derivative 9 was subjected to the Stille cross-coupling reaction with vinyltributyltin under catalysis of Pd(t-Bu3P)2 in THF to afford 9-vinyl-9-deazapurine derivative 10 (Scheme 3) in a 69% yield. These are the optimized reaction conditions as various protecting groups at the N-7 position and several other conditions were evaluated as well. For example, the use of other palladium catalysts during the Stille cross-coupling reaction of compound 9 with vinyltributyltin led to complex reaction mixtures containing various ratios of the desired product 10, the homocoupling product, starting compound 9, as well as the dehalogenated starting compound. Utilization of the bromo analogue of compound 9, i.e. 9-bromo-6-chloro-9-deazapurine, as starting compound also significantly reduced yields of the desired intermediate 10.
Scheme 3.
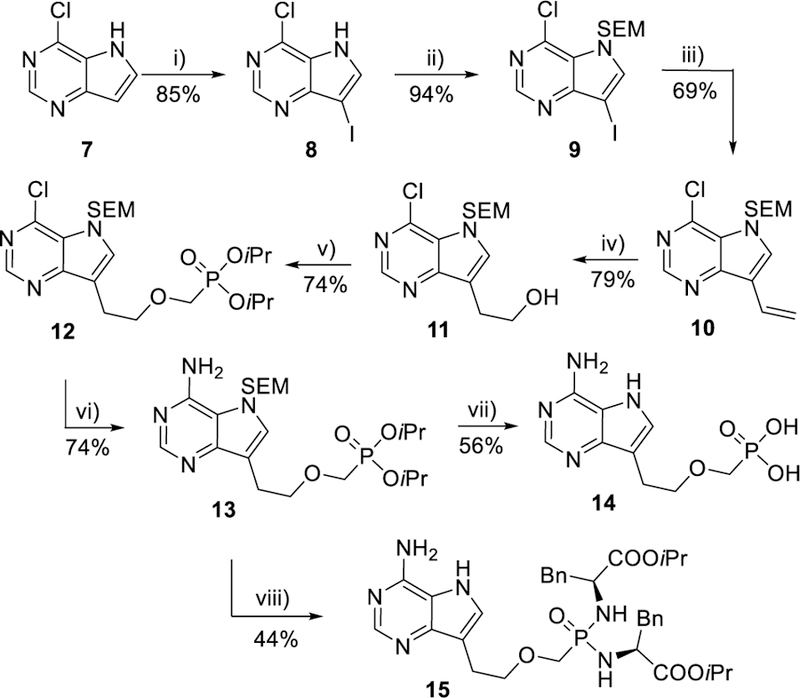
Synthesis of 9-deazapurine derivatives 14 and 15. Reagents and conditions: i) NIS, THF, rt; ii) (CH3)3Si(CH2)2OCH2Cl, NaH, DMF, rt; iii) CH2=CHSnBu3, Pd(t-Bu3P)2, THF, rt; iv) 9-BBN, THF, 0 °C to rt; then aq. NaBO3; v) n-BuLi, CF3SO2OCH2P(O)(OiPr)2, THF, −78 °C; vi) EtOH/NH3, 100 °C; vii) HCl 2 eq, H2O,130 °C; viii) TMSBr/TMSI, pyridine, rt; then (L)-NH2CH(Bn)COOiPr·HCl, PPh3, Aldrithiol-2, pyridine, Et3N, 70 °C.
Desired 9-(2-hydroxyethyl)-9-deazapurine derivative 11 (Scheme 3) was obtained in a 79% yield by hydroboration51 of compound 10, followed by in situ oxidation with aqueous sodium perborate. The subsequent alkylation of compound 11 to give phosphonate 12 was accomplished by modified alkylation conditions52 using CF3SO2OCH2P(O)(OiPr)2 in the presence of n-BuLi at low temperature. Obtained phosphonate 12 was treated with ethanolic ammonia to give protected 9-deazaadenine intermediate 13 in a good yield. The simultaneous removal of SEM and isopropyl ester groups from 13 using microwave-assisted hydrolysis with aqueous HCl53 yielded free phosphonic acid 14 in a 56% yield, while treatment of 13 by the standard procedure48 (with an addition of TMSI to remove SEM protecting group) afforded desired bisamidate prodrug 15 (Scheme 3) in a 44% yield.
Secondly, we focused on the synthesis of the 8-aza-7-deazapurine and 8-azapurine analogues, compounds 18a and 18b (Scheme 4), respectively. Diisopropyl ester 17a was synthesized from 8-aza-7-deazaadenine (16a) in a 53% yield using an analogous procedure as above (heating of diisopropyl [(2-chloroethoxy)methyl]phosphonate49 with preformed sodium salt of the nucleobase in DMF at 100 °C) reported previously for the preparation of the corresponding phosphonate diethyl ester.54 Similarly, alkylation of 8-azaadenine (16b) with the same alkylating agent in DMF using DBU as a base afforded desired product 17b in a 32% yield (together with 32% of the corresponding 8-regioisomer).55 Finally, phosphonate diesters 17a and 17b were converted to bisamidates 18a and 18b by the standard procedure48 (Scheme 4) in moderate yields.
Scheme 4.
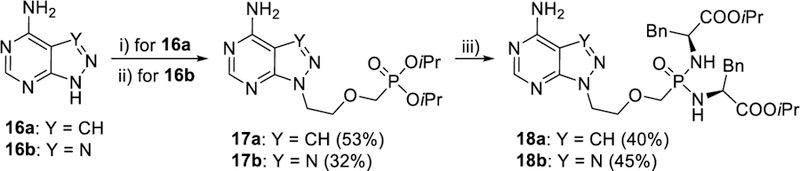
Synthesis of compounds 18a and 18b. Reagents and conditions: i) NaH, DMF, 80 °C, then Cl(CH2)2OCH2P(O)(OiPr)2, DMF, 100 °C; ii) Cl(CH2)2OCH2P(O)(OiPr)2, DBU, DMF, 100 °C; iii) TMSBr, pyridine, rt; then (L)-NH2CH(Bn)COOiPr·HCl, PPh3, Aldrithiol-2, pyridine, Et3N, 70 °C.
We have decided to extend our structure-activity relationship study for ANPs bearing other non-purine bases that are able to mimic the adenine moiety and preserve the exocyclic “6-aminopurine” group. Thus, thieno[3,2-d]pyrimidine, quinazoline, and pyrrolo[2,1-f][1,2,4]triazine were selected as suitable starting materials. The common feature of these heterocycles is their 9-deaza character. Thus, the above reported methodology for the preparation of 9-deaza-PMEA prodrug 15 (Scheme 3) was employed for the synthesis of target compounds.
Treatment of commercially available 7-bromo-4-chloro-thieno[3,2-d]pyrimidine (19, Scheme 5) with vinyltributyltin under the above developed Stille cross-coupling reaction conditions (catalysis with Pd(t-Bu3P)2 in THF) afforded only traces of the desired vinyl derivative 20, but an addition of CuI increased the yield of 20 to 50%. Tandem hydroboration and oxidation of vinyl compound 20 gave 2-hydroxyethyl derivative 21 in a good yield, as well as the subsequent alkylation of the hydroxyl group with CF3SO2OCH2P(O)(OiPr)2 in the presence of BuLi at −78 °C to yield phosphonate 22 (Scheme 5). Bisamidate prodrug 24 was obtained in good yield by ammonolysis of chloro derivative 22 (to give 23 in a 66% yield), followed by treatment of phosphonate 23 by the standard procedure.48
Scheme 5.
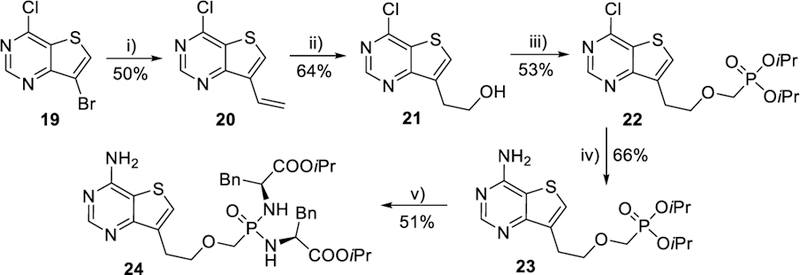
Synthesis of thieno[3,2-d]pyrimidine analogue 24. Reagents and conditions: i) CH2=CHSnBu3, Pd(t-Bu3P)2, CuI, dioxane, 120 °C; ii) 1) 9-BBN, THF, 0 °C to rt, 2) aq. NaBO3; iii) n-BuLi, CF3SO2OCH2P(O)(OiPr)2, THF, −78 °C; iv) EtOH/NH3, 100 °C; v) TMSBr, pyridine, rt; then (L)-NH2CH(Bn)COOiPr·HCl, PPh3, Aldrithiol-2, pyridine, Et3N, 70 °C.
Starting 8-bromoquinazolin-4(3H)-one (25, Scheme 6) was first treated with 2,4-dimethoxybenzylamine, BOP reagent, and DBU at elevated temperature to afford 2,4-dimethoxybenzyl (DMB)-protected 8-bromo-4-aminochinazoline 26. Compound 26 was then converted step by step into vinyl derivative 27 (Stille cross-coupling with vinyltributyltin, Pd(t-Bu3P)2 and CuI in N-methylpyrrolidone), into 2-hydroxyethyl compound 28 (hydroboration51 and in-situ oxidation), and into phosphonate 29 (alkylation with CF3SO2OCH2P(O)(OiPr)2). DMB deprotection with TFA in DCM (to give 30 in a 49% yield) followed by standard bisamidate formation48 yielded final analogue 31 (Scheme 6) in a good overall yield.
Scheme 6.
Synthesis of 4-aminoquinazoline analogue 31. i) BOP reagent, DBU, DMF, CH3CN, 2,4-dimethoxybenzylamine, 80 °C; ii) CH2=CHSnBu3, Pd(t-Bu3P)2, CuI, dioxane, 120 °C; iii) 9-BBN, 0 °C to RT; then aq. NaBO3; iv) n-BuLi, CF3SO2OCH2P(O)(OiPr)2, THF, −78 °C; v) CF3COOH, CH2Cl2, rt; vi) TMSBr, pyridine, rt; then (L)-NH2CH(Bn)COOiPr·HCl, PPh3, Aldrithiol-2, pyridine, Et3N, 70 °C.
Commercially available 4-chloropyrrolo[2,1-f][1,2,4]triazine (32, Scheme 7) was transformed, using microwave-assisted treatment with 2,4-dimethoxybenzylamine and Et3N in absolute ethanol, into DMB-protected derivative 33, which was then iodinated with NIS to give compound 34 in a high yield. 4-Aminopyrrolo[2,1-f][1,2,4]triazine derivative 39 (Scheme 7) was prepared in a good overall yield starting from the iodo derivative 34 in analogy to the step by step synthetic methodology described for compound 31 (Scheme 6).
Scheme 7.
Synthesis of 4-aminopyrrolo[2,1-f][1,2,4]triazine derivative 39. i) Et3N, EtOH, 2,4-dimethoxybenzylamine, 120 °C MW; ii) NIS, CH3CN, RT; iii) CH2=CHSnBu3, Pd(t-Bu3P)2, CuI, NMP, 120 °C; iv) 9-BBN, THF, 0 °C to RT; then aq. NaBO3; v) n-BuLi, CF3SO2OCH2P(O)(OiPr)2, THF, −78 °C; vi) CF3COOH, CH2Cl2, rt; vii) TMSBr, pyridine, rt then (L)-NH2CH(Bn)COOiPr·HCl, PPh3, Aldrithiol-2, pyridine, Et3N, 70 °C.
Finally, for enzymatic assays, triphosphate analogues 40a, 40b, and 41 (Scheme 8) were prepared from compounds 5a, 17a, and 14, respectively, employing the standard morpholidate methodology reported by Holý and Rosenberg.56
Scheme 8.
Synthesis of phosphonodiphosphates 40a, 40b, and 41. Reagents and conditions: i) TMSBr/CH3CN; ii) DCC, morpholine, tBuOH, H2O, reflux; iii) (Bu3N)2P2O7, Bu3N, DMF.
Inhibition of ACT in the cell-based assay.
All prepared bisamidates 2a, 2b, 6a-6e, 15, 18a-18b, 24, 31, and 39 were tested for their ability to inhibit ACT activity in J774A.1 macrophage cells (Table 1). For comparison, bis(POM)PMEA (I) and PMEA bisamidate III (Fig. 1) were used as reference compounds. Murine macrophage cells J774A.1 were preincubated with various concentrations of tested prodrugs and subsequently exposed to B. pertussis ACT. The cells were lysed and the cAMP content was determined. Compounds that exhibited inhibitory activity in the low micromolar and sub-micromolar range (Table 1) were also evaluated for their effects on the viability of J774A.1 cells under the same conditions of the cAMP assay, in order to exclude false positives due to potential cytotoxic effects of the compounds.
Table 1.
ACT inhibition and cytotoxic effects of the base-modified PMEA derivatives measured in J774A.1 cells.
| Compound | IC50 (μM)[a] | Viability (%)[b] |
|---|---|---|
| I | 0.006 ± 0.001 | 59 |
| III | 0.147 ± 0.034 | 93 |
| 2a | >10 | ND[c] |
| 2b | >10 | ND |
| 6a | 0.051 ± 0.008 | 120 |
| 6b | 0.134 ± 0.019 | 90 |
| 6c | 1.268 ± 0.222 | 89 |
| 6d | 3.780 ± 0.360 | 105 |
| 6e | 2.185 ± 0.454 | 106 |
| 15 | >10 | ND |
| 18a | 0.016 ± 0.004 | 109 |
| 18b | 0.208 ± 0.088 | 101 |
| 24 | 0.196 ± 0.037 | 84 |
| 31 | >10 | ND |
| 39 | 1.235 ± 0.603 | 96 |
Data represent the mean ± SD of at least three independent experiments. IC50 = concentration of a compound causing a 50% decrease in ACT-induced cAMP accumulation.
Data represent the percentage of cell viability at a fixed prodrug concentration (10 µM) versus untreated control.
ND: not determined.
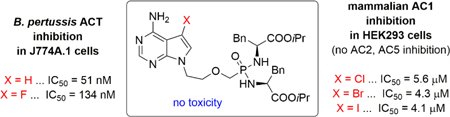
All prepared bisamidates, except derivatives 2a, 2b, 15, and 31, inhibited ACT in low micromolar to submicromolar range. Three bisamidates (6a, 6b, and 18a) were more potent ACT inhibitors than the parent PMEA derivative III (Table 1). The most potent ACT inhibitor within the bisamidate prodrug series in the macrophage cell-based assay was compound 18a, the 8-aza-7-deazapurine derivative of PMEA, with a IC50 value of 16 nM (Table 1). Compound 18a is about an order of magnitude more potent ACT inhibitor than parent PMEA derivative III and, thus, as a promising ACT inhibitor, warrants further biological and pharmacological evaluation.
Deaza-modifications of the pyrimidine part of the purine moiety, as seen in compounds 2a and 2b, as well as the replacement of the adenine moiety with 4-aminoquinazoline base in derivative 31 led to a substantial loss of inhibitory activity. On the other hand, modification of the imidazole part of the purine scaffold, as seen in compounds 6a, 6b, and 18a, seemed to be well-tolerated by the bacterial AC enzyme and led to an increase in potency compared to parent PMEA derivative III. Interestingly, novel 9-deaza-PMEA derivative 15 did not exhibit any ACT inhibitory effects (IC50 value >10 μM, Table 1). As the key structural difference between compound 15 and parent PMEA analogue III is the presence of the acidic hydrogen atom at the N-7 position, compound 15 (in the form of diphosphate 41) was selected for further evaluation at the enzymatic level (see cell-free assay results).
Finally, all bisamidates evaluated in the cell-based assay exhibited improved cytotoxicity profiles compared to adefovir dipivoxil (bis(POM)PMEA, I, Fig. 1, Table 1) with most of them being non-toxic under the cAMP assay conditions.
Inhibition of ACT activity in a cell-free assay.
Selected compounds, i.e. two most active derivatives 6a and 18a (Table 1), as well as novel 9-deazapurine analogue 15, were prepared in the form of corresponding phosphonodiphosphates 40a, 40b and 41, respectively, representing the metabolically active species. Their direct anti-AC activity was assessed and compared with that of parent PMEApp (II, Fig. 1). The compounds were tested on two commercially available B. pertussis ACTs (from ENZO and Sigma), recombinantly expressed in E. coli, and on EF from Bacillus anthracis (Table 2). Compounds 40a and 40b were equally or more potent inhibitors of all enzymes tested compared to PMEApp (II) with IC50 values ranging from 0.5 to 21 nM. In the case of the ACT enzyme from Sigma, the 7-deaza derivative of PMEApp, i.e. compound 40a, was found to be about 30 times more potent than PMEApp (II). Similarly, the novel 9-deazapurine derivative of PMEApp (II), compound 41, was shown to be nearly equipotent compared to compounds 40a and 40b, as well as to parent PMEApp (II, Table 2). Thus, the apparent lack of inhibitory effect of bisamidate 15 (analogue of 41) in the cell-based assay (Table 1) is speculated to be due to the inefficient phosphorylation (by cellular kinases) of intermediate phosphonate 14 (released from bisamidate 15 in the cell) to the biologically active species 41.
Table 2.
IC50 values of selected ANP-diphosphates for ACT and EF.
| Compound | IC50 (μM)[a] |
||
|---|---|---|---|
| ACT Enzo | ACT Sigma | EF | |
| II | 13.6 ± 4.7 | 16.0 ± 0.1 | 11.5 ± 0.6 |
| 40a | 4.08 ± 0.75 | 0.51 ± 0.12 | 2.54 ± 0.65 |
| 40b | 20.8 ± 0.1 | 12.7 ± 0.3 | 5.28 ± 1.33 |
| 41 | 13.3 ± 2.3 | 9.32 ± 2.61 | 20.9 ± 1.8 |
Data represent the mean ± SD of at least three independent experiments.
Molecular modelling of the most potent inhibitor for all studied bacterial adenylate cyclases, 7-deaza-PMEApp (40a), was performed using the recently reinterpreted57 crystal structure of adenylate cyclase domain (ACD) from B. pertussis ACT with calmodulin (CaM) and PMEApp (PDB ID:1ZOT).41 The docking revealed almost identical binding mode for both PMEApp (II) and compound 40a (Fig. 3). The improved binding potential of 40a is speculated to be due to the higher electron density of the pyrrol moiety when compared to the imidazole ring in PMEApp and, thus, stronger C-H – π interaction between Asn304 and the pyrrol moiety. Moreover, the hydrogen at position C-7 of 40a is placed nearby (2.45 Å) the carbonyl group of Gly299 suggesting that their direct interaction may also be important (Fig. 3).
Figure 3.
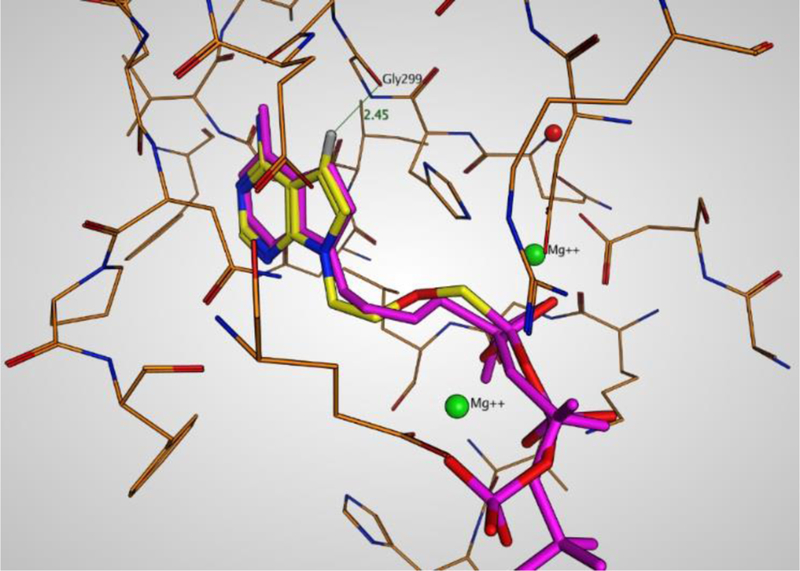
Docking study of 7-deaza-PMEApp (compound 40a) to adenylate cyclase domain of ACT in comparison with PMEApp. Picture description: Purple structure – optimized position of parent PMEApp (II). Yellow structure - docking results for compound 40a. Red dot - Mg902 changed to a water molecule; Green dots - Mg901 and Mg903 ions.
