

Abstract
BACKGROUND:
Genomic profiling of colorectal cancer aims to identify actionable somatic mutations, but can also discover incidental germline findings.
OBJECTIVE:
To report the detection of pathogenic germline variants that confer heritable cancer predisposition.
DESIGN:
Retrospective.
SETTINGS:
A tertiary-referral institution.
PATIENTS:
Between 2012 and 2015, 1000 patients with advanced cancer underwent targeted exome sequencing of a 202-gene panel. The subgroup of 151 patients with advanced colorectal cancer who underwent matched tumor-normal (blood) sequencing formed our study cohort.
INTERVENTIONS:
Germline variants in 46 genes associated with hereditary cancer predisposition were classified according to a defined algorithm based on in silico predictions of pathogenicity. Patients with presumed pathogenic variants were examined for type of mutation as well as clinical, pedigree, and clinical genetic testing data.
MAIN OUTCOME MEASURE:
Detection of pathogenic germline variants
RESULTS:
1910 distinct germline variants were observed in 151 patients. After filtering, 15 (9.9%) pathogenic germline variants were found in 15 patients, arising from 9 genes of varying penetrance for colorectal cancer (APC [2; 13%], ATM [1; 6%], BRCA1 [2; 13%], CDH1 [2; 13%], CHEK2 [4; 27%], MSH2 [1; 7%], MSH6 [1; 7%], NF2 [1; 7%], and TP53 [1; 7%]). Patients with pathogenic variants were diagnosed at a younger age than those without (median 45 vs. 52 years, p=0.03). Of the 15 patients, 7 (46.7%) patients with variants in low/moderate penetrant genes for colorectal cancer, would likely have not been tested based on clinical and pedigree criteria, where 2 harbored clinically actionable variants (CDH1, and NF2, 28.5% of 7).
LIMITATIONS:
Small sample size, advanced-stage patients
CONCLUSIONS:
Tumor-normal sequencing can incidentally discover clinically unsuspected germline variants that confer cancer predisposition in 9.9% of advanced colorectal cancer patients. Precision medicine should integrate clinical cancer genetics to inform and interpret the actionability of germline variants and to provide follow-up care to mutation carriers. See Video Abstract at http://links.lww.com/DCR/Axxx.
Keywords: Colorectal cancer, Hereditary cancer, Gene panel testing, Genetic testing, Germline variant, Tumor sequencing
INTRODUCTION
Precision medicine and tumor genomic profiling are being increasingly performed for cancer patients. These efforts have the potential to direct therapy, predict response, and define prognosis. Available tumor sequencing strategies range from directed single-gene assays, to targeted mutation testing of a gene panel, to whole-exome or whole-genome sequencing. The two general approaches for genomic profiling have been tumor-only sequencing or tumor-normal sequencing, with the latter approach being preferred and recommended to help avoid misinterpretation of results.1,2 Tumor-normal sequencing utilizes matched blood or saliva specimen, and germline DNA is sequenced in the background as a genomic reference for tumor sequencing.
Profiling of germline DNA in this process opens the possibility for identifying germline variants. These have been termed incidental or secondary findings as they were unrelated to the original indication for testing (i.e. tumor mutation profiling) but are of medical value.3 When presumed pathogenic germline variants (PGVs) that confer heritable predisposition to cancer are incidentally identified, clinical genetics care should follow and should include genetic risk assessment, clinical confirmatory testing, and follow-up cancer preventive care for the proband and at-risk relatives.4 Indeed, recent studies have suggested that large-scale or whole-exome tumor-normal sequencing could lead to the increased identification of patients who carry germline mutations, when compared to identification through traditional targeted germline testing based on clinical and pedigree criteria.5
Among patients with metastatic CRC, current guidelines have highlighted the benefits of mutation profiling for KRAS/NRAS, BRAF and DNA mismatch repair (MMR) genes.6 Increasingly, multi-gene panel testing or whole exome sequencing is replacing targeted mutation testing of individual genes.7 Because a familial or hereditary component can be reported in nearly 25% of all CRCs,8 the potential for identifying incidental PGVs during tumor-normal sequencing can be significant. However, currently, clinicians who order tumor sequencing are often not aware of the potential for secondary germline findings, and/or do not have the infrastructure and support needed to provide follow-up clinical genetics care based on such findings. In this study, we aimed to examine germline variant detection in patients with advanced CRC who underwent tumor-normal sequencing, and to correlate the molecular, clinical and pedigree features of patients with presumed PGVs. We hypothesized that tumor-normal sequencing can identify more PGVs than traditional germline testing based on clinical and pedigree criteria, and a partnership with clinical genetics expertise is needed for clinical interpretation.
