

Abstract
The central role of Ca2+ signaling in the development of functional immunity and tolerance is well established. These signals are initiated by antigen binding to cognate receptors on lymphocytes that trigger store operated Ca2+ entry (SOCE). The underlying mechanism of SOCE in lymphocytes involves TCR and BCR mediated activation of Stromal Interaction Molecule 1 and 2 (STIM1/2) molecules embedded in the ER membrane leading to their activation of Orai channels in the plasma membrane. STIM/Orai dependent Ca2+ signals guide key antigen induced lymphocyte development and function principally through direct regulation of Ca2+ dependent transcription factors. The role of Ca2+ signaling in NFAT activation and signaling is well known and has been studied extensively, but a wide appreciation and mechanistic understanding of how Ca2+ signals also shape the activation and specificity of NF-κB dependent gene expression has lagged. Here we discuss and interpret what is known about Ca2+ dependent mechanisms of NF-kB activation, including what is known and the gaps in our understanding of how these signals control lymphocyte development and function.
Graphical abstract

1. A central role for STIM/Orai mediated Ca2+ entry in lymphocytes
The central role for calcium (Ca2+) signaling in lymphocytes has been appreciated for 30 years. However, our understanding of the regulation and specific mechanisms of Ca2+ action have advanced more rapidly since the identification of the Stromal Interaction Molecule 1 and 2 proteins (STIM1/2) and Orai channels nearly a decade ago. STIM and Orai are expressed in many if not most cell types and although the mechanisms and biological consequences of this store operated Ca2+ entry (SOCE) pathway vary widely, in virtually all instances STIMs play a pivotal role. Triggering STIM1 and STIM2 begins with Ca2+ dissociation from their EF hand domains within the endoplasmic reticulum (ER) lumen. Stimulus-induced decreases in ER Ca2+ then promote its dissociation from STIM trigger its conformational activation. The resulting physical interaction with Orai channels initiate their activation and Ca2+ entry across the plasma membrane. In lymphocytes STIM1 plays a dominant role in antigen-receptor (TCR and BCR)-induced Orai1 activation; while STIM2 controls cytoplasmic Ca2+ levels under resting conditions [1–4].
Through the analysis of patient samples and the development of new genetic models, the mechanisms controlling SOCE and its central role in lymphocyte development and activation have been firmly established. Humans with spontaneous inactivating mutations in STIM1 or ORAI1 and mice in which both Stim1 and Stim2 are deleted develop autoimmune diseases and a spectrum of immune deficiencies [5, 6]. These phenotypes reflect a cell-intrinsic requirement for STIM/Orai-mediated Ca2+ entry in the development of regulatory T cells in the thymus and in the activation and function of peripheral T cells. Both are essential for the maintenance of immune tolerance and this crucial layer of immune regulation is severely dysregulated in the absence of functional STIM or Orai proteins.
Prior to the identification of STIM and Orai, a conceptual framework for understanding how Ca2+ controls lymphocyte fates had been established. These early studies revealed that variations in the affinity and avidity of antigen binding to the T cell receptor (TCR) and B cell receptor (BCR) are encoded as quantitatively discrete patterns of intracellular Ca2+ signaling and that distinct dynamics drive alternative lymphocyte fates and functions [7–9]. While our understanding of how dynamic Ca2+ signals are generated is limited, new details have emerged recently about the complex mechanisms that decode and translate dynamic and quantitatively distinct changes in Ca2+ concentration into unique transcriptionally driven fate-specific programs of gene expression.
Two key pro-inflammatory transcription factors central to this function are Nuclear Factor of Activated T cells (NFAT) and Nuclear Factor Kappa B (NF-κB). At rest, both are localized primarily in the cytoplasm in transcriptionally inactive forms. NFAT is activated by Ca2+/calcineurin (CaN)-dependent de-phosphorylation that exposes a nuclear localization domain to promote its entry into the nucleus, DNA binding, and transcriptional activation. CaN-dependent NFAT phosphorylation is the only Ca2+-regulated checkpoint in NFAT activation and it has long-served as a paradigm for Ca2+-regulated transcriptional control [10–12]. By contrast, recognition of the important role, and a comprehensive understanding of the mechanisms and consequences of Ca2+ control of NF-κB activation have lagged. Indeed, this control appears to be more complex as studies in our laboratory and by others have revealed multiple Ca2+-regulated checkpoints in its activation and specificity. Therefore, the primary objective of this review is to highlight the known and emerging mechanisms by which STIM/Orai-mediated Ca2+ signals control NF-κB activity in lymphocytes and to discuss the immunological consequences of this level of transcriptional control.
2. Quantitative features of Ca2+ signals control lymphocyte fates and functions
The initial recognition of the central role of Ca2+ in T cell development and differentiation nearly 30 years ago hinged on the development of fluorescent Ca2+ indicators [13]. These probes helped reveal that variations in the affinity and/or avidity of antigen binding are encoded as quantitatively and qualitatively distinct Ca2+ signals that drive fate decisions of multipotent thymic T lymphocytes. In general terms, higher affinity/avidity antigen binding leads to higher input/amplitude Ca2+ signals than low affinity/avidity antigen binding [14–19]. More recent studies of mice with defects in the expression of Stim1 and Stim2 have established how such fate specific TCR-induced Ca2+ signals are generated [20–22]. Specifically, the loss of Ca2+ entry in Stim1Stim2 double knock-out (STIM DKO) mice leads to defects in antigen-induced death of autoreactive cells by negative selection, in the development of natural regulatory T lymphocytes (nTregs) that play a key role preventing autoimmunity, and also in regulatory B cell function [20–23].
Interestingly, the development of T lymphocytes that provide protective immunity by positive selection is not measurably altered in STIM DKO mice. This suggests that “weaker” signals, such as low amplitude/input, shorter duration, or sporadic spikes in Ca2+ are sufficient to drive this fate choice [14, 20, 21, 24]. These weaker signals could be generated in the absence of Ca2+ entry via STIM/Orai by TCR-mediated Ca2+ release from intracellular stores, which is intact in STIM and Orai defective lymphocytes [21]. Indeed, in situ 2-photon measurements of Ca2+ dynamics in lymphocytes exposed to low affinity, positively selecting antigen in the intact thymic cortex, and studies of Ca2+ signals induced by agonist and altered peptides with a range of affinities for a transgenic TCR support this mechanism [14, 15, 24, 25]. Simply stated, low affinity TCR agonists promote positive selection in part by initiating brief low amplitude and/or sporadic Ca2+ spikes; whereas, high affinity interactions that induce negative selection or the development of nTregs produce higher input and/or longer duration signals.
Together, these studies raise the intriguing possibility that fate specification by Ca2+ is not simply a function of steady state changes in its mean amplitude or duration, but that more complex waveforms or more efficient dynamics, of similar or distinct input, might drive alternative transcriptionally driven cell fates [26–29]. Indeed, we demonstrated that the mean amplitude of Ca2+ signals varies with the avidity of TCR engagement and costimulatory (CD28) receptor signaling, and that distinct patterns of signaling are triggered by stimuli that induce distinct or alternative cell fate decisions [18]. Altogether, these studies support the concept that antigen-induced fates are encoded in both the frequency and amplitude-modulated patterns of Ca2+ signaling rather than variations in steady-state Ca2+ amplitude.
3. Decoding Calcium Dynamics in Lymphocytes
Dynamic Ca2+ signals including low frequency spikes, persistent oscillations, or sustained steady-state elevations can only have physiological relevance if cellular targets exist to decode these dynamics into distinct functional outcomes. Among the targets capable of decoding these signals in lymphocytes are Ca2+-regulated transcription factors including NFAT, JNK, and NF-κB (see Figure 1, Graphical Abstract). Most importantly, each of these is optimally tuned to a different dynamic. For example, efficient NFAT activation requires a sustained increase in Ca2+ concentration, whereas NF-κB and JNK can be selectively activated by one or a few transient cytoplasmic Ca2+ “spikes” [30–32]. Remarkably, a single Ca2+ spike is sufficient to initiate IκBα degradation and the release of heterodimers containing the canonical NF-κB proteins p65 and c-Rel, to facilitate their nuclear translocation and transcriptional activation [30].
Figure 1. (Graphical Abstract): Antigen receptor induced Ca2+ dynamics tune the patterns. of transcriptional activation.