Inhibition of mammalian ACs.
All prepared ANPs in the bisamidate form were also evaluated as potential inhibitors of mammalian adenylate cyclases (mACs). Specifically, the enzyme isoforms AC1, AC2, and AC5, representing the three major mAC subfamilies, were tested to explore the potential selectivity of studied compounds for bacterial ACs over mACs (Table 3). Most of the compounds failed to markedly inhibit any of the mACs tested, revealing promising selectivity for the bacterial ACs. Several compounds even seemed to potentiate AC2 (e.g. I, 6a, 6c, 6d, 18a, 18b, and 24) and/or AC5 (e.g. I, 6d, 6e, and 31) at 30 µM.
Table 3.
Mammalian AC1, AC2 and AC5 inhibition with base-modified ANPs at 30 µM measured in HEK293 cells.
| Compound | Response of Control (%)[a] |
||
|---|---|---|---|
| AC1 | AC2 | AC5 | |
| I | 177 ± 9 | 391 ± 30 | 345 ± 24 |
| III | 64 ± 9 | 200 ± 45 | 99 ± 8 |
| 2a | 88 ± 6 | 149 ± 15 | 98 ± 4 |
| 2b | 88 ± 15 | 107 ± 26 | 77 ± 5 |
| 6a | 66 ± 15 | 207 ± 39 | 139 ± 14 |
| 6b | 48 ± 12 | 176 ± 16 | 108 ± 8 |
| 6c | 23 ± 4 | 236 ± 39 | 182 ± 10 |
| 6d | 24 ± 5 | 238 ± 49 | 215 ± 17 |
| 6e | 22 ± 6 | 182 ± 50 | 229 ± 33 |
| 15 | 62 ± 9 | 189 ± 27 | 134 ± 15 |
| 18a | 71 ± 11 | 223 ± 37 | 147 ± 2 |
| 18b | 79 ± 18 | 308 ± 12 | 176 ± 17 |
| 24 | 63 ± 5 | 262 ± 46 | 178 ± 30 |
| 31 | 56 ± 6 | 197 ± 12 | 220 ± 13 |
| 39 | 56 ± 7 | 174 ± 3 | 172 ± 12 |
| SQ22536[b] | 71 ± 6 | 52 ± 1 | 67 ± 12 |
| NKY80[c] | 74 ± 10 | 123 ± 27 | 54 ± 5 |
| SKF83566[d] | 82 ± 9 | 4 ± 4 | 103 ± 14 |
Data are the mean ± SEM relative to the control response (100%) of at least two independent experiments. Values greater than 100% represent a potentiated response.
SQ22536 and NKY80 are non-selective P-site inhibitors.
SKF83566 is a selective inhibitor of AC2.
A number of compounds appeared to selectively inhibit AC1 (e.g. 6b, 6c, 6d, 6e, 31, and 39, Table 3). Interestingly, 7-halo-7-deazapurine analogues 6c, 6d, and 6e were found to be the most efficacious AC1 inhibitors from the whole series, therefore, IC50 values were also determined for these compounds (Fig. 4, Table 4). Moreover, the potentiation of these compounds at AC2 was only evident at the highest concentration tested.
Figure 4.
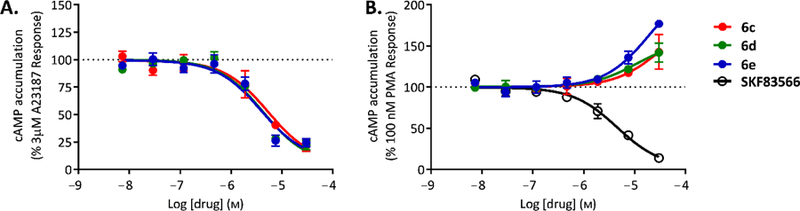
Dose response curves of compounds 6c, 6d, and 6e at AC1 and AC2. Increasing concentrations of the compounds were added to HEK293 cells expressing AC1 (A) or AC2 (B) followed by selective AC stimulation using the calcium ionophore A23187 or the phorbol ester PMA as indicated. SKF83566 is a selective inhibitor of AC2. Data shown represent the mean and SEM of three independent experiments conducted in duplicate.
Table 4.
IC50 values and % activity on AC1 and AC2 in HEK293 cells.
| Compound | AC1 | AC2 | ||
|---|---|---|---|---|
| % Response[a] | IC50 (µM)[b] | % Response | IC50 (µM) | |
| 6c | 22 ±5 | 5.6 ±1.7 | 143 ±21 | ND[c] |
| 6d | 22 ±4 | 4.3 ±0.5 | 142 ±11 | ND |
| 6e | 25 ±4 | 4.1 ±1.1 | 177 ±3 | ND |
| SKF83566 | ND | ND | 14 ±5 | 5.0 ±1.6 |
Percentages shown correspond to % mAC activity at 30 μM inhibitor normalized to the control response (no inhibitor). Values greater than 100% represent a potentiated response.
Values are shown as mean and SEM of three independent experiments.
ND, not determined.
All three 7-halo-7-deazapurine analogues 6c, 6d, and 6e were selective AC1 inhibitors (no AC2 and AC5 inhibition was observed) with potency in low µM range (IC50 values of 4.1–5.6 µM, Table 4). AC1 belongs to a group of mACs stimulated by calcium in a calmodulin-dependent manner, and AC1 inhibitors have a great potential for treating neuropathic and inflammatory pain.58,59 Thus, compounds 6c, 6d, and 6e warrant further study with AC1 knockout mice and their development as potent and selective AC1 inhibitors. Furthermore, the inhibitory activity of these 7-halo-7-deazapurine analogues on both the bacterial ACs and mammalian AC1 may suggest a shared binding domain, as both the bacterial ACs and the mammalian AC1 isoforms are also regulated by calcium/calmodulin.
Conclusions
A series of 13 acyclic nucleoside phosphonates (ANPs) bearing 2-(phosphonomethoxy)ethyl (PME) moiety in the form of bisamidate prodrug was synthesized as potential inhibitors of adenylate cyclases (ACs) of Bordetella pertussis (ACT) and Bacillus anthracis (EF). The prepared compounds are characterized by the replacement of the purine moiety of adefovir (PMEA) with various bicyclic heterocycles, namely 7H-pyrrolo[2,3-d]pyrimidine (7-deazapurine), 1H-pyrazolo[3,4-d]pyrimidine (8-aza-7-deazapurine), 3H-[1,2,3]triazolo[4,5-d]pyrimidine (8-azaapurine), 5H-pyrrolo[3,2-d]pyrimidine (9-deazapurine), thieno[3,2-d]pyrimidine, quinazoline, and pyrrolo[2,1-f][1,2,4]triazine (4-aza-7,9-dideazapurine). Bisamidate prodrugs with L-phenylalanine isopropyl ester were used based on their significantly improved stability in plasma and decreased cytotoxicity compared to the original adefovir dipivoxil (bis(POM)PMEA).43
Prepared compounds, with the exception of four derivatives (2a, 2b, 15, and 31), are potent inhibitors of adenylate cyclase toxin from Bordetella pertussis (ACT) in the J774A.1 macrophage cell-based assay. The SAR study suggested that structural modification of the pyrimidine part of the molecule is not tolerated but aza-, deaza-, and even 7-thia-modifications in the imidazole part of the molecule seem to preserve or even improve potency. An additional substitution of the C-7 position of 7-deaza derivative 6a by halogen atom led to decrease of inhibitory activity. Compound 18a, 8-aza-7-deazapurine derivative of PMEA, was identified as the most potent ACT inhibitor in the series with 16 nM inhibitory potency towards ACT, substantial selectivity over mammalian ACs, and no observed cytotoxicity. As such, the compound represents a promising lead structure for further pharmacological evaluation in the mouse model of pertussis or anthrax.
The two most active ANP bisamidates identified in the macrophage cell-based assay, 7-deaza-PMEA derivative 6a and 8-aza-7-deaza-PMEA derivative 18a, were also prepared as ANP-diphosphates 40a and 40b, respectively, for the evaluation of their direct interaction with the proteins of interest in a cell-free assay. Compounds 40a and 40b were equally or more potent inhibitors of bacterial adenylate cyclases tested (ACT Enzo, ACT Sigma, and EF) compared to parent PMEApp (II). In fact, compound 40a, was up to 30 times more potent than PMEApp on ACT from B.pertussis.
Synthesis of novel 9-deaza-PMEA (14) was designed and executed. Although its bisamidate analogue 15 was not a particularly efficient ACT inhibitor in the murine macrophage cell-based assay, 9-deaza-PMEApp (41) exhibited similar potency to PMEApp (II) on all three bacterial ACs tested in vitro. The lack of the potency of compound 15 in the cellular assay is speculated to be due to its inefficient intracellular transformation into the active nucleoside triphosphate analogue 41.
Finally, several of the prepared compounds were discovered to inhibit mammalian AC1 with a considerable efficacy and a noticeable selectivity over AC2 and AC5. The 7-halo-7-deazapurine derivatives 6c, 6d, and 6e were selective AC1 inhibitors with IC50 values in the range 4.1–5.6 µM. These compounds may represent promising lead structures for further optimization of their structure and for potential development of new agents for the treatment of human neurological and/or inflammatory diseases.
Experimental Section
Chemistry:
Unless otherwise stated, solvents were evaporated at 40 °C/2 kPa and the compounds were dried over P2O5 at 2 kPa. The microwave-assisted reactions were carried out in CEM Discover (Explorer) microwave apparatus. Chemicals and reagents were obtained from commercial sources (Sigma–Aldrich and Fluorochem Ltd.), bis(POM)PMEA was obtained from SANTIAGO company. Solvents were dried by standard procedures. Pyridine was stored over molecular sieves (4 Å). Tetrahydrofuran (THF) was freshly distilled from LiAlH4 pellets under Ar. TLC was performed on plates of Kieselgel 60 F254 (Merck). NMR spectra were recorded on Bruker Avance 500 (1H at 500 MHz, 13C at 125.8 MHz) spectrometer with TMS as internal standard or referenced to the residual solvent signal. Mass spectra were measured on UPLC-MS (Waters SQD-2), and HR-MS were taken on a LTQ Orbitrap XL spectrometer using electrospray ionization (ESI). Preparative HPLC purifications were performed on columns packed with 10 μm C18 reversed phase resin (Phenomenex Gemini 10 μm 21 × 250 mm) on Waters Delta 600 chromatography system in ca. 200 mg batches of mixtures using gradient MeOH/H2O as eluent. Flash chromatography on normal phase and on reversed phase was performed on Reveleris Flash Chromatography System. The deionization was performed on column Redisep®Rf GOLD C18 Teledyne ISCO. Preparative HPLC purification of triphosphate analogues was performed on a column packed with POROS® HQ 50 mm (50mL) with use of a gradient of TEAB in water (0.05–0.5 M). The purity of target compounds was determined by HPLC (H2O-CH3CN, linear gradient) and was higher than 95%.
Method A. General procedure for synthesis of bis-(L-phenylalanine ethyl ester) prodrugs of phosphonates:48
TMSBr (1 mL) was added to the corresponding phosphonate diester (1.0 mmol) dissolved in dry pyridine (10 mL). The reaction mixture was stirred at RT overnight. Volatiles were removed, and the moisture sensitive product was permanently kept under Ar. Solid isopropyl ester L-phenylalanine hydrochloride (0.97 g, 4.0 mmol) was added to the intermediate, followed by dry pyridine (8 mL) and dry Et3N (2 mL) under Ar. The mixture was preheated to 70 °C and freshly prepared solution of Aldrithiol-2 (1.37 g, 6.2 mmol) and triphenylphosphine (1.64 g, 6.2 mmol) in pyridine (10 mL) was added. The resulting mixture was stirred at 70 °C for 72 h. Reaction mixture was evaporated in vacuo and the residue was purified by column chromatography (0–100% MeOH in a mixture of Hexane:EtOAc, 6:4) followed by C18 reversed phase column chromatography (0–100% MeOH in water).
Method B. General procedure for alkylation of modified nucleobases:
Diisopropyl [(2-chloroethoxy)methyl]phosphonate49 (2.0 g, 7.9 mmol) was added to the solution of halogenated nucleobase (5.9 mmol) in DMSO (12 mL) under argon at RT. The reaction mixture was warmed up to 80 °C, Cs2CO3 (1.25 g, 3.9 mmol) was added, and the resulting mixture was stirred at 80 °C. The mixture was cooled down, dissolved in Et2O (100 mL) and extracted with 80% aqueous solution of sat. NH4Cl (3 × 30 mL). Organic phase was dried over Na2SO4. Volatiles were removed in vacuo and the residue was purified by flash column chromatography (MeOH:CHCl3, 0–10% gradient).
Method C. General procedure for hydroboration of vinyl compounds:51
9-BBN (0.5 M in THF) (16 mL, 8.0 mmol) was added dropwise to the solution of vinyl compound (4.0 mmol) in THF (10 mL) at 0 °C under argon within 15 min. The reaction mixture was allowed to warm up to RT and stirred at RT for 3 h. NaBO3.4 H2O (2.4 g) in water (50 mL) was added and the mixture was stirred for additional 3 h. The reaction mixture was filtered, CHCl3 (50 mL) was added, the organic phase was washed with 10% aqueous NaCl (2 × 25 mL) and brine (25 mL), and then dried over Na2SO4. Volatiles were removed in vacuo and the residue was purified by column chromatography.
Method D. General procedure for alkylation of 2-hydroxyethyl derivatives with CF3SO2CH2P(O)(OiPr)2:
A solution of BuLi (2.5 M in hexane, 1.42 mL, 3.55 mmol) was added dropwise to the corresponding 2-hydroxyethyl compound (2.8 mmol) in THF (23 mL) under argon at −78 °C and the mixture was stirred at −78 °C for 10 min. CF3SO2CH2P(O)(OiPr)2 (2.24 g, 6.8 mmol) in THF (5 mL) was added and the reaction mixture was stirred at −78 °C for 15 min. The resulting mixture was allowed to warm up to −40 °C (in approx. 3 h) and a mixture of sat. NH4Cl in H2O (1:1, 25 mL) was added, followed by EtOAc (150 mL). The mixture was washed with H2O (50 mL) and brine (50 mL) and then dried over Na2SO4. Volatiles were removed and the residue was purified by C18 reverse-phase chromatography (H2O:MeOH, 0–100%).
Method E. General procedure for preparation of phosphonate diphosphates:56
A solution of dicyclohexylcarbodiimide (0.44 g, 2.2 mmol) in t-BuOH (4 mL) was added dropwise (2 h) to the refluxing mixture of corresponding phosphonic acid (0.5 mmol) and morpholine (0.19 mL) in 50% aqueous t-BuOH (16 mL). The reaction mixture was refluxed overnight, cooled down, and concentrated to half-volume in vacuo (bath max. 35 °C). The mixture was diluted with H2O (10 mL) and extracted with Et2O (3 × 10mL). The aqueous layer was evaporated in vacuo and codistilled with EtOH and toluene. The residue was dissolved in DMF (4.5 mL) and Bu3N (0.35 mL, 1.5 mmol) and tributylammoniumpyrophosphate (0.33 g, 1.5 mmol) were added. The solution was stirred at RT overnight. The reaction mixture was pour into Et2O. The solid was filtered off, washed with Et2O and dissolved in 1 M aqueous TEAB (5 mL). Volatiles were removed in vacuo (bath 30 °C), the residue was dissolved in 0.05 M aqueous TEAB (5 mL) and applied on column POROS 50 HQ and eluted by TEAB (0.05–1 M gradient). The corresponding eluate was evaporated in vacuo, codistilled with H2O (3 × 5 mL) and applied on column of Dowex (50 × 8) (Na+ cycle). The UV absorbing fraction was lyophilized.
9-{2-[(Diisopropoxyphosphoryl)methoxy]ethyl}−1-deazaadenine (1a):
Compound 1a was synthesized from 1-deazaadenine according to the reported procedure.46,47
9-{2-[(Diisopropoxyphosphoryl)methoxy]ethyl}−3-deazaadenine (1b):
Diisopropyl ((2-chloroethoxy)methyl)phosphonate (1.93 g, 7.5 mmol) was added to the reaction mixture (preheated at 80 °C for 1 h) of NaH (60 % susp. in mineral oil, 0.3 g, 7.5 mmol) and 3-deazaadenine (7.5 mmol) in DMF (150 mL). The resulting mixture was stirred at 100 °C overnight. The clear solution was cooled down and volatiles were removed in vacuo. The residue was codistilled with toluene and purified by column chromatography (MeOH:CHCl3, 0–10%) to give 1b (1.64 g, 62%) as yellowish oil. ESI-MS, m/z (%): 357 [M+H+] (100). 1H NMR (DMSO-d6): δ 1.12 (d, J(CH3, CH) = 6.2 Hz, 6H, CH3); 1.16 (d, J(CH3, CH) = 6.2 Hz, 6H, CH3); 3.76 (d, J(CH2, P) = 8.3 Hz, 2H, P-CH2); 3.84 (m, 2H, H-2'); 4.34 (m, 2H, H-1'); 4.47 (m, 2H, CHiPr); 6.09 (brs, 2H, NH2); 6.81 (d, J(3, 2) = 5.8 Hz, 1H, H-3); 7.65 (d, J(2, 3) = 5.8 Hz, 1H, H-2); 8.01 (s, 1H, H-8). 13C NMR (DMSO-d6): δ 23.79 (d, J(C-C-O-P) = 4.5 Hz, CH3iPr); 23.92 (d, J(C-C-O-P) = 3.8 Hz, CH3iPr); 44.20 (C-1'); 64.87 (d, J(C-P) = 164.1 Hz, CH2-P); 70.32 (d, J(C-O-P) = 6.4 Hz, CHiPr); 70.91 (d, J(2'-P) = 11.6 Hz, C-2'); 96.80 (C-3); 126.75 (C-5); 138.55 (C-4); 140.30 (C-2); 141.93 (C-8); 152.49 (C-6). HR-MS (ESI+): m/z [M + H]+ calculated for: C15H26N4O4P, 357.1686, found: 357.1687.
Bis(L-phenylalanine isopropyl ester) prodrug of 9-[2-(phosphorylmethoxy)ethyl]-1-deazaadenine (2a):
Treatment of 1a (0.17 g, 0.60 mmol) by Method A afforded 2a (0.21 g, 60%) as a colourless foam. ESI-MS, m/z (%): 651 [M+H+] (100). 1H NMR (DMSO-d6): δ 1.01, 1.07, 1.11 and 1.16 (d, J(CH3, CH) = 6.3 Hz, 12H, CH3); 2.71–2.89 (m, 4H, -CH2Ph); 3.21 (dd, J(CH2a, P) = 8.4 Hz, J(gem) = 13.2 Hz, 1H, P-CH2a); 3.30 (dd, J(CH2b, P) = 7.8 Hz, J(gem) = 13.2 Hz, 1H, P-CH2b); 3.68 (t, J(2', 1') = 5.3 Hz, 2H, H-2'); 3.83–4.01 m, 2H, NH-CH); 4.13 (t, J(NH, CH) = J(NH, P) = 11.3 Hz, 1H, CH-NH); 4.25 (t, J(1', 2') = 5.3 Hz, 2H, H-1'); 4.45 (t, J(NH, CH) = J(NH, P) = 11.5 Hz, 1H, CH-NH); 4.78 a 4.82 (2x sept, 1H, J(CH3, CH) = 6.3 Hz, CHiPr); 6.28 (brs, 2H, NH2); 7.08 (m, 10H, Ph-ortho, meta, para); 6.34 (d, J(2, 1) = 5.5 Hz, 1H, H-2); 7.82 (d, J(2, 1) = 5.5 Hz, 1H, H-2); 8.03 (s, 1H, H-8). 13C NMR (DMSO-d6): δ 21.47, 21.54, 21.60 a 21.67 (CH3iPr); 39.90 (CH2Bn); 42.42 (C-1'); 54.02 a 54.24 (NH-CH); 67.48 (d, J(C-P) = 135.3 Hz, CH2-P); 67.98 a 68.10 (CHiPr); 70.61 (d, J(2'-P) = 11.4 Hz, C-2'); 102.12 (C-1); 122.88 (C-5); 126.58 a 126.64 (Ph-para); 128.22 a 128.26 (Ph-meta); 129.65 (Ph-ortho); 137.22 a 136.29 (ipso); 140.85 (C-8); 144.66 (C-2); 146.95 (C-6); 147.44 (C-4); 172.34 and 172.47 m (COO). HR-MS (ESI+): m/z [M + H]+ calculated for: C33H44N6O6P, 651.3055, found: 651.3055.