METHODS
Study population
Between 2012 and 2015, 1000 adult patients with advanced cancer were referred at the discretion of their treating oncologists and enrolled in an institutional review board (IRB)-approved protocol for personalized cancer therapy using next-generation somatic mutation sequencing (Clearinghouse study; NCT01772771). A companion germline protocol enrolled patients for optional blood collection and germline testing.9 This study focuses on 151 patients with metastatic CRC who underwent sequencing of both tumor and germline DNA. Although the general results of the entire cohort had been previously described,9 we herein provide a detailed germline analysis of the CRC patients. As previously described,9 germline findings of pathogenic significance were reviewed by a Return of Incidental Findings committee, and communicated with the treating clinician.10 Patients were offered the opportunity to undergo clinical genetic risk assessment, further testing, and enrollment in the Familial High-risk GI Cancer Clinic (Figure 1).
Figure 1.

A model for partnered care integrating precision medicine tumor genomic profiling with clinical genetics care, for pathogenic germline variants incidentally detected among advanced colorectal cancer patients
Patient demographics, clinical treatments, and tumor characteristics were retrospectively reviewed. Personal and family history, clinical genetic counseling, and CLIA (Clinical Laboratory Improvement Amendments) confirmatory germline testing were noted. Clinical actionability of PGV was based on the guidelines of the National Comprehensive Cancer Network (NCCN).11
Genomic sequencing and bioinformatic analyses
Genomic DNA was extracted from tumor and blood for targeted exome sequencing of 202 cancer-related genes as described previously.9,12
We focused on determining the pathogenicity of variants arising from 46 genes known to be associated with hereditary cancer predisposition (Table 1). We a priori pooled 25 genes from the updated list of 59 medically actionable genes as recommended by the American College of Medical Genetics and Genomics (ACMG)3 and 21 additional genes from commonly tested on multiplex germline cancer panels in commercial Clinical Laboratory Improvement Amendments (CLIA)-certified labs (Table 1).
Table 1.
Germline variants in 46 hereditary cancer genes observed in 151 patients with advanced colorectal cancer.
| 46 Hereditary Cancer Genes Examined |
No. Of Patients With At Least One Germline Variant In This Gene |
No. Of Patients With A Presumed Pathogenic Germline Variant in This Gene |
|---|---|---|
| 26 (56.5%) genes with at least one germline variant | 15 | |
| APC | 143 | 2 |
| ATM | 145 | 1 |
| BAP1 | 4 | 0 |
| BRCA1 | 102 | 2 |
| BRCA2 | 143 | 0 |
| CDH1 | 3 | 2 |
| CDK4 | 1 | 0 |
| CHEK2 | 6 | 4 |
| MEN1 | 143 | 0 |
| MITF | 3 | 0 |
| MLH1 | 75 | 0 |
| MSH2 | 17 | 1 |
| MSH6 | 71 | 1 |
| NF1 | 3 | 0 |
| NF2 | 2 | 1 |
| PALB2 | 37 | 0 |
| RB1 | 5 | 0 |
| RET | 139 | 0 |
| SMAD4 | 1 | 0 |
| SMARCA4 | 4 | 0 |
| STK11 | 6 | 0 |
| TGFBR2 | 1 | 0 |
| TP53 | 124 | 1 |
| TSC1 | 48 | 0 |
| TSC2 | 23 | 0 |
| VHL | 2 | 0 |
| 20 (43.5%) genes with no germline variant | ||
| BMPR1A, BRIP1, CDKN2A, FANCC, FH, FLCN, MET, MRE11A, MUTYH, NBN, PMS2, PRKAR1A, PTEN, RAD50, SDHA, SDHB, SDHC, SDHD, WT1, XRCC2 |
Genes where a pathogenic germline variant was identified are bolded.
Variant classification and pathogenicity calling
Variants were filtered by removing frequent variants, defined by population allele frequency > 1% in 1000 Genomes (http://browser.1000genomes.org/index.html),13 and by presence in more than 4 patients in our cohort (Figure 2). They were then prioritized into a 5-tier classification using specific standard terminology: ‘pathogenic’, ‘likely pathogenic’, ‘uncertain significance’, ‘likely benign’, and ‘benign’, per recommendation of the ACMG and similar to our previous study.14 Pathogenic variants are considered to be disease-causing based on predicted functions of their resulting protein products. Tier 1 variants typically resulted in a nonsense (stop gain) or a frameshift mutation and are expected to be pathogenic. Tiers 2–4 variants typically resulted in missense mutations and their pathogenicity was determined based on three in silico prediction programs (Condel-http://bg.upf.edu/fannsdb/;15 PolyPhen-http://genetics.bwh.harvard.edu/pph/data/;16 and SIFT-http://sift.jcvi.org/).17 Variants classified to Tier 2 had deleterious predictions in both Condel and SIFT and probably damaging predictions in PolyPhen; those to Tier 3 had probably damaging predictions in two of three algorithms; those to Tier 4 had only one probably or possibly damaging prediction. Other variants were classified into Tier 5. Pathogenicity of individual variants was checked in three existing variant databases ClinVar,18 Breast Cancer Information Core database (BIC),19 and InSiGHT variant database.20 For novel variants with no established pathogenicity, we relied on the literature for predicted impact on the protein/domain structure and function.21 Conflicting interpretations were resolved according to more recent published clinical reports.
