

Abstract
Insomnia is a common disorder linked with adverse long-term medical and psychiatric outcomes. The underlying pathophysiological processes and causal relationships of insomnia with disease are poorly understood. Here we identify 57 loci for self-reported insomnia symptoms in the UK Biobank (n = 453,379) and confirm their impact on self-reported insomnia symptoms in the HUNT study (n = 14,923 cases, 47,610 controls), physician-diagnosed insomnia in Partners Biobank (n = 2,217 cases, 14,240 controls), and accelerometer-derived measures of sleep efficiency and sleep duration in the UK Biobank (n = 83,726). Our results suggest enrichment of genes involved in ubiquitin-mediated proteolysis and of genes expressed in multiple brain regions, skeletal muscle, and adrenal gland. Evidence of shared genetic factors is found between frequent insomnia symptoms and restless legs syndrome, aging, cardio-metabolic, behavioral, psychiatric and reproductive traits. Evidence is found for a possible causal link between insomnia symptoms and coronary artery disease, depressive symptoms and subjective well-being.
Editorial summary:
Genome-wide association analyses identify 57 loci associated with insomnia symptoms and evidence of shared genetic architecture between insomnia and cardio-metabolic, behavioral, psychiatric and reproductive traits.
Insomnia disorder, persistent difficulty in initiating or maintaining sleep, and corresponding daytime dysfunction, occurs in 10–20% of the population1. Up to one-third of the population experience transient insomnia symptoms at any given time2. Longitudinal studies suggest that insomnia increases the risk for developing anxiety disorders, alcohol abuse, major depression, and cardio-metabolic disease3. However, little is known about underlying pathophysiologic mechanisms. Cognitive-behavioral therapies are the recommended first-line treatment, but many barriers to treatment exist4,5. Common drug treatments target synaptic neurotransmission (via GABAergic pathways), hypothalamic neuropeptides (via hypocretin/orexin), cortical arousal (via histamine receptors), or the melatonin system, but these drugs have variable effectiveness, may be habit forming, and have side effects6,7. Therefore, it is necessary to develop new personalized therapeutic strategies. Model organism studies have identified genes involved in sleep processes8–13. Family-based heritability estimates suggest that insomnia has a genetic component (22%–25%)14. Recent GWAS reported four loci for insomnia symptoms15,16, but insights into underlying biological pathways and causal genetic links with disease are limited.
UK Biobank participants of European ancestry (n = 453,379) self-reported insomnia symptoms to the question “do you have trouble falling asleep at night or do you wake up in the middle of the night?”. In this sample, 29% of individuals self-reported frequent insomnia symptoms (“usually”), with a higher prevalence in women (32% vs. 24%) and in older participants, shift workers, and those with shorter self-reported sleep duration (Supplementary Table 1).
We performed two parallel GWAS: (i) frequent insomnia symptoms (“never/rarely” vs. “usually” insomnia symptoms, n = 129,270 cases and 108,357 controls); (ii) any insomnia symptoms (“never/rarely” vs. “sometimes”/”usually” insomnia symptoms, n = 345,022 cases and 108,357 controls), adjusting for age and sex using 14,661,600 genotyped and imputed genetic variants across the autosomes and genotyped variants on the X chromosome. We identified 57 association signals explaining 1% of the variance (Fig. 1, Supplementary Table 2, and Supplementary Figs. 1 and 2). Of these, 20 loci were identified in both analyses, 28 loci in analysis of frequent insomnia symptoms only, and 9 in analysis of any insomnia symptoms only (Supplementary Table 2). Conditional analyses identified no secondary association signals. The 57 associations were independent of insomnia risk factors, as sensitivity analyses adjusting for body mass index (BMI), lifestyle, caffeine consumption, and depression or recent stress did not notably alter the magnitude or direction of effect estimates (Supplementary Table 3). The MEIS1 association signal identified in the interim release of the UK Biobank was confirmed in the remainder of the UK Biobank sample (n = 75,508 cases of frequent insomnia symptoms and 64,403 controls; rs113851554 T, OR (95% CI) = 1.19 (1.15–1.23), P = 1.5 × 10−21), and nominal replication was seen for previously reported CYCL1 (P = 9.0 × 10−3). The TMEM132E and SCFD2 signals showed concordant direction of effect in both UK Biobank sub-samples, but were not significant, perhaps reflecting selection bias in the interim release sample17. No other findings from previous candidate gene association studies or smaller GWAS were confirmed (Supplementary Table 4).
Figure 1 ∣. Manhattan plots for genome-wide association analyses of insomnia.
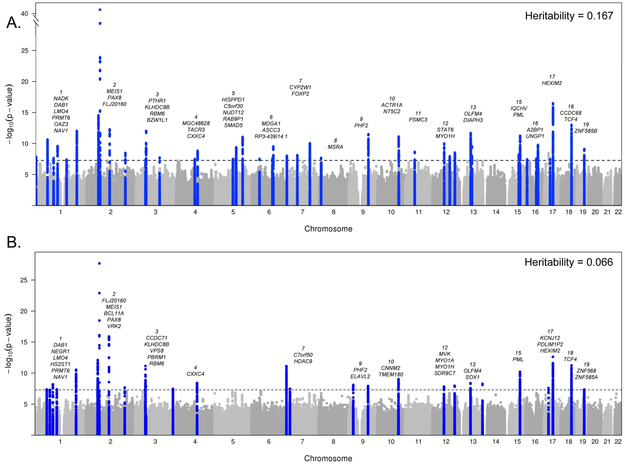
a, Frequent insomnia symptoms (ncases = 129,270, ncontrols = 108,352). b, Any insomnia symptoms (ncases = 345,022, ncontrols = 108,352). Dotted line is genome-wide significant (P = 5 × 10−8) results of linear mixed models adjusted for age, sex, principal components of ancestry, and genotyping array. SNP-based heritability estimates were calculated using BOLT-REML variance components analysis. Chromosomes are annotated with the nearest gene to each association signal.
Secondary GWAS excluding current shift workers or individuals reporting hypnotic, anti-anxiolytic or psychiatric medication usage, and/or with selected chronic diseases or psychiatric illnesses (excluding n = 76,470 participants) revealed strong pair-wise genetic correlation to the primary GWAS (rg ~ 1) and did not identify additional association signals (Supplementary Figs. 1–3). Thus, biological processes underlying pathophysiology of insomnia symptoms may be common between the general population and those with co-morbidities, in accordance with the recent clinical reclassification of primary and secondary insomnia diagnoses into an insomnia disorder18.
The prevalence of insomnia symptoms varies by sex; therefore, we performed secondary sex stratified GWAS (Supplementary Table 5). As described previously15,16, the genetic architecture for frequent insomnia symptoms differed by sex, with a genetic correlation between the stratified GWAS of rg = 0.807 (Supplementary Fig. 3). We found 13 additional loci (8 in women and 5 in men), although there were no genome-wide significant sex interactions. Effects in women were not modified by menopausal status.