Bis(L-phenylalanine isopropyl ester) prodrug of 9-[2-(phosphorylmethoxy)ethyl]-3-deazaadenine (2b):
Treatment of 1 (0.35 g, 1.0 mmol) by Method A afforded 2b (0.11 g, 16%) as a colourless foam. ESI-MS, m/z (%): 651 [M+H+] (100). 1H NMR (DMSO-d6): δ 1.00–1.17, (m, 12H, CH3); 2.67–2.88 (m, 4H, -CH2Ph); 3.16–3.43 (m, 2H, P-CH2); 3.64 (m, 2H, H-2'); 3.83–4.00 m, 2H, NH-CH); 4.10 (m, 1H, CH-NH); 4.25 (m, 2H, H-1'); 4.41 (m, 1H, CH-NH); 4.73–4.86 (m, 2H, CHiPr); 6.08 (brs, 2H, NH2); 6.79 (d, J(3, 2) = 5.8 Hz, 1H, H-3); 7.05–7.26 (m, 10H, Ph-ortho, meta, para); 7.65 (d, J(2, 3) = 5.8 Hz, 1H, H-2); 8.03 (s, 1H, H-8). 13C NMR (DMSO-d6): δ 21.47, 21.54, 21.61 a 21.67 (CH3iPr); 39.90 (CH2Bn); 44.31 (C-1'); 54.01 and 54.22 (NH-CH); 67.57 (d, J(C-P) = 135.0 Hz, CH2-P); 67.98 and 68.12 (CHiPr); 70.86 (d, J(2'-P) = 11.0 Hz, C-2'); 96.72 (C-3); 126.57 and 126.65 (Ph-para); 126.72 (C-5); 128.20 and 128.26 (Ph-meta); 129.62 (Ph-ortho); 137.17 and 137.27 (ipso); 138.58 (C-4); 140.37 (C-2); 142.00 (C-8); 152.50 (C-6); 172.31–172.52 m (COO). HR-MS (ESI+): m/z [M + H]+ calculated for: C33H44N6O6P, 651.3055, found: 651.3055.
9-{2-[(Diisopropoxyphosphoryl)methoxy]ethyl}−6-chloro-7-deazapurine (4a):
For the synthesis of 4a look at the preparation of compound 5a.
9-{2-[(Diisopropoxyphosphoryl)methoxy]ethyl}−7-fluoro-6-chloro-7-deazapurine (4b):
Treatment of 3b (2.0 g, 11.7 mmol) by Method B (4 h at 80 °C) and chromatography (MeOH:CHCl3, 5:95) afforded 4b (2.77 g, 60%) as a yellowish oil. ESI-MS, m/z (%): 394 [M+H+] (100). 1H NMR (CDCl3): δ 1.25 (d, J(CH3, CH) = 6.2 Hz, 6H, CH3); 1.29 (d, J(CH3, CH) = 6.2 Hz, 6H, CH3); 3.70 (d, J(H-C-P) = 8.4 Hz, 2H, P-CH2); 3.89 (t, J(1', 2') = 4.9 Hz, 2H, H-1'); 4.44 (t, J(2', 1') = 4.9 Hz, 2H, H-2'); 4.61–4.75 (m, 2H, CHiPr); 7.24 (d, J(8, F) = 2.6 Hz, 1H, H-8); 8.58 (s, 1H, H-2). 13C NMR (CDCl3): δ 23.89 (d J(C-C-O-P) = 4.6 Hz, CH3iPr); 23.97 (d J(C-C-O-P) = 4.0 Hz, CH3iPr); 44.13 (C-1'); 65.91 (d, J(C-P) = 168.2 Hz, CH2-P); 71.15 (d, J(C-O-P) = 6.8 Hz, CHiPr); 71.64 (d, J(2'-P) = 10.7 Hz, C-2'); 106.69 (d, J(5, F) = 13.7 Hz, C-5); 113.34 (d, J(8, F) = 26.2 Hz, C-8); 140.62 (d, J(C-F) 251.6 Hz, C-7); 146.22 (C-4); 150.40 (C-6); 151.12 (C-2). HR-MS (ESI+): m/z [M + H]+ calculated for: C15H23ClFN3O4P, 394.1093, found: 394.1090.
6,7-Dichloro-9-{2-[(diisopropoxyphosphoryl)methoxy]ethyl}−7-deazapurine (4c):
Treatment of 3c (1.0 g, 5.3 mmol) by Method B (5 h at 80 °C) and chromatography (MeOH:CHCl3, 5:95) afforded 4c (1.83 g, 84%) as a yellowish oil. ESI-MS, m/z (%): 410 [M+H+] (100). 1H NMR (DMSO-d6): δ 1.08 (d, J(CH3, CH) = 6.2 Hz, 6H, CH3); 1.14 (d, J(CH3, CH) = 6.2 Hz, 6H, CH3); 3.75 (d, J(H-C-P) = 8.4 Hz, 2H, P-CH2); 3.90 (t, J(1', 2') = 5.0 Hz, 2H, H-1'); 4.39–4.46 (m, 2H, CHiPr); 4.45–4.47 (m, 2H, H-2'); 7.95 (s, 1H, H-8); 8.67 (s, 1H, H-2). 13C NMR (DMSO-d6): δ 23.38 (d J(C-C-O-P) = 4.5 Hz, CH3iPr); 23.56 (d, J(C-C-O-P) = 3.8 Hz, CH3iPr); 43.96 (C-1'); 64.36 (d, J(C-P) = 164.1 Hz, CH2-P); 69.95 (d, J(C-O-P) = 6.4 Hz, CHiPr); 70.21 (d, J(2'-P) = 11.7 Hz, C-2'); 100.97 (C-5); 112.59 (C-7); 128.89 (C-8); 149.45 and 149.84 (C-4, 6); 150.83 (C-2). HR-MS (ESI+): m/z [M + H]+ calculated for: C15H23Cl2N3O4P, 410.0799, found: 410.0798.
7-Bromo-9-{2-[(diisopropoxyphosphoryl)methoxy]ethyl}−6-chloro-7-deazapurine (4d):
Treatment of 3d (1.0 g, 4.3 mmol) by Method B (4 h at 80 °C) and chromatography (MeOH:CHCl3, 6:94) afforded 4d (1.67 g, 85%) as a colourless oil. ESI-MS, m/z (%): 454 [M+H+] (100). 1H NMR (DMSO-d6): δ 1.08 (d, J(CH3, CH) = 6.2 Hz, 6H, CH3); 1.14 (d, J(CH3, CH) = 6.2 Hz, 6H, CH3); 3.76 (d, J(H-C-P) = 8.4 Hz, 2H, P-CH2); 3.90 (t, J(1', 2') = 5.0 Hz, 2H, H-1'); 4.38–4.46 (m, 2H, CHiPr); 4.45–4.48 (m, 2H, H-2'); 7.98 (s, 1H, H-8); 8.67 (s, 1H, H-2). 13C NMR (DMSO-d6): δ 23.40 (d, J(C-C-O-P) = 4.5 Hz, CH3iPr); 23.57 (d, J(C-C-O-P) = 3.8 Hz, CH3iPr); 44.05 (C-1'); 64.37 (d, J(C-P) = 163.99 Hz, CH2-P); 69.96 (d, J(C-O-P) = 6.4 Hz, CHiPr); 70.22 (d, J(2'-P) = 11.8 Hz, C-2'); 85.23 (C-7); 113.78 (C-5); 131.38 (C-8); 150.92 and 151.15 (C-4 and C-6); 155.96 (C-2). HR-MS (ESI+): m/z [M + H]+ calculated for: C15H23BrClN3O4P, 454.0293, found: 454.0293.
9-{2-[(Diisopropoxyphosphoryl)methoxy]ethyl}−6-chloro-7-iodo-7-deazapurine (4e):
Treatment of 3e (1.5 g, 5.3 mmol) by Method B (5 h at 80 °C) and chromatography (MeOH:CHCl3, 5:95) afforded 4e (1.92 g, 72%) as a yellowish oil. ESI-MS, m/z (%): 502 [M+H+] (10); 524 [M+Na+] (100). 1H NMR (DMSO-d6): δ 1.08 (d, J(CH3, CH) = 6.2 Hz, 6H, CH3); 1.14 (d, J(CH3, CH) = 6.2 Hz, 6H, CH3); 3.80 (d, J(H-C-P) = 8.3 Hz, 2H, P-CH2); 3.72–3.77 (m, 2H, H-1'); 4.40–4.49 (m, 2H, H-2'); 4.56–4.64 (m, 2H, CHiPr); 7.97 (s, 1H, H-8); 8.64 (s, 1H, H-2). 13C NMR (DMSO-d6): δ 24.22 (d, J(C-C-O-P) = 4.6 Hz, CH3iPr); 24.34 (d, J(C-C-O-P) = 3.7 Hz, CH3iPr); 43.68 (C-1'); 65.08 (d, J(C-P) = 164.4 Hz, CH2-P); 70.70 (d, J(C-O-P) = 6.4 Hz, CHiPr); 72.74 (d, J(2'-P) = 11.9 Hz, C-2'); 51.80 (C-7); 116.61 (C-5); 137.18 (C-8); 150.80 (C-2); 151.08 and 151.34 (C-4 and C-6). HR-MS (ESI+): m/z [M + Na]+ calculated for: C15H22ClIN3O4PNa, 523.9973, found: 523.9974.
9-{2-[(Diisopropoxyphosphoryl)methoxy]ethyl}−7-deazaadenine (5a):
Diisopropyl ((2-chloroethoxy)methyl) phosphonate49 (13.6 g, 52.0 mmol) was added to the preheated (80 °C) reaction mixture of 3a (6.0 g, 39.0 mmol) and Cs2CO3 (8.2 g, 25.4 mmol) in DMSO (90 mL). The resulting mixture was stirred at 80 °C for 5 h and then cooled down to RT. Aqueous solution of NH4Cl (20%, 300 mL) was added and the mixture was washed with Et2O (3 × 100 mL). The combined organic layers were concentrated and codistilled with toluene (3 × 25 mL) to give crude 4a. A mixture of crude 4a in ethanolic ammonia solution (40 mL) was heated at 100 °C for 24 h. The solvent was removed in vacuo and the residue was applied on Dowex (50 × 8) and washed with 30% aqueous MeOH solution successively. The column was then washed with 2.5% aqueous ammonia solution, the UV absorbing fraction was evaporated in vacuo and repurified on C18 column (H2O:MeOH, 0–100%) to give 5a (5.48 g, 39%) as a yellowish oil. The analytical data are in agreement with published data.49
9-{2-[(Diisopropoxyphosphoryl)methoxy]ethyl}−7-fluoro-7-deazaadenine (5b):
Compound 4b (1.92 g, 4.87 mmol) was dissolved in EtOH/NH3 (30 mL) and stirred at 100 °C for 16 h. The volatiles were removed and the residue was purified by C18 reversed phase chromatography (H2O:MeOH 0–100%) to give 5b (0.89 g, 49%) as a yellowish oil. ESI-MS, m/z (%): 375 [M+H+] (100). 1H NMR (CDCl3): δ 1.27 (d, J(CH3, CH) = 6.2 Hz, 6H, CH3); 1.30 (d, J(CH3, CH) = 6.2 Hz, 6H, CH3); 3.71 (d, J(H-C-P) = 8.5 Hz, 2H, P-CH2); 3.88 (t, J(1', 2') = 5.1 Hz, 2H, H-1'); 4.33 (t, J(2', 1') = 5.1 Hz, 2H, H-2'); 4.63–4.75 (m, 2H, CHiPr); 5.41 (brs, 2H, NH2); 6.88 (d, J(8, F) = 2.5 Hz, 1H, H-8); 8.23 (s, 1H, H-2). 13C NMR (CDCl3): δ 23.91 (d, J(C-C-O-P) = 4.7 Hz, CH3iPr); 24.01 (d, J(C-C-O-P) = 3.9 Hz, CH3iPr); 43.74 (C-1'); 65.97 (d, J(C-P) = 167.9 Hz, CH2-P); 71.11 (d, J(C-O-P) = 6.7 Hz, CHiPr); 72.11 (d, J(2'-P) = 11.2 Hz, C-2'); 93.19 (d, J(5, F) = 14.9 Hz, C-5); 107.98 (d, J(8, F) = 26.4 Hz, C-8); 142.52 (d, J(C, F) = 244.1 Hz, C-7); 145.65 (d, J(4, F) = 2.5 Hz, C-4); 152.66 (C-2); 155.43 (d, J(6, F) = 2.9 Hz, C-6). HR-MS (ESI+): m/z [M + H]+ calculated for: C15H25N4O4FP, 375.1592, found: 375.1591.
9-{2-[(Diisopropoxyphosphoryl)methoxy]ethyl}−7-chloro-7-deazaadenine (5c):
Compound 4c (1.6 g, 3.9 mmol) was dissolved in EtOH/NH3 (50 mL) and stirred at 100 °C for 16 h. The volatiles were removed and the residue was purified by C18 reversed phase chromatography (H2O:MeOH 0–100%) to give 5c (1.00 g, 66%) as a colourless oil. ESI-MS, m/z (%): 391 [M+H+] (100). 1H NMR (DMSO-d6): δ 1.13 (d, J(CH3, CH) = 6.2 Hz, 6H, CH3); 1.17 (d, J(CH3, CH) = 6.2 Hz, 6H, CH3); 3.75 (d, J(H-C-P) = 8.5 Hz, 2H, P-CH2); 3.83 (m, 2H, H-2'); 4.27 (m, 2H, H-1'); ); 4.83 (d of septets, J(H-C-O-P) = 7.7 Hz, J(CH, CH3) = 6.2 Hz, 2H, CHiPr); 6.81 (brs, 2H, NH2); 7.34 (s, 1H, H-8); 8.08 (s, 1H, H-2). 13C NMR (DMSO-d6): δ 23.76 (d, J(C-C-O-P) = 4.5 Hz, (CH3iPr); 23.92 (d, J(C-C-O-P) = 3.8 Hz, CH3iPr); 43.37 (C-1'); 64.65 (d, J(C-P) = 163.9 Hz, CH2-P); 70.34 (d, J(C-O-P) = 6.4 Hz, CHiPr); 70.94 (d, J(2'-P) = 12.1 Hz, C-2'); 99.65 (C-5); 101.11 (C-7); 122.17 (C-8); 148.73 (C-4); 152.48 (C-2); 156.81 (C-6). HR-MS (ESI+): m/z [M + H]+ calculated for: C15H25N4O4ClP, 391.1297, found: 391.1298.
9-{2-[(Diisopropoxyphosphoryl)methoxy]ethyl}−7-bromo-7-deazaadenine (5d):
Compound 4d (1.5 g, 3.3 mmol), was dissolved in EtOH/NH3 (40 mL) and stirred at 100 °C for 16 h. The volatiles were removed and the residue was purified by C18 reversed phase chromatography (H2O:MeOH 0–100%) to give 5d (1.17 g, 81%) as a colourless oil. ESI-MS, m/z (%): 435 [M+H+] (100). 1H NMR (DMSO-d6): δ 1.13 (d, J(CH3, CH) = 6.2 Hz, 6H, CH3); 1.17 (d, J(CH3, CH) = 6.2 Hz, 6H, CH3); 3.75 (d, J(H-C-P) = 8.5 Hz, 2H, P-CH2); 3.83 (m, 2H, H-2'); 4.27 (m, 2H, H-1'); 4.47 (d of septets, J(H-C-O-P) = 7.7 Hz, J(CH, CH3) = 6.2 Hz, 2H, CHiPr); 6.71 (brs, 2H, NH2); 7.39 (s, 1H, H-8); 8.09 (s, 1H, H-2). 13C NMR (DMSO-d6): δ 23.77 (d J(C-C-O-P) = 4.5 Hz, CH3iPr); 23.93 (d, J(C-C-O-P) = 3.8 Hz, CH3iPr); 43.47 (C-1'); 64.66 (d, J(C-P) = 164.0 Hz, CH2-P); 70.34 (d, J(C-O-P) = 6.4 Hz, CHiPr); 70.95 (d, J(2'-P) = 12.0 Hz, C-2'); 85.00 (C-7); 100.85 (C-5); 124.66 (C-8); 149.24 (C-4); 152.40 (C-2); 157.06 (C-6). HR-MS (ESI+): m/z [M + H]+ calculated for: C15H25N4O4BrP, 435.0791, found: 435.0792.
9-{2-[(Diisopropoxyphosphoryl)methoxy]ethyl}−7-iodo-7-deazaadenine (5e):
Compound 4e (1.92 g, 3.8 mmol), was dissolved in EtOH/NH3 (30 mL) and stirred at 100 °C for 16 h. The solvent was removed and the residue was purified by C18 reversed phase chromatography (H2O:MeOH 0–100%) to give 5e (1.70 g, 92%) as a yellowish oil. ESI-MS, m/z (%): 483 [M+H+] (67); 505 [M+Na+] (100). 1H NMR (DMSO-d6): δ 1.13 (d, J(CH3, CH) = 6.2 Hz, 6H, CH3); 1.18 (d, J(CH3, CH) = 6.2 Hz, 6H, CH3); 3.75 (d, J(H-C-P) = 8.5 Hz, 2H, P-CH2); 3.83 (t, J(1', 2') = 5.2 Hz, 2H, H-1'); 4.29 (t, J(2', 1') = 5.2 Hz, 2H, H-2'); 4.48 (d of septets, J(H-C-O-P) = 7.7 Hz, J(CH, CH3) = 6.2 Hz, 2H, CHiPr); 6.59 (brs, 2H, NH2); 7.42 (s, 1H, H-8); 8.09 (s, 1H, H-2). 13C NMR (DMSO-d6): δ 23.52 (d, J(C-C-O-P) = 4.5 Hz, CH3iPr); 23.67 (d, J(C-C-O-P) = 3.7 Hz, CH3iPr); 43.25 (C-1'); 49.47 (C-7); 64.39 (d, J(C-P) = 163.9 Hz, CH2-P); 70.04 (d, J(C-O-P) = 6.4 Hz, CHiPr); 70.70 (d, J(2'-P) = 12.1 Hz, C-2'); 102.76 (C-5); 129.65 (C-8); 149.56 (C-4); 151.61 (C-2); 156.98 (C-6). HR-MS (ESI+): m/z [M + H]+ calculated for: C15H25N4O4IP, 483.0653, found: 483.0654.