Figure 2.
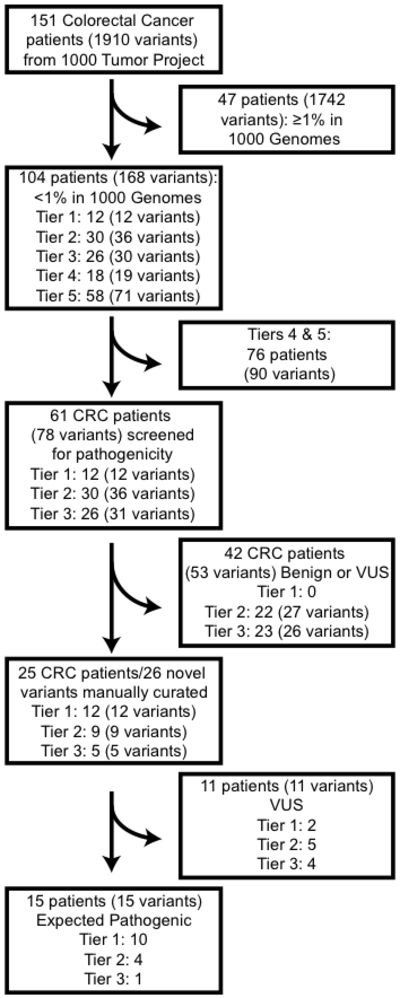
Flow chart of variants and patients. Initially 151 patients with 1910 distinct variants were screened for presumed pathogenic germline variants. Variants with low population frequencies (<1%) were tiered using three in silico prediction tools. Variants in Tiers 1-3 (i.e. pathogenic/probably pathogenic calls by at least 2 tools) were further characterized for pathogenicity.
Statistical Analyses
Standard descriptive statistics and SPSS Statistics 23 (IBM) were utilized. Student’s T-Test was used for comparisons of continuous variables. P values less than 0.05 were considered statistically significant.
RESULTS
Patient characteristics
The median age of our study cohort was 52.5 years, with 44.4% having been diagnosed younger than age 50. The majority of the tumors were located in the distal colon and rectum (Table 2).
Table 2.
Patient Characteristics
| Characteristic | All Patients (n=151) |
Patients with Pathogenic Germline Variants (n=15) |
|---|---|---|
| Age at diagnosis (median, range; years) | 52.5 (22-78) | 45 (22-69) |
| Age (years) | ||
| < 50 | 67 (44.4%) | 10 (66.7%) |
| 50-65 | 67 (44.4%) | 4 (26.7%) |
| > 65 | 17 (11.2%) | 1 (6.7%) |
| Race | ||
| Non-Hispanic White | 111 (73.5%) | 14 (93.3%) |
| Non-Hispanic Black | 17 (11.3%) | 0 |
| Hispanic | 10 (6.6%) | 1 (6.7%) |
| Other | 13 (8.6%) | 0 |
| Sex | ||
| Male | 75 (49.7%) | 11 (73.3%) |
| Female | 76 (50.3%) | 4 (26.7%) |
| Tumor location | ||
| Proximal colon | 42 (27.8%) | 7 (41.2%) |
| Distal colon | 76 (50.3%) | 4 (23.5%) |
| Rectum | 32 (21.2%) | 6 (35.3%) |
| Metastatic Disease | ||
| 1 site | 78 (51.7%) | 9 (60%) |
| 2 sites | 50 (33.1%) | 5 (33.3%) |
| > 2 sites | 23 (15.2%) | 1 (6.7%) |
Variant filtering and pathogenicity classification
A total of 1910 distinct germline variants were identified in 151 patients, with the majority of patients harboring more than 1 germline variant (Table 1). Among the 46 genes analyzed, at least one germline variant was identified in 26 (53.6%) genes, and no germline variant was identified in 20 (43.5%; Table 1).
After filtering and tiering, we focused on 78 Tier 1–3 variants in 61 patients (Figure 2) Among these, 53 variants were benign or of uncertain significance (VUS) and further filtered. The remaining 26 variants were individually examined for pathogenicity. Two had been reported as known pathogenic. The remaining variants of previously unknown pathogenicity were expected to be pathogenic in 13 and VUS in 11 (Figure 2).