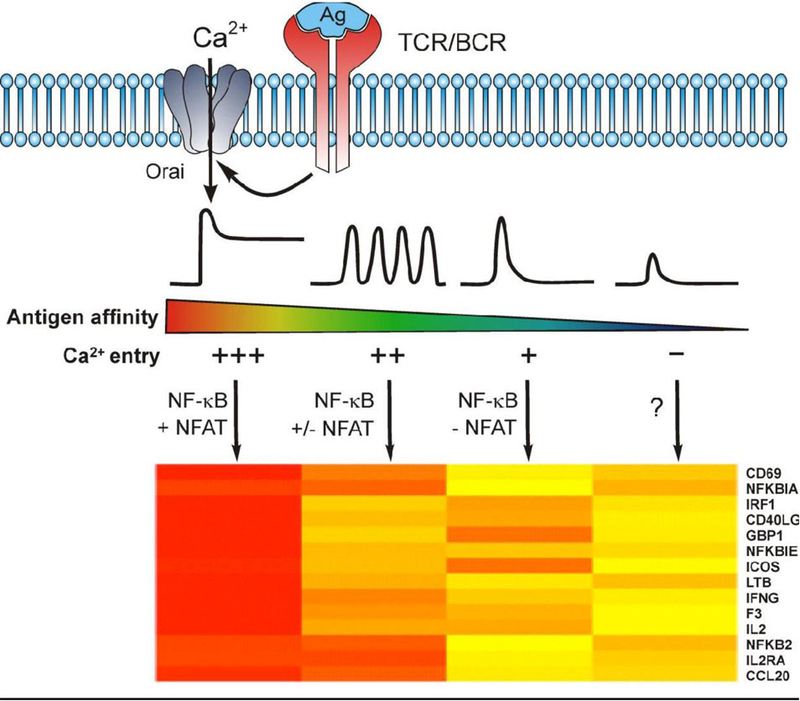
This schematic proposes how variations in antigen receptor induced Ca2+ dynamics elicit distinct patterns of gene expression depicted by a heatmap of differential gene expression. Work over the past 30 years has established that the strength of antigen receptor stimulation is encoded as quantitatively distinct patterns of Ca2+ signaling, and that these can each initiate a distinct transcriptionally driven fate of lymphocytes by activating Ca2+ dependent transcription factors. The key pro-inflammatory transcription factors NF-κB and NFAT, which exhibit differences in Ca2+ sensitive activation, decode differences in the strength of antigen stimulation into distinct patterns of transcriptional activation. High affinity antigen binding causes sustained STIM/Orai-dependent Ca2+ entry and these signals activate both NF-κB and NFAT. Intermediate antigen affinity/avidity interactions cause distinct patterns of signaling, possibly Ca2+ oscillations, that preferentially activate NF-κB and depending on the frequency can potentially activate NFAT. Low affinity/avidity interactions that cause a significant but transient elevation in cytoplasmic Ca2+ leading to the selective activation of NF-κB signaling and NF-κB dependent target gene expression. Finally, in the absence of Ca2+ entry, low amplitude, transient elevations in cytoplasmic Ca2+ may be insufficient for the activation of either NF-κB or NFAT.
This differential Ca2+-dependent tuning of transcription factors has important biological implications for T cells developing in the thymus and in the fate choices and functions of peripheral T and B lymphocytes. Moreover, different lymphocyte subsets may decode signals of similar phenotype differently. For example, engagement of the B cell receptor (BCR) on naïve B cells triggers sustained high amplitude Ca2+ signals that activate NFAT, NF-κB, JNK, and ERK; whereas, engagement of the BCR on tolerized B cells produces low amplitude Ca2+ “oscillations” that activate only NFAT and ERK [33]. These studies highlight several important mechanistic points. First, Ca2+ dynamics are subject to stimulus and developmental or differentiation stage-specific control. Second, variations in stimulus strength can induce distinct patterns of Ca2+ signaling at any given differentiation or developmental state. Lastly, physiologically relevant differences in Ca2+ dynamics can be decoded by Ca2+-sensitive transcription factors such as NFAT and NF-κB to direct or dictate transcriptionally controlled fate choices of lymphocytes.
Remarkably, the nature or phenotype of physiologically relevant Ca2+ signals remain largely unexplored. However, with the recent advent of genetically encoded Ca2+ indicators and multi-photon imaging, it is technologically feasible to interrogate these signals within the native milieu of intact lymphoid organs. Going forward, these tools can be exploited to visualize Ca2+ signals within lymphoid organs throughout the full time-course of lymphocyte development and differentiation. Studies using organic Ca2+ indicators have revealed a wide dynamic range of Ca2+ signals can be generated in vitro [14, 24, 34], we do not know the phenotype of these signals over a relevant time course of differentiation in vivo. More difficult studies are also required to understand how different dynamics are generated. For example, spikes may be generated by sporadic release and reuptake of Ca2+ from intracellular stores without the need for ion flow across the plasma membrane. Alternatively, fluctuations in ER release and uptake that activate and deactivate STIM and Orai may underlie larger amplitude, longer duration or more sustained trains of Ca2+ spikes [31, 35–38].
Despite our limited understanding of the mechanisms by which Ca2+ dynamics are generated in vivo, in vitro studies have established how quantitatively distinct Ca2+ signals can drive different transcriptional outcomes. Elegant studies that utilized a “Ca2+ clamp” perfusion setup to generate Ca2+ signals of distinct and defined steady state amplitudes or spike frequencies within lymphocytes revealed that NFAT and NF-κB are tuned to distinct Ca2+ dynamics. For example, high amplitude steady-state signals or high frequency spikes (> 2mHz) are required to activate NFAT and Oct/OAP (and also activate NF-κB); whereas, low frequency Ca2+ spikes (< 2mHz) only activate NF-κB. A further insight of this work was that Ca2+ oscillations reduce the effective Ca2+ threshold for activating these transcription factors, allowing for more efficient signaling at low levels of stimulation [35]. Indeed, this mechanism was validated in concurrent studies [39] in which photo-activation of intracellular “caged” InsP3 was used to generate Ca2+ spikes of different frequencies in lymphocytes. Together, these independent approaches helped to advance a new paradigm by which Ca2+ alone can drive distinct transcriptionally driven fates of lymphocytes.
4. Multiple Ca2+ Regulated checkpoints control NF-κB activity and specificity
Although the mechanism of Ca2+ dependent NFAT activation was established over 25 years ago [40], our understanding of how Ca2+ regulates NF-κB activity and function, including the basis for its distinct Ca2+ sensitivity, continues to evolve. In the case of canonical NF-κB signaling, inhibitory kappa-B (IκB) proteins sequester hetero-/homo-dimers of p65, c-Rel and p50 proteins in the cytoplasm [41, 42]. Antigen receptor coupled phospholipases (PLCγ−1 and PLCγ−2 in T cells and B cells respectively) hydrolyze plasma membrane phosphatidylinositol 4,5-bisphosphate (PIP2) to form diacylglycerol (DAG) and Inositol 1,4,5 trisphosphate (IP3). IP3 activates IP3 receptor/channels in the ER membrane and the resulting loss of ER Ca2+ activates STIM1 and STIM2 to initiate Orai-mediated Ca2+ entry. DAG, in some instances in conjunction with Ca2+, activates one of several protein kinase C (PKC) isoforms that phosphorylate CARMA1, promoting its association with Bcl10 and MALT1 to form the CBM complex [43, 44]. The CBM complex in turn activates the IκB kinase (IKK) complex comprised of IKKα, IKKβ, and NEMO [45]. IKKβ mediated phosphorylation of IκB bound to p65/p50 and c-Rel/p50 targets it for proteasomal degradation, freeing p65/p50 and c-Rel/p50 dimers, which migrate to the nucleus where they can initiate gene expression. This framework of NF-κB signaling in T and B cells is very well established, yet the mechanisms by which Ca2+ controls NF-κB activity are more complex and not fully resolved. The extent of known mechanisms (Figure 2) by which Ca2+ regulates proximal and distal checkpoints in antigen receptor induced NF-κB activation are described below.
Figure 2: Ca2+ regulated checkpoints in canonical NF-κB signaling in lymphocytes.

A schematic representation of known mechanisms by which Ca2+ regulates the activation of NF-κB dependent gene expression. In resting lymphocytes, homo- and heterodimers composed of NF-κB p50, p65, and c-Rel proteins are sequestered in the cytoplasm by IκB proteins including IκBα. BCR or TCR engagement activates a series of protein tyrosine kinases (PTKs) that subsequently activate PLCγ isoforms, cleaving PtdIns(4,5)P2 to generate InsP3 (IP3) and DAG. IP3 binds to IP3 receptors (IP3R) channels allowing release of Ca2+ from the endoplasmic reticulum (ER). The resulting decrease in ER [Ca2+] causes oligomerization of STIM1 proteins and activation of plasma membrane Orai1 channels facilitating extracellular Ca2+ entry and a significant or sustained elevation in cytoplasmic Ca2+. In B cells, Ca2+ and DAG activation of PKCβ and in T cells, DAG activation of PKCθ controls assembly and activity of the CARMA-BCL10-MALT1 (CBM) complex, which is required for antigen receptor induced activation of the IKK complex (NEMO-IKKα-IKKβ). Furthermore, PKCα, the CaM dependent phosphatase Calcineurin (CaN), and calmodulin (CaM)-dependent kinase II (CaMKII) each also regulate CBM complex formation by controlling the phosphorylation levels of these proteins. Ca2+ also plays a negative regulatory role via CaM activation that can directly bind to BCL10 to disrupt the CBM complex, activate CaMKII that phosphorylates BCL10 on S138 or binds to NF-κB to block nuclear translocation (all depicted by red lines). NEMO recruitment to the CBM complex facilitates full activation of the IKK complex, which phosphorylates IκB proteins, marking them for ubiquitylation and proteasomal degradation. Both p65 and c-Rel can undergo Ca2+ dependent (e.g. Ser536 phosphorylation, CaM binding) and independent post-translational modifications that regulate their nuclear localization, transactivation, and target gene binding potential. Thus, Ca2+ dependent mechanisms that regulate the IκB protein degradation and expression and those that modify the post-translational landscape of NF-κB proteins cooperate to regulate gene expression.