Bis(L-phenylalanine isopropyl ester) prodrug of 9-[2-(phosphorylmethoxy)ethyl]-7-deazaadenine (6a):
Treatment of 5a (0.35 g, 0.98 mmol) by Method A afforded 6a (0.38 g, 60%) as a whitish amorphous solid. ESI-MS, m/z (%): 651 [M+H+] (100). 1H NMR (DMSO-d6): δ 1.15, 1.19, 1.20 and 1.24 (4 x d, J(CH3, CH) = 6.3 Hz, 12H, CH3); 2.76 (dd, J(CH2, CH) = 6.9 Hz, J(gem) = 13.7 Hz, 1H, CH2aPh); 2.81 (dd, J(NH, CH) = 11.1 Hz, J(NH, P) = 12.6 Hz, 1H, P-NH); 2.82 (dd, J(CH2a, Ph) = 7.7 Hz, J(gem) = 13.7 Hz, 1H, CH2aPh); 2.93 (dd, J(CH2, CH) = 5.7 Hz, J(gem) = 13.7 Hz, 1H, CH2bPh); 3.00 (dd, J(CH2, CH) = 5.7 Hz, J(gem) = 13.6 Hz, 1H, CH2bPh); 3.14 (bt, J(NH, CH) = J(NH, P) = 10.7 Hz, 1H, NH); 3.17 (dd, J(CH2a, P) = 9.2 Hz, J(gem) = 12.9 Hz, 1H, P-CH2a); 3.26 (dd, J(CH2b, P) = 8.5 Hz, J(gem) = 12.9 Hz, 1H, P-CH2b); 3.65 (m, 2H, H-2'); 4.04 (dddd, J(CH, CH2b) = 5.7 Hz, J(CH, CH2a) = 6.9 Hz, J(CH, P) = 9.0 Hz, J(CH, NH) = 11.0 Hz, 1H, CH-NH); 4.16 (dddd, J(CH, CH2b) = 5.7 Hz, J(CH, CH2a) = 7.7 Hz, J(CH, P) = 9.5 Hz, J(CH, NH) = 11.0 Hz, 1H, CH-NH); 4.29 (m, 2H, H-1'); 4.94 and 4.98 (2x sept, 1H, CHiPr); 5.24 (brs, 2H, NH2); 6.27 (d, J(6, 5) = 3.6 Hz, 1H, H-7); 6.99 (d, J(8, 7) = 3.6 Hz, 1H, H-8); 7.05 and 7.11 (2x m, 4H, o-Bn); 7.16–7.27 (m, 6H, m,p-Bn); 8.30 (s, 1H, H-2). 13C NMR (DMSO-d6): δ 21.62, 21.70, 21.71 and 21.79 (CH3iPr); 40.61 (d, J(C-P) = 4.0 Hz, CH2Bn); 40.80 (d, J(C-P) = 5.3 Hz, CH2Bn); 44.26 (C-1'); 53.62 and 54.02 (NH-CH); 67.95 (d, J(C-P) = 136.6 Hz, CH2-P); 69.07 and 69.12 (CHiPr); 71.96 (d, J(2'-P) = 13.4 Hz, C-2'); 97.40 (C-7); 103.14 (C-5); 125.99 (C-8); 126.84 and 126.88 (Ph-para); 128.32 and 128.37 (Ph-meta); 129.62 and 129.66 (Ph-ortho); 136.29 and 136.52 (ipso); 150.13 (C-4); 151.68 (C-2); 156.63 (C-6); 172.37 and 172.54 (m, COO). HR-MS (ESI+): m/z [M + H]+ calculated for: C33H44N6O6P, 651.3055, found: 651.3054.
Bis(L-phenylalanine isopropyl ester) prodrug of 9-[2-(phosphorylmethoxy)ethyl]-7-fluoro-7-deazaadenine (6b):
Treatment of 5b (0.35 g, 0.93 mmol) by Method A afforded 6b (0.32 g, 51%) as a whitish amorphous solid. ESI-MS, m/z (%): 669 [M+H+] (50); 691 [M+Na+] (100). 1H NMR (DMSO-d6): δ 1.01, 1.06, 1.11 and 1.16 (4 x d, J(CH3, CH) = 6.3 Hz, 12H, CH3); 2.73 (dd, J(gem) = 13.4 Hz, J(CH2, CH) = 7.1 Hz, 2H, PhCH2); 2.77–2.87 (m, 4H, PhCH2); 3.22 (dd, J(CH2a, P) = 8.3 Hz, J(gem) = 13.1 Hz, 1H, P-CH2a); 3.26 (dd, J(CH2b, P) = 8.0 Hz, J(gem) = 13.1 Hz, 1H, P-CH2b); 3.62 (brd, J(2', 1') = 5.1 Hz, 2H, H-2'); 3.87 (ddt, J(CH, NH) = 10.5 Hz, J(CH, P) = 9.0 Hz, J(CH, CH2) = 7.0 Hz, 1H, CH-NH); 3.93 (ddt, J(CH, NH) = 10.8 Hz, J(CH, P) = 9.3 Hz, J(CH, CH2) = 7.0 Hz, 1H, CH-NH); 4.12 (dd, J(NH, P) = 12.0 Hz, J(NH, CH) = 10.6 Hz, 1H, P-NH); 4.17 (brtd, J(1', 2'a) = J(1', 2'b) = 5.0 Hz, Jl.r. = 3.7 Hz, 2H, H-1'); 4.41 (dd, J(NH, P) = 11.9 Hz, J(NH, CH) = 10.8 Hz, 1H, P-NH); 4.77 (sept, J(CH, CH3) = 6.3 Hz, 2H, CHiPr); 4.82 (sept, J(CH, CH3) = 6.3 Hz, 2H, CHiPr); 6.91 (brs, 2H, NH2); 7.10 (m, 2H, Ph-ortho); 7.14 (d, J(8, F) = 2.3 Hz, 1H, H-8); 7.15 (m, 2H, Ph-ortho); 7.17–7.27 (m, 6H, Ph-meta, para); 8.05 (s, 1H, H-2). 13C NMR (DMSO-d6): δ 21.47, 21.54, 21.59 and 21.67 (CH3iPr); 40.20 (CH2Ph); 43.02 (C-1'); 54.05 and 54.19 (NH-CH); 67.52 (d, J(C-P) = 135.1 Hz, CH2-P); 67.99 and 68.12 (CHiPr); 71.11 (d, J(2'-P) = 11.9 Hz, C-2'); 92.02 (d, J(2'-P) = 15.0 Hz, C-5); 107.51 (d, J(C, F) = 26.0 Hz, C-8); 126.61 and 126.65 (Ph-para); 128.22 and 128.25 (Ph-meta); 129.64 and 129.65 (Ph-ortho); 137.20 and 137.30 (ipso); 141.95 (d, J(C, F) = 243.5 Hz, C-7); 145.43 (d, J(4, F) = 2.9 Hz, C-4); 152.73 (C-2); 155.96 (C-6); 172.33 and 172.46 J(C-C-N-P) = 5.1 Hz and J(C-C-N-P) = 2.9 Hz, COO). HR-MS (ESI+): m/z [M + H]+ calculated for: C33H42FN6O6P, 669.2960, found: 669.2958.
Bis(L-phenylalanine isopropyl ester) prodrug of 9-[2-(phosphorylmethoxy)ethyl]-7-chloro-7-deazaadenine (6c):
Treatment of 5c (0.50 g, 1.3 mmol) by Method A afforded 6c (0.49 g, 56%) as a whitish amorphous solid. ESI-MS, m/z (%): 685 [M+H+] (100). 1H NMR (DMSO-d6): δ 1.00, 1.06, 1.11 and 1.16 (4 x d, J(CH3, CH) = 6.3 Hz, 12H, CH3); 2.71–2.88 (m, 4H, PhCH2); 3.19–3.29 (m, 2H, P-CH2); 3.63 (m, 2H, H-2'); 3.83–3.96 (m, 2H, CH-NH); 4.11 (m, 1H, NH); 4.21 (m. 2H, H-1'); 4.41 (m, 1H, NH); 4.77 (sept, J(CH, CH3) = 6.3 Hz, 2H, CHiPr); 4.82 (sept, J(CH, CH3) = 6.3 Hz, 2H, CHiPr); 6.78 (brs, 2H, NH2); 7.07 (m, 10 H, Ph-ortho, meta, para); 7.36 (s, 1H, H-8); 8.09 (s, 1H, H-2). 13C NMR (DMSO-d6): δ 21.47, 21.54, 21.60 and 21.67 (CH3iPr); 40.20 (CH2Ph); 43.46 (C-1'); 54.05 and 54.20 (NH-CH); 67.48 (d, J(C-P) = 135.2 Hz, CH2-P); 67.98 and 68.12 (CHiPr); 70.93 (d, J(2'-P) = 11.5 Hz, C-2'); 99.68 (C-5); 101.17 (C-7); 122.26 (C-8); 126.60 and 126.65 (Ph-para); 128.21 and 128.25 (Ph-meta); 129.63 and 129.64 (Ph-ortho); 137.20 and 137.30 (ipso); 148.70 (C-4); 152.66 (C-2); 156.92 (C-6); 172.32–172.47 m (COO). HR-MS (ESI+): m/z [M + H]+ calculated for: C33H43N6O6ClP, 685.2665, found: 685.2667.
Bis(L-phenylalanine isopropyl ester) prodrug of 9-[2-(phosphorylmethoxy)ethyl]-7-bromo-7-deazaadenine (6d):
Treatment of 5d (0.50 g, 1.2 mmol) by Method A afforded 6d (0.31 g, 37%) as a whitish amorphous solid. ESI-MS, m/z (%): 729 [M+H+] (100). 1H NMR (DMSO-d6): δ 1.00, 1.06, 1.11 and 1.16 (4 x d, J(CH3, CH) = 6.2 Hz, 12H, CH3); 2.71–2.88 (m, 4H, PhCH2); 3.18–3.29 (m, 2H, P-CH2); 3.64 (m, 2H, H-2'); 3.83–3.96 (m, 2H, CH-NH); 4.10 (m, 1H, P-NH); 4.22 (m, 2H, H-1'); 4.41 (m, 1H, P-NH); 4.77 (sept, J(CH, CH3) = 6.3 Hz, 2H, CHiPr); 4.82 (sept, J(CH, CH3) = 6.3 Hz, 2H, CHiPr); 6.71 (brs, 2H, NH2); 7.08–7.26 (m, 10H, Ph-ortho, meta, para); 7.42 (s, 1H, H-8); 8.10 (s, 1H, H-2). 13C NMR (DMSO-d6): δ 21.47, 21.55, 21.61 and 21.68 (CH3iPr); 40.21 (CH2Ph); 43.58 (C-1'); 54.05 and 54.21 (NH-CH); 67.47 (d, J(C-P) = 135.3 Hz, CH2-P); 67.98 and 68.12 (CHiPr); 70.91 (d, J(2'-P) = 11.4 Hz, C-2'); 85.12 (C-7); 100.87 (C-5); 124.77 (C-8); 126.60 and 126.65 (Ph-para); 128.21 and 128.25 (Ph-meta); 129.25 and 129.64 (Ph-ortho); 137.20 and 137.30 (ipso); 149.19 (C-4); 152.47 (C-2); 157.09 (C-6); 172.32–172.47 m (COO). HR-MS (ESI+): m/z [M + H]+ calculated for: C33H43BrN6O6P, 729.2160, found: 729.2163.
Bis(L-phenylalanine isopropyl ester) prodrug of 9-[2-(phosphorylmethoxy)ethyl]-7-iodo-7-deazaadenine (6e):
Treatment of 5e (0.83 g, 1.7 mmol) by Method A afforded 6e (0.48 g, 62%) as a whitish amorphous solid. ESI-MS, m/z (%): 777 [M+H+] (100). 1H NMR (DMSO-d6): δ 1.01, 1.06, 1.11 and 1.16 (d, J(CH3, CH) = 6.3 Hz, 12H, CH3); 2.71–2.73 (m, 4H, PhCH2); 3.20 (dd, J(CH2b, P) = 8.2 Hz, J(gem) = 13.2 Hz, 1H, P-CH2b); 3.27 (dd, J(CH2a, P) = 7.9 Hz, J(gem) = 13.2 Hz, 1H, P-CH2a); 3.64 (m, 2H, H-2'); 3.83–3.96 (m, 2H, CH-NH); 4.08 (m, 1H, P-NH); 4.22 (m, 2H, H-1'); 4.40 (m, 1H, P-NH); 4.78 (sept, J(CH, CH3) = 6.2 Hz, 2H, CHiPr); 4.82 (sept, J(CH, CH3) = 6.2 Hz, 2H, CHiPr); 6.59 (brs, 2H, NH2); 7.07–7.25 (m, 10H, Ph-ortho, meta, para); 7.44 (s, 1H, H-8); 8.10 (s, 1H, H-2). 13C NMR (DMSO-d6): δ 21.49, 21.56, 21.62 and 21.69 (CH3iPr); 40.21 (CH2Ph); 43.65 (C-1'); 49.92 (C-7); 54.05 and 54.21 (NH-CH); 67.46 (d, J(C-P) = 135.4 Hz, CH2-P); 67.98 and 68.12 (CHiPr); 70.95 (d, J(2'-P) = 11.4 Hz, C-2'); 103.08 (C-5); 126.59 and 126.64 (Ph-para); 128.21 and 128.25 (Ph-meta); 129.65 (Ph-ortho); 130.00 (C-8); 137.19 and 137.29 (ipso); 149.83 (C-4); 151.98 (C-2); 157.32 (C-6); 172.32–172.47 m (COO). HR-MS (ESI+): m/z [M + H]+ calculated for: C33H43IN6O6P, 777.2021, found: 777.2022.
6-Chloro-9-iodo-9-deazapurine (8):50
N-Iodosuccinimide (8.06 g, 35.8 mmol) was added to a solution of 6-chloro-9-deazapurine 7 (5.0 g, 32.6 mmol) in dry THF (70 mL) under Ar at RT. The reaction mixture was strirred at RT for 1 h. Volatiles were removed and the residue was dissolved in CHCl3 (250 mL) and washed with H2O (100 mL), 10% Na2S2O3 (100 mL), brine (100 mL) and then dried over Na2SO4. The solvent was removed in vacuo to give 8 (7.70 g, 85%) as white solid, which was used in the next step without further purification The analytical data are identical with reported data.50,60
6-Chloro-9-iodo-7-((2-(trimethylsilyl)ethoxy)methyl)-9-deazapurine (9):
NaH (60% susp. in mineral oil, 1.01 g, 27.5 mmol) was added to a solution of 8 (5.64 g, 20.2 mmol) in DMF (75 mL) under Ar at RT. After 40 min, SEMCl (5.36 mL, 30.2 mmol) was added to the mixture at RT. The resulting mixture was stirred at RT overnight, then sat. NH4Cl (5 mL) was added. Solvents were removed, the residue was dissolved in EtOAc (300 mL), extracted with H2O (2 × 50 mL) and brine (50 mL) and then dried over Na2SO4. Volatiles were removed and the residue crystalized from hexan/EtOAc to give 9 (7.8 g, 94%) as a white amorphous solid. ESI-MS, m/z (%): 410 [M+H+] (100). 1H NMR (DMSO-d6): −0.12 (s, 9H, CH3); 0.79 (t, J(CH2, CH2) = 7.8 Hz, 2H, Si-CH2); 3.50 (t, J(CH2, CH2) = 7.8 Hz, 2H, Si-CH2-CH2); 5.76 (s, 2H, N-CH2); 8.42 (s, 1H, H-8); 8.75 (s, 1H, H-2). 13C NMR (DMSO-d6): δ −1.57 (CH3); 16.90 (Si-CH2); 58.90 (C-9); 65.27 (Si-CH2-CH2 ); 76.72 (N-CH2); 123.30 (C-5); 141.89 (C-6); 142.29 (C-8); 150.24 (C-2); 152.67 (C-4). HR-MS (ESI+): m/z [M + H]+ calculated for: C12H18ClIN3OSi, 409.9947, found: 409.9945.
6-Chloro-9-vinyl-7-((2-(trimethylsilyl)ethoxy)methyl)-9-deazapurine (10):
Tributyl(vinyl)tin (1.94 mL, 6.6 mmol) was added to a solution of 9 (2.3 g, 5.6 mmol) and Pd(t-Bu3P)2 (0.1 g, 0.2 mmol) in dioxane (20 mL) under Ar. The resulting mixture was stirred at RT for 24 h, cooled to RT and sat. NH4Cl (5 mL) and EDTA (10 mL) were added. After 15 min, EtOAc (250 mL) was added to the stirred slurry. The resulting mixture was filtered over Cellite. The organic layer was separated and washed with sat. EDTA solution (50 mL), H2O (50 mL), brine (50 mL) and then dried over Na2SO4. Volatiles were removed in vacuo, and the residue was purified by flash chromatography (Hex:EtOAc 0–70%) to give 10 (1.2 g, 69%) as a yellowish oil. ESI-MS, m/z (%): 310 [M+H+] (100). 1H NMR (DMSO-d6): δ −0.12 (s, 9H, CH3); 0.80 (t, J(CH2, CH2) = 7.8 Hz, 2H, Si-CH2); 3.50 (t, J(CH2, CH2) = 7.8 Hz, 2H, Si-CH2-CH2); 5.34 (dd, J(H-C=C-H) = 11.3 Hz, J(gem) = 2.2 Hz 1H, CH2=CH); 5.76 (s, 2H, N-CH2); 6.30 (dd, J(H-C=C-H) = 17.7 Hz, J(gem) = 2.2 Hz, 1H, CH2=CH); 6.83 (dd, J(H-C=C-H) = 17.7 Hz, J(H-C=C-H) = 11.4 Hz, 1H, CH2=CH); 8.28 (s, 1H, H-8); 8.74 (s, 1H, H-2). 13C NMR (DMSO-d6): δ −1.25 (CH3); 17.22 (Si-CH2); 65.49 (Si-CH2-CH2); 76.88 (N-CH2); 114.11 (C-9); 115.18 (CH2=CH); 123.98 (C-5); 126.51 (CH2=CH); 137.53 (C-8); 142.18 (C-6); 150.30 (C-2); 150.49 (C-4). HR-MS (ESI+): m/z [M + H]+ calculated for: C14H21ClN3OSi, 310.1137, found: 310.1134.
6-Chloro-7-((2-(trimethylsilyl)ethoxy)methyl)-9-(2-hydroxyethyl)-9-deazapurine (11):
Treatment of 10 (1.2 g, 3.9 mmol) by Method C afforded 11 (1.0 g, 79%) as yellowish oil. ESI-MS, m/z (%): 350 [M+Na+] (100). 1H NMR (DMSO-d6): δ −0.12 (s, 9H, CH3); 0.80 (t, J(CH2, CH2) = 7.8 Hz, 2H, Si-CH2); 2.88 (t, J(CH2, CH2) = 7.0 Hz, 2H, H-1'); 3.47 (t, J(CH2, CH2) = 7.8 Hz, 2H, Si-CH2-CH2); 3.68 (dt, J(CH2, CH2) = 7.0 Hz, J = 5.4 Hz, H-2'); 5.74 (s, 2H, N-CH2); 8.00 (s, 1H, H-8); 8.66 (s, 1H, H-2). 13C NMR (DMSO-d6): δ −1.56 (CH3); 16.98 (Si-CH2); 26.87 (C-1'); 60.50 (C-2'); 65.00 (Si-CH2-CH2 ); 76.21 (N-CH2); 113.07 (C-9); 122.93 (C-5); 137.12 (C-8); 141.32 (C-6); 149.13 (C-2); 151.76 (C-4). HR-MS (ESI+): m/z [M + H]+ calculated for: C14H22ClN3O2Si, 350.1062, found: 350.1066.