Presumed pathogenic germline variants: molecular and clinical characteristics
The 15 presumed PGV were identified in 15 patients (9.9%). Germline mutations were identified in 9 genes (APC [2 mutations; 13%], ATM [1; 6%], BRCA1 [2; 13%], CDH1 [2; 13%], CHEK2 [4; 27%], MSH2 [1; 7%], MSH6 [1; 7%], NF2 [1; 7%], and TP53 [1; 7%]; Table 1). They included 10 nonsense (stop) or frameshift mutations (Tier 1), 4 missense variants from Tier 2, and 1 missense variant from Tier 3. Genomic locations of mutations and domains in protein products of the 9 genes are depicted in Figure 3.
Figure 3.
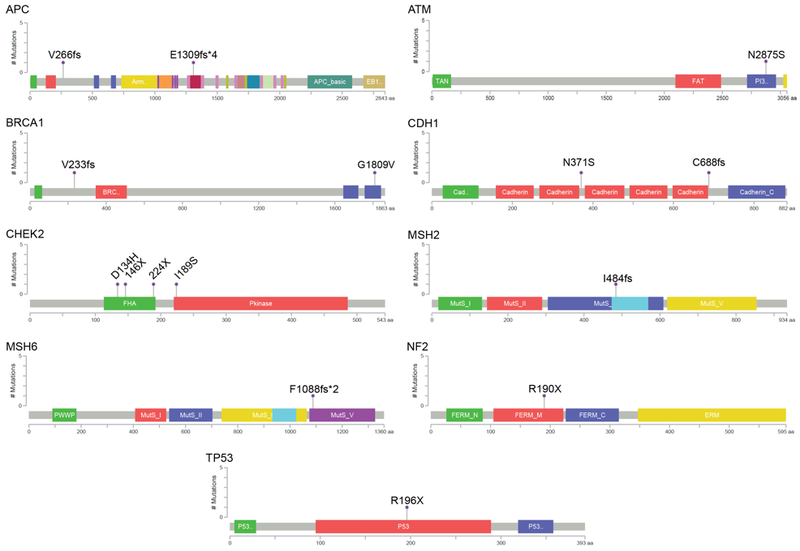
Presumed pathogenic germline variants. The locations of mutations and domains in the encoded protein products (shown by different colors) for the 15 presumed pathogenic variants are shown in lollipop plots. On the graph of each gene, the x axis reflects the number of amino acid residues, and the y axis represents the total number of mutations identified. Frameshift and stop gain mutations were classified in Tier 1, and were found in the following genes (as denoted by chromosomal locations): APC (5q22.2), BRCA1 (17q21.31), CDH1 (16q22.1), CHEK2 (22q12.1), MSH2(2p21-p16.3), MSH6 (2p16.3), NF2 (22q12.2), TP53 (17p13.1). Missense mutations were classified into Tiers 2 and 3, and were found in the following genes (as denoted by chromosomal locations): ATM (11q22.3), BRCA1 (17q21.31), CDH1 (16q22.1), CHEK2 (22q12.1).
Clinical genetics care
The median age of CRC for patients with a PGV was 45 years (range 22–69 years), which was significantly younger than that for patients without a PGV (52 years, range: 25–78; p=0.03). Thirteen (86.7%) patients had a family history of cancer, with 9 (60%) having a CRC in a first or second degree relative (Table 3).
Table 3.