Self-report of insomnia symptoms has limitations, including recall bias and lack of granularity19. Therefore, we sought replication of association signals in the HUNT population study with self-reported insomnia symptoms (n = 14,923 cases, 47,610 controls; Supplementary Table 6)20 and the Partners Biobank with physician-diagnosed insomnia (n = 2,217 cases, 14,240 controls). Replication was observed for the MEIS1 variant in both cohorts, and a genetic risk score (GRS) using our 57 variants and weighted by the effect estimates from GWAS of frequent insomnia symptoms (provided in Supplementary Table 2) was also associated with insomnia symptoms in HUNT (OR (95%CI) = 1.015 (1.01–1.02) per allele, P = 2.71 × 10−11) and the Partners Biobank (OR (95%CI) 1.017 (1.007–1.027) per allele, P = 8.88 × 10−4) (Table 1 and Supplementary Table 8). A meta-analysis of the UK Biobank, HUNT, and Partners Biobank studies shows consistency across all three cohorts (Supplementary Table 2). Next, to investigate impact of genetic variants on objective sleep patterns, we tested the 57 lead variants for association with 8 activity-monitor sleep measures in a subset of the UK Biobank participants of European ancestry who had undergone up to 7 days of wrist-worn accelerometry (n = 84,745; Supplementary Table 6). The lead MEIS1 risk variant was associated with a higher number of sleep episodes, indicating an interrupted sleep pattern, lower sleep efficiency, shorter sleep duration and later sleep timing (P < 0.0008; Supplementary Table 9). The GRS was associated with reduced sleep efficiency (difference = −0.04 (0.01) % per allele; P = 4 × 10−14), shorter sleep duration (difference = −0.25 (0.035) minutes per allele; P = 8 × 10−13), and greater day-to-day variability in sleep duration (difference = 0.077 (0.025) minutes per allele; P = 0.0017) but not with the number of sleep episodes or diurnal inactivity duration (Table 1 and Supplementary Table 9).
Table 1 ∣.
A risk score of genetic variants for self-reported insomnia symptoms (57 SNPs) associates with self-reported insomnia symptoms in the HUNT study, physician diagnosed insomnia in the Partners Biobank, and activity-monitor based measures of sleep fragmentation, timing and duration from 7 day accelerometry in the UK Biobank
| Frequent insomnia symptoms Weighted GRS |
Frequent insomnia symptoms Weighted GRS excluding MEIS1 |
||||||
|---|---|---|---|---|---|---|---|
| Trait | OR | 95% CI | P | OR [95% CI] | 95% CI | P | |
| HUNT Study (n = 14,923 cases, 47,610 controls) | Self-reported insomnia symptoms | 1.015 | 1.01-1.02 | 2.71×10−11 | 1.0130 | 1.009-1.018 | 2.32×10−9 |
| Partners Biobank (n = 2,217 cases, 14,240 controls) | Physician diagnosed insomnia | 1.017 | 1.01–1.03 | 8.88×10−4 | 1.0120 | 1.002-1.022 | 0.015 |
| Beta | SE | P | Beta (SE) | SE | P | ||
| Sleep fragmentation measures | |||||||
| UK Biobank 7-day accelerometry (n = 84,745) | Sleep efficiency % | −0.0400 | 0.010 | 4.21×10−14 | −0.0300 | 0.001 | 1.4×10−11 |
| Number of sleep bouts (n) | 0.0053 | 0.002 | 0.030 | 0.0044 | 0.003 | 0.073 | |
| Sleep duration measures | |||||||
| Sleep duration (minutes) | −0.2460 | 0.036 | 8.39×10−13 | −0.2210 | 0.001 | 2.07×10−10 | |
| Sleep duration standard deviation (minutes) | 0.0780 | 0.024 | 1.72×10−3 | 0.0750 | 0.001 | 2.24×10−3 | |
| Sleep timing measures | |||||||
| Midpoint of 5h daily period of minimum activity (L5 timing) (minutes) | 0.0960 | 0.042 | 0.025 | 0.0580 | 0.001 | 0.184 | |
| Midpoint of 10 daily period of maximum activity (M10 timing) (minutes) | 0.0420 | 0.048 | 0.429 | 0.0300 | 0.001 | 0.537 | |
| Diurnal inactivity duration (minutes) | 0.0060 | 0.030 | 0.901 | 0.0080 | 0.001 | 0.784 | |
| Sleep midpoint (minutes) | −0.0124 | 0.024 | 0.975 | −0.0070 | 0.001 | 0.745 | |
Tested using a risk score weighted by effect of SNP on self-reported frequent insomnia symptoms in UK Biobank sample (Supplementary Table 2) in the gtx package in R or PLINK.
SE, standard error; OR, odds ratio per risk allele; Beta, effect per risk allele; CI, 95% confidence interval.
In order to gain insight into the probable causal variants underlying the 57 association signals, we performed fine-mapping in PICS21 and identified 38 variants with a causal probability of 0.20 or greater (Supplementary Table 10 and Supplementary Fig. 4)22. This list includes missense variants in NAD kinase NADK (N262K) and MDGA1 (L61P), as well as rs324017, a SNP within the gene encoding transcriptional repressor NAB1 (EGR-1 binding protein) that is predicted to disrupt a binding site for EGR1 (Supplementary Table 10), a transcription factor involved in response to stress23 and synaptic plasticity during REM sleep23.
The 57 insomnia symptoms loci lie in genomic regions encompassing up to 236 genes, and a summary of annotations for signals or genes within each locus is shown in Supplementary Table 11. Association signals at 14 loci overlapped with NHGRI GWAS signals (r2 > 0.7 in 1KG CEU) for one or more complex traits, with the insomnia symptoms risk allele associated with a greater risk of restless legs syndrome, schizophrenia, Tourette’s syndrome, or obsessive-compulsive disorder, and associated with higher systolic blood pressure, greater carotid plaque burden, lower age at menarche and influencing adiposity traits, height, and educational attainment. Notably, experimental studies in mouse or fly models have implicated five genes at three associated loci in sleep regulation (DVL1, LRP1, NR1H3, PRKAR2A, and SEMA3F). Genes within 16 loci are known drug targets.
Two lead SNPs were associated with one or more of 3,144 human brain structure and function traits assessed in the UK Biobank (P < 2.8 × 10−7, n = 9,707; Oxford Brain Imaging Genetics Server)24, including rs1544637 (within a transcript LOC642659) with several large white matter tracts and with cingulate gyrus morphometry, which has previously been connected to insomnia25, and rs62158170 (near PAX8) with resting-state fMRI networks (Supplementary Fig. 5).