4.1. Calcineurin
CaN is a Ca2+ and calmodulin (see below) regulated serine/threonine phosphatase whose activity controls several steps of TCR- and BCR-mediated lymphocyte activation [46, 47]. CaN is a target of inhibition by a cyclophilin-cyclosporine A (CsA) complex and a FKBP12-FK506 complex [46, 47] and both CsA and FK506 are routinely used to assess the role of CaN in cell signaling. Indeed, initial evidence for Ca2+-dependent regulation of NF-κB in the early 1990s was inferred from the inhibitory effect observed for CsA on NFAT, AP-1, NF-κB, and SV40 binding to regulatory sequences in an IL-2 enhancer construct [48] and on NF-κB binding to the HIV enhancer [49]. The similar inhibition of NF-κB activation by two distinct CaN inhibitors, provides additional but indirect evidence that Ca2+ regulates NF-κB activity [50].
The impact of CaN inhibitors on NF-κB binding to promotor elements prompted more detailed mechanistic studies to understand how Ca2+ and CaN control NF-κB activity. Interestingly, expression of a constitutively active catalytic subunit of CaN or calcineurin A (CnA) in T cells, in the absence of any other signals, activates NF-κB upstream of the IKKβ but not IKKα of the IKK complex [51–53]. Moreover, in T lymphocytes with defective Ca2+ entry, constitutively active CnA rescues the block in NF-κB activation [51, 54] indicative of a role for Ca2+/CaN in efficient CBM complex formation [55]. Thus, PKC-mediated phosphorylation of CARMA1 regulates its membrane association while Ca2+/CaN-dependent de-phosphorylation of MALT1-associated BCL10 promotes its interaction with CARMA1 leading to assembly and activation of the CBM complex [55].
4.2. Calmodulin
Calmodulin (CaM) is another ubiquitous Ca2+ binding protein that regulates a wide range of processes in lymphocytes and literally every other cell type. The exquisite Ca2+ sensitivity and specificity of CaM is imparted by four Ca2+-binding EF hand domains, N- and C-terminal E and F helixes respectively, and a centrally positioned Ca2+-coordinating loop [56]. Following Ca2+ dependent activation, CaM regulates many kinases and phosphatases including CaN. Direct binding of CaM to BCL10 has been established (Edin et al, 2010) and its association with other members of the CBM or IKK complex has been inferred based on the effects of CaM inhibitors [57]. A role for CaM-dependent kinases in the formation and activity of the CBM complex has been described (see below). Finally, CaM directly binds to both p65 and c-Rel in activated T cells [58] and while this binding inhibits the nuclear translocation of c-Rel, p65 translocation, DNA binding and transcriptional activity remain intact. Thus, Ca2+ can promote transcriptionally driven fates of lymphocytes by tuning distinct steps that control the activation and nuclear localization of distinct NF-κB proteins. Interestingly, while CaM inhibitors block CaN-dependent NF-κB activation, these drugs also reduce CaN-independent steps of activation suggesting additional, as yet unknown, Ca2+/CaM-dependent mechanisms control NF-κB activity [57].
4.3. Calmodulin kinase II
Calmodulin kinases (CaMK) are Ser/Thr kinases whose activity is regulated by Ca2+ and CaM. CaMKII has separate CaM binding, catalytic, and auto-inhibitory domains and at basal Ca2+ levels, the auto-inhibitory domain blocks substrate binding to the catalytic domain [59, 60]. Elevations in cytoplasmic Ca2+ that promote CaM binding to CaMKII disrupt interactions between the auto-inhibitory and catalytic domains to trigger kinase activation [60]. Importantly, autophosphorylation of CaMKII leads to sustained CaM-independent activity that persists beyond the duration of an activating calcium signal [61].
CaMKII has been implicated in proximal steps of NF-κB signaling where it phosphorylates the CBM components CARMA1 and BCL10; although, evidence for a physiologically role for CaMKII in IKK activation largely relies upon pharmacological studies. For example, the CaMKII selective inhibitor KN93 attenuates antigen receptor mediated IκBα phosphorylation and degradation, and NF-κB transcriptional activation [62]. Conversely, a constitutively activate, CaM-independent CaMKII mutant rescues IκBα degradation in T lymphocytes [62]. CaMKII-dependent control of IKK activation may reflect its ability to phosphorylate CARMA1 on Ser109 to promote interactions between CARMA1 and BCL10 [63], and to phosphorylate several residues on BCL10 that modulate CBM complex activity [64]. Moreover, its phosphorylation of BCL10 on Thr91 positively regulates BCL10 interactions with CARMA1 and specifically counteracts the inhibitory effects of CaM binding to BCL10 [64]. BCL10 Thr91 phosphorylation also promotes Lys63-linked poly-ubiquitination of BCL10 to drive subsequent IKK and NF-κB activation [64]. Furthermore, CaMKII-mediated phosphorylation of BCL10 on Ser48 also supports CBM complex formation and IKK activation by an as yet unknown mechanism [64]. Finally, while CaMKII mediated phosphorylation typically promotes TCR-induced NF-κB activity, some phospho-targets of CaMKII negatively regulate NF-κB activation. For example, CaMKII-dependent phosphorylation of BCL10 at Ser138 disrupts the BCL10-MALT1 interaction leading to dissociation of the CBM complex and downregulation of IKK activity [65].
Resolving the complex mechanisms by which CaMKII, CaM, and other kinases and phosphatases control IKK activity is an important prerequisite to understanding the interplay between these Ca2+ regulated factors that fine tune NF-κB activity. Indeed, computational modeling in neurons suggests that CaMKII-dependent activation dominates over CaN-mediated inhibition as the frequency of Ca2+ spike increases, but this remains to be tested in lymphocytes [66]. Especially intriguing is the possibility that CaMKII can sum multiple subthreshold Ca2+ spikes to initiate NF-κB activation, and this may be a mechanism by which low affinity antigens selectively activate NF-κB over NFAT. Finally, as CaMKII splice variants are tuned to different Ca2+ spike frequencies [67], expression of distinct isoforms in different lymphocyte populations may dictate transcriptional specificity. Consequently, it remains critical to define the frequency to which specific CaMKII isoforms expressed in lymphocyte subpopulations are tuned, to identify the precise nature of the Ca2+ signals in these populations, and then to determine if CaMKII isoforms couple specific Ca2+ dynamics to distinct transcriptionally driven fates in each lymphocyte population.
4.4. Protein Kinase C
The requisite role for Protein Kinase C (PKC) in NF-κB activation in lymphocytes was first identified over 20 years ago [68] and numerous studies have since dissected how specific isoforms control cell-specific activation of NF-κB [69]. The PKC family includes both Ca2+-dependent and -independent isoforms. The conventional PKC isoforms (α1, β1, β2, and γ) are regulated by diacylglycerol (DAG), Ca2+, and phospholipids, the novel PKC isoforms (δ, ε, η, and θ) regulated by DAG and phospholipids, and the atypical PKC isoforms (ζ and λ) are DAG and Ca2+ insensitive [70]. The PKC isoforms expressed in T cells include PKCθ, PKCα, and PKCβ while in B cells PKCβ, PKCλ, and PKCζ have important functions [69].
Both PKCα and PKCβ have been implicated in Ca2+-dependent NF-κB activation in lymphocytes. In T cells, conventional PKCα and PKCβ1 are activated by TCR engagement, and the PKCα and PKCβ1 selective inhibitor Gö6976 diminished NF-κB activity [71]. PKCα appears to play an early role in T cell activation as it and PKCθ translocate to the plasma membrane within minutes of activation; whereas, PKCβ1 does not localize to the plasma membrane over this timeframe [72]. Interestingly, in vitro kinase assays suggest that even a catalytically active version of PKCα requires a Ca2+ signal to induce IKK activity and NF-κB-dependent transcription; although, the implications of this control by Ca2+ requires further investigation [71]. PKCs may cooperate to control NF-κB activity as PKCθ initiates CBM complex formation and activation, whereas PKCα appears to modulate IKK activity with delayed kinetics [71]. Differences in the Ca2+ dependent regulation of NF-κB activity in distinct lymphocyte populations also reflect differences in antigen receptor coupling to IKK. For example, Ca2+ independent PKCθ regulates proximal steps of TCR signaling, whereas Ca2+ sensitive PKCβ is the principle component of the BCR “signalosome” required for recruitment and activation of the IKK complex [73]. Indeed, in B cells, Ca2+ entry rapidly regulates the extent of BCR-induced PKCβ membrane localization and this controls the efficiency of CBM and IKK complex activation [74]. The predominant role of PKCβ in BCR-induced NF-κB activation implies that Ca2+ is also involved in this proximal step; however, to date this has not been directly verified.
4.5. Post-translational modifications control NF-κB transcriptional activity and specificity
While TCR- and BCR-induced activation of the CBM and IKK complexes initiate canonical NF-κB activation by inducing the degradation of IκB proteins, additional crucial mechanisms distal to the release of p50/p65 and p50/c-Rel tune their transcriptional activation and specificity. A multitude of studies have established that stimulus specific phosphorylation of serine and threonine residues in p65 and c-Rel regulate their activity. Phosphorylation critically regulates the kinetics, extent of nuclear localization [54, 75, 76], nuclear DNA interactions [77, 78], transcriptional activation [79–82] and the specificity of gene expression [83]. Additional modifications including acetylation, methylation and O-GlcNAcylation also control transcriptional competency, promoter accessibility, and specificity [78, 79, 84–88]. Importantly however, we know relatively little about the role of Ca2+ in regulating these essential post-translational events required for p65 and c-Rel activity.