Clinical details of 15 patients with presumed pathogenic germline variants
| Patient | Age at diagnosis |
Tumor Location |
Tier | Gene | cDNA change | Protein change | Variant type | Classification | Family history criteria |
Cancer in FDR | Cancer in SDR | Tumor MMR status |
Meets clinical criteria for clinical genetic testing? |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 24 | Rectum | 1 | APC | c.798delC | V266fs | Frameshift | EP | Bethesda | Colorectal | Colorectal | - | Yes |
| 2 | 28 | Left | 1 | APC | c.3921-3925delAAAAG | E1309fs4 | Frameshift | KP | Bethesda | Colorectal | Yes | ||
| 3 | 48 | Rectum | 1 | BRCA1 | c.697-698delGT | V233fs | Frameshift | KP | Bethesda | Ovarian | Colorectal | - | Yes |
| 4 | 56 | Left | 1 | CDH1 | c2062-2063delTG | C688fs | Frameshift | EP | No | Breast Prostate | - | - | Yes (panel) |
| 5 | 62 | Right | 1 | CHEK2 | c.672delC | 224 | Frameshift | EP | No | - | - | - | No |
| 6 | 43 | Right | 1 | CHEK2 | c.437delC | 146 | Frameshift- | EP | Amsterdam II | Renal | Colorectal | - | No |
| 7 | 38 | Rectum | 1 | MSH2 | c.1452-1455delAATG | I484fs | Frameshift | EP | Bethesda | - | Colorectal | - | Yes |
| 8 | 45 | Right | 1 | MSH6 | c.3254delC | F1088fs2 | Frameshift- | EP | Bethesda | - | Colorectal | Deficient: loss of MSH6 | Yes |
| 9 | 47 | Right | 1 | NF2 | c.568C>T | R190X | Nonsense | EP | Bethesda | Prostate | Lung; Breast; Prostate |
Proficient | No |
| 10 | 22 | Rectum | 1 | TP53 | c.586C>T | R196X | Nonsense | EP | Bethesda | - | Breast | Proficient | Yes (panel) |
| 11 | 69 | Right | 2 | ATM | c.8624A>G | N2875S | Missense | EP | No | Colorectal | - | Proficient | No |
| 12 | 54 | Left | 2 | BRCA1 | C.5426G>T | G1809V | Missense | EP | No | Colorectal; Breast/Ovarian; Bone | Gastric,/Lung; Pancreas; Lung | Proficient | Yes |
| 13 | 40 | Right | 2 | CDH1 | C.1112A>G | N371S | Missense | EP | Bethesda | - | Pancreas | Proficient | No |
| 14 | 50 | Left | 2 | CHEK2 | c.566T>GI | I189S | Missense | EP | No | Prostate | - | Proficient | No |
| 15 | 41 | Right | 3 | CHEK2 | c.400G>C | D134H | Missense | EP | Bethesda | - | Colorectal (2), Brain | Proficient | No |
EP: Expected pathogenic
KP: Known pathogenic
FDR: First degree relative
SDR: Second degree relative
Overall, 3 patients with mutations in APC, BRCA1, and NF2, had been referred and completed clinical genetic testing. If all 15 had been referred for genetic counseling, 8 patients would have been recommended to undergo clinical genetic testing based on age of CRC diagnosis, personal and family history. Thus, 8 PGVs (53.3% of 15) were felt to be likely identifiable through either phenotype-directed testing or CRC-specific multigene panel testing.11 On the other hand, clinical criteria were unlikely to have triggered testing to identify the remaining 7 (46.7% of 15) PGVs that arose from low to moderate penetrance genes for CRC: ATM (1),CDH1 (1), CHECK2 (4), and NF2 (1). Nonetheless, two of these were actionable cancer predisposition genes (CDH1 and NF2, 28.5% of 7;Table 3).11
DISCUSSION
Tumor genomic profiling is often performed to identify actionable somatic mutations in CRC, most commonly for RAS, BRAF and MMR gene mutations.6 However, it can also discover incidental germline variants, when germline DNA is being sequenced in the background as a control. In this study, we analyzed germline data from 151 advanced CRC patients undergoing tumor-normal sequencing. We incidentally found that 15 (9.9%) patients harbored a PGV in 9 genes of varying penetrance for CRC. Traditional genetic risk assessment would likely have missed 7 variants (43% of 15 PGVs, 4.6% of 151 patients) from genes with low to moderate penetrance for CRC, where 2 (28.5%) of the missed PGVs were clinically actionable mutations (in CDH1 and NF2 genes). Thus, clinicians should be aware that when a tumor-normal sequencing is performed for somatic mutation profiling for CRC, PGVs associated with hereditary cancer predisposition can be identified in genes of both high and low penetrance for CRC. Therefore, an infrastructure for clinical genetics care is needed to inform and interpret the clinical actionability of germline variants and to provide follow-up care of mutation carriers and family members.