Gene-based tests26 identified 135 associated genes (P ≤ 2.29 × 10−6; Supplementary Table 12). These genes are enriched for expression in brain regions9 (Supplementary Table 13), including the cerebellum (P = 1.3 × 10−6), frontal cortex (P = 1.3 × 10−5), anterior cingulate cortex (P = 1.7 × 10−5), hypothalamus (P = 2.2 × 10−5), basal ganglia (P = 7.0 × 10−4), amygdala (P = 3.4 × 10−4), and hippocampus (P = 8.4 × 10−4), in line with previous research linking these brain regions to insomnia25,27 and mouse single-cell studies linking insomnia GWAS hits to specific populations of brain cells28. Integration of gene expression data with GWAS using transcriptome-wide association analyses29 identified 24 genes where insomnia-SNPs influence gene expression in one or more of 14 tissue types tested, including eight brain regions, muscles, peripheral nerves, whole blood, pituitary, thyroid, and adrenal gland tissue (Supplementary Table 16).
SNP-based heritability of frequent insomnia symptoms was estimated at h2 = 16.7%30. Partitioning of heritability across tissue types31,32 confirmed enrichment in the central nervous system, adrenal/pancreas tissue, and skeletal muscle (P < 10−5) (Supplementary Table 17). Partitioning across functional class implicated activation and repression of enhancers (Supplementary Table 17). Pathway and ontology analyses26,33,34 reveal a significant role for ubiquitin mediated proteolysis (Pbonf = 0.04; Supplementary Table 14), similar to findings in recent GWAS of RLS35, and suggestive roles for phototransduction and muscle tissue development, across several databases (Supplementary Tables 14 and 15). Evidence from model organisms supports the link between Cullin-3 mediated ubiquitination and sleep/circadian rhythms36–39. Furthermore, the restless legs syndrome (RLS) gene BTBD9 has been implicated as a substrate adaptor for E3 ubiquitin ligases40. We find no evidence for enrichment of neurotransmission receptor or biosynthesis genes known to be involved in the regulation of sleep (e.g. GABA, glutamate, adenosine).
We investigated the genetic link between frequent insomnia symptoms and other behavioral and/or disease states. Based on previous links between RLS and insomnia symptoms15,16, we tested a GRS of 20 SNPs for RLS35 and found association with frequent insomnia symptoms (OR = 1.03 (1.02–1.04) per RLS risk allele, P = 2.57 × 10−57), driven partly by the MEIS1 region (GRS excluding MEIS1 region OR = 1.02 (1.02–1.03) per RLS risk allele, P = 2.06 × 10−31)(Supplementary Table 18 and Supplementary Fig. 6). We also tested a GRS of our 57 insomnia SNPs in RLS and found an association with RLS (OR = 1.39 (1.34–1.44), P = 1.70 × 10−80) driven partly by the MEIS1 signal (GRS excluding MEIS1 region OR = 1.17 (1.13–1.21), P = 2.56 × 10−20) (Supplementary Table 18). We also observe a positive genetic correlation between insomnia and RLS (rg = 0.291, P = 6.33 × 10−12). We performed BUHMBOX analysis to distinguish between pleiotropy and heterogeneity. BUHMBOX41 analyses using all 20 RLS SNPs points towards heterogeneity (P = 4.09 × 10−6), most likely due to undiagnosed RLS cases misclassified as insomnia. However, when we exclude the MEIS1 locus, we detect a possible shared genetic basis not explained by heterogeneity (P = 0.137).
To test the proportion of variance frequent insomnia symptoms shares with other traits based on genetic overlap, we performed genetic correlation analyses with 233 traits with public GWAS summary statistics31,32,42,43. We found strong positive genetic correlations (P ≤ 2 × 10−3) between frequent insomnia symptoms and adiposity traits, coronary artery disease, number of children ever born, neuroticism, smoking behavior, and depressive symptoms and disorder. Strong negative genetic correlations were observed with self-reported sleep duration, subjective well-being, cognitive measures, proxy longevity measures, and the remaining reproductive traits (Fig. 2 and Supplementary Table 19). These genetic links persisted with GWAS excluding subjects with chronic and psychiatric illnesses (Supplementary Table 19), indicating a relationship not driven by the presence of concomitant conditions.
Figure 2 ∣. Shared genetic architecture between frequent insomnia symptoms and behavioral and disease traits.
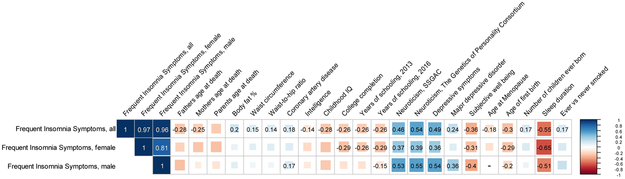
LD-score regression estimates of genetic correlation (rG) of frequent insomnia symptoms are compared with the summary statistics from 224 publicly available genome-wide association studies of psychiatric and metabolic disorders, immune diseases, and other traits of natural variation. Blue, positive genetic correlation; red, negative genetic correlation, rG values displayed for significant correlations. Larger squares correspond to more significant P-values. Genetic correlations that are significantly different from zero after Bonferroni correction are shown on the plot (after Bonferroni correction, P-value cut-off is 0.0002). All genetic correlations in this report can be found in tabular form in Supplementary Table 19. IQ, intelligence quotient.
To test for causal links between frequent insomnia symptoms and seven clusters of genetically correlated traits, we performed Mendelian randomization (MR) analyses mostly within a two-sample summary data framework. Using the inverse variance (IVW) method44 (Fig. 3 and Supplementary Table 20), we found evidence of a causal association (P < 0.001) between frequent insomnia symptoms and prevalent coronary artery disease (CAD; OR (95%CI) = 2.15 (1.38–3.35)) reduced subjective well-being (difference in mean SD units of −0.29 (s.e. 0.06)), greater depressive symptoms (difference in mean SD units of 0.42 (s.e. 0.08)); all associations had a consistent direction and similar magnitude of effect in sensitivity analyses using MR-Egger45 and weighted median methods (Fig. 3 and Supplementary Table 20)46. We found similar results using effect estimates from GWAS excluding those with preexisting conditions. We validated the causal association between insomnia and CAD with one-sample MR in the UK Biobank (n = 23,980 cases and 361,706 controls, OR (95%CI) = 2.95 (2.18–3.99), P = 2.3 × 10−12) (Supplementary Table 20 and Supplementary Fig. 7). Bidirectional MR identified no evidence of reverse causality. The one-sample MR causal association between insomnia status and CAD in UK Biobank is consistent with the robust empirical evidence seen in prospective studies and meta-analyses3,47.
Figure 3 ∣. Causal relationships of insomnia symptoms.