Of the numerous p65 phospho-acceptor sites identified, Ser536 is the only residue currently known to undergo Ca2+-dependent modification. Initial studies of interleukin receptor associated kinase (IRAK1) signaling found that CaMKII-mediated phosphorylation of p65 S536 regulates its transactivation potential in lymphoblastoid lines [89]. CaMKIV mediated phosphorylation of p65 in its transactivation domain has also been implicated in its activation and recruitment of the transcriptional co-activator cAMP-response protein binding protein (CREB), but the target residue of this phosphorylation has not been identified [90]. Finally, our recent studies directly established that TCR-induced STIM/Orai mediated Ca2+ entry regulates p65 Ser536 phosphorylation to control its nuclear retention, promoter binding, and transcriptional activity [54]. Furthermore, we established that PKCα contributes to this Ca2+ dependent phosphorylation that critically regulates NF-κB transcriptional activation and specificity. Given the large number of phospho-acceptors identified on both p65 and c-Rel [91], further work is needed to fully dissect which of these may be targets of Ca2+ dependent modification and how each post-translational modification fine tunes NF-κB driven gene expression.
4.6. Ca2+-dependent control of NF-κB protein expression
Dynamic control of NF-κB activity by Ca2+-dependent mechanisms is central to lymphocyte activation. However, de novo transcription of NF-κB proteins, including p100, p50, and c-Rel, and pathway mediators including IκB proteins, is critical for maintaining oscillatory patterns of NF-κB signaling and persistent activation of non-canonical NF-κB signaling (discussed below). In this regard NF-κB driven expression of the Nfkbia gene encoding IκBα exerts crucial feedback inhibition on the activity of NF-κB. Notably, we established that STIM/Orai-dependent Ca2+ entry is required for efficient induction of Nfkbia gene expression following TCR [54] and BCR engagement (unpublished data). Thus Ca2+ controls the activation but also exerts feedback inhibition on NF-κB activity in lymphocytes.
The level of basal and induced expression of individual NF-κB proteins including p65, c-Rel, and p50 represents another level of control that varies across tissues and within cell types. For example, c-Rel expression is largely limited to the hematopoietic compartment and is only significantly expressed in some specific types of B and T cell subsets [92, 93]. While mature B cells and regulatory T cells (and their precursors and progenitors (see Figure 3) express relatively high amounts of c-Rel, other T and B cell subsets only express significant amounts of c-Rel following activation. Interestingly, unlike Rela or Nfkb1 (the genes encoding p65 and p105:p50 respectively), Rel (the gene encoding c-Rel) expression increases substantially following BCR or TCR engagement and this is strongly regulated by Ca2+-dependent mechanisms [94, 95]. Indeed, both NF-κB and NFAT regulate de novo Rel expression highlighting the absolute Ca2+-dependence of antigen receptor driven Rel transcription in lymphocytes [94, 95].
Figure 3: STIM/Orai dependent regulation of nTreg differentiation by NF-κB and NFAT signaling.
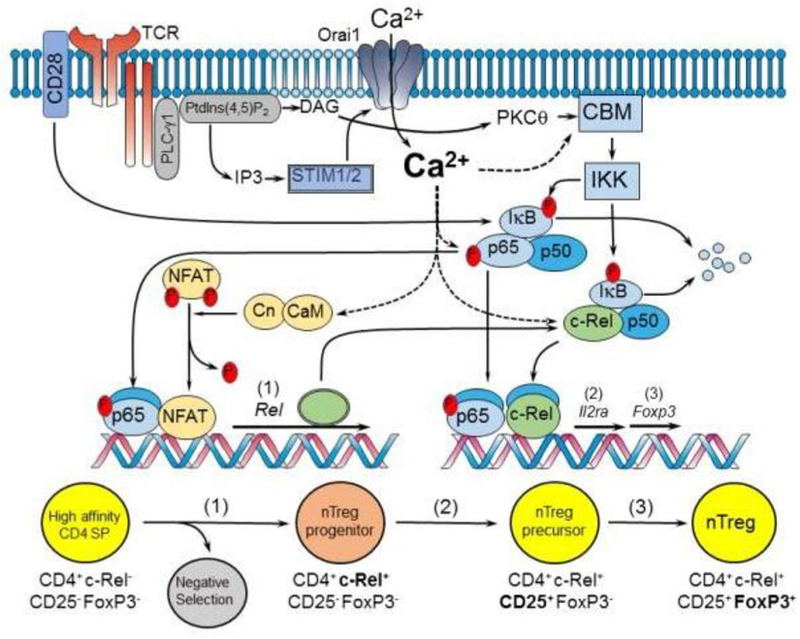
A schematic and simplified representation depicting Ca2+ dependent mechanisms that control the generation of natural regulatory T cells from CD4SP thymocytes. The upstream events leading to full NF-κB activation are described in Fig.2. NFAT is activated by Ca2+-dependent calcineurin (Cn) mediated dephosphorylation. Intermediate to high affinity interactions with self-antigen drive the upregulation of c-Rel (1) and survival (2) of a select proportion of T cells through NFAT and NF-κB dependent mechanisms. A population of c-Rel expressing thymocytes (nTreg progenitors) then upregulate CD25 expression (3) to generate nTreg precursors through Ca2+ and c-Rel dependent mechanisms. Sufficient binding of cytokines including IL2 drive upregulation of Foxp3 (4) in nTreg precursors generating functional CD4+c-Rel+CD25+Foxp3+ T cells capable of inhibiting autoreactive or excessive inflammatory immune reactions.
4.7. Ca2+ Regulation of Non-Canonical NF-κB Signaling in Lymphocytes
NF-κB activation following antigen receptor engagement in T and B cells occurs via Ca2+ control of the canonical NF-κB pathway. As described above, Ca2+ activation of CaN controls formation of the CBM complex. Downstream of the CBM complex NEMO-dependent IKKβ activation leading to phosphorylation and degradation of IκB proteins and liberation of p65 and c-Rel containing NF-κB dimers [96]. Calcium also plays a role in canonical NF-κB activation in response to genotoxic stress [97], but the mechanism underlying this response remains unclear, and it is not known whether Ca2+ regulates this pathway in lymphocytes. However, a distinct NF-κB signaling mechanism termed the “non-canonical pathway” also controls key steps of lymphocyte development, fate and function [98]. The non-canonical pathway is not activated by BCR or TCR engagement but rather by a subset of TNF receptor family members expressed on the surface of B and / or T cells including BAFFR, LTβR, CD40, CD30, Ox40, TWEAK and RANK [98]. Non-canonical NF-κB signaling in stromal cells regulates the formation and development of secondary lymph nodes and B cell survival, maturation, and homeostasis [99, 100] [101–106], and is also crucial for the generation and function of subsets of antigen-primed T cell effector and memory cells [107, 108] [109] [110]. To date, the role of Ca2+ in the non-canonical NF-κB pathway has not been directly addressed; although, several lines of evidence suggest that Ca2+ may control the activity of non-canonical NF-κB signaling. Non-canonical NF-κB signaling does not involve NEMO or IKKβ but instead requires activation of IKKα by the upstream kinase NF-κB Inducing Kinase (NIK) [111–114]. NIK is constitutively expressed in unstimulated cells but its recruitment to a complex containing TRAF2, TRAF3 and cellular inhibitor of apoptosis (cIAP) 1 and 2 causes its TRAF3-dependent ubiquitination and proteasomal degradation [115]. Receptor ligation leads to auto-degradation of the TRAF2/TRAF3/cIAP complex resulting in NIK stabilization and accumulation, and subsequent IKKα activation [114, 115]. Active IKKα phosphorylates the NF-κB protein p100 that exists as a heterodimer with RelB which is sequestered in the cytoplasm. Phosphorylation of p100 leads to its proteasomal processing to p52, which migrates to the nucleus with RelB as the non-canonical p52:RelB NF-κB dimer that binds target genes to regulate their expression [111, 112]
A role for Ca2+ in non-canonical NF-κB signaling can be inferred from studies describing CaN dependent regulation of key components of the pathway. A recent study found that multiple isoforms of the CaN phosphatase subunit A (CnA) bind directly to, and inhibit the activity of NIK [116], LTβR- and TWEAK-induced p100 processing to p52, and RelB:p52 nuclear localization in fibroblasts. In this same study, CnA was also found to associate with TRAF3 suggesting a role for CaN in regulating the stability and activity of NIK. In another study, the regulatory B subunit of calcineurin (CnB) was found to associate with TRAF3 leading to CaN degradation, presumably via the ubiquitin ligase activity of TRAF3 [117]. In Jurkat T cells, overexpression of TRAF3 induced the loss of CaN and inhibited NFAT dependent IL-2 expression [117]. Thus, in addition to stabilizing NIK and activating non-canonical NF-κB, receptor-induced TRAF3 degradation may also stabilized CaN expression and promote NFAT activation. Additional studies are needed to elucidate the precise mechanism underlying the inhibition of NIK activity by CaN, and the functional relationship between CaN and TRAF3. Also, whether these mechanisms play a significant role in non-canonical NF-κB activation in lymphocytes needs to be definitively established. Nevertheless, these findings indicate direct cross-talk between Ca2+ signaling and the non-canonical NF-κB pathway via the activity of CaN that warrant further investigation.