The convergence of somatic and germline mutational profiling is not a novel concept in CRC. For example, Lynch Syndrome (LS) is the most common inherited CRC syndrome, and universal testing of CRCs for microsatellite instability is an established approach to identify patients who may harbor germline mutations or LS.8 While targeted germline mutation testing of specific MMR genes can confirm LS diagnosis, germline sequencing on a larger scale can lead to finding germline variants whose clinical significance and actionability may be challenging to interpret. In this study, we focused on data from tumor-normal genomic profiling and defined a structured approach to analyzing germline DNA data. We identified potentially pathogenic mutations in 15 patients (10%). Patients in this cohort underwent germline testing in an “unselected” fashion, i.e. simply due to their participation in tumor profiling,22 regardless of clinical criteria such as age, personal history, family history, or tumor MSI phenotype.23–26 Previous series that have similarly included large cohorts of patients with advanced cancers of various organs, had reported overall PGV detection rates of 3–17.5%.1,5,9,27 This range likely arose from the heterogeneous case mix and algorithms of variant classification in the different series. In our series, variant classification and pathogenicity determination algorithms were defined a priori to data analysis, followed standard recommendations, and were consistent with that used in the largest PGV analysis study reported to date.3,27–29 Based on the finding that patients with PGVs were significantly younger than those without, it is likely that if additional selection criteria were applied (such as age younger than 50), the yield of PGV detection would have been even higher.26
Our study highlighted the incremental detection of PGVs through germline sequencing of multiple genes. Traditional clinical genetics risk assessment and counseling focused on specific highly-penetrant genes relevant to the cancer type examined. Accumulating experience with multiplex germline gene panels that are not cancer-type specific25,26,30 have shown that pathogenic mutations in genes previously unanticipated to be associated with hereditary CRC could be found. For example, mutations have been detected in the BRCA1 gene, supporting that atypical Hereditary Breast Ovarian Cancer (HBOC) phenotype may include CRC,25,26,31 and in ATM and CHEK2 genes, suggesting that these genes may confer CRC risk.26,30,32 In our study, germline DNA sequencing on a large gene panel incidentally identified 46.7% more PGVs than if testing were directed by phenotype or by personal and family history clinical criteria. The 15 PGVs identified arose from 9 genes with varying penetrance for CRC, including highly penetrant genes such as APC, MSH2, MSH6, TP53, moderately penetrant ones such as BRCA1 and CHEK2, as well as those with low penetrance for CRC: ATM, CDH1, NF2.11 Variants in low penetrance genes for CRC are most likely to be missed through clinical criteria-based testing, since classical clinical genetic testing would likely to have detected PGVs in high penetrance genes, and use of CRC-specific multiplex germline gene panel would have likely detected those in moderate penetrance genes.24 It is important to note that among the PGVs from low-penetrant genes for CRC, 2 (28.5% of 7) of the would-have-been-missed variants were clinically actionable because they are highly penetrant genes for other cancer predisposing syndromes (CDH1 and NF2).
Analysis and interpretation of germline DNA sequencing requires a standardized process, that involves both pathogenicity calling and also clinical actionability interpretation. While the ACMG guidelines21,29 provide a useful framework, we identified 26 variants from Tiers 1–3 that required further individual curation to determine their pathogenicity (Figure 2). Current efforts for filtering based on variant frequency and on in silico determination of a variant’s effect on the protein product were supplemented by careful search of the existing literature and by construction of lollipop diagrams (Figure 3). These bioinformatics algorithms established in the research setting will need to be developed into standardized clinical pipelines if tumor-normal sequencing were to be performed for clinical care. Increased sharing of clinically annotated germline variant data over time will assist in future efforts in pathogenicity calling. Additionally, a partnership with clinical genetics expertise is critical for the interpretation for clinical actionability and for translating germline findings to clinical management decisions.2 Currently, the return of incidental germline findings at our institution involves a dedicated committee including genetic counselors, clinical genetics physicians, molecular pathologists, and genomic profiling team who reviews the pathogenicity call of each variant. The treating clinician is contacted, and referral to clinical genetic counseling is recommended. The patient is assessed at our Gastrointestinal Familial High-risk clinic, where personalized clinical genetics care recommendations are made.9 Indeed, practice guidelines for surveillance and preventive strategies are well established for PGVs in highly penetrant genes,33,34 but are less clear for those in moderate- or low-penetrance genes.11 The advanced stage at cancer diagnosis, the limited expected life expectancy, the age of the patient, family cancer history, and the presence or absence of other at-risk relatives are additional factors that are considered in order to provide personalized clinical cancer genetics care recommendations.34 Therefore, our study illustrates key components of a precision medicine program, specifically those that would be required for the management of germline DNA sequencing data: genomic sequencing, bioinformatics analysis of variants and pathogenicity calling, return of incidental germline results, clinical genetic testing and follow-up care (Figure 1). While these infrastructures have been initiated in the research setting, they need to become programmatically supported in the clinical setting. Future research in cancer care delivery, implementation, as well as clinical and psychosocial impact of genomic profiling is needed.