9-{2-[(Diisopropoxyphosphoryl)methoxy]ethyl}−6-chloro-7-((2-(trimethylsilyl)ethoxy)methyl)-9-deazapurine (12):
Treatment of 11 (3.0 g, 9.2 mmol) by Method D afforded recovered starting material 11 (1.40 g, 46%) and 12 (2.38 g, 51%) as yellowish oils. ESI-MS, m/z (%): 506 [M+H+] (75); 528 [M+Na+] (100). 1H NMR (DMSO-d6): δ −0.11 (s, 9H, CH3); 0.80 (m, 2H, Si-CH2); 1.16 (d, J(CH3, CH) = 6.2 Hz, 6H, CH3); 1.20 (d, J(CH3, CH) = 6.2 Hz, 6H, CH3); 2.98 (td, J(1', 2') = 6.8 Hz, J(1', 8) = 0.8 Hz, 2H, H-1'); 3.49 (m, 2H, Si-CH2-CH2); 3.75 (d, J(H-C-P) = 8.3 Hz, 2H, P-CH2); 3.82 (t, J(2', 1') = 6.8 Hz, 2H, H-2'); 4.54 (d septet, J(H-C-O-P) = 7.7 Hz, J(CH, CH3) = 6.2 Hz, 2H, CHiPr); 5.73 (s, 2H, N-CH2); 8.02 (t, J(8, 1') = 0.8 Hz, 1H, H-8); 8.67 (s, 1H, H-2). 13C NMR (DMSO-d6): δ −1.28 (Si-CH3); 17.26 (Si-CH2); 23.57 (C-1'); 23.78 (d, J(C-C-O-P) = 4.5 Hz, CH3iPr); 23.94 (d, J(C-C-O-P) = 3.8 Hz, CH3iPr); 64.79 (d, J(C-P) = 164.7 Hz, CH2-P); 65.34 (Si-CH2-CH2 ); 70.24 (d, J(C-O-P) = 6.4 Hz, CHiPr); 71.85 (d, J(2'-P) = 11.8 Hz, C-2'); 76.55 (N-CH2); 112.61 (C-9); 123.22 (C-5); 137.39 (C-8); 141.64 (C-6); 149.51 (C-2); 151.92 (C-4). HR-MS (ESI+): m/z [M + H]+ calculated for: C21H38ClN3O5PSi, 506.2001, found: 506.2004.
9-{2-[(Diisopropoxyphosphoryl)methoxy]ethyl}−7-((2-(trimethylsilyl)-ethoxy)methyl)-9-deazaadenine (13):
Compound 12 (1.0 g, 2.0 mmol), was dissolved ina EtOH/NH3 mixture (40 mL) and stirred at 100 °C for 16 h. Volatiles were removed and the residue was purified by C18 reversed phase chromatography (H2O:MeOH 0–100%) to give 13 (0.71 g, 74%) as a white amorphous solid. ESI-MS, m/z (%): 487 [M+H+] (100). 1H NMR (DMSO-d6): δ −0.06 (s, 9H, CH3); 0.85 (m, 2H, Si-CH2); 1.19 (d, J(CH3, CH) = 6.2 Hz, 6H, CH3); 1.22 (d, J(CH3, CH) = 6.2 Hz, 6H, CH3); 2.86 (td, J(1', 2') = 7.0 Hz, J(1', 8) = 0.8 Hz, 2H, H-1'); 3.49 (m, 2H, Si-CH2-CH2); 3.74 (d, J(H-C-P) = 8.3 Hz, 2H, P-CH2); 3.76 (t, J(2', 1') = 7.1 Hz, 2H, H-2'); 4.56 (d septet, J(H-C-O-P) = 7.7 Hz, J(CH, CH3) = 6.2 Hz, 2H, CHiPr); 5.54 (s, 2H, N-CH2); 6.58 (brs, 2H, NH2); 7.46 (t, J(8, 1') = 0.8 Hz, 1H, H-8); 8.67 (s, 1H, H-2). 13C NMR (DMSO-d6): δ −1.27 (Si-CH3); 17.27 (Si-CH2); 23.82 (C-1'); 23.82 (d J(C-C-O-P) = 4.3 Hz, CH3iPr); 23.96 (d, J(C-C-O-P) = 3.8 Hz, CH3iPr); 64.79 (d, J(C-P) = 164.6 Hz, CH2-P); 64.89 (Si-CH2-CH2 ); 70.23 (d, J(C-O-P) = 6.4 Hz, CHiPr); 72.32 (d, J(2'-P) = 11.8 Hz, C-2'); 76.90 (N-CH2); 111.10 (C-9); 114.21 (C-5); 131.45 (C-8); 148.57 (C-4); 150.40 (C-2); 150.97 (C-6). HR-MS (ESI+): m/z [M + H]+ calculated for: C21H40N4O5PSi, 487.2500, found: 487.2495.
9-[2-(Phosphonomethoxy)ethyl]-9-deazaadenine (14):
Compound 13 (0.5 g, 1 mmol) was suspended in H2O (20 mL) and conc. HCl (0.46 mL, 5.15 mmol). The reaction mixture was heated to 130 °C for 40 min in microwave reactor. Volatiles were removed and the residue was codistilled with H2O (2 × 5 mL) and dissolved in 3% aq. NH3 (1 mL). The solution was applied on Dowex 1 × 2 (acetate form) column and eluted with H2O and grad. 0.05 M-1M aq. acetic acid. The product containing UV-absorbing fractions were collected, evaporated and codistilled with water (2 × 5 mL). Crystallization from H2O gave 14 (0.16 g, 56%) as white crystals, m.p. >250 °C (dec.). ESI-MS, m/z (%): 273 [M+H+] (81); 295 [M+Na+] (100). 1H NMR (DMSO-d6): δ 2.94 (td, J(1', 2') = 6.9 Hz, J(1', 8) = 0.8 Hz, 2H, H-1'); 3.53 (d, J(H-C-P) = 8.6 Hz, 2H, P-CH2); 3.82 (t, J(2', 1') = 7.1 Hz, 2H, H-2'); 7.39 (t, J(8, 1') = 0.8 Hz, 1H, H-8); 8.05 (s, 1H, H-2). 13C NMR (DMSO-d6): δ 24.11 (C-1'); 69.36 (d, J(C-P) = 150.2 Hz, CH2-P); 72.86 (d, J(2'-P) = 10.1 Hz, C-2'); 111.95 (C-9); 114.83 (C-5); 129.87 (C-8); 145.46 (C-4); 149.77 (C-2); 150.99 (C-6). HR-MS (ESI+): m/z [M + H]+ calculated for: C9H14N4O4P, 273.0747, found: 273.0744.
Bis(L-phenylalanine isopropyl ester) prodrug of 9-[2-(phosphorylmethoxy)ethyl]-9-deazaadenine (15):
Bromotrimethyl-silane (1.0 mL) was added to 13 (0.5 g, 1 mmol) in dry pyridine (10 mL) and the resulting mixture was stirred at RT overnight. Iodotrimethylsilane (1.0 mL) was added and the reaction mixture was stirred for additional 2 h. Subsequent treatment by Method A afforded 15 (0.29 g, 44%) as a whitish amorphous solid. ESI-MS, m/z (%): 651 [M+H+] (100). 1H NMR (DMSO-d6): δ 1.03, 1.08, 1.12 and 1.17 (d, J(CH3, CH) = 6.2 Hz, 12H, CH3); 2.75–2.91 (m, 6H, CH2Ph, H-1'); 3.24–3.33 (m, 2H, P-CH2); 3.59 (m, 2H, H-2'); 3.89 and 3.96 (2x m, 2H, NH-CH); 4.08 (dd, J(NH-P) = 12.1 Hz, J(NH-CH) = 10.5 Hz, P-NH); 4.40 (dd, J(NH-P) = 11.8 Hz, J(NH-CH) = 10.7 Hz, P-NH); 4.80 (sept, J(CH, CH3) = 6.3 Hz, 2H, CHiPr); 4.83 (sept, J(CH, CH3) = 6.3 Hz, 2H, CHiPr); 6.62 (brs, 2H, NH2); 7.12–7.27 (m, 10H, Ph); 7.32 (dt, J(8, 7) = 2.8 Hz, J(8, 1') = 0.8 Hz, 1H, H-8); 8.09 (s, 1H, H-2); 10.65 (brs, 1H, H-7). 13C NMR (DMSO-d6): δ 21.48, 21.55, 21.61 and 21.67 (CH3iPr); 24.14 (C-1'); 39.90 (CH2Ph); 54.10 and 54.11 (NH-CH); 67.57 (d, J(C-P) = 134.5 Hz, CH2-P); 67.98 and 68.11 (CHiPr); 72.61 (d, J(2'-P) = 11.9 Hz, C-2'); 111.34 (C-9); 113.38 (C-5); 125.98 (C-8); 126.63 (Ph-para); 128.24 (Ph-meta); 129.65 (Ph-ortho); 137.15 and 137.32 (ipso); 146.04 (C-4); 150.05 (C-2); 150.46 (C-6); 172.27–172.49 m, (COO). HR-MS (ESI+): m/z [M + H]+ calculated for: C33H44N6O6P, 651.3055, found: 651.3053.
9-{2-[(Diisopropoxyphosphoryl)methoxy]ethyl}−7-deaza-8-azaadenine (17a):
Treatment of 6-amino-7-deaza-8-azapurine (16a, 1.0 g, 7.4 mmol) according to the lit.54 afforded 17a (1.4 g, 53%) as a yellowish oil. ESI-MS, m/z (%): 358 [M+H+] (100). 1H NMR (DMSO-d6): δ 1.20 (d, J(CH3, CH) = 6.2 Hz, 6H, CH3); 1.25 (d, J(CH3, CH) = 6.2 Hz, 6H, CH3); 3.73 (d, J(CH2, P) = 8.5 Hz, 2H, P-CH2); 4.07 (t, J(1', 2') = 5.6 Hz, 2H, H-1'); 4.59 (t, J(2', 1') = 5.6 Hz, 2H, H-2'); 4.68–4.56 (m, 2H, CHiPr); 6.07 (brs, 2H, NH2); 7.97 (s, 1H, H-8); 8.35 (s, 1H, H-2). 13C NMR (DMSO-d6): δ 23.81, 23.86, 23.98 and 24.01 (CH3iPr); 46.48 (C-1'); 65.74 (d, J(C-P) = 167.3 Hz, CH2-P); 70.89 (d, J(2'-P) = 11.3 Hz, C-2'); 71.16 (d, J(C-O-P) = 6.6 Hz, CHiPr); 100.65 (C-5); 131.11 (C-7); 153.92 (C-4); 155.87 (C-2); 157.61 (C-6). HR-MS (ESI+): m/z [M + H]+ calculated for: C14H25N5O4P, 358.1639, found: 358.1639.
9-{2-[(Diisopropoxyphosphoryl)methoxy]ethyl}−8-azaadenine (17b):
Diisopropyl ((2-chloroethoxy)methyl)phosphonate55 (2.65 g, 10.3 mmol) dissolved in DMF (4 mL) was added to a preheated (80 °C) reaction mixture of 8-azaadenine (16a, 1.0 g, 7.3 mmol) and DBU (2.63 mL, 17.2 mmol) in DMF (8 mL). The resulting mixture was heated to 100 °C overnight. The clear solution was cooled down and volatiles were removed in vacuo. The residue was codistilled with toluene (2 × 5 mL) and purified by column chromatography EtOH:CHCl3 (0–10%) to give 17b (0.85 g, 32%) and the corresponding 8-regioisomer (0.85 g, 32%) as yellowish oils. Compound 17b: ESI-MS, m/z (%): 359 [M+H+] (11); 381 [M+Na+] (100). 1H NMR (DMSO-d6): δ 1.04 (d, J(CH3, CH) = 6.2 Hz, 6H, CH3); 1.11 (d, J(CH3, CH) = 6.2 Hz, 6H, CH3); 3.74 (d, J(H-C-P) = 8.4 Hz, 2H, P-CH2); 4.06 (m, 2H, H-2'); 4.40 (d septet, J(CH, CH3) = 6.2 Hz, J(H-C-O-P) = 7.7 Hz, 2H, CHiPr); 4.71 (m, 2H, H-1'); 8.28 (s, 1H, H-2); 8.06 and 8.41 (brs, 2H, NH2). 13C NMR (DMSO-d6): δ 23.66 (d J(C-C-O-P) = 4.6 Hz, CH3iPr); 23.87 (d J(C-C-O-P) = 3.8 Hz, CH3iPr); 46.03 (C-1'); 64.66 (d, J(C-P) = 163.7 Hz, CH2-P); 69.87 (d, J(2'-P) = 12.1 Hz, C-2'); 70.33 (d, J(C-O-P) = 6.4 Hz, C-2'); 123.98 (C-5); 149.33 (C-4); 156.40 (C-6); 156.83 (C-2). HR-MS (ESI+): m/z [M + H]+ calculated for: C13H24N6O4P, 359.1591, found: 359.1592.
Bis(L-phenylalanine isopropyl ester) prodrug of 9-[2-(phosphorylmethoxy)ethyl]-7-deaza-8-azaadenine (18a):
Treatment of 17a (0.70 g, 2.0 mmol) by Method A afforded 18a (0.51 g, 40%) as a whitish amorphous solid. ESI-MS, m/z (%): 652 [M+H+] (48); 674 [M+Na+] (100). 1H NMR (DMSO-d6): δ 1.00, 1.05, 1.11 and 1.15 (4 x d, J(CH3, CH) = 6.3 Hz, 12H, CH3); 2.71 (dd, J(gem) = 13.4 Hz, J(CH2-CH) = 8.2 Hz, 1H, PhCH2); 2.74–2.86 (m, 3H, PhCH2); 3.23 (dd, J(CH2b, P) = 8.0 Hz, J(gem) = 13.3 Hz, 1H, P-CH2b); 3.27 (dd, J(CH2a, P) = 8.3 Hz, J(gem) = 13.3 Hz, 1H, P-CH2a); 3.77 (m, 2H, H-2'); 3.83 (m, 1H, CH-NH); 3.91 (m, 1H, CH-NH); 4.04 (dd, J(NH, P) = 12.0 Hz, J(NH, CH) = 10.7 Hz, 1H, P-NH); 4.30–4.45 (m, 3H, H-1', P-NH); 4.77 (sept, J(CH, CH3) = 6.3 Hz, 2H, CHiPr); 4.80 (sept, J(CH, CH3) = 6.3 Hz, 2H, CHiPr); 7.08 and 7.15 (2x m, 2H, Ph-ortho); 7.16–7.28 (m, 6H, Ph-meta, para); 7.70 (brm, 2H, NH2); 8.09 (s, 1H, H-8); 8.16 (s, 1H, H-2). 13C NMR (DMSO-d6): δ 21.47, 21.54, 21.61 and 21.67 (CH3iPr); 40.10 and 40.20 (CH2Ph); 45.79 (C-1'); 54.09 and 54.14 (NH-CH); 67.30 (d, J(C-P) = 135.0 Hz, CH2-P); 67.98 and 68.09 (CHiPr); 70.24 (d, J(2'-P) = 11.4 Hz, C-2'); 100.25 (C-5); 126.62 and 126.65 (Ph-para); 128.24 and 128.25 (Ph-meta); 129.65 (Ph-ortho); 132.28 (C-7); 137.15 and 137.30 (ipso); 153.49 (C-4); 156.12 (C-2); 158.28 (C-6); 172.25 (d, J(C-C-N-P) = 5.1 Hz, COO); 172.43 (d, J(C-C-N-P) = 3.0 Hz, COO). HR-MS (ESI+): m/z [M + H]+ calculated for: C32H43N7O6P, 652.3007, found: 652.3007.
Bis(L-phenylalanine isopropyl ester) prodrug of 9-[2-(phosphorylmethoxy)ethyl]-8-azaadenine (18b):
Treatment of 17b (0.46 g, 1.3 mmol) by Method A afforded 18b (0.38 g, 45%) as a whitish amorphous solid. ESI-MS, m/z (%): 653 [M+H+] (10); 675 [M+Na+] (100). 1H NMR (DMSO-d6): δ 1.00, 1.05, 1.10 and 1.14 (4 x d, J(CH3, CH) = 6.3 Hz, 12H, CH3); 2.68–2.85 (m, 4H, PhCH2); 3.24–3.33 (m, 2H, P-CH2); 3.79–3.96 (m, 4H, H-2', CH-NH); 4.02 (m, 1H, P-NH); 4.38 (m, 1H, P-NH); 3.65 (m, 2H, H-2'); 4.72–4.81 (m, 2H, CHiPr); 7.06–7.26 (m, 10H, Ph-ortho, meta, para); 8.26 (s, 1H, H-2); 8.07 and 8.40 (2x brs, 1H, NH2). 13C NMR (DMSO-d6): δ 21.45, 21.52, 21.59 and 21.65 (CH3iPr); 40.16 (CH2Ph); 45.98 (C-1'); 54.05 and 54.09 (NH-CH); 67.35 (d, J(C-P) = 135.6 Hz, CH2-P); 67.97 and 68.08 (CHiPr); 69.85 (d, J(2'-P) = 10.9 Hz, C-2'); 124.01 (C-5); 126.59 and 126.64 (Ph-para); 128.21 and 128.24 (Ph-meta); 129.62 (Ph-ortho); 137.10 and 137.27 (ipso); 149.29 (C-4); 156.43 (C-6); 156.93 (C-2); 172.21 (d, J(C-C-N-P) = 5.2 Hz, COO); 172.40 (d, J(C-C-N-P) = 2.9 Hz, COO). HR-MS (ESI+): m/z [M + H]+ calculated for: C31H42N8O6P, 653.2959, found: 653.6962.
4-Chloro-7-vinylthieno[3,2-d]pyrimidine (20):
Tributyl(vinyl)tin (7.7 mL, 26.3 mmol) was added to a solution of 7-bromo-4-chlorothieno[3,2-d]pyrimidine (19, 4.19 g, 16.9 mmol), Pd(t-Bu3P)2 (0.37 g, 0.72 mmol) and CuI (0.48 g, 2.5 mmol) in dioxane (54 mL) under Ar. The resulting mixture was heated to 120 °C for 24 h, cooled to RT and aq. solution of KF (4.0 g in 30 mL) was added. After 15 min, EtOAc (100 mL) was added to the resulting stirred slurry. The resulting mixture was filtered over Cellite after additional 15 min. The organic layer was separated and washed with sat. EDTA solution (50 mL), H2O (50 mL), brine (30 mL) and then dried over Na2SO4. Volatiles were removed in vacuo, and the residue was purified by flash chromatography (grad. Hex:CHCl3 0–100% and subsequent grad. MeOH v CHCl3 0–10%) to give 20 (2.33 g, 50%) as a yellow oil. ESI-MS, m/z (%): 195 [M+] (100). 1H NMR (DMSO-d6): δ ppm 5.55 (dd, J(vic) = 11.3 Hz, J(gem) = 1.7 Hz, 1H, CH2b); 6.48 (dd, J(vic) = 17.8 Hz, J(gem) = 1.7 Hz, 1H, CH2a); 7.04 (ddm, J(vic) = 17.8 Hz, J(vic) = 11.3 Hz, 1H, CH=); 8.62 (s, 1H, H-8); 9.11 (s, 1H, H-2). 13C NMR (DMSO-d6): δ 118.47 (CH2=CH-); 127.80 (CH2=CH-); 131.02 (C-5); 133.95 (C-9); 135.42 (C-8); 154.19 (C-6); 154.53 (C-2); 159.34 (C-4). HR-MS (EI+): m/z [M]+ calculated for: C8H5N2SCl, 195.9862, found: 195.9861.
4-Chloro-7-(2-hydroxyethyl)thieno[3,2-d]pyrimidine (21):
Treatment of 20 (1.3 g, 6.7 mmol) by Method C (additional 9-BBN (26.8 mL, 13.4 mmol) was added after 3 h at RT and the reaction mixture was stirred overnight; column chromatography in grad. MeOH:CHCl3 0–10%) afforded 21 (0.91 g, 64%) as a yellowish oil. ESI-MS, m/z (%): 215 [M+H+] (100). 1H NMR (DMSO-d6): δ 3.04 (td, J(1', 2') = 6.7 Hz, J(1', 8) = 1.0 Hz, 2H, H-1'); 3.74 (t, J(2', 1') = 6.7 Hz, 2H, H-2'); 4.80 (brs, 1H, OH); 8.27 (t, J(8, 1') = 1.0 Hz, 1H, H-8); 9.05 (s, 1H, H-2). 13C NMR (DMSO-d6): δ 31.01 (C-1'); 60.04 (C-2'); 130.46 (C-5); 135.13 (C-8); 135.47 (C-9); 154.03 (C-6); 154.21 (C-2); 160.98 (C-4). HR-MS (ESI+): m/z [M + H]+ calculated for: C8H8N2OClS, 215.0040, found: 215.0040.