Finally, Ca2+ also plays an indirect role in non-canonical NF-κB signaling by regulating the expression of p100, which is encoded by the canonical NF-κB regulated gene Nfkb2. BCR and TCR activation of canonical NF-κB is dependent upon STIM/Orai regulated Ca2+ entry, which drives p100 expression [54]. Moreover, in B cells, basal p100 levels are maintained by tonic BCR signaling and is increased following antigen recognition [118]. This increase following BCR engagement replenishes the cellular pool of p100 required to maintain BAFF-mediated processing to p52 and non-canonical NF-κB activity. Thus, Ca2+ control of canonical NF-κB by antigen receptor engagement is crucial for the appropriate regulation of non-canonical NF-κB activity in lymphocytes. As non-canonical NF-κB is activated by receptors including BAFF, CD40 and LTβR that ultimately shape the developmental fate of B and T cells, further exploration of the mechanisms by which Ca2+ regulates non-canonical signaling is required.
5. Ca2+ regulation of NF-κB signaling in immunity and tolerance
While work to date has provided a framework for understanding key Ca2+ regulated checkpoints in NF-κB signaling, the field must also develop deeper mechanistic insight into how these pathways coordinate the developmental fates and functions of lymphocytes. This is particularly important, as the significant overlap in the phenotypes of lymphocytes lacking key mediators of Ca2+ or NF-κB signaling strongly supports a central regulatory role for Ca2+-dependent control of NF-κB in the balance between effective immunity and the loss of self-tolerance.
5.1. STIM and NF-κB in nTreg development and function
T cell development is a complex and highly regulated process that begins when bone marrow derived lymphoid precursors emigrate to the thymus to undergo a series of TCR-dependent selection events. During this selection process, the affinity of a TCR expressed on T cell progenitors for self-antigens dictates the “strength” of the TCR induced Ca2+ signal generated, and this determines the fate of each developing thymocyte [119]. CD4/CD8 double positive (DP) thymocytes whose TCR recognizes self-MHC that present low affinity self-antigens receive a survival signal and are positively selected. These cells then down regulate either CD4 or CD8 and migrate to the periphery as naïve T cells. DP and single positive (SP) thymocytes whose TCR exhibit high affinity for self-antigens are induced to undergo negative selection by apoptosis. This tolerogenic process eliminates self-reactive TCRs from the immune repertoire before they enter the peripheral circulation. In parallel, a small but consequential population of thymocytes that express TCRs with intermediate to high affinity for self-antigens [119, 120] escapes negative selection and differentiates into subpopulations of “agonist selected” T cells that potently suppress autoimmunity [121]. These subpopulations of T cells including natural regulatory T cells (nTreg), invariant natural killer cells (iNKT), and TCRαβ+CD8αα+ intestinal intraepithelial lymphocytes (iIELS), also critically regulate the balance between functional immunity and tolerance.
While nTregs only account for 5–10% of peripheral T cells, they play a dominant role in the maintenance of immune homeostasis and prevention of autoimmune diseases by suppressing or dampening autoreactive immune responses [25]. Indeed, humans and mice with missense mutations in the FOXP3 gene, which is a master regulator of nTreg development and function, develop a lethal autoimmune disease called immunodysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome [122]. nTreg development from CD4 SP thymocytes requires they transition through a series of discrete developmental checkpoints before they acquire Foxp3 expression (Fig. 3). TCR-dependent signals first initiate the expression of c-Rel to form nTreg progenitors, and subsequently, c-Rel and other transcription factors drive expression of the high affinity IL-2 receptor (CD25) to generate nTreg precursors [123–125]. The subsequent expression of Foxp3, a transcription factor whose expression coordinates Treg development and suppressive function, is driven by IL-2 [126, 127]. Importantly, both Ca2+ and NF-κB control these key developmental transitions as well as the function and stability of Tregs.
The remarkably selective role for STIM mediated Ca2+ entry in nTreg development was first revealed in studies of mice with a conditional deletion of Stim1 and Stim2 in T cells [21]. In STIM DKO mice, positive selection proceeds without any measurable alteration. In contrast, the loss of Ca2+ entry impacts both negative selection and agonist selected T cell development [22], suggesting that TCR-induced Ca2+ entry is required for driving fates of cells that encounter high affinity antigen. The block in nTreg development likely precedes CD25 expression as nTreg precursors (CD4+CD25+Foxp3-) also fail to develop efficiently [20–22]. Furthermore, while exogenous IL-2 can drive a modest increase in Treg numbers in STIM DKO cells, these “rescued” Tregs are functionally impaired and unable to suppress T cell proliferation, suggesting an ineffective or partial commitment of CD4+CD25+ precursors to the Treg lineage [20]. Together, these findings indicate that STIM/Orai-dependent Ca2+ entry guides nTreg differentiation and identity. Moreover, defective nTreg development in STIM DKO mice also reflects decreased activation of NFAT activation and binding to the Foxp3 promoter and cis-regulatory element 1 (CNS1) to drive Foxp3 expression [128, 129]. Surprisingly though, defective NFAT signaling alone may not account for the entire loss of nTregs in STIM DKO mice as NFAT1/NFAT2 DKO mice exhibit a partial nTreg deficit (50%) while NFAT2/NFAT4 DKO mice have no defect at all [130]. Together, these findings suggest that additional STIM-dependent mechanisms regulate nTreg differentiation.
A clue to the identity of these additional mechanisms is suggested by studies of mice lacking molecules required for TCR-induced NF-κB activation [131]. Mice with deficiencies in Prkcq (gene encoding PKCθ), BCL10, Ikk2 (gene encoding IKKβ), or Ikbkg (gene encoding NEMO) each harbor a similar nTreg deficit [124, 131–135]. However, strikingly low numbers of nTregs develop in mice lacking c-Rel whereas, loss of p65 leads to a more modest (~50%) reduction [136]. Indeed, c-Rel and p65 both bind to the Foxp3 promoter and coordinate its activity and the activity of its CNS3 enhancer element. The requirement for c-Rel relates in part to its crucial pioneering role in the assembly of an enhanceosome at the Foxp3 locus [137]. However, c-Rel deficient mice also exhibit a developmental block that precedes CD25 expression as these animals have a significant decrease in CD25+ nTreg progenitors [125, 136, 137]. Interestingly, development of iNKT cells, which also selected on high affinity self-antigen like nTregs and are deficient in STIM DKO mice, is similarly impaired in mice with a conditional deletion of Rela (p65 protein) in thymocytes [131, 138]. The similar requirement for p65 and Ca2+ further supports the idea that Ca2+ and NF-κB dependent mechanisms regulates the development of cells with high affinity for cell antigen.
While these previous studies underscore the import role of Ca2+ in nTreg development, at present, we can only speculate exactly how this occurs (see Figure 3). However, an important clue might be found in the quantitatively similar defect in nTregs observed in STIM DKO mice, proximal NF-κB signaling molecule-deficient mice, and c-Rel KO mice. This parallel phenotype raises the intriguing possibility that Ca2+ regulates nTreg development in part via control of c-Rel-dependent gene expression. In addition to its regulation of preexisting NF-κB complexes in T cells, Ca2+ also promotes de novo c-Rel expression through NFAT and NF-κB-dependent mechanisms [94, 95, 139, 140]. Not surprisingly, the Rel promoter has multiple NF-κB and NFAT binding sites and these appear to be functionally relevant as inhibition of CaN prevents TCR-induced Rel upregulation [95]. Importantly, de novo Rel expression plays an important role in maintaining sustained nuclear c-Rel accumulation [141]. Thus, in the context of nTreg development, high input Ca2+ signals, triggered only by intermediate to high affinity antigens, induce NFAT and NF-κB activation as well as de novo c-Rel expression. c-Rel then directly induces expression of genes that drive nTreg development, but also modifies the chromatin landscape to allow for the subsequent induction of Foxp3 expression by cytokines [137].
Figure 4: STIM/Orai and NF-κB dependent regulation of B lymphocyte activation, survival, and function.

BCR triggered STIM/Orai dependent Ca2+ entry critically regulates B cell activation and survival. In the absence of BCR induced Ca2+ signals or strong co-stimulation, B cells undergo apoptosis through undefined but likely NF-κB dependent mechanisms. Appropriate BCR and costimulatory activation rescues cells from apoptosis and initiates their growth and metabolic programing. STIM/Orai dependent transcriptional induction of key metabolic transcriptional regulators and NF-κB/NFAT target genes Myc, Hif1a, and Irf4 is critical to the survival and function of mature B cells. Upregulation of Myc, Hif1-α, and Irf4 orchestrates the induction of critical glycolytic intermediates and facilitate the reprogramming of lymphocyte metabolism from oxidative phosphorylation to glycolysis. These metabolic changes support rapid cell growth, DNA replication, and cell division. Additional roles for STIM/Orai dependent regulation of B cell function are evident in regulatory B cells (Bregs). Bregs are defined by their expression of suppressive cytokines including IL-10 and TGF-β and in the absence of STIM/Orai mediated calcium entry exhibit defective NF-κB and NFAT dependent Il10 expression.