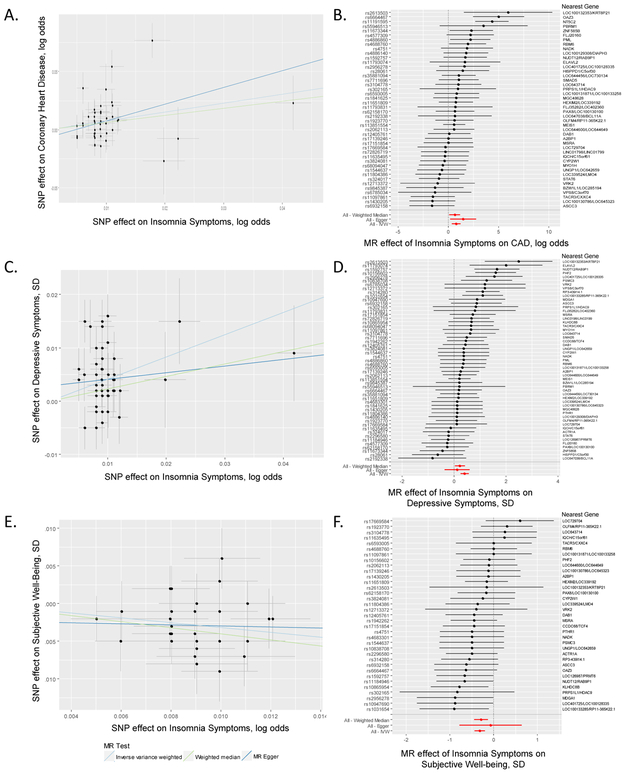
a-f, Association between single nucleotide polymorphisms associated with frequent insomnia symptoms and CAD (a), subjective well-being (c), and depressive symptoms (e). Per allele associations with risk plotted against per allele associations with frequent insomnia symptom risk (vertical and horizontal black lines around points show 95% confidence interval for each polymorphism) results are shown for three different MR association tests. Forest plots show the estimate of the effect of genetically increased insomnia risk on CAD (b), depressive symptoms (d), and subjective well-being (f) as assessed for each SNP. Nearest gene(s) is displayed to the right of plot. Also shown for each SNP is the 95% confidence interval (gray line segment) of the estimate and the Inverse Variance MR, MR-Egger, and Weighted Median MR results in red. Sample size of each GWAS used in the MR analyses is as follows: frequent insomnia symptoms (ncases = 129,270, ncontrols = 108,352), CAD (ncases = 60,801, ncontrols = 123,504), subjective well-being (n = 298,420), and depressive symptoms (n = 161,460).
This study provides a comprehensive description of the genetic architecture of frequent or persistent insomnia symptoms, pointing to putative causal variants and candidate genes, pathways and tissues for functional studies. Further, we define physiological correlates for insomnia symptoms and meaningful clinical links, including a causal link with CAD.
Methods
UK Biobank population and study design.
Study participants were from the UK Biobank study, described in detail elsewhere48. In brief, the UK Biobank is a prospective study of >500,000 people living in the United Kingdom. All people in the National Health Service registry aged 40–69 and living <25 miles from a study center were invited to participate between 2006–2010. In total, 503,325 participants (5%) were recruited from over 9.2 million mailed invitations. Self-reported baseline data were collected by questionnaire and anthropometric assessments were performed. For the current analysis, individuals of non-white ethnicity (n = 48,667) were excluded to avoid confounding effects. A subset analysis was also performed excluding UK Biobank subjects from the interim release and their relatives (exclusion n = 190,216).
Insomnia and covariate measures.
Study subjects self-reported insomnia symptoms, depression, medication use, age, and sex on a touch-screen questionnaire at baseline assessment. Height and weight were measured at baseline. To assess insomnia symptoms, subjects were asked, “Do you have trouble falling asleep at night or do you wake up in the middle of the night?” with responses “never/rarely”, “sometimes”, “usually”, “prefer not to answer”. Subjects who responded “Prefer not to answer” (n = 637) were set to missing. We undertook two GWAS, one in which insomnia symptoms were dichotomized into controls (“never/rarely”) and cases with any symptoms (“sometimes” and “usually”), and the second in which participants were dichotomized into controls (“never/rarely”) or frequent insomnia symptoms (“usually”), with those reporting “sometimes” excluded. Additional covariates used in sensitivity analyses included BMI, self-reported and prevalent sleep apnea diagnosis, area deprivation index, alcohol intake, snoring, nap behavior, smoking status, menopause status, weekly physical activity, tea and coffee intake, depression, and extreme stress. BMI was calculated from measured height and weight at baseline visit. Prevalent sleep apnea cases were defined based on primary or secondary ICD10 diagnosis code (G47.3; n = 391 cases) at the time of baseline assessment. Social deprivation for participant area of residence at the time of recruitment was represented by the Townsend deprivation index, based on national census data immediately preceding participation in the UK Biobank. The Townsend deprivation index was log transformed for use in the analyses. Alcohol intake was self-reported in response to the question “About how often do you drink alcohol?” with answers ranging from “daily or almost daily” to “never”. Snoring was reported in answer to the question “Does your partner or a close relative or friend complain about your snoring?”. Nap behavior was self-reported in response to the question “Do you have a nap during the day” with answers “never/rarely”, “sometimes”, or “usually”. Smoking status was self-reported as past smoking behavior and current smoking behavior, and classified into “current”, “past”, or “never” smoked. Menopause status was reported in answer to the question “Have you had your menopause (periods stopped)?” with answers “yes”, “no”, “not sure – had a hysterectomy”, “not sure – other reason”. Weekly physical activity in total metabolic equivalents per week, total MET-h/week, was calculated using self-reported estimates of type and duration of physical activity. Tea and coffee intake was estimated using the questions “How many cups of tea do you drink each day? (Include black and green tea)” and “How many cups of coffee do you drink each day? (Include decaffeinated coffee),” respectively, with responses in cups/day. Employment status was self-reported in response to the following question “Which of the following describes your current situation?” with responses “in paid employment or self-employed”, “retired”, “looking after home and/or family”, “unable to work because of sickness or disability”, “unemployed”, doing unpair or voluntary work”, “full or part-time student”, “none of the above”, “prefer not to answer”. Marital status was derived from self-reported household occupancy and relatedness data as follows: married/partner was derived from those reporting husband/wife/partner in household with >1 person reported to live in household. Depression was reported in answer to the question “How often did you feel down, depressed or hopeless mood in last 2 weeks?” (cases, n = 4,242 based on answers “more than half the days”, or “nearly every day”). Extreme stress was assessed with the question “In the last two years have you experiences any of the following (you can select more than one answer” with responses “serious illness, injury or assault to yourself”, “serious illness, injury or assault of a close relative”, “death of a close relative”, “death of a spouse or partner”, “marital separation/divorce”, “financial difficulties”, “none of the above”. A secondary GWAS further excluded shift workers, sleep and psychiatric medication users, and subjects with chronic and psychiatric illness (n = 76,470). Cases of psychiatric disorder was determined using any of the following definitions (derived from Howard et al.): (i) ICD-10 codes for major depressive disorder (F32, F33), bipolar disorder (F30, F31), schizophrenia (F20-F29), autism (F84.0, F84.3, F84.5), intellectual disability (F70.0, F70.1, F70.9), anxiety disorder (F40 - F43), multiple personality disorder (F44.8), or mood disorder (F30 - F39); (ii) Self-reported antidepressant, antipsychotic, or anxiolytic use at nurse-led interview; (iii) Self-reported depression, major depressive disorder, bipolar disorder, or schizophrenia at nurse-led interview at baseline visit; (iv) “Broad depression”: responded yes to the question “Have you ever seen a general practitioner (GP) for nerves, anxiety, tension or depression?” and yes to either, “have you ever had a time when you were feeling depressed or down for at least a whole week,” or, “Have you ever had a time when you were uninterested in things or unable to enjoy the things you used to for at least a whole week?” lasting for more than 1 week; (v) Questionnaire-assessed bipolar disorder (Smith algorithm) responded yes to the question, “Have you ever had a period of time lasting at least two days when you were feeling so good, “high”, excited or “hyper” that other people thought you were not your normal self or you were so “hyper” that you got into trouble?” or “Have you ever had a period of time lasting at least two days when you were so irritable that you found yourself shouting at people or starting fights or arguments?” for a duration of at least one week and with at least 3 manic/hyper symptoms49. A list of sleep medications can be found in the Supplementary Note.