4-Chloro-7-{2-[(diisopropoxyphosphoryl)methoxy]ethyl}thieno[3,2-d]pyrimidine (22):
Treatment of 21 (0.60 g, 2.8 mmol) by Method D afforded 22 (0.58 g, 53%) as yellowish oil, which was used in the next reaction step without further purification and characterization.
4-Amino-7-{2-[(diisopropoxyphosphoryl)methoxy]ethyl}thieno[3,2-d]pyrimidine (23):
Compound 22 (0.86 g, 2.2 mmol) was dissolved in EtOH/NH3 (50 mL) and stirred at 100 °C for 24 h. The solvent was removed and the residue was purified by C18 reversed phase chromatography (H2O:MeOH 0–100%) to give 23 (0.54 g, 66%) as a yellowish oil. ESI-MS, m/z (%): 374 [M+H+] (36); 396 [M+Na+] (100). 1H NMR (DMSO-d6): δ 1.17 (d, J(CH3, CH) = 6.2 Hz, 6H, CH3); 1.20 (d, J(CH3, CH) = 6.2 Hz, 6H, CH3); 3.02 (td, J(1', 2') = 6.7 Hz, J(1', 8) = 1.0 Hz, 2H, H-1'); 3.75 (d, J(H-C-P) = 8.4 Hz, 2H, P-CH2); 3.82 (t, J(2', 1') = 6.7 Hz, 2H, H-2'); 4.54 (d septet, J(H-C-O-P) = 7.7 Hz, J(CH, CH3) = 6.2 Hz, 2H, CHiPr); 7.38 (brs, 2H, NH2); 7.82 (t, J(8, 1') = 1.0 Hz, 1H, H-8); 8.38 (s, 1H, H-2). 13C NMR (DMSO-d6): δ 23.84 (d, J(C-C-O-P) = 4.5 Hz, CH3iPr); 23.98 (d, J(C-C-O-P) = 3.7 Hz, (CH3iPr); 27.46 (C-1'); 64.73 (d, J(C-P) = 164.4 Hz, CH2-P); 70.28 (d, J(C-O-P) = 6.3 Hz, CHiPr); 71.46 (d, J(C-O-C-P) = 12.3 Hz, C-2'); 114.19 (C-5); 128.96 (C-8); 133.83 (C-9); 154.69 (C-2); 158.56 (C-4); 158.66 (C-6). HR-MS (ESI+): m/z [M + H]+ calculated for: C15H25N3O4PS, 374.1298, found: 374.1298.
Bis(L-phenylalanine isopropyl ester) prodrug of (4-aminothieno[3,2-d]pyrimidin-7-yl)ethoxy)methyl)phosphonic acid (24):
Treatment of 23 (0.27 g, 0.7 mmol) by Method A afforded 24 (0.25 g, 51%) as a whitish amorphous solid. ESI-MS, m/z (%): 668 [M+H+] (35); 690 [M+Na+] (100). 1H NMR (DMSO-d6): δ 1.02, 1.07, 1.12 and 1.16 (4 x d, J(CH3, CH) = 6.3 Hz, 12H, CH3); 2.74–2.90 (m, 4H, PhCH2); 2.97 (td, J(CH2-CH2) = 6.7 Hz, J(1', 8) = 0.9 Hz, 2H, H-1'); 3.23–3.32 (m, 2H, P-CH2); 3.64 (t, J(2', 1') = 7.0 Hz, 2H, H-2'); 3.85–4.00 (m, 2H, NH-CH); 4.10 (dd, J(NH-P) = 11.9 Hz, J(NH-CH) = 10.5 Hz, 1H, P-NH); 4.42 (m, 1H, P-NH); 4.79 (sept, J(CH, CH3) = 6.3 Hz, 2H, CHiPr); 4.82 (sept, J(CH, CH3) = 6.3 Hz, 2H, CHiPr); 7.10–7.26 (m, 10H, Ph-ortho, meta, para); 7.38 (brs, 2H, NH2); 7.81 (t, J(8, CH2) = 1.0 Hz, 1H, H-8); 8.39 (s, 1H, H-2). 13C NMR (DMSO-d6): δ 21.50, 21.56, 21.63 and 21.69 (CH3iPr); 27.43 (C-1'); 39.53 (CH2Ph); 54.11 and 54.13 (NH-CH); 67.55 (d, J(C-P) = 135.3 Hz, CH2-P); 68.01 and 68.14 (CHiPr); 71.31 (d, J(2'-P) = 12.2 Hz, C-2'); 114.17 (C-5); 126.63 and 126.65 (Ph-para); 128.23 and 128.25 (Ph-meta); 128.96 (C-8); 129.66 and 129.67 (Ph-ortho); 133.90 (C-9); 137.17 and 137.32 (ipso); 154.71 (C-2); 158.56 and 158.66 (C-4 and C-6); 172.32 (d, J(C-C-N-P) = 5.3 Hz, COO); 172.50 (d, J(C-C-N-P) = 3.0 Hz, COO). HR-MS (ESI+): m/z [M + H]+ calculated for: C33H43N5O6PS, 668.2666, found: 668.2667.
8-Bromo-4-(2,4-dimethoxybenzyl)aminoquinazoline (26):
DBU (3.4 mL, 22.7 mmol) was added to a solution of 8-bromo-4-oxochinazoline 25 (2.0 g, 8.9 mmol) and BOP (6.0 g, 13.6 mmol) in CH3CN (60 mL) under Ar. The mixture was stirred at RT for 10 min and then preheated to 80 °C. 2,4-Dimethoxybenzylamin (4 mL, 26.4 mmol) was added and the mixture was stirred at 80 °C overnight. Volatiles were removed after cooling down. The residue was dissolved in EtOAc (300 mL) and washed with brine (60 mL) and dried by Na2SO4. The residue was purified by flash chromatography (grad. MeOH-CHCl3 0–10%) to give 26 (1.73 g, yield 55%) as a white amorphous solid. ESI-MS, m/z (%): 374 [M+H+] (100). 1H NMR (DMSO-d6): δ 3,72 (s, 3H, 4'-OCH3); 3.82 (s, 3H, 2'-OCH3); 4.67 (bd, J(CH2-NH) = 5.6 Hz, 2H, PhCH2); 6.44 (dd, J(5', 6') = 8.4 Hz, J(5', 3') = 2.4 Hz, 1H, H-5'); 6.58 (d, J(3', 5') = 2.4 Hz, 1H, H-3'); 7.08 (d, J(6', 5') = 8.4 Hz, 1H, H-6'); 7.42 (dd, J(6, 5) = 8.3 Hz, J(6, 7) = 7.6 Hz, 1H, H-6); 8.12 (dd, J(7, 6) = 7.6 Hz, J(7, 5) = 1.1 Hz, 1H, H-7); 8.36 (dd, J(5, 6) = 8.4 Hz, J(5, 7) = 1.1 Hz, 1H, H-5); 8.51 (s, 1H, H-2); 8.76 (brt, J(NH-CH2) = 5.7 Hz, 1H, NH). 13C NMR (DMSO-d6): δ 39.14 (PhCH2); 55.37 (4'-OCH3); 55.62 (2'-OCH3); 98.46 (C-3'); 104.47 (C-5'); 116.50 (C-4a); 118.46 (C-1'); 122.87 (C-8); 122.90 (C-5); 126.44 (C-6); 128.48 (C-6'); 136.21 (C-7); 146.74 (C-8a); 156.20 (C-2); 157.97 (C-2'); 159.80 and 159.88 (C-4' and C-4). HR-MS (ESI+): m/z [M + H]+ calculated for: C17H17N3O2Br, 374.0499, found: 374.0499.
4-(2,4-Dimethoxybenzyl)amino-8-vinylquinazoline (27):
Tributyl(vinyl)tin (1.9 mL, 6.6 mmol) was added to the solution of 26 (2.0 g, 5.3 mmol), CuI (0.31 g, 1.6 mmol) and Pd(t-Bu3P)2 (0.1 g, 0.2 mmol) in NMP (40 mL) under Ar. The resulting mixture was heated at 120 °C for 24 h, then cooled to RT and an aqueous solution of KF (2.0 g in 15 mL) was added. After 15 min, EtOAc (120 mL) was added to the resulting slurry. After 15 min, the resulting mixture was filtered over Cellite. The organic layer was separated and washed with a sat. EDTA solution (50 mL), H2O (50 mL), brine (50 mL) and then dried over Na2SO4. Volatiles were removed in vacuo, and the residue was purified by flash chromatography (grad. Hex:CHCl3 0–100% and grad. MeOH in CHCl3 0–10%) to give 27 (0.76 g, 44%) as a white oil. ESI-MS, m/z (%): 322 [M+H+] (100). 1H NMR (DMSO-d6): δ 3.72 (s, 3H, 4'-OCH3); 3.82 (s, 3H, 2'-OCH3); 4.66 (bd, J(CH2-NH) = 5.7 Hz, 2H, PhCH2); 5.41 (dd, J(vic) = 11.2 Hz, J(gem) = 1.5 Hz, 1H, CH2b); 6.00 (dd, J(vic) = 18.0 Hz, J(gem) = 1.5 Hz, 1H, CH2a); 6.43 (dd, J(5', 6') = 8.4 Hz, J(5', 3') = 2.4 Hz, 1H, H-5'); 6.57 (d, J(3', 5') = 2.4 Hz, 1H, H-3'); 7.07 (dm, J(6', 5') = 8.4 Hz, 1H, H-6'); 7.50 (ddd, J(6, 5) = 8.3 Hz, J(6, 7) = 7.4 Hz, J(6, 8-CH) = 0.5 Hz, 1H, H-6); 7.76 (ddm, J(vic) = 18.0 Hz, J(vic) = 11.2 Hz, 1H, CH=); 8.05 (ddd, J(7, 6) = 7.4 Hz, J(7, 5) = 1.3 Hz, J(7,8-CH) = 0.5 Hz, 1H, H-7); 8.27 (dd, J(5, 6) = 8.4 Hz, J(5, 7) = 1.3 Hz, 1H, H-5); 8.46 (s, 1H, H-2); 8.58 (brt, J(NH-CH2) = 5.8 Hz, 1H, NH). 13C NMR (DMSO-d6): δ 38.85 (PhCH2); 55.37 (4'-OCH3); 55.62 (2'-OCH3); 98.44 (C-3'); 104.44 (C-5'); 115.26 (C-4a); 115.84 (CH2=CH-); 118.80 (C-1'); 122.55 (C-5); 125.40 (C-6); 128.05 (C-7); 128.32 (C-6'); 132.28 (CH2=CH-); 134.07 (C-8); 146.56 (C-8a); 154.78 (C-2); 157.93 (C-2'); 159.81 and 159.84 (C-4' a C-4). HR-MS (ESI+): m/z [M + H]+ calculated for: C19H20N3O2, 322.1550, found: 322.1550.
4-(2,4-Dimethoxybenzyl)amino-8-(2-hydroxyethyl)quinazoline (28):
Treatment of 27 (0.7 g, 2.2 mmol) by Method C (column chromatography in gradient MeOH:CHCl3 0–10%) afforded 28 (0.27 g, 44%) as a yellowish oil. ESI-MS, m/z (%): 340 [M+H+] (100). 1H NMR (DMSO-d6): δ 3.19 (t, J(1', 2') = 7.1 Hz, 2H, H-1'); 3.67 (t, J(2', 1') = 7.1 Hz, 2H, H-2'); 3.72 (s, 3H, 4''-OCH3); 3.82 (s, 3H, 2''-OCH3); 4.65 (bd, J(CH2-NH) = 5.8 Hz, 2H, PhCH2); 4.74 (brs, 1H, OH); 6.43 (dd, J(5'', 6'') = 8.4 Hz, J(5'', 3'') = 2.4 Hz, 1H, H-5''); 6.57 (d, J(3'', 5'') = 2.4 Hz, 1H, H-3''); 7.05 (dm, J(6'', 5'') = 8.4 Hz, 1H, H-6''); 7.41 (dd, J(6, 5) = 8.3 Hz, J(6, 7) = 7.2 Hz, 1H, H-6); 7.64 (dm, J(7, 6) = 7.2 Hz, 1H, H-7); 8.18 (dd, J(5, 6) = 8.5 Hz, J(5, 7) = 1.4 Hz, 1H, H-5); 8.45 (s, 1H, H-2); 8.52 (brt, J(NH-CH2) = 5.8 Hz, 1H, NH). 13C NMR (DMSO-d6): δ 34.94 (C-1'); 38.79 (PhCH2); 55.36 (4''-OCH3); 55.61 (2''-OCH3); 61.72 (C-2'); 98.42 (C-3''); 104.43 (C-5''); 114.95 (C-4a); 118.93 (C-1''); 120.86 (C-5); 125.23 (C-6); 128.19 (C-6''); 133.08 (C-7); 136.94 (C-8); 147.91 (C-8a); 154.53 (C-2); 157.89 (C-2''); 159.76 (C-4''); 160.04 (C-4). HR-MS (ESI+): m/z [M + H]+ calculated for: C19H22N3O3, 340.1656, found: 340.1656.
4-(2,4-Dimethoxybenzyl)amino-8-{2-[(diisopropoxyphosphoryl)-methoxy]ethyl}quinazoline (29):
Treatment of 28 (0.27 g, 0.8 mmol) by Method D afforded 29 (0.17 g, 42%) as a yellowish oil. ESI-MS, m/z (%): 518 [M+H+] (100). 1H NMR (DMSO-d6): δ 1.16 (d, J(CH3, CH) = 6.2 Hz, 6H, CH3); 1.19 (d, J(CH3, CH) = 6.2 Hz, 6H, CH3); 3.29 (t, J(1', 2') = 6.7 Hz, 2H, H-1'); 3.72 (s, 3H, 4''-OCH3); 3.73 (d, J(H-C-P) = 8.5 Hz, 2H, P-CH2); 3.80 (t, J(CH2, CH2) = 6.7 Hz, 2H, H-2'); 3.82 (s, 3H, 2''-OCH3); 4.52 (d septet, J(H-C-O-P) = 7.6 Hz, J(CH, CH3) = 6.2 Hz, 2H, CHiPr); 4.66 (m, 2H, PhCH2); 6.42 (dd, J(5'', 6'') = 8.4 Hz, J(5'', 3'') = 2.4 Hz, 1H, H-5''); 6.57 (d, J(3'', 5'') = 2.4 Hz, 1H, H-3''); 7.05 (d, J(6'', 5'') = 8.4 Hz, 1H, H-6''); 7.42 (dd, J(6, 5) = 8.2 Hz, J(6, 7) = 7.3 Hz, 1H, H-6); 7.67 (dd, J(7, 6) = 7.1 Hz, J(7, 5) = 0.5 Hz, 1H, H-7); 8.19 (dd, J(5, 6) = 8.3 Hz, J(5, 7) = 1.0 Hz, 1H, H-5); 8.45 (s, 1H, H-2); 8.52 (brt, J(NH-CH2) = 5.7 Hz, 1H, NH). 13C NMR (DMSO-d6): δ 23.55 (d J(C-C-O-P) = 4.5 Hz, CH3iPr); 23.68 (d J(C-C-O-P) = 3.7 Hz, CH3iPr); 30.65 (C-1'); 38.49 (PhCH2); 55.06 (4''-OCH3); 55.31 (2''-OCH3); 64.48 (d, J(C-P) = 164.5 Hz, CH2-P); 69.93 (d, J(C-O-P) = 6.4 Hz, CHiPr); 72.43 (d, J(C-O-C-P) = 12.3 Hz, C-2'); 98.13 (C-3''); 104.12 (C-5''); 114.66 (C-4a); 118.62 (C-1''); 120.80 (C-5); 124.81 (C-6); 127.89 (C-6''); 132.67 (C-7); 135.65 (C-8); 147.57 (C-8a); 154.29 (C-2); 157.87 (C-2''); 159.46 and 159.70 (C-4''a C-4). HR-MS (ESI+): m/z [M + H]+ calculated for: C26H37N6O3P, 518.2415, found: 518.2414.
4-Amino-8-{2-[(diisopropoxyphosphoryl)methoxy]ethyl}quinazoline (30):
Compound 29 (0.4 g, 0.8 mmol) was dissolved in a mixture of CH2Cl2 (1 mL) and CF3COOH (1 mL). The reaction mixture was stirred at RT overnight, then aq. NH3 (1 mL) and CHCl3 (150 mL) were added. The organic layer was washed with H2O (50ml) and brine (50 mL). Volatiles were removed and the residue was purified by flash chromatography (gradient MeOH:CHCl3 0–10%) to give 30 (0.14 g, (49%) as a yellowish solid ESI-MS, m/z (%): 368 [M+H+] (100). 1H NMR (DMSO-d6): δ 1.16 (d, J(CH3, CH) = 6.2 Hz, 6H, CH3); 1.19 (d, J(CH3, CH) = 6.2 Hz, 6H, CH3); 3.27 (t, J(1', 2') = 6.8 Hz, 2H, H-1'); 3.73 (d, J(H-C-P) = 8.4 Hz, 2H, P-CH2); 3.80 (t, J(2', 1') = 6.8 Hz, 2H, H-2'); 4.52 (d septet, J(H-C-O-P) = 7.7 Hz, J(CH, CH3) = 6.2 Hz, 2H, CHiPr); 7.38 (dd, J(6, 5) = 8.3 Hz, J(6, 7) = 7.2 Hz, 1H, H-6); 7.65 (dm, J(7, 6) = 7.2 Hz, 1H, H-7); 7.71 (brs, 2H, NH2); 8.06 (dd, J(5, 6) = 8.3 Hz, J(5, 7) = 1.4 Hz, 1H, H-5); 8.41 (s, 1H, H-2). 13C NMR (DMSO-d6): δ 23.85 (d, J(C-C-O-P) = 4.6 Hz, CH3iPr); 23.99 (d, J(C-C-O-P) = 3.7 Hz, CH3iPr); 30.88 (C-1'); 64.76 (d, J(C-P) = 164.4 Hz, CH2-P); 70.25 (d, J(C-O-P) = 6.4 Hz, CHiPr); 72.70 (d, J(2'-P) = 12.3 Hz, C-2'); 114.36 (C-4a); 121.93 (C-5); 124.89 (C-6); 133.19 (C-7); 135.73 (C-8); 148.36 (C-8a); 154.92 (C-2); 162.29 (C-4). HR-MS (ESI+): m/z [M + H]+ calculated for: C17H27N3O4P, 368.1734, found: 368.1734.
Bis(L-phenylalanine isopropyl ester) prodrug of (4-aminoquinazoline-8-yl)ethoxy)methyl)phosphonic acid (31):
Treatment of 30 (0.14 g, 0.38 mmol) by Method A afforded 31 (0.12 g, 46%) as a whitish amorphous solid. ESI-MS, m/z (%): 662 [M+H+] (100). 1H NMR (DMSO-d6): δ 1.03, 1.07, 1.12 and 1.16 (4 x d, J(CH3, CH) = 6.2 Hz, 12H, CH3); 2.75–2.90 (m, 4H, PhCH2); 3.18–3.33 (m, 4H, P-CH2, H-1'); 3.59–3.66 (m, 2H, H-2'); 3.82–3.98 (m, 2H, NH-CH); 4.05 (m, 1H, P-NH); 4.37 (m, 1H, P-NH); 4.79 (sept, J(CH, CH3) = 6.3 Hz, 2H, CHiPr); 4.82 (sept, J(CH, CH3) = 6.3 Hz, 2H, CHiPr); 7.10–7.25 (m, 10H, Ph-ortho, meta, para); 7.34 (dd, J(6, 5) = 8.3 Hz, J(6, 7) = 7.2 Hz, 1H, H-6); 7.61 (dd, J(7, 6) = 7.2 Hz, J(7, 5) = 1.4 Hz, 1H, H-7); 7.71 (brs, 2H, NH2); 8.06 (dd, J(5, 6) = 8.4 Hz, J(5, 7) = 1.4 Hz, 1H, H-5); 8.42 (s, 1H, H-2). 13C NMR (DMSO-d6): δ 21.50, 21.56, 21.62 and 21.68 (CH3iPr); 30.92 (C-1'); 40.22 (CH2Ph); 54.04 and 54.13 (NH-CH); 67.66 (d, J(C-P) = 135.6 Hz, CH2-P); 68.01 and 68.14 (CHiPr); 72.59 (d, J(C-O-C-P) = 12.3 Hz, C-2'); 114.38 (C-4a); 121.93 (C-5); 124.91 (C-6); 126.62 and 126.63 (Ph-para); 128.22 and 128.23 (Ph-meta); 129.65 and 129.67 (Ph-ortho); 133.08 (C-7); 135.73 (C-8); 137.13 and 137.33 (ipso); 148.35 (C-8a); 154.94 (C-2); 162.30 (C-4); 172.26 (d, J(C-C-N-P) = 5.4 Hz, COO); 172.47 (d, J(C-C-N-P) = 3.0 Hz, COO). HR-MS (ESI+): m/z [M + H]+ calculated for: C32H47N4O9P, 662.3075, found: 662.3081.