In addition to a role in nTreg development, STIM/Orai-dependent Ca2+ entry also plays an important role in nTreg function. While some CD25+Foxp3+ cells do develop in STIM DKO mice, these cells express significantly less CD25, CD122, and Foxp3 than normal nTregs and are functionally impaired [20]. Administration of exogenous IL-2 modest increases Treg numbers in STIM DKO mice and their relative expression of Foxp3 but STIM deficient Tregs still fail to suppress proliferation of responder T cells. This functional deficit likely reflects decreased expression of inhibitory proteins required for suppression, including CTLA-4, LAG-3, and TIGIT [20–22]. However, this defect is not likely due to a block in NFAT activation, as existing Tregs do not require NFAT to suppress T cell proliferation [142]. Indeed, deletion of p65 and c-Rel in Tregs using a Tamoxifen-inducible Foxp3 Cre-ERT2 transgene [143] revealed that canonical NF-κB signaling is required to maintain Treg identity and notably Foxp3, Il2ra (CD25), Ctla4, Ikzf2 (Helios), and Tnfrsf18 (Gitr) expression. Moreover, canonical NF-κB is required for the suppressive functions of Tregs as Rela + Rel null Tregs fail to prevent the development of colitis [143]. Together, these studies have established the critical role played by Ca2+ and NF-κB in tolerance but also highlight the need to obtain a more comprehensive understanding of how Ca2+ regulates p65 and c-Rel activity to control of nTreg development, and more generally to understand mechanisms by which dynamic Ca2+ signals control the transcriptionally driven fates of multipotent lymphocytes.
5.2. STIM and NF-κB in T helper cell fate and function
Following development in the thymus, naïve CD4+ T cells migrate to the periphery and recirculate via the blood, spleen and lymph nodes in search for their cognate antigen. Once activated, the fate of naïve CD4+ T cells depends on a number of factors, but most importantly, the cytokines present during activation [144–146]. Indeed, activation of naïve T cells in the presence of IL-12 and the absence of IL-4 generates Th1 cells, which regulate immunity to intracellular pathogens through the production of IFNγ, IL-2, and TNFα [147]. Furthermore, naïve T cell activation in the presence of IL-4 and IL-2 generates Th2 cells, which orchestrate immunity to helminth parasites, B cell proliferation, and B cell class switching through their production of IL-4 and IL-5 [148, 149]. While many factors have been implicated in the differentiation and function of these distinct T cell subsets, STIM/Orai dependent Ca2+ signals, NFAT, and NF-κB, each appear to play essential roles.
Much of our insight into the role of Ca2+ entry in peripheral T cell development has been gleaned from studies of STIM DKO mice and patients with inactivating mutations in STIM1 or ORAI1. Remarkably, in mice with a conditional loss of Stim1 and Stim2 in CD4 T cells, peripheral T cells numbers and proportions are normal in young (4–8 weeks) animals but thereafter increase, due to uncontrolled lymphoproliferation resulting from a loss of regulatory T cells [21]. Despite the apparent normal development of conventional T cells, severe impairments in T cell function including proliferation and cytokine production become apparent following T cell activation [21, 150]. Consistent with these findings, in vitro stimulation assays with Stim1 and Stim2 single KO T cells revealed that Ca2+ entry is required for efficient expression of IFNγ and IL-4 under Th1 and Th2 polarizing conditions [21]. Interestingly, both NFAT and NF-κB proteins are required for optimal production of IFNγ and IL-4. Loss of either NFAT1 or c-Rel causes decreased IFNγ production and Th1 functionality [151, 152] while loss of NFAT2 or p50 causes defective Th2 differentiation [153–155]. Together, these findings suggest that defective NFAT and NF-κB signaling may underlie skewed Th1 and Th2 development and function observed in STIM DKO mice. However, additional studies are needed to determine what role Ca2+ dependent NF-κB activation plays in the specification and function of Th1 and Th2 subsets.
Human patients with inactivating mutations in STIM1 or ORAI1 also exhibit defective humoral immunity including decreased pathogen specific antibody production and the production of autoantibodies [156]. Interestingly, these humoral defects may not result from B cell intrinsic defects in antibody production as B cells from Stim1fl/flStim2fl/fl x Mb1-Cre mice produce normal amounts and isotypes of antibodies upon immunization whereas Stim1fl/flStim2fl/fl x CD4-Cre do not [23]. Instead, decreased pathogen specific antibody production and humoral autoimmunity occurs as a result of defects in the differentiation and function of T follicular helper (Tfh) cells and T follicular regulatory (Tfr) cells, respectively [150]. Tfh cells play a vital role in promoting antibody production from B cells by providing critical T cell help through CD40L and cytokines including IL-4 and IL-21 while Tfr cells limit the germinal center reaction through expression of inhibitory molecules including CTLA-4 and IL-10 [150, 157, 158]. While aberrant Tfr and Tfh development has been attributed to defective NFAT activity, loss of individual NFAT proteins fail to recapitulate the Tfh defect [159, 160]. Interestingly, Rel−/− mice also have defects in humoral immunity and c-Rel has been shown to regulate the differentiation of Tfh cells and the production of IL-21 [161]. Furthermore, control of IRF4, a pioneering transcription factor critical for Tfh differentiation is critically regulated by NF-κB signaling [162]. These findings are only suggestive but motivate additional investigation of the role of Ca2+ dependent NF-κB activity in the differentiation and function of Tfh cells.
5.3. STIM and NF-κB in lymphocyte growth and metabolism
A recent study established that STIM/Orai-dependent Ca2+ signaling controls T cell metabolism and their metabolic reprogramming following TCR stimulation [163]. Importantly, a Ca2+-entry dependent switch from oxidative phosphorylation to glycolysis is critical for T cell activation and differentiation (Figure 4). Indeed, TCR-induced increases in glucose uptake reflect STIM dependent increases in GLUT1 and GLUT3 expression [163]. Furthermore, Ca2+ entry drives the expression of many glycolytic enzymes, and this reprogramming likely reflects Ca2+-dependent induction of the metabolic master regulators Myc, Hif1a, and Irf4. Intriguingly, while Hif1a and Irf4 expression are directly regulated by NFAT, Myc induction is CaN-dependent but expression in not decreased in NFAT1/2−/− T cells [163]. Importantly, Myc activity in lymphocytes is largely regulated by transcriptional activation of the Myc promoter by NF-κB binding. Together, these data raise the significant possibility that Myc expression and Myc targets in T cells are induced by NFAT-independent, Ca2+/CaN-dependent transcription factors such as NF-κB. Indeed, in B cells, c-Rel and p50 are required for Myc expression [164] while in T cells, p65 and c-Rel control Myc expression and cell growth [95]. Going forward, it will be important to quantify NF-kB activity and p65 and c-Rel Myc promoter binding during activation of STIM DKO naïve T cells to determine the impact of Ca2+ dependent NF-κB activity on Myc dependent metabolic reprogramming.
5.4. STIM and NF-κB in B lymphocyte survival and function
B cells develop throughout vertebrate life and like T cells, they express an antigen receptor whose functionality and specificity determines the fate of each clone. BCR diversity is generated by rearrangement of immunoglobulin heavy and light chain genes [165] and successful rearrangement in pro-B and pre-B cells, respectively, yields immature B cells. An immature population of transitional B cells, then leave the bone marrow and enter the peripheral circulation. In the spleen, T1 and T2 transitional cells, further differentiate into mature B cells [166]. All pre-T2 populations are induced to die when their BCR is engaged by self-antigen and this culls autoreactive clones. Engagement of the BCR on mature B cells; however, initiates proliferation and further differentiation into marginal zone B cells, responsible for T cell-dependent and independent immunity, and follicular B cells that are involved in T cell-dependent immune responses [165].
Interestingly, BCR engagement at almost every stage of development triggers a STIM-dependent Ca2+ signal. Yet, this signal does not appear to be necessary for development or antibody production, as B cells develop normally in STIM DKO mice [23, 167]. Nonetheless, B cell survival ex vivo requires STIM/Orai-dependent Ca2+ entry and this is evident from an increase in the rate and extent of BCR-induced death of STIM DKO B cells [23, 167] (Figure 4). Importantly, rescue from this Bim-mediated mitochondrial-dependent apoptotic death requires efficient BCR induced NF-κB activity [168]. Indeed, transitional and/or follicular B cells lacking Rel or Rela are more susceptible to BCR-induced death in vitro [165] and loss of c-Rel and p65 [169, 170] or IKKα and IKKβ [171] also block follicular B cell survival in vivo. This rescue from BCR-induced apoptosis reflects the critical role of c-Rel in the expression of anti-apoptotic molecules Bcl-xL and A1, which counteract Bim-dependent apoptosis [172–174]. Indeed, c-Rel deficient B cells undergo rapid BCR-induced death, and the extent and kinetics of death are similar to that of STIM DKO B cells. The similarity in phenotypes of STIM DKO and c-Rel deficient B cells raises the intriguing possibility that Ca2+ control of c-Rel expression and activity drives expression of molecules required for B cell survival. Consistent with this mechanism, CD40, which activates both canonical and non-canonical NF-κB signaling and drives Bcl-xL expression, significantly increases STIM DKO B cell survival and proliferation [23].