A description of coronary artery disease and activity monitor derived measures of sleep in the UK Biobank can be found in the Supplementary Note.
Genotyping and quality control.
Phenotype data is available for 502,631 subjects in the UK Biobank. Genotyping was performed by the UK Biobank, and genotyping, quality control, and imputation procedures are described in detail here50. In brief, blood, saliva, and urine was collected from participants, and DNA was extracted from the buffy coat samples. Participant DNA was genotyped on two arrays, UK BiLEVE and UKB Axiom with >95% common content and genotypes for ~800,000 autosomal SNPs were imputed to two reference panels. Genotypes were called using Affymetrix Power Tools software. Sample and SNPs for quality control were selected from a set of 489,212 samples across 812,428 unique markers. Sample QC was conducted using 605,876 high quality autosomal markers. Samples were removed for high missingness or heterozygosity (968 samples) and sex chromosome abnormalities (652 samples). Genotypes for 488,377 samples passed sample QC (~99.9% of total samples). Marker based QC measures were tested in the European ancestry subset (n = 463,844), which was identified based on principal components of ancestry. SNPs were tested for batch effects (197 SNPs/batch), plate effects (284 SNPs/batch), Hardy-Weinberg equilibrium (572 SNPs/batch), sex effects (45 SNPs/batch), array effects (5417 SNPs), and discordance across control replicates (622 on UK BiLEVE Axiom array and 632 UK Biobank Axiom array) (P < 10−12 or <95% for all tests). For each batch (106 batches total), markers that failed at least one test were set to missing. Before imputation, 805,426 SNPs pass QC in at least one batch (>99% of the array content). Population structure was captured by principal component analysis on the samples using a subset of high quality (missingness <1.5%), high frequency SNPs (>2.5%) (~100,000 SNPs) and identified the sub-sample of white British descent. We further clustered subjects into four ancestry clusters using K-means clustering on the principal components, identifying 453,964 subjects of European ancestry. Imputation of autosomal SNPs was performed to UK10K haplotype, 1000 Genomes Phase 3, and Haplotype Reference Consortium (HRC) with the current analysis using only those SNPs imputed to the HRC reference panel. Autosomal SNPs were pre-phased using SHAPEIT351 and imputed using IMPUTE4. In total ~96 million SNPs were imputed. Related individuals were identified by estimating kinship coefficients for all pairs of samples, using only markers weakly informative of ancestral background. In total, there are 107,162 related pairs comprised of 147,731 individuals related to at least one other subject in the UK Biobank.
Genome-wide association analysis.
Genetic association analysis across the autosomes was performed in related subjects of European ancestry (n = 453,964) using BOLT-LMM30 linear mixed models and an additive genetic model adjusted for age, sex, 10 PCs, genotyping array and genetic correlation matrix with a maximum per SNP missingness of 10% and per sample missingness of 40%. We used a genome-wide significance threshold of 5 × 10−8 for each GWAS. Genetic association analysis was also performed in unrelated subjects of white British ancestry (n = 337,545) using PLINK52 logistic regression and an additive genetic model adjusted for age, sex, 10 PCs and genotyping array to determine SNP effects on self-reported insomnia symptoms. We used a hard-call genotype threshold of 0.1, SNP imputation quality threshold of 0.80, and a MAF threshold of 0.001. Genetic association analysis for the X chromosome was performed using the genotyped markers on the X chromosome with the additional –sex flag in PLINK. We are 80% powered to detect a relative difference of 4% or more (i.e. an OR of 1.04 or 0.96 assuming a MAF 0.1, P = 5 × 10−8). Sex-specific GWAS were performed in PLINK 1.9 using logistic regression stratified by sex adjusting for age, 10 principal components of ancestry, and genotyping array. We used a hard-call genotype threshold of 0.1, SNP imputation quality threshold of 0.80, and a MAF threshold of 0.001. SNP x sex interactions (13 tests) and SNP x menopause interaction in females (13 tests) were tested for genome-wide significant signals with the significance threshold defined by Bonferroni correction. SNP-based trait heritability was calculated as the proportion of trait variance due to additive genetic factors measured in this study using BOLT-REML30, to leverage the power of raw genotype data together with low frequency variants (MAF ≥ 0.001). Additional independent risk loci were identified using the approximate conditional and joint association method implemented in GCTA (GCTA-COJO)53. Fixed effects meta-analysis was performed using METAL54 with the standard error scheme.
Sensitivity analyses on top signals.
Follow-up analyses on genome-wide significant loci in the primary analyses included covariate sensitivity analysis individually adjusting for sleep apnea, coffee/tea intake, physical activity, severe stress, depression, psychiatric medication use, socio-economic status, smoking, employment and marital status, and snoring, or BMI in addition to baseline adjustments for age, sex, 10 PCs and genotyping array. Sensitivity and sex-specific analyses were conducted only in unrelated subjects of white British ancestry.
Gene, pathway and tissue-enrichment analyses.