4-(2,4-Dimethoxybenzyl)aminopyrrolo[2,1-f][1,2,4]triazine (33):
2,4-Dimethoxybenzylamine (4.62 mL, 18.5 mmol) was added to a solution of 4-chloro-aminopyrrolo[2,1-f][1,2,4]triazine (32, 1.75 g, 11.4 mmol) in abs. EtOH (21 mL) and Et3N (3.5 mL). The resulting mixture was heated at 120 °C for 1 h in microwave reactor. Volatiles were removed and the residue was purified on C18 RP in (H2O:MeOH 0–100%) to give 33 (2.76 g, 85%) as an amorphous solid. ESI-MS, m/z (%): 285 [M+H+] (45); 307 [M+Na+] (100). 1H NMR (DMSO-d6): δ 3.73 (s, 3H, 4'-OCH3); 3.81 (s, 3H, 2'-OCH3); 4.60 (d, J(NH-CH2) = 5.7 Hz, 2H, PhCH2); 6.47 (dd, J(5', 6') = 8.4 Hz, J(5', 3') = 2.4 Hz, 1H, H-5'); 6.58 (d, J(3', 5') = 2.4 Hz, 1H, H-3'); 6.60 (dd, J(6, 5) = 4.3 Hz, J(6, 7) = 2.6 Hz, 1H, H-6); 6.96 (dd, J(5, 6) = 4.3 Hz, J(5, 7) = 1.6 Hz, 1H, H-5); 7.11 (d, J(6', 5') = 8.3 Hz, 2H, H-6'); 7.60 (dd, J(7, 6) = 2.6 Hz, J(7, 5) = 1.6 Hz, 1H, H-7); 7.84 (s, 1H, H-2); 8.48 (brt, J(NH-CH2) = 5.7 Hz, 1H, NH). 13C NMR (DMSO-d6): δ 37.69 (PhCH2); 55.10 (4'-OCH3); 55.34 (2'-OCH3); 98.18 (C-3'); 100.73 (C-5); 104.26 (C-5'); 109.88 (C-6); 114.50 (C-4a); 117.97 (C-7); 118.37 (C-1'); 128.55 (C-6'); 147.46 (C-2); 153.78 (C-4); 157.63 (C-2'); 159.68 (C-4'). HR-MS (ESI+): m/z [M + H]+ calculated for: C15H17N4O2, 285.1346, found: 285.1347.
4-(2,4-Dimethoxybenzyl)amino-7-iodo-pyrrolo[2,1-f][1,2,4]triazine (34):
N-Iodosuccinimide (3.1 g, 13.8 mmol) was added to the solution of 33 (3.6 g, 12.6 mmol) in dry CH3CN (40 mL) cooled to 0 °C under Ar. The reaction mixture was strirred at 0 °C for 1 h. Volatiles were removed and the residue was dissolved in CHCl3 (250 mL) and washed with H2O (100 mL), 10% Na2S2O3 (100 mL), brine (100mL) and then dried over Na2SO4. The solvent was removed in vacuo and the residue was purified by flash chromatography (grad. MeOH:CHCl3 0–10%) to give 34 (4.0 g, 77%) as a yellowish oil. ESI-MS, m/z (%): 411 [M+H+] (65); 433 [M+Na+] (100). 1H NMR (DMSO-d6): δ 3.73 (s, 3H, 4'-OCH3); 3.80 (s, 3H, 2'-OCH3); 4.60 (d, J(NH-CH2) = 5.7 Hz, 2H, PhCH2); 6.46 (dd, J(5', 6') = 8.4 Hz, J(5', 3') = 2.4 Hz, 1H, H-5'); 6.57 (d, J(3', 5') = 2.4 Hz, 1H, H-3'); 6.83 (d, J(6, 5) = 4.4 Hz, 1H, H-6); 7.09–7.11 (m, 2H, H-5 and 6'); 7.97 (s, 1H, H-2); 8.57 (brt, J(NH-CH2) = 5.7 Hz, 1H, NH). 13C NMR (DMSO-d6): δ 38.20 (PhCH2); 55.41 (4'-OCH3); 55.65 (2'-OCH3); 71.71 (C-7); 98.49 (C-3'); 103.74 (C-5); 104.57 (C-5'); 118.04 (C-4a); 118.44 (C-1'); 118.47 (C-6); 128.94 (C-6'); 148.46 (C-2); 153.76 (C-4); 157.96 (C-2'); 160.04 (C-4'). HR-MS (ESI+): m/z [M + H]+ calculated for: C15H16N4O2I, 411.0313, found: 411.0315.
4-(2,4-Dimethoxybenzyl)amino-7-vinyl-pyrrolo[2,1-f][1,2,4]triazine (35):
Tributyl(vinyl)tin (1.4 mL, 4.8 mmol) was added to the solution of 34 (1.58 g, 5.6 mmol), CuI (87 mg, 0.45 mmol) and Pd(t-Bu3P)2 (0.1 g, 0.2 mmol) in NMP (30 mL) under Ar. The resulting mixture was heated at 120 °C for 5 h, then cooled to RT and an aqueous solution of KF (1.3 g in 10 mL) was added. After 15 min, EtOAc (120 mL) was added and resulting slurry was stirred for 15 min. The mixture was filtered over Cellite, the organic layer was separated and washed with a sat. EDTA solution (50 mL), H2O (50 mL), brine (50 mL) and then dried over Na2SO4. Volatiles were removed in vacuo, and the residue was purified by flash chromatography ((Hex:CHCl3(1:1) together with grad MeOH 1–10%) to give 35 (1.08 g, 90%) as a yellowish oil. ESI-MS, m/z (%): 311 [M+H+] (65); 333 [M+Na+] (100). 1H NMR (DMSO-d6): δ 3.73 (s, 3H, 4'-OCH3); 3.81 (s, 3H, 2'-OCH3); 4.60 (bd, J(CH2-NH) = 5.7 Hz, 2H, PhCH2); 5.27 (dd, J(vic) = 11.5 Hz, J(gem) = 1.7 Hz, 1H, CH2b); 5.93 (dd, J(vic) = 17.8 Hz, J(gem) = 1.7 Hz, 1H, CH2a); 6.47 (dd, J(5', 6') = 8.4 Hz, J(5', 3') = 2.4 Hz, 1H, H-5'); 6.58 (d, J(3', 5') = 2.4 Hz, 1H, H-3'); 7.10 (d, J(6', 5') = 8.4 Hz, 1H, H-6'); 6.87–7.02 (m, 2H, H-5 and H-6); 7.93 (s, 1H, H-2); 8.49 (brt, J(NH-CH2) = 5.7 Hz, 1H, NH). 13C NMR (DMSO-d6): δ 37.74 (PhCH2); 55.10 (4'-OCH3); 55.34 (2'-OCH3); 98.18 (C-3'); 101.49 (C-5); 104.27 (C-5'); 108.26 (C-6); 113.18 (CH=CH2); 115.07 (C-4a); 118.31 (C-1'); 123.95 (CH=CH2); 128.18 (C-7); 128.58 (C-6'); 147.64 (C-2); 153.76 (C-4); 157.64 (C-2'); 159.70 (C-4'). HR-MS (ESI+): m/z [M + H]+ calculated for: C17H19N4O2, 311.1503, found: 311.1503.
4-(2,4-Dimethoxybenzyl)amino-7-(2-hydroxyethyl)-pyrrolo[2,1-f][1,2,4]triazine (36):
Treatment of 35 (1.0 g, 3.2 mmol) by Method C (column chromatography in grad. MeOH:CHCl3 0–10%) afforded 36 (0.56 g, 53%) as a yellowish oil. ESI-MS, m/z (%): 329 [M+H+] (30); 351 [M+Na+] (100). 1H NMR (DMSO-d6): δ 3.00 (t, J(1', 2') = 7.1 Hz, 2H, H-1'); 3.68 (m, 2H, H-2'); 3.73 (s, 3H, 4''-OCH3); 3.81 (s, 3H, 2''-OCH3); 4.59 (d, J(NH-CH2) = 5.8 Hz, 2H, PhCH2); 4.73 (brt, J(OH, CH2) = 5.4 Hz, 1H, OH); 6.45 (d, J(6, 5) = 4.3 Hz, 1H, H-6); 6.46 (dd, J(5'', 6'') = 8.3 Hz, J(5'', 3'') = 2.4 Hz, 1H, H-5''); 6.57 (d, J(3'', 5'') = 2.4 Hz, 1H, H-3''); 6.60 (d, J(5, 6) = 4.3 Hz, 1H, H-5); 7.08 (d, J(6'', 5'') = 8.3 Hz, 2H, H-6''); 7.85 (s, 1H, H-2); 8.36 (brt, J(NH-CH2) = 5.8 Hz, 1H, NH). 13C NMR (DMSO-d6): δ 29.02 (C-1'); 37.88 (PhCH2); 55.40 (4''-OCH3); 55.63 (2''-OCH3); 59.44 (C-2'); 98.44 (C-3''); 100.37 (C-5); 104.54 (C-5''); 109.45 (C-6); 114.25 (C-4a); 118.86 (C-1''); 128.05 (C-7); 128.68 (C-6''); 147.34 (C-2); 154.11 (C-4); 157.78 (C-2''); 159.92 (C-4''). HR-MS (ESI+): m/z [M + H]+ calculated for: C17H21N4O3, 329.1608, found: 329.1608.
4-(2,4-Dimethoxybenzyl)amino-7-{2-[(diisopropoxyphosphoryl)-methoxy]ethyl}pyrrolo[2,1-f][1,2,4]triazine (37):
Treatment of 36 (0.53 g, 1.7 mmol) by Method D afforded 37 (0.85 g, 98%) as a yellowish oil. ESI-MS, m/z (%): 507 [M+H+] (100). 1H NMR (DMSO-d6): δ 1.18 (d, J(CH3, CH) = 6.2 Hz, 6H, CH3); 1.21 (d, J(CH3, CH) = 6.2 Hz, 6H, CH3); 3.10 (t, J(1', 2') = 6.7 Hz, 2H, H-1'); 3.73 (s, 3H, 4''-OCH3); 3.75 (d, J(H-C-P) = 8.4 Hz, 2H, P-CH2); 3.80 (t, J(CH2, CH2) = 6.7 Hz, 2H, H-2'); 3.82 (s, 3H, 2''-OCH3); 4.54 (d septet, J(H-C-O-P) = 7.7 Hz, J(CH, CH3) = 6.2 Hz, 2H, CHiPr); 4.59 (m, 2H, PhCH2); 6.45 (dd, J(5'', 6'') = 8.4 Hz, J(5'', 3'') = 2.4 Hz, 1H, H-5''); 6.49 (d, J(6, 5) = 4.3 Hz, 1H, H-6); 6.57 (d, J(3'', 5'') = 2.4 Hz, 1H, H-3''); 6.90 (d, J(5, 6) = 4.3 Hz, 1H, H-5); 7.08 (dt, J(6'', 5'') = 8.4 Hz, J(6'', CH2) = 0.8 Hz, 2H, H-6''); 7.86 (s, 1H, H-2); 8.37 (brt, J(NH-CH2) = 5.9 Hz, 1H, NH). 13C NMR (DMSO-d6): δ 23.85 (d, J(C-C-O-P) = 4.5 Hz, CH3iPr); 23.99 (d J(C-C-O-P) = 3.7 Hz, CH3iPr); 25.37 (C-1'); 37.88 (PhCH2); 55.38 (4''-OCH3); 55.62 (2''-OCH3); 64.78 (d, J(C-P) = 164.4 Hz, CH2-P); 70.28 (d, J(C-O-P) = 6.4 Hz, CHiPr); 70.54 (d, J(C-O-C-P) = 12.1 Hz, C-2'); 98.43 (C-3''); 100.40 (C-5); 104.51 (C-5''); 109.55 (C-6);114.36 (C-4a); 118.80 (C-1''); 127.18 (C-7); 128.67 (C-6''); 147.37 (C-2); 154.06 (C-4); 157.87 (C-2''); 159.92 (C-4''). HR-MS (ESI+): m/z [M + H]+ calculated for: C24H36N4O6P, 507.2367, found: 507.2367.
4-Amino-7-{2-[(diisopropoxyphosphoryl)methoxy]ethyl}pyrrolo[2,1-f][1,2,4]triazine (38):
Compound 37 (0.7 g, 1.4 mmol) was dissolved in a mixture of CH2Cl2 (10 mL) and CF3COOH (10 mL). The reaction mixture was stirred at RT for 3 h, cooled with ice and aq. NH3 (10 mL) and CHCl3 (150 mL) were added. The organic layer was washed with H2O (50ml) and brine (50 mL). Volatiles were removed and the residue was purified by flash chromatography (grad. MeOH:CHCl3 0–10%) to give 38 (0.3 g, 61%) as a white amorphous solid. ESI-MS, m/z (%): 357 [M+H+] (100). 1H NMR (DMSO-d6): δ 1.18 (d, J(CH3, CH) = 6.2 Hz, 6H, CH3); 1.21 (d, J(CH3, CH) = 6.2 Hz, 6H, CH3); 3.10 (t, J(1', 2') = 6.7 Hz, 2H, H-1'); 3.75 (d, J(H-C-P) = 8.3 Hz, 2H, P-CH2); 3.80 (t, J(2', 1') = 6.7 Hz, 2H, H-2'); 4.54 (d septet, J(H-C-O-P) = 7.7 Hz, J(CH, CH3) = 6.2 Hz, 2H, CHiPr); 6.49 (d, J(6, 5) = 4.3 Hz, 1H, H-6); 6.80 (d, J(5, 6) = 4.3 Hz, 1H, H-5); 7.58 (brs, 2H, NH2); 7.81 (s, 1H, H-2). 13C NMR (DMSO-d6): δ 23.85 (d J(C-C-O-P) = 4.5 Hz, CH3iPr); 24.00 (d, J(C-C-O-P) = 3.7 Hz, CH3iPr); 25.39 (C-1'); 64.79 (d, J(C-P) = 164.5 Hz, CH2-P); 70.30 (d, J(C-O-P) = 6.4 Hz, CHiPr); 70.50 (d, J(C-O-C-P) = 12.2 Hz, C-2'); 100.85 (C-5); 109.69 (C-6);114.13 (C-4a); 127.23 (C-7); 147.84 (C-2); 155.68 (C-4). HR-MS (ESI+): m/z [M + H]+ calculated for: C15H26N4O4P, 357.1686, found: 357.1687.
Bis(L-phenylalanine isopropyl ester) prodrug of 2-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)ethoxy)methyl)phosphonic acid (39):
Treatment of 38 (0.29 g, 0.8 mmol) by Method A afforded 39 (0.27 g, 50%) as a whitish amorphous solid. ESI-MS, m/z (%): 651 [M+H+] (100). 1H NMR (DMSO-d6): δ 1.03, 1.07, 1.13 and 1.16 (d, J(CH3, CH) = 6.2 Hz, 12H, CH3); 2.74–2.90 (m, 4H, CH2Ph); 3.01–3.08 (m, 2H, H-1'); 3.22–3.34 (m, 2H, P-CH2); 3.59–3.66 (m, 2H, H-2'); 3.85–3.98 (2x m, 2H, NH-CH); 4.06 (m, 1H, P-NH); 4.38 (m, 1H, P-NH); 4.79 (sept, J(CH, CH3) = 6.3 Hz, 1H, CHiPr); 4.82 (sept, J(CH, CH3) = 6.3 Hz, 1H, CHiPr); 6.47 (d, J(6, 5) = 4.3 Hz, 1H, H-6); 6.81 (d, J(5, 6) = 4.3 Hz, 1H, H-5); 7.10–7.26 (m, 10H, Ph-ortho, meta, para); 7.59 (s, 2H, NH2); 7.81 (s, 1H, H-2). 13C NMR (DMSO-d6): δ 21.49, 21.55, 21.62 and 21.68 (CH3iPr); 25.42 (C-1'); 40.10 (CH2Ph); 54.09 and 54.13 (NH-CH); 67.61 (d, J(C-P) = 135.8 Hz, CH2-P); 68.00 and 68.13 (CHiPr); 70.33 (d, J(2'-P) = 12.1 Hz, C-2'); 100.93 (C-5); 109.62 (C-6); 114.13 (C-4a); 126.63 and 126.64 (Ph-para); 127.20 (C-7); 128.22 and 128.24 (Ph-meta); 129.66 and 129.66 (Ph-ortho); 137.16 and 137.32 (ipso); 147.86 (C-2); 155.68 (C-4); 172.28 (d, J(C-C-N-P) = 5.3 Hz, COO); 172.46 (d, J(C-C-N-P) = 3.0 Hz, COO). HR-MS (ESI+): m/z [M + H]+ calculated for: C33H44N6O6P, 651.3055, found: 651.3050.
(7-Deazaadenine-9-yl)ethoxy)methyl)phosphonic acid diphosphate (40a):
Compound 5a (0.204 g, 0.57 mmol) was converted into the free phosphonate (0.15 g, 0.55 mmol) according to the reported procedure,49 and subsequent treatment by Method E afforded 40a (90 mg, 38%) as an amorphous solid after lyophilization. ESI-MS, m/z (%): 431 [M-] (100). 1H NMR (D2O): δ 3.85 (d, J(H-C-P) = 8.7 Hz, 2H, P-CH2); 3.98 (t, J(2', 1') = 5.1 Hz, 2H, H-2'); 4.36 (t, J(1', 2') = 5.1 Hz, 2H, H-1'); 6.55 (d, J(7, 8) = 3.6 Hz, 1H, H-7); 7.38 (d, J(8, 7) = 3.6 Hz, 1H, H-8); 8.07 (s, 1H, H-2). 13C NMR (D2O): δ 44.90 (C-1'); 67.03 (d, J(C-P) = 164.5 Hz, CH2-P); 71.57 (d, J(2'-P) = 10.9 Hz, C-2'); 100.63 (C-7); 102.44 (C-5); 128.73 (C-8); 145.10 (C-2); 147.31 (C-4); 153.10 (C-6). 31P NMR (D2O) −22.41 (m, Pβ); −9.30 (m, Pα ); 8.80 (d, Pγ).
(8-Aza-7-deazaadenine-9-yl)ethoxy)methyl)phosphonic acid diphosphate (40b):
Compound 17a (0.28 g, 0.8 mmol) was converted into the free phosphonate (0.16 g, 0.58 mmol) according to the reported procedure,54 and subsequent treatment by Method E afforded 40b (110 mg, 45%) as an amorphous solid after lyophilization. ESI-MS, m/z (%): 432 [M-] (100). 1H NMR (D2O): δ 3.76 (d, J(H-C-P) = 8.0 Hz, 2H, P-CH2); 4.07 (t, J(2', 1') = 5.3 Hz, 2H, H-2'); 4.56 (t, J(1', 2') = 5.3 Hz, 2H, H-1'); 8.12 (s, 1H, H-7); 8.21 (s, 1H, H-2). 13C NMR (D2O): δ 47.36 (C-1'); 67.34 (d, J(C-P) = 162.5 Hz, CH2-P); 71.18 (d, J(2'-P) = 9.1 Hz, C-2'); 101.08 (C-5); 133.72 (C-7); 152.95 (C-4); 156.32 (C-2); 158.96 (C-6). 31P NMR (D2O) −22.03 (m, Pβ); −8.89 (m, Pα ); 9.37 (d, J(P-O-P) = 26.2 Hz, Pγ).