Our study has several limitations. First, we analyzed germline sequencing data from a relatively small cohort of patients with metastatic CRC who were enrolled to a precision medicine protocol where the main goal was to examine somatic mutations for personalized treatment options as previously described.9,12 Therefore, the reported PGV frequency cannot be generalized to all patients with CRC. More specifically, we have analyzed direct germline sequencing data acquired through a tumor-normal sequencing approach and thus our findings are not generalizable to personalized medicine approaches where tumor-only sequencing is performed. Currently, tumor-only sequencing for well-defined activating mutations in a limited panel of oncogenes is more commonly available, because it can be done without the need for a matched germline sample, at lower cost and faster turnaround.7 While the probability of finding a mutation that confers germline cancer susceptibility with limited tumor-only sequencing is low, it is expected to increase as larger numbers of genes are sequenced more comprehensively in the future. Indeed, large panels, whole exome, or genome-scale sequencing require tumor-normal analyses, to help distinguish between somatic versus germline origins for mutations and to avoid misinterpretation of results.1,2,5,7 Secondly, we were limited by the retrospective design of our study. Only 3 of the patients identified to have a PGV through our analysis had undergone confirmatory clinical germline mutation testing in the CLIA-environment. As previously reported, common reasons for not undergoing clinical genetics testing have included: failure to refer to genetics, failure to meet clinical testing criteria for insurance coverage, patient refusal, and appointment no-show.9 While guidelines for germline variant tiering and pathogenicity calling exist, the specific bioinformatics analysis algorithms can differ among testing agencies and can change over time based on available variant databases and literature.3,28 Therefore, PGVs identified in the research setting should undergo confirmatory testing in the CLIA-environment, before any clinical actions are instituted. Taken together, these findings highlight a need for strong partnership and improved transition of care between precision medicine research efforts and clinical genetics care (Figure 1). Finally, although our calls of pathogenicity are based on the recommendations of the ACMG and standardized algorithms utilizing data from available literature and variant databases, conducting specific validation studies including segregation analyses and functional analyses27 was beyond the scope of this study.
CONCLUSION
We present herein incidental findings from germline DNA sequencing that was performed as a part of a tumor-normal genomic profiling panel in patients with advanced CRC. A small but important fraction (9.9%) of the patients had PGVs in genes associated with hereditary predisposition to CRC or to other cancers. Nearly half (4.6%) of these would have been missed by clinical criteria-based genetic testing, and 28.6% of the missed mutations were clinically actionable. Tumor profiling with concurrent germline analysis can lead to discovery of PGVs in both expected and previously unanticipated genes. Precision medicine should integrate clinical cancer genetics to inform and interpret the actionability of germline variants and to provide follow-up care to mutation carriers.
Supplementary Material
ACKNOWLEDGMENTS
The authors are grateful to Amanda Cuddy, MPH and Sarah Bannon M.S., C.G.C., for providing organization and clinical genetics care through the University of Texas MD Anderson High Risk Familial GI Cancer Clinic.
Funding/Support: This study was supported in part by The University of Texas MD Anderson Cancer Center Clinical Innovator Award (to YNYou), The Cancer Prevention Education Award (to K Chang, R25T CA057730), The Cancer Prevention and Research Institute of Texas (RP1100584), the Sheikh Khalifa Bin Zayed Al Nahyan Institute for Personalized Cancer Therapy, and the MD Anderson Cancer Center Support Grant (P30 CA016672).
Footnotes
Financial Disclosure: The authors report no proprietary or commercial interest in any product mentioned or concept discussed in this article.
Presented as a podium presentation at the American Society of Colorectal Surgeons Annual Meeting, Nashville, TN, May 19–23, 2018
REFERENCES
- 1.Jones S, Anagnostou V, Lytle K, et al. Personalized genomic analyses for cancer mutation discovery and interpretation. Sci Transl Med. 2015;7:283ra53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bombard Y, Robson M, Offit K. Revealing the incidentalome when targeting the tumor genome. JAMA. 2013;310:795–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kalia SS, Adelman K, Bale SJ, et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): a policy statement of the American College of Medical Genetics and Genomics. Genet Med. 2017;19:249–255. [DOI] [PubMed] [Google Scholar]
- 4.Robson ME, Bradbury AR, Arun B, et al. American Society of Clinical Oncology Policy Statement Update: genetic and genomic testing for cancer susceptibility. J Clin Oncol. 2015;33:3660–3667. [DOI] [PubMed] [Google Scholar]
- 5.Mandelker D, Zhang L, Kemel Y, et al. Mutation detection in patients with advanced cancer by universal sequencing of cancer-related genes in tumor and normal DNA vs guideline-based germline testing. JAMA. 2017;318:825–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sepulveda AR, Hamilton SR, Allegra CJ, et al. Molecular biomarkers for the evaluation of colorectal cancer: guideline from the American Society for Clinical Pathology, College of American Pathologists, Association for Molecular Pathology, and the American Society of Clinical Oncology. J Clin Oncol. 2017;35:1453–1486. [DOI] [PubMed] [Google Scholar]