Gene-based analysis was performed using PASCAL26. Tissue enrichment analysis was conducted using FUMA55. Enrichment for pathways and ontologies was performed in EnrichR33,34 using the human genome as the reference set and a minimum number of 2 genes per category. A genetic risk score for RLS was tested using the weighted genetic risk score calculated by summing the products of the RLS risk allele count for 20 genome-wide significant SNPs multiplied by the scaled RLS effect reported by Schormair et al.35 using the summary statistics from our frequent insomnia symptom GWAS using the GTX package in R56. Integrative transcriptome-wide association analyses with GWAS were performed using the FUSION TWAS package29 with weights generated from gene expression in 8 brain regions and 6 tissues from the GTEX consortium (v6), and SNPs common to the 1000 Genomes LD reference panel and our frequent insomnia symptoms GWAS summary statistics. Tissues for TWAS testing were selected from the FUMA tissue enrichment analyses and here we present significant results that survive Bonferroni correction for the number of genes tested per tissue and for all 14 tissues. In the results table, we show the number of individuals per tissue type used to generate expression weights, the total number of genes expressed per tissue, gene symbol for significant gene, rsID for the best GWAS and eQTL SNPs in the reference panel, and the TWAS P value. GWAS and eQTL SNPs analyzed include only those SNPs present in the 1000 Genomes LD reference panel used by the FUSION TWAS software; therefore, the best GWAS SNP listed in this table may be in strong linkage disequilibrium with, but not identical to, the lead GWAS SNP for hits in Supplementary Table 2.
Heterogeneity analysis.
Analyses to distinguish pleiotropy and heterogeneity between insomnia and RLS was performed using Breaking Up Heterogeneous Mixture Based On Cross-locus correlations (BUHMBOX)41. BUHMBOX analyses tests the presence of heterogeneity between two traits. BUHMBOX was performed in the insomnia GWAS using all 20 RLS SNPs and weights reported by Schormair et al.35 and in the RLS GWAS using all 57 SNPs reported in Supplementary Table 2. Additional sensitivity analyses were performed excluding SNPs in the MEIS1 region.
Genetic correlation analyses.
Post-GWAS genome-wide genetic correlation analysis of LD Score Regression (LDSC)32,42,43 using LDHub was conducted using all UK Biobank SNPs also found in HapMap3 and included publicly available data from 224 published genome-wide association studies, with a significance threshold of P = 0.002 after Bonferroni correction for all tests performed. LDSC estimates genetic correlation between two traits from summary statistics (ranging from −1 to 1) using the fact that the GWAS effect-size estimate for each SNP incorporates effects of all SNPs in LD with that SNP, SNPs with high LD have higher X2 statistics than SNPs with low LD, and a similar relationship is observed when single study test statistics are replaced with the product of z-scores from two studies of traits with some correlation. Furthermore, genetic correlation is possible between case/control studies and quantitative traits, as well as within these trait types. We performed partitioning of heritability using the 8 pre-computed cell-type regions, and 25 pre-computed functional annotations available through LDSC, which were curated from large-scale robust datasets31. Enrichment both in the functional regions and in an expanded region (+500 bp) around each functional class was calculated in order to prevent the estimates from being biased upward by enrichment in nearby regions. The multiple testing threshhold for the partitioning of heritability was determined using the conservative Bonferroni correction (P of 0.05/25 classes). Summary GWAS statistics will be made available at the UK Biobank web portal.
Mendelian randomization analyses.
MR analysis was carried out using MR-Base57, using the inverse variance weighted approach as our main analysis method44, and MR-Egger45 and weighted median estimation46 as sensitivity analyses. MR results may be biased by horizontal pleiotropy, i.e. where the genetic variants that are robustly related to the exposure of interest (here frequent insomnia symptoms) independently influence levels of a causal risk factor for the outcome. IVW assumes that there is no horizontal pleiotropy. MR-Egger provides unbiased causal estimates even if all of the genetic instruments have horizontal pleiotropic effects, but it assumes that the association of genetic instruments with risk factor is not correlated with any pleiotropic genetic instrument associations with outcome. The weighted median approach is valid if less than 50% of the weight is pleiotropic (i.e. no single SNP that contributes 50% of the weight or a number of SNPs that together contribute 50% should be invalid because of horizontal pleiotropy. Given these different assumptions, if all three methods are broadly consistent, this strengthens our causal inference. For most of our MR analyses, we used two-sample MR, in which, for all 57 of the insomnia GWAS hits identified in this study, we looked for the per allele difference in odds (binary outcomes) or means (continuous) with outcomes from summary publicly available data in the MR-Base platform. Results are therefore a measure of ‘any insomnia’ and sample 1 is UK Biobank (our GWAS) and sample 2 a number of different GWAS consortia covering the outcomes we explored (Supplementary Table 17). For all four of the longevity outcomes and as follow up for CAD, we used one-sample MR with the SNP-outcome associations also obtained from UK Biobank. If we could not find one of the 57 SNPs in the outcome database we substituted for a proxy where possible, LD proxies are defined using 1000 Genomes European sample with r2 > 0.8. The number of SNPs used in each MR analysis varies by outcome from 11 to 53 because of some SNPs (or proxies for them) not being located in the outcome GWAS (Table 1).
Primary association analyses of the 57 genome-wide significant insomnia SNPs with CAD were performed in Hail (https://github.com/hail-is/hail) using imputed genotype dosages and a logistic regression model adjusting for age at first visit, sex, genotyping array, and the first 10 principal components of ancestry. A total of 23,980 CAD cases were compared to 388,326 referents. For Mendelian randomization, a fixed-effects inverse-variance weighted meta-analysis was performed of the SNP-specific association estimates with CAD, aligning each insomnia SNP allele/beta coefficient to “increased risk of insomnia.” Sensitivity analyses were performed excluding the variants with the strongest effect estimate and/or widest confidence intervals to account for SNP heterogeneity.
Replication cohort.
Replication cohort sample ascertainment, phenotype definition, genotyping, quality control, imputation, and analyses are described in the Supplementary Note.
Reporting summary.
An accompanying Life Sciences Reporting Summary is published along with this manuscript.
Data availability.
GWAS summary statistics are available at the Sleep Disorders Knowledge Portal data download page (http://sleepdisordergenetics.org/informational/data).