(9-Deazaadenine-9-yl)ethoxy)methyl)phosphonic acid diphosphate (41):
Treatment of 14 (0.12 g, 0.44 mmol) by Method E afforded 41 (61 mg, 32%) as a whitish amorphous solid after lyophilization. ESI-MS, m/z (%): 431 [M-] (100). 1H NMR (D2O): δ 2.95 (t, J(1', 2') = 5.9 Hz, 2H, H-1'); 3.83 (t, J(2', 1') = 6.0 Hz, 2H, H-2'); 3.88 (d, J(H-C-P) = 8.6 Hz, 2H, P-CH2); 7.51 (s, 1H, H-8); 8.31 (s, 1H, H-2). 13C NMR (D2O): δ 24.61 (C-1'); 67.11 (d, J(C-P) = 162.5 Hz, CH2-P); 73.54 (d, J(2'-P) = 11.8 Hz, C-2'); 110.04 (C-9); 113.19 (C-5); 131.67 (C-8); 135.41 (C-4); 145.50 (C-2); 151.92 (C-6). 31P NMR (D2O) −21.82 (dd, J(P-O-P) = 25.4 Hz, J(P-O-P) = 19.8 Hz Pβ); −7.84 (d, J(P-O-P) = 19.8 Hz Pα); 9.81 (d, J(P-O-P) = 25.4 Hz, Pγ).
Inhibition of ACT – cell free assay:
AC enzymatic activity was evaluated by determining the conversion of [3H] ATP to [3H]cAMP. Each assay mixture contained 3 μM BSA, 20 mM HEPES (pH 7.4), 10 mM MnCl2, 1 mM EDTA, 1 μM CaCl2, 0.1 mM cold ATP, 20 μCi [2,8-3H]ATP (ARC, St. Louis, MO, USA; specific activity 20 Ci/mmol), 1.2 μM calmodulin and tested compound at a concentration of 0 – 100 μM. Inhibition of AC activity was evaluated towards three different commercially available bacterial adenylate cyclases: ACT (Sigma), specific activity 65 μmol/min/mg, ACT (Enzo), specific activity 115 μmol/min/mg and finally, EF-A (LBL), specific activity 830 μmol/min/mg, with the final enzyme concentration of 1.1 nM , 0.67 nM and 0.12 nM, respectively. The incubation was carried out for 30 min at 30 °C in a reaction volume of 50 μL. A 2 μL aliquot of the assay mixture was spotted onto a polyethyleneimine-cellulose TLC plate, and developed in 4M LiCl:1 M acetic acid (1:4). After developing the TLC plate, the spots containing ATP and cAMP were quantified using Radio-TLC scanner RITA (RAYTEST, Germany) equipped with the evaluation software GINA STAR TLC. Inhibition rates were calculated from the percentage of [3H]ATP to [3H]cAMP conversion. Ki values were calculated using the Graphpad Prism 5 software (San Diego, CA, USA). All assays were performed in duplicate with three independent experiments, and results are presented as mean values ± SD.
Inhibition of ACT – cell based assay:
J774A.1 cells were seeded in a 96-well plate at 5×104 cells per well, and left to attach overnight. Prior to the experiment, cells were washed with HBSS (135 mM NaCl, 5.9 mM KCl, 1.5 mM CaCl2, 1.2 mM MgCl2, 25 mM glucose, 10 mM HEPES [pH 7.4]) and pre-incubated with the tested compounds at concentrations of 0.001–30 μM for 5 h. After that, cells were exposed to ACT (2 nM) from B.pertussis (Enzo Life Sciences, Palo Alto, CA; SA=115 μmol/min/mg) for 30 min. Finally, the cAMP content was determined by using the CatchPointcAMP immunoassay kit (Molecular Devices, Wokingham, UK). Briefly, the reaction was terminated by the addition of lysis buffer to the treated cells (50 μL per well), the cellular content was extracted by shaking the plate at 250 rpm for 10 min. The plate was then centrifuged to remove cell debris, the supernatant was transferred to the immunoassay plate, and immunoassays were carried out according to the manufacturer’s instructions. Fluorescence signal was acquired using an Infinite M1000 plate reader (Tecan Systems Inc., San Jose, CA, USA).
Effect on the viability of J774A.1 cells:
J774A.1 cells were plated onto white 96-well assay plates at 5×104 cells per well and allowed to attach overnight. Cells were then washed with HBSS and treated with 10 μM compounds for 5 h. Cell viability was then assessed with the Cell Titer-Glo Luminescent Cell Viability assay (Promega, Madison, WI, USA) according to the manufacturer’s instructions. Measurement of luminescence signal was performed by use of a GENios micro plate reader (Tecan Systems). Data are expressed as a percentage of the viability of control (untreated) cells.
Assays with mAC. Materials:
3-Isobutyl-1-methylxanthine (IBMX), and A23187 were purchased from Sigma-Aldrich (St. Louis, MO). Phorbol 12-myristate 13-acetate (PMA), and forskolin (FSK) were purchased from Tocris Bioscience (Ellisville, MO). Opti-MEM, antibiotic-antimycotic 100x solution, and Dulbecco’s modified Eagle’s medium (DMEM) were purchased from Life technologies (Grand Island, NY). FetalClone I serum and bovine calf serum, were purchased from Hyclone (Logan, UT). G418 was purchased from InvivoGen (San Diego, CA). The homogenous time-resolved fluorescence (HTRF) cAMP kits were purchased from Cisbio Bioassays (Bedford, MA). The tested compounds were prepared as 50 mM stocks in DMSO, and stored at −20 °C.
Compound Screening (30 µM) and Concentration Response Curves:
Cryopreserved HEK293 cells stably expressing human AC1, human AC2, or human AC5 were thawed rapidly at 37 °C, and resuspended in 10mL of Opti-MEM. Cells were centrifuged at 500 g for 5 min. The supernatant was aspirated, and the cell pellet was resuspended in 10 mL of Opti-MEM for a second centrifugation step. The cells were plated in a 384-well plate, and incubated for 1 h in a humidified incubator at 37 °C and 5% CO2. Cells were then incubated with each corresponding compound at 30 µM or a concentration response curve as indicated for 30 min, followed by the addition of each cell line’s corresponding selective stimulant diluted in Opti-MEM and 500 µM IBMX (AC1: 3 µM A23187, AC2: 100 nM PMA and AC5: 1 µM FSK). The stimulation was terminated by the addition of equal parts of the Cisbio HTRF kit reagents, cAMP-d2 and the anti-cAMP cryptate conjugate. After 1 h of incubation, the homogenous time-resolved fluorescence energy transfer (HTR-FRET) was measured using a Synergy4 (BioTek) fluorescence plate reader (excitation filter: 330/80 nm and emission filters: 620/10 nm and 665/8 nm). The resulting cAMP concentrations were calculated in GraphPad Prism by interpolating the 620/665 nm fluorescence ratio values from a cAMP standard curve in parallel. Compound concentration response curves were also analyzed using GraphPad Prism.
Molecular Modelling:
Recently reinterpreted57 crystal structure of adenylate cyclase domain (ACD) from Bordetella pertussis adenylate cyclase toxin (ACT) with calmodulin (CaM) and bound PMEApp (PDB ID:1ZOT, resolution 2.2 Å)41 was used for molecular modelling study. The pre-processed protein was prepared with MOE Structure Preparation and Protonate 3D tools with the default setup. 7-Deaza-PMEApp (compound 40a) was properly protonated and minimized to RMS gradient of 0.001 kcal/mol. For docking study, the Template docking protocol was selected with substructure setup where all atoms of the acyclic moiety (purine base was excluded) of the PMEApp were selected as the Query in the setup. Bond rotation of ligands was allowed and default placement and refinement methods were used with 50 retained structures after the placement and 30 retained structures after the refinement. For all calculations Amber12:EHT mixed force-field was used with R-Field solvent model.
Acknowledgements
This work was supported by the subvention for development of research organization (Czech Academy of Sciences, Institute of Organic Chemistry and Biochemistry, no. RVO 61388963) and by the Ministry of Interior of the Czech Republic (no. VG20102015046). We acknowledge the financial support of the Czech Science Foundation (P208/12/G016), US National Institutes of Health (NIH) (MH101673) and MCMP Research Enhancement Award. This work was also supported by the Ministry of Education, Youth and Sports from the Large Infrastructures for Research, Experimental Development and Innovations project “IT4Innovations National Supercomputing Center – LM2015070” and NPU I project No. LO1302.
References:
- [1].Friedman RL, Nordensson K, Wilson L, Akporiaye ET, Yocum DE, Infect. Immun 1992, 60, 4578–4585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Yeung KHT, Duclos P, Nelson EAS, Hutubessy RCW, Lancet Infect. Dis 2017, 17, 974–980. [DOI] [PubMed] [Google Scholar]
- [3].Paddock CD, Sanden GN, Cherry JD, Gal AA, Langston C, Tatti KM, Wu KK-H, Goldsmith CS, Greer PW, Montague JL, Eliason MT, Holman RC, Guarner J, Shieh W-JW, Zaki SR, Clin. Infect. Dis 2008, 47, 328–338. [DOI] [PubMed] [Google Scholar]
- [4].Kilgore PE, Salim AM, Zervos MJ, Schmitt H-J, Clin. Microbiol. Rev 2016, 29, 449–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Tiwari T, Murphy TV, Moran J, Morb. Mortal. Wkly. Rep 2005, 54, 1–16. [Google Scholar]
- [6].Wood N, McIntyre P, Paediatr. Respir. Rev 2008, 9, 201–212. [DOI] [PubMed] [Google Scholar]
- [7].Bartkus JM, Juni BA, Ehresmann K, Miller CA, Sanden GN, Cassiday PK, Saubolle M, Lee B, Long J, Harrison AR, Besser JM, J. Clin. Microbiol 2003, 41, 1167–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Davies J, Davies D, Microbiol. Mol. Biol. Rev 2010, 74, 417–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Weiss AA, Hewlett EL, Myers GA, Falkow S, J. Infect. Dis 1984, 150, 219–222. [DOI] [PubMed] [Google Scholar]
- [10].Carbonetti NH, Future Microbiol 2010, 5, 455–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Fedele G, Bianco M, Ausiello CM, Archivum Immunologiae et Therapiae Experimentalis 2013, 445–457. [DOI] [PubMed]
- [12].Weiss AA, Mary GMS, Infect. Immun 1989, 57, 3757–3764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Goodwin MSM, Weiss AA, Infect. Immun 1990, 58, 3445–3447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Gross MK, Au DC, Smith AL, Storm DR, Proc. Natl. Acad. Sci. U. S. A 1992, 89, 4898–4902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Bumba L, Masin J, Fiser R, Sebo P, PLoS Pathog 2010, 6, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Basler M, Knapp O, Masin J, Fiser R, Maier E, Benz R, Sebo P, Osicka R, J. Biol. Chem 2007, 282, 12419–12429. [DOI] [PubMed] [Google Scholar]
- [17].Osičková A, Osička R, Maier E, Benz R, Šebo P, J. Biol. Chem 1999, 274, 37644–37650. [PubMed] [Google Scholar]
- [18].Ladant D, Ullmann A, Trends Microbiol 1999, 172–176. [DOI] [PubMed]
- (19).Vojtova J, Kamanova J, Sebo P, Curr. Opin. Microbiol 2006, 9, 69–75. [DOI] [PubMed] [Google Scholar]
- [20].Shrivastava R, Miller JF, Curr. Opin. Microbiol 2009, 12, 88–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Confer D, Eaton J, Science 1982, 217, 948–950. [DOI] [PubMed] [Google Scholar]
- [22].Friedman RL, Fiederlein RL, Glasser L, Galgiani JN, Infect. Immun 1987, 55, 135–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Pearson RD, Symes P, Conboy M, Weiss AA, Hewlett EL, J. Immunol 1987, 139, 2749–2754. [PubMed] [Google Scholar]
- [24].Weingart CL, Weiss AA, Society 2000, 68, 1735–1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Ross PJ, Lavelle EC, Mills KHG, Boyd AP, Infect. Immun 2004, 72, 1568–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Spensieri F, Fedele G, Fazio C, Nasso M, Stefanelli P, Mastrantonio P, Ausiello CM, Infect. Immun 2006, 74, 2831–2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].De Clercq E, Holý A, Rosenberg I, Sakuma T, Balzatini J, Maudgal PC, Nature 1986, 323, 464–467. [DOI] [PubMed] [Google Scholar]
- [28].De Clercq E, Holý A, Nat. Rev. Drug Discov 2005, 4, 928–940. [DOI] [PubMed] [Google Scholar]
- [29].Reiser H, Wang J, Chong L, Watkins WJ, Ray AS, Shibata R, Birkus G, Cihlar T, Wu S, Li B, Liu X, Henne LN, Wolfgang GHI, Desai M, Rhodes GR, Fridland A, Lee WA, Plunkett W, Vail D, Thamm DH, Jeraj R, Tumas DB, Clin. Cancer Res 2008, 14, 2824–2832. [DOI] [PubMed] [Google Scholar]
- [30].Zídek Z, Potměšil P, Holý A, Toxicol. Appl. Pharmacol 2003, 192, 246–253. [DOI] [PubMed] [Google Scholar]
- [31].Česnek M, Hocková D, Holý A, Dračínský M, Baszczyňski O, De Jersey J, Keough DT, Guddat LW, Bioorg. Med. Chem 2012, 20, 1076–1089. [DOI] [PubMed] [Google Scholar]
- [32].Keough DT, Hocková D, Holý A, Naesens LMJ, Skinner-Adams TS, De Jersey J, Guddat LW, J. Med. Chem 2009, 52, 4391–4399. [DOI] [PubMed] [Google Scholar]
- [33].Hocková D, Keough DT, Janeba Z, Wang TH, De Jersey J, Guddat LW, J. Med. Chem 2012, 55, 6209–6223. [DOI] [PubMed] [Google Scholar]
- [34].Keough DT, Špaček P, Hocková D, Tichý T, Vrbková S, Slavětínká L, Janeba Z, Naesens L, Edstein MD, Chavchich M, Wang TH, De Jersey J, Guddat LW, J. Med. Chem 2013, 56, 2513–2526. [DOI] [PubMed] [Google Scholar]
- [35].Keough DT, Hocková D, Janeba Z, Wang TH, Naesens L, Edstein MD, Chavchich M, Guddat LW, J. Med. Chem 2015, 58, 827–846. [DOI] [PubMed] [Google Scholar]
- [36].Keough DT, Hocková D, Rejman D, Špaček P, Vrbková S, Krečmerová M, Eng WS, Jans H, West NP, Naesens LMJ, De Jersey J, Guddat LW, J. Med. Chem 2013, 56, 6967–6984. [DOI] [PubMed] [Google Scholar]
- [37].Česnek M, Holý A, Masojídková M, Kmoníčková E, Zídek Z, Bioorg. Med. Chem 2008, 16, 965–980. [DOI] [PubMed] [Google Scholar]
- [38].Zídek Z, Franková D, Holý A, Antimicrob. Agents Chemother 2001, 45, 3381–3386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Zídek Z, Franková D, Holý A, Int. J. Immunopharmacol 2000, 22, 1121–1129. [DOI] [PubMed] [Google Scholar]
- [40].Potměšil P, Krečmerová M, Kmoníčková E, Holý A, Zídek Z, Eur. J. Pharmacol 2006, 540, 191–199. [DOI] [PubMed] [Google Scholar]
- [41].Guo Q, Shen Y, Lee Y-S, Gibbs CS, Mrksich M, Tang W-J, EMBO J 2005, 24, 3190–3201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Shen Y, Zhukovskaya NL, Zimmer MI, Soelaiman S, Bergson P, Wang C-R, Gibbs CS, Tang W-J, Proc. Natl. Acad. Sci. U. S. A 2004, 101, 3242–3247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Šmídková M, Dvořáková A, Tloust’ová E, Česnek M, Janeba Z, Mertlíková-Kaiserová H, Antimicrob. Agents Chemother 2014, 58, 664–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Česnek M, Jansa P, Šmídková M, Mertlíková-Kaiserová H, Dračínský M, Brust TF, Pávek P, Trejtnar F, Watts VJ, Janeba Z, ChemMedChem 2015, 10, 1351–1364. [DOI] [PubMed] [Google Scholar]
- [45].Göttle M, Dove S, Steindel P, Shen Y, Tang W-J, Geduhn J, König B, Seifert R, Mol. Pharmacol 2007, 72, 526–535. [DOI] [PubMed] [Google Scholar]
- [46].Dvořáková H, Holý A, Snoeck R, Balzarini J, De Clercq E, Collect. Czech. Chem. Commun 1990, 55, S113–S116. [Google Scholar]
- [47].Dvořáková H, Holý A, Collect. Czech. Chem. Commun 1993, 58, 1419–1429. [Google Scholar]
- [48].Jansa P, Baszczyňski O, Dračínský M, Votruba I, Zídek Z, Bahador G, Stepan G, Cihlar T, Mackman R, Holý A, Janeba Z, Eur. J. Med. Chem 2011, 46, 3748–3754. [DOI] [PubMed] [Google Scholar]
- [49].Holý A, Günter J, Dvořáková H, Masojídková M, Andrei G, Snoeck R, Balzarini J, De Clercq E, J. Med. Chem 1999, 42, 2064–2086. [DOI] [PubMed] [Google Scholar]
- [50].Kaiser MM, Baszczyňski O, Hocková D, Poštová-Slavětínská L, Dračínský M, Keough DT, Guddat LW, Janeba Z, ChemMedChem 2017, 12, 1133–1141. [DOI] [PubMed] [Google Scholar]
- [51].Bambuch V, Otmar M, Pohl R, Masojídková M, Holý A, Tetrahedron 2007, 63, 1589–1601. [Google Scholar]
- [52].Herdewijn P, Česnek M, Heterocycles 2010, 82, 663–687. [Google Scholar]
- [53].Jansa P, Baszczyňski O, Procházková E, Dračínský M, Janeba Z, Green Chem 2012, 14, 2282–2288. [Google Scholar]
- [54].Holý A, Rosenberg I, Dvořáková H, Collect. Czech. Chem. Commun 1989, 54, 2190–2210. [Google Scholar]
- [55].Holý A, Dvořáková H, Jindřich J, Masojídková M, Buděšínský M, Balzarini J, Andrei G, De Clercq E, J. Med. Chem 1996, 39, 4073–4088. [DOI] [PubMed] [Google Scholar]
- [56].Holý A, Rosenberg I, Collect. Czech. Chem. Commun 1987, 52, 2801–2809. [Google Scholar]
- [57].Frydrych J, Skácel J, Šmídková M, Mertlíková-Kaiserová H, Dračínský M, Gnanasekaran R, Lepšík M, Soto-Velasquez M, Watts VJ, Janeba Z, ChemMedChem 2018, 13, 199–206. [DOI] [PubMed] [Google Scholar]
- [58].Wang H, Xu H, Wu LJ, Kim SS, Chen T, Koga K, Descalzi G, Gong B, Vadakkan KI, Zhang X, Kaang BK, Zhuo M, Sci. Transl. Med 2011, 3, 65ra3. [DOI] [PubMed] [Google Scholar]
- [59].Brust TF, Alongkronrusmee D, Soto-Velasquez M, Baldwin TA, Ye Z, Dai M, Dessauer CW, van Rijn RM, Watts VJ, Sci. Signal 2017, 10, eaah5381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Klečka M, Pohl R, Čejka J, Hocek M, Org. Biomol. Chem 2013, 11, 5189–5193. [DOI] [PubMed] [Google Scholar]