Another important role identified for STIM/Orai-dependent Ca2+ signals in B cells is control of IL-10 production. Interestingly, IL-10 and TGF-β are anti-inflammatory cytokines produced by regulatory B cells (Bregs) [175]. A defect in Ca2+ dependent IL-10 production by B cells may drive the pathophysiology of experimental autoimmune encephalomyelitis (EAE), which is exacerbated in STIM DKO mice [23]. While previous studies suggest that IL-10 production is controlled by NFAT1, this conclusion was based upon the incorrect assumption that calcineurin selectively inhibits NFAT activation in lymphocytes. However, given that NF-κB activation and de novo c-Rel expression are both depend on CaN activity, it is critical to reassess the Ca2+-dependent NF-κB activity in IL-10 production and the pathogenesis of autoimmune disease [23]. As p65 [176], c-Rel [177], and p50 [178] regulate Il10 expression in lymphocytes and macrophages, more detailed studies of NF-κB signaling in STIM DKO B cells are needed to clarify the role of STIM/Orai-dependent Ca2+ entry in Breg function.
6. Concluding remarks
This review focuses on the mechanisms and consequences of Ca2+ dependent activation of the proinflammatory transcription factor NF-κB. The significance of this signaling pathway stems from the critical regulatory role played by both Ca2+ and NF-κB in the generation of effective immunity but also the remarkable phenotypic concordance between mice deficient in proteins that regulate each pathway. While there is ample evidence that Ca2+ regulates NF-κB activity and specificity, much remains to be learned mechanistically. We present our perspective in this review, which is supported by a broad literature, that NF-κB possess a wide dynamic range that bestows upon it the capacity to decode complex and varied Ca2+ waveforms, including those to which NFAT is not sensitive, into specific and distinct transcriptionally driven fates. In conjunction and cooperation with NFAT, NF-κB proteins decode information generated upon antigen binding and costimulatory receptor engagement, into appropriate developmental and fates of lymphocytes. It is hoped that this review highlights all the complex aspects of this signaling mechanism but also illuminates important gaps in our understanding of Ca2+ dependent control of NF-κB activation and function.
Highlights.
Antigen binding to lymphocyte antigen receptors triggers STIM and Orai mediated Ca2+ entry
The dynamics of antigen induced cytoplasmic [Ca2+] changes reflect the affinity and avidity of antigen binding and the extent of costimulatory receptor engagement.
Quantitative features of antigen induced calcium signals dictate the fates and functions of multipotent lymphocytes.
Dynamic Ca2+ signals generated in lymphocytes are decoded by immune-modulatory transcription factors including NF-κB and NFAT, each of which are tuned to distinct Ca2+ dynamics.
Ca2+ dependent regulation of the phosphorylation status of multiple proteins that control NF-κB activation, and of NF-κB proteins themselves, direct the transcriptionally driven fates of lymphocytes.
Acknowledgements
This work was funded by R56AI125415 awarded to Bruce Freedman and Michael May, R01 AI060921 awarded to Bruce Freedman, and NIH R01AR066567 awarded to Michael May
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Roos J, DiGregorio PJ, Yeromin AV, Ohlsen K, Lioudyno M, Zhang S, et al. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J. Cell Biol 2005;169(3):435–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liou J, Kim ML, Heo WD, Jones JT, Myers JW, Ferrell JE Jr., et al. STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr. Biol 2005;15(13):1235–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vig M, Peinelt C, Beck A, Koomoa DL, Rabah D, Koblan-Huberson M, et al. CRACM1 is a plasma membrane protein essential for store-operated Ca2+ entry. Science 2006;312(5777):1220–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Feske S, Gwack Y, Prakriya M, Srikanth S, Puppel SH, Tanasa B, et al. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature 2006;441(7090):179–85. [DOI] [PubMed] [Google Scholar]
- 5.Feske S, Picard C, Fischer A. Immunodeficiency due to mutations in ORAI1 and STIM1. Clin. Immunol 2010;135(2):169–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shaw PJ, Feske S. Regulation of lymphocyte function by ORAI and STIM proteins in infection and autoimmunity. J. Physiol 2012;590(Pt 17): 4157–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hogan PG, Lewis RS, Rao A. Molecular basis of calcium signaling in lymphocytes: STIM and ORAI. Annu. Rev. Immunol 2010;28:491–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Feske S, Skolnik EY, Prakriya M. Ion channels and transporters in lymphocyte function and immunity. Nat. Rev. Immunol 2012;12(7):532–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vig M, Kinet JP. Calcium signaling in immune cells. Nat. Immunol 2009;10(1):21–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Oh-Hora M Calcium signaling in the development and function of T-lineage cells. Immunol. Rev 2009;231(1):210–24. [DOI] [PubMed] [Google Scholar]
- 11.Shaw KT, Ho AM, Raghavan A, Kim J, Jain J, Park J, et al. Immunosuppressive drugs prevent a rapid dephosphorylation of transcription factor NFAT1 in stimulated immune cells. Proc Natl Acad Sci U S A 1995;92(24):11205–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Okamura H, Aramburu J, Garcia-Rodriguez C, Viola JP, Raghavan A, Tahiliani M, et al. Concerted dephosphorylation of the transcription factor NFAT1 induces a conformational switch that regulates transcriptional activity. Molecular Cell 2000;6(3):539–50. [DOI] [PubMed] [Google Scholar]
- 13.Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem 1985;260:3440–50. [PubMed] [Google Scholar]
- 14.Melichar HJ, Ross JO, Herzmark P, Hogquist KA, Robey EA. Distinct temporal patterns of T cell receptor signaling during positive versus negative selection in situ. Sci. Signal 2013;6(297):ra92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ebert PJ, Ehrlich LI, Davis MM. Low ligand requirement for deletion and lack of synapses in positive selection enforce the gauntlet of thymic T cell maturation. Immunity 2008;29(5):734–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ross JO, Melichar HJ, Au-Yeung BB, Herzmark P, Weiss A, Robey EA. Distinct phases in the positive selection of CD8+ T cells distinguished by intrathymic migration and T-cell receptor signaling patterns. Proceedings of the National Academy of Sciences of the United States of America 2014;111(25):E2550–E8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Daniels MA, Teixeiro E, Gill J, Hausmann B, Roubaty D, Holmberg K, et al. Thymic selection threshold defined by compartmentalization of Ras/MAPK signalling. Nature 2006;444(7120):724–9. [DOI] [PubMed] [Google Scholar]
- 18.Freedman BD, Liu QH, Somersan S, Kotlikoff MI, Punt JA. Receptor avidity and costimulation specify the intracellular Ca2+ signaling pattern in CD4(+)CD8(+) thymocytes. J. Exp. Med 1999;190(7):943–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nakayama T, Ueda Y, Yamada H, Shores EW, Singer A, June CH. In vivo calcium elevations in thymocytes with T cell receptors that are specific for self ligands. Science 1992;257:96–9. [DOI] [PubMed] [Google Scholar]
- 20.Oh-hora M, Komatsu N, Pishyareh M, Feske S, Hori S, Taniguchi M, et al. Agonist-Selected T Cell Development Requires Strong T Cell Receptor Signaling and Store-Operated Calcium Entry. Immunity 2013;38(5):881–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Oh-Hora M, Yamashita M, Hogan PG, Sharma S, Lamperti E, Chung W, et al. Dual functions for the endoplasmic reticulum calcium sensors STIM1 and STIM2 in T cell activation and tolerance. Nat. Immunol 2008;9(4):432–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McDonald BD, Bunker JJ, Erickson SA, Oh-Hora M, Bendelac A. Crossreactive alphabeta T Cell Receptors Are the Predominant Targets of Thymocyte Negative Selection. Immunity 2015;43(5):859–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Matsumoto M, Fujii Y, Baba A, Hikida M, Kurosaki T, Baba Y. The calcium sensors STIM1 and STIM2 control B cell regulatory function through interleukin-10 production. Immunity 2011;34(5):703–14. [DOI] [PubMed] [Google Scholar]
- 24.Bhakta NR, Oh DY, Lewis RS. Calcium oscillations regulate thymocyte motility during positive selection in the three-dimensional thymic environment. Nature Immunology 2005;6(2):143–51. [DOI] [PubMed] [Google Scholar]