Supplementary Material
Acknowledgements
This research has been conducted using the UK Biobank Resource (UK Biobank application number 6818 and 9072). We would like to thank the participants and researchers from the UK Biobank who contributed or collected data. This work was supported by NIH grants R01DK107859 (R.S.), R21HL121728 (R.S.), F32DK102323 (J.M.L.), R01HL113338 (J.M.L., S.R. and R.S.), R01DK102696 (R.S. and F.S.), NHLBI R35 35HL135818 (S.R. and R.S), R01DK105072 (R.S. and F.S.), T32HL007567 (J.M.L.), K01HL136884 (J.M.L.), The MGH Research Scholar Fund (R.S), HG003054 (X.Z.), The University of Manchester (Research Infrastructure Fund), the Wellcome Trust (salary support for D.W.R. and A.L.), UK Medical Research Council MC_UU_12013/5 (D.A.L.), UK Medical Research Council MC_UU_00011/6 (D.A.L.), and UK National Institute of Health Research NF-SI-0611–10196 (D.A.L.). A.R.W. and T.M.F. are supported by a European Research Council grant (SZ-245 50371-GLUCOSEGENES-FP7-IDEAS-ERC). S.E.J. is funded by the Medical Research Council (MR/M005070/1). M.N.W. is supported by the Wellcome Trust Institutional Strategic Support Award (WT097835MF). The following groups provided summary statistics to LDHub and MR-base: ADIPOGen (Adiponectin genetics consortium), C4D (Coronary Artery Disease Genetics Consortium), CARDIoGRAM (Coronary ARtery DIsease Genome wide Replication and Meta-analysis), CKDGen (Chronic Kidney Disease Genetics consortium), dbGAP (database of Genotypes and Phenotypes), DIAGRAM (DIAbetes Genetics Replication And Meta-analysis), ENIGMA (Enhancing Neuro Imaging Genetics through Meta Analysis), EAGLE (EArly Genetics & Lifecourse Epidemiology Eczema Consortium, excluding 23andMe), EGG (Early Growth Genetics Consortium), GABRIEL (A Multidisciplinary Study to Identify the Genetic and Environmental Causes of Asthma in the European Community), GCAN (Genetic Consortium for Anorexia Nervosa), GEFOS (GEnetic Factors for OSteoporosis Consortium), GIANT (Genetic Investigation of ANthropometric Traits), GIS (Genetics of Iron Status consortium), GLGC (Global Lipids Genetics Consortium), GPC (Genetics of Personality Consortium), GUGC (Global Urate and Gout consortium), HaemGen (haemotological and platelet traits genetics consortium), HRgene (Heart Rate consortium), IIBDGC (International Inflammatory Bowel Disease Genetics Consortium), ILCCO (International Lung Cancer Consortium), IMSGC (International Multiple Sclerosis Genetic Consortium), MAGIC (Meta-Analyses of Glucose and Insulin-related traits Consortium), MESA (Multi-Ethnic Study of Atherosclerosis), PGC (Psychiatric Genomics Consortium), Project MinE consortium, ReproGen (Reproductive Genetics Consortium), SSGAC (Social Science Genetics Association Consortium) and TAG (Tobacco and Genetics Consortium), TRICL (Transdisciplinary Research in Cancer of the Lung consortium), UK Biobank. The Nord-Trøndelag Health Study (The HUNT Study) is a collaboration between HUNT Research Centre (Faculty of Medicine, NTNU, Norwegian University of Science and Technology), Nord-Trøndelag County Council, Central Norway Health Authority, and the Norwegian Institute of Public Health. We are grateful for the contributions from He Zhang and Hyun Min Kang. We also acknowledge the support given to us by the Genotyping core and Jin Chen. The K.G. Jebesen center for genetic epidemiology is financed by Stiftelsen Kristian Gerhard Jebsen, Faculty of Medicine and Health Sciences Norwegian University of Science and Technology (NTNU) and Central Norway Regional Health Authority. Ben Michael Brumpton and Linn Strand received research grants from The Liaison Committee for education, research and innovation in Central Norway.
Footnotes
Competing Interests
J.W.W. is a consultant for Merck and Flex Pharma. He receives royalties from UpToDate. He has received speaker fees and travel support from Otsuka. He has received research grants from UCB Pharma, NeuroMetrix, NIMH, the RLS Foundation, and Luitpold Pharma. F.A.J.L.S. has received lecture fees from Bayer HealthCare, Sentara HealthCare, Philips, Vanda Pharmaceuticals, and Pfizer. D.A.L. has received funding from Medtronic and Roche Diagnostics for biomarker research unrelated to this study. M.K.R. has acted as a consultant for GSK, Novo Nordisk, Roche and MSD, and also participated in advisory board meetings on their behalf. M.K.R. has received lecture fees from MSD and grant support from Novo Nordisk, MSD and GSK.
Contributor Information
HUNT All In Sleep:
Amy E. Martinsen, Anne H. Skogholt, Ben Brumpton, Bendik S. Winsvold, Børge Sivertsen, Cristen J. Willer, Daniela Bragantini, Håvard Kallestad, Imre Janszky, Ismail C. Guzey, John-Anker Zwart, Jonas B. Nielsen, Kristian B. Nilsen, Kristian Hveem, Lars Fritsche, Linda M. Pedersen, Linn B. Strand, Maiken E. Gabrielsen, Marianne B. Johnsen, Marie U. Lie, Morten Engstrøm, Trond Sand, and Wei Zhou
References
- 1.Morin CM & Benca R Chronic insomnia. Lancet Lond. Engl 379, 1129–1141 (2012). [DOI] [PubMed] [Google Scholar]
- 2.Morin CM et al. Insomnia disorder. Nat. Rev. Dis. Primer 1, 15026 (2015). [DOI] [PubMed] [Google Scholar]
- 3.Hoevenaar-Blom MP, Spijkerman AMW, Kromhout D, van den Berg JF & Verschuren WMM Sleep duration and sleep quality in relation to 12-year cardiovascular disease incidence: the MORGEN study. Sleep 34, 1487–1492 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Riemann D et al. European guideline for the diagnosis and treatment of insomnia. J. Sleep Res 26, 675–700 (2017). [DOI] [PubMed] [Google Scholar]
- 5.Qaseem A, Barry MJ, Kansagara D & for the Clinical Guidelines Committee of the American College of Physicians. Nonpharmacologic versus pharmacologic treatment of adult patients with major depressive disorder: A clinical practice guideline from the American College of Physicians. Ann. Intern. Med 164, 350 (2016). [DOI] [PubMed] [Google Scholar]
- 6.Sateia MJ, Buysse DJ, Krystal AD, Neubauer DN & Heald JL Clinical practice guideline for the pharmacologic treatment of chronic insomnia in adults: An American Academy of Sleep Medicine clinical practice guideline. J. Clin. Sleep Med. JCSM Off. Publ. Am. Acad. Sleep Med 13, 307–349 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kapil V, Green JL, Le Lait C, Wood DM & Dargan PI Misuse of benzodiazepines and Z-drugs in the UK. Br. J. Psychiatry J. Ment. Sci 205, 407–408 (2014). [DOI] [PubMed] [Google Scholar]
- 8.Naylor E et al. The Circadian clock mutation alters sleep homeostasis in the mouse. J. Neurosci 20, 8138–8143 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cirelli C The genetic and molecular regulation of sleep: from fruit flies to humans. Nat. Rev. Neurosci 10, 549–560 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Allada R, Cirelli C & Sehgal A Molecular mechanisms of sleep homeostasis in flies and mammals. Cold Spring Harb. Perspect. Biol 9, a027730 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pimentel D et al. Operation of a homeostatic sleep switch. Nature 536, 333–337 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Funato H et al. Forward-genetics analysis of sleep in randomly mutagenized mice. Nature 539, 378–383 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chung S et al. Identification of preoptic sleep neurons using retrograde labelling and gene profiling. Nature 545, 477–481 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lind MJ & Gehrman PR Genetic pathways to insomnia. Brain Sci. 6, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lane JM et al. Genome-wide association analyses of sleep disturbance traits identify new loci and highlight shared genetics with neuropsychiatric and metabolic traits. Nat. Genet 49, 274–281 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hammerschlag AR et al. Genome-wide association analysis of insomnia complaints identifies risk genes and genetic overlap with psychiatric and metabolic traits. Nat. Genet 49, 1584–1592 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wain LV et al. Novel insights into the genetics of smoking behaviour, lung function, and chronic obstructive pulmonary disease (UK BiLEVE): a genetic association study in UK Biobank. Lancet Respir. Med 3, 769–781 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vahia VN Diagnostic and statistical manual of mental disorders 5: A quick glance. Indian J. Psychiatry 55, 220–223 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Benjamins JS et al. Insomnia heterogeneity: Characteristics to consider for data-driven multivariate subtyping. Sleep Med. Rev 36, 71–81 (2017). [DOI] [PubMed] [Google Scholar]