- 7.Borad MJ, LoRusso PM. Twenty-first century precision medicine in oncology: genomic profiling in patients with cancer. Mayo Clin Proc. 2017;92:1583–1591. [DOI] [PubMed] [Google Scholar]
- 8.Jasperson KW, Tuohy TM, Neklason DW, Burt RW. Hereditary and familial colon cancer. Gastroenterology. 2010;138:2044–2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Meric-Bernstam F, Brusco L, Daniels M, et al. Incidental germline variants in 1000 advanced cancers on a prospective somatic genomic profiling protocol. Ann Oncol. 2016;27:795–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brusco LL, Wathoo C, Mills Shaw KR, et al. Physician interpretation of genomic test results and treatment selection. Cancer. 2018;124:966–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.The National Comprehensive Cancer Network Clinical Practice Guidelines in Oncology. Genetic/familial high-risk assessment: colorectal. www.nccn.org. Accessed March 31, 2018. [Google Scholar]
- 12.Chen K, Meric-Bernstam F, Zhao H, et al. Clinical actionability enhanced through deep targeted sequencing of solid tumors. Clin Chem. 2015;61:544–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Auton A, Brooks LD, Durbin RM, et al. ; 1000 Genomes Project Consortium. A global reference for human genetic variation. Nature. 2015;526:68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Borras E, San Lucas FA, Chang K, et al. Genomic landscape of colorectal mucosa and adenomas. Cancer Prev Res (Phila). 2016;9:417–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.González-Pérez A, López-Bigas N. Improving the assessment of the outcome of nonsynonymous SNVs with a consensus deleteriousness score, Condel. Am J Hum Genet. 2011;88:440–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ramensky V, Bork P, Sunyaev S. Human non-synonymous SNPs: server and survey. Nucleic Acids Res. 2002;30:3894–3900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4:1073–1081. [DOI] [PubMed] [Google Scholar]
- 18.Harrison SM, Riggs ER, Maglott DR, et al. Using ClinVar as a resource to support variant interpretation. Curr Protoc Hum Genet. 2016;89:8.16.1–18.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Szabo C, Masiello A, Ryan JF, Brody LC. The breast cancer information core: database design, structure, and scope. Hum Mutat. 2000;16:123–131. [DOI] [PubMed] [Google Scholar]
- 20.Thompson BA, Spurdle AB, Plazzer JP, et al. Application of a 5-tiered scheme for standardized classification of 2,360 unique mismatch repair gene variants in the InSiGHT locus-specific database. Nat Genet. 2014;46:107–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Richards S, Aziz N, Bale S, et al. ; ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Meric-Bernstam F, Brusco L, Shaw K, et al. Feasibility of large-scale genomic testing to facilitate enrollment onto genomically matched clinical trials. J Clin Oncol. 2015;33:2753–2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pritchard CC, Mateo J, Walsh MF, et al. Inherited DNA-repair gene mutations in men with metastatic prostate cancer. N Engl J Med. 2016;375:443–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mork ME, You YN, Ying J, et al. High prevalence of hereditary cancer syndromes in adolescents and young adults with colorectal cancer. J Clin Oncol. 2015;33:3544–3549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yurgelun MB, Allen B, Kaldate RR, et al. Identification of a Variety of Mutations in Cancer Predisposition Genes in Patients With Suspected Lynch Syndrome. Gastroenterology. 2015;149:604–613 e620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pearlman R, Frankel WL, Swanson B, et al. ; Ohio Colorectal Cancer Prevention Initiative Study Group. Prevalence and spectrum of germline cancer susceptibility gene mutations among patients with early-onset colorectal cancer. JAMA Oncol. 2017;3:464–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang KL, Mashl RJ, Wu Y, et al. Pathogenic Germline Variants in 10,389 Adult Cancers. Cell. 2018;173:355–370 e314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Green RC, Berg JS, Grody WW, et al. ; American College of Medical Genetics and Genomics. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet Med. 2013;15:565–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maxwell KN, Hart SN, Vijai J, et al. Evaluation of ACMG-Guideline-based variant classification of cancer susceptibility and non-cancer-associated genes in families affected by breast cancer. Am J Hum Genet. 2016;98:801–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rohlin A, Rambech E, Kvist A, et al. Expanding the genotype-phenotype spectrum in hereditary colorectal cancer by gene panel testing. Fam Cancer. 2017;16:195–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Susswein LR, Marshall ML, Nusbaum R, et al. Pathogenic and likely pathogenic variant prevalence among the first 10,000 patients referred for next-generation cancer panel testing. Genet Med. 2016;18:823–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shirts BH, Casadei S, Jacobson AL, et al. Improving performance of multigene panels for genomic analysis of cancer predisposition. Genet Med. 2016;18:974–981. [DOI] [PubMed] [Google Scholar]
- 33.Syngal S, Brand RE, Church JM, Giardiello FM, Hampel HL, Burt RW; American College of Gastroenterology. ACG clinical guideline: genetic testing and management of hereditary gastrointestinal cancer syndromes. Am J Gastroenterol. 2015;110:223–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tung N, Domchek SM, Stadler Z, et al. Counselling framework for moderate-penetrance cancer-susceptibility mutations. Nat Rev Clin Oncol. 2016;13:581–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.