- 20.Krokstad S et al. Cohort profile: the HUNT Study, Norway. Int. J. Epidemiol 42, 968–977 (2013). [DOI] [PubMed] [Google Scholar]
- 21.Farh KK-H et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature 518, 337–343 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang F & Lupski JR Non-coding genetic variants in human disease. Hum. Mol. Genet 24, R102–110 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Duclot F & Kabbaj M The role of early growth response 1 (EGR1) in brain plasticity and neuropsychiatric disorders. Front. Behav. Neurosci 11, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Elliott L et al. Genome-wide association studies of brain imaging phenotypes in UK Biobank. Nature 562, 210–216 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Winkelman JW et al. Increased Rostral Anterior Cingulate Cortex Volume in Chronic Primary Insomnia. Sleep 36, 991–998 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lamparter D, Marbach D, Rueedi R, Kutalik Z & Bergmann S Fast and Rigorous Computation of Gene and Pathway Scores from SNP-Based Summary Statistics. PLoS Comput. Biol 12, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vetrivelan R, Qiu M-H, Chang C & Lu J Role of Basal Ganglia in Sleep–Wake Regulation: Neural Circuitry and Clinical Significance. Front. Neuroanat 4, (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jansen PR et al. Genome-wide analysis of insomnia (N=1,331,010) identifies novel loci and functional pathways. bioRxiv 214973 (2018). doi: 10.1101/214973 [DOI] [PubMed] [Google Scholar]
- 29.Gusev A et al. Integrative approaches for large-scale transcriptome-wide association studies. Nat. Genet 48, 245–252 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Loh P-R et al. Efficient Bayesian mixed-model analysis increases association power in large cohorts. Nat. Genet 47, 284–290 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Finucane HK et al. Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat. Genet (2015). doi: 10.1038/ng.3404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bulik-Sullivan BK et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet 47, 291–295 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kuleshov MV et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 44, W90–W97 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen EY et al. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 14, 128 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schormair B et al. Identification of novel risk loci for restless legs syndrome in genome-wide association studies in individuals of European ancestry: a meta-analysis. Lancet Neurol. 16, 898–907 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stavropoulos N & Young MW Insomniac and Cullin-3 regulate sleep and wakefulness in Drosophila. Neuron 72, 964–976 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Livingston WS et al. Improved sleep in military personnel is associated with changes in the expression of inflammatory genes and improvement in depression symptoms. Front. Psychiatry 6, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Freeman AAH, Mandilaras K, Missirlis F & Sanyal S An emerging role for Cullin-3 mediated ubiquitination in sleep and circadian rhythm. Fly (Austin) 7, 39–43 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Anafi RC et al. Sleep is not just for the brain: transcriptional responses to sleep in peripheral tissues. BMC Genomics 14, 362 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Trotti LM Restless Legs Syndrome and Sleep-Related Movement Disorders. Contin. Minneap. Minn 23, 1005–1016 (2017). [DOI] [PubMed] [Google Scholar]
- 41.Han B et al. A method to decipher pleiotropy by detecting underlying heterogeneity driven by hidden subgroups applied to autoimmune and neuropsychiatric diseases. Nat. Genet 48, 803–810 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zheng J et al. LD Hub: a centralized database and web interface to perform LD score regression that maximizes the potential of summary level GWAS data for SNP heritability and genetic correlation analysis. Bioinformatics 33, 272–279 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bulik-Sullivan B et al. An atlas of genetic correlations across human diseases and traits. Nat. Genet (2015). doi: 10.1038/ng.3406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Burgess S, Butterworth A & Thompson SG Mendelian randomization analysis with multiple genetic variants using summarized data. Genet. Epidemiol 37, 658–665 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bowden J, Davey Smith G & Burgess S Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int. J. Epidemiol 44, 512–525 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bowden J, Davey Smith G, Haycock PC & Burgess S Consistent estimation in Mendelian Randomization with some invalid instruments using a weighted median estimator. Genet. Epidemiol 40, 304–314 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Javaheri S & Redline S Insomnia and risk of cardiovascular disease. Chest 152, 435–444 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sudlow C et al. UK biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med. 12, e1001779 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Smith DJ et al. Prevalence and characteristics of probable Major Depression and Bipolar Disorder within UK Biobank: Cross-sectional study of 172,751 participants. PLoS ONE 8, e75362 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bycroft C et al. The UK Biobank resource with deep phenotyping and genomic data. Nature 562, 203–209 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.O’Connell J et al. Haplotype estimation for biobank-scale data sets. Nature Genet. 48, 817–820 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Purcell S et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81, 559–575 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yang J et al. Conditional and joint multiple-SNP analysis of GWAS summary statistics identifies additional variants influencing complex traits. Nat. Genet 44, 369–375 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Willer CJ, Li Y & Abecasis GR METAL: fast and efficient meta-analysis of genomewide association scans. Bioinforma. Oxf. Engl 26, 2190–2191 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Watanabe K, Taskesen E, van Bochoven A & Posthuma D Functional mapping and annotation of genetic associations with FUMA. Nat. Commun 8, 1826 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.R Development Core Team. R: A language and environment for statistical computing R Found. Stat. Comput Vienna, ISBN 3–900051-07–0 (URL http://www.R-project.org/). [Google Scholar]
- 57.Hemani G et al. The MR-Base platform supports systematic causal inference across the human phenome. eLife 7, e34408 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
GWAS summary statistics are available at the Sleep Disorders Knowledge Portal data download page (http://sleepdisordergenetics.org/informational/data).