- 25.Stritesky GL, Jameson SC, Hogquist KA. Selection of self-reactive T cells in the thymus. Annu. Rev. Immunol 2012;30:95–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tomida T, Hirose K, Takizawa A, Shibasaki F, Iino M. NFAT functions as a working memory of Ca2+ signals in decoding Ca2+ oscillation. EMBO J 2003;22(15):3825–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Song S, Li J, Zhu L, Cai L, Xu Q, Ling C, et al. Irregular Ca2+ oscillations regulate transcription via cumulative spike duration and spike amplitude. J. Biol. Chem 2012;287(48):40246–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhu L, Song S, Pi Y, Yu Y, She W, Ye H, et al. Cumulated Ca2+ spike duration underlies Ca2+ oscillation frequency-regulated NFkappaB transcriptional activity. J. Cell Sci 2011;124(Pt 15):2591–601. [DOI] [PubMed] [Google Scholar]
- 29.Smedler E, Uhlen P. Frequency decoding of calcium oscillations. Bba-Gen Subjects 2014;1840(3):964–9. [DOI] [PubMed] [Google Scholar]
- 30.Dolmetsch RE, Lewis RS, Goodnow CC, Healy JI. Differential activation of transcription factors induced by Ca2+ response amplitude and duration. Nature 1997;386(6627):855–8. [DOI] [PubMed] [Google Scholar]
- 31.Dolmetsch RE, Lewis RS. Signaling between intracellular Ca2+ stores and depletion-activated Ca2+ channels generates [Ca2+]i oscillations in T lymphocytes. J. Gen. Physiol 1994;103(3):365–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jiang T, Sweeney G, Rudolf MT, Klip A, Traynor-Kaplan A, Tsien RY. Membrane-permeant esters of phosphatidylinositol 3,4,5-trisphosphate. J. Biol. Chem 1998;273(18):11017–24. [DOI] [PubMed] [Google Scholar]
- 33.Healy JI, Dolmetsch RE, Timmerman LA, Cyster JG, Thomas ML, Crabtree GR, et al. Different nuclear signals are activated by the B cell receptor during positive versus negative signaling. Immunity 1997;6(4):419–28. [DOI] [PubMed] [Google Scholar]
- 34.Bhakta NR, Lewis RS. Real-time measurement of signaling and motility during T cell development in the thymus. Semin. Immunol 2005;17(6):411–20. [DOI] [PubMed] [Google Scholar]
- 35.Dolmetsch RE, Xu K, Lewis RS. Calcium oscillations increase the efficiency and specificity of gene expression. Nature 1998;392(6679):933–6. [DOI] [PubMed] [Google Scholar]
- 36.Putney JW, Bird GS. Cytoplasmic calcium oscillations and store-operated calcium influx. J Physiol-London 2008;586(13):3055–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Thiel M, Lis A, Penner R. STIM2 drives Ca2+ oscillations through store-operated Ca2+ entry caused by mild store depletion. J Physiol-London 2013;591(6):1433–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lewis RS, Cahalan MD. Mitogen-induced oscillations of cytosolic Ca2+ and transmembrane Ca2+ current in human leukemic T cells. Cell. Regul 1989;1(1):99–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li W, Llopis J, Whitney M, Zlokarnik G, Tsien RY. Cell-permeant caged InsP3 ester shows that Ca2+ spike frequency can optimize gene expression. Nature 1998;392(6679):936–41. [DOI] [PubMed] [Google Scholar]
- 40.Flanagan WM, Corthesy B, Bram RJ, Crabtree GR. Nuclear association of a T-cell transcription factor blocked by FK-506 and cyclosporin A. Nature 1991;352(6338):803–7. [DOI] [PubMed] [Google Scholar]
- 41.Hayden MS, West AP, Ghosh S. NF-kappaB and the immune response. Oncogene 2006;25(51):6758–80. [DOI] [PubMed] [Google Scholar]
- 42.Ghosh S, May MJ, Kopp EB. NF-kappa B and Rel proteins: evolutionarily conserved mediators of immune responses. AnnuRevImmunol 1998;16:225–60. [DOI] [PubMed] [Google Scholar]
- 43.Altman A, Villalba M. Protein kinase C-theta (PKCtheta): it’s all about location, location, location. Immunol. Rev 2003;192:53–63. [DOI] [PubMed] [Google Scholar]
- 44.Villalba M, Coudronniere N, Deckert M, Teixeiro E, Mas P, Altman A. A novel functional interaction between Vav and PKCtheta is required for TCR-induced T cell activation. Immunity 2000;12(2):151–60. [DOI] [PubMed] [Google Scholar]
- 45.Hayden MS, Ghosh S. Shared principles in NF-kappaB signaling. Cell 2008;132(3):344–62. [DOI] [PubMed] [Google Scholar]
- 46.Clipstone NA, Crabtree GR. Identification of calcineurin as a key signalling enzyme in T-lymphocyte activation. Nature 1992;357(6380):695–7. [DOI] [PubMed] [Google Scholar]
- 47.Liu J, Farmer JD, Jr., Lane WS, Friedman J, Weissman I, Schreiber SL. Calcineurin is a common target of cyclophilin-cyclosporin A and FKBP-FK506 complexes. Cell 1991;66(4):807–15. [DOI] [PubMed] [Google Scholar]
- 48.Emmel EA, Verweij CL, Durand DB, Higgins KM, Lacy E, Crabtree GR. Cyclosporin A specifically inhibits function of nuclear proteins involved in T cell activation. Science 1989;246(4937):1617–20. [DOI] [PubMed] [Google Scholar]
- 49.Albrecht Schmidt LH, Siebenlist U, Inducible Nuclear Factor Binding to the KB Elements of the HumanImmunodeficiency Virus Enhancer in T Cells Can Be Blocked by Cyclosporin A in a Signal-Dependent Manner. Journal of Virology 1990;64(8):4037–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mattila PS, Ullman KS, Fiering S, Emmel EA, McCutcheon M, Crabtree GR, et al. The actions of cyclosporin A and FK506 suggest a novel step in the activation of T lymphocytes. EMBO J 1990;9(13):4425–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Frantz B, Nordby EC, Bren G, Steffan N, Paya CV, Kincaid RL, et al. Calcineurin acts in synergy with PMA to inactivate I kappa B/MAD3, an inhibitor of NF-kappa B. EMBO J 1994;13(4):861–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Steffan NM, Bren GD, Frantz B, Tocci MJ, O’Neill EA, Paya CV. Regulation of IkB alpha phosphorylation by PKC- and Ca2+-dependent signal transduction pathways. Journal of Immunol 1995;155(10):4685–91. [PubMed] [Google Scholar]
- 53.Trushin SA, Pennington KN, Algeciras-Schimnich A, Paya CV. Protein kinase C and calcineurin synergize to activate IkappaB kinase and NF-kappaB in T lymphocytes. J. Biol. Chem 1999;274(33):22923–31. [DOI] [PubMed] [Google Scholar]
- 54.Liu X, Berry CT, Ruthel G, Madara JJ, MacGillivray K, Gray CM, et al. T cell receptor-induced NF-kappaB signaling and transcriptional activation are regulated by STIM1- and Orai1-mediated calcium entry. J Biol Chem 2016: 291(16):8440– 8452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Palkowitsch L, Marienfeld U, Brunner C, Eitelhuber A, Krappmann D, Marienfeld RB. The Ca2+-dependent phosphatase calcineurin controls the formation of the Carma1-Bcl10-Malt1 complex during T cell receptor-induced NF-kappaB activation. J. Biol. Chem 2011;286(9):7522–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chin D, Means AR. Calmodulin: a prototypical calcium sensor. Trends in cell biology 2000;10(8):322–8. [DOI] [PubMed] [Google Scholar]
- 57.Hughes K, Antonsson A, Grundstrom T. Calmodulin dependence of NFkappaB activation. FEBS Lett 1998;441(1):132–6. [DOI] [PubMed] [Google Scholar]
- 58.Antonsson A, Hughes K, Edin S, Grundstrom T. Regulation of c-Rel nuclear localization by binding of Ca2+/calmodulin. Mol. Cell. Biol 2003;23(4):1418–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Braun AP, Schulman H. The multifunctional calcium/calmodulin-dependent protein kinase: from form to function. Annu. Rev. Physiol 1995;57:417–45:417–45. [DOI] [PubMed] [Google Scholar]
- 60.Swulius MT, Waxham MN. Ca2+/Calmodulin-dependent Protein Kinases. Cell. Mol. Life Sci 2008;65(17):2637–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hanson PI, Meyer T, Stryer L, Schulman H. Dual role of calmodulin in autophosphorylation of multifunctional CaM kinase may underlie decoding of calcium signals. Neuron 1994;12(5):943–56. [DOI] [PubMed] [Google Scholar]
- 62.Hughes K, Edin S, Antonsson A, Grundstrom T. Calmodulin-dependent kinase II mediates T cell receptor/CD3- and phorbol ester-induced activation of IkappaB kinase. J. Biol. Chem 2001;276(38):36008–13. [DOI] [PubMed] [Google Scholar]
- 63.Ishiguro K, Green T, Rapley J, Wachtel H, Giallourakis C, Landry A, et al. Ca2+/calmodulin-dependent protein kinase II is a modulator of CARMA1-mediated NF-kappaB activation. Mol. Cell. Biol 2006;26(14):5497–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Oruganti SR, Edin S, Grundstrom C, Grundstrom T. CaMKII targets Bcl10 in T-cell receptor induced activation of NF-kappaB. MolImmunol 2011;48(12–13):1448–60. [DOI] [PubMed] [Google Scholar]
- 65.Ishiguro K, Ando T, Goto H, Xavier R. Bcl10 is phosphorylated on Ser138 by Ca2+/calmodulin-dependent protein kinase II. MolImmunol 2007;44(8):2095–100. [DOI] [PubMed] [Google Scholar]
- 66.Li L, Stefan MI, Le NN. Calcium input frequency, duration and amplitude differentially modulate the relative activation of calcineurin and CaMKII. PLoSOne 2012;7(9):e43810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.DeKoninck P, Schulman H. Sensitivity of CaM kinase II to the frequency of Ca2+ oscillations. Science 1998;279(5348):227–30. [DOI] [PubMed] [Google Scholar]
- 68.Williams DH, Woodrow M, Cantrell DA, Murray EJ. Protein kinase C is not a downstream effector of p21ras in activated T cells. European journal of immunology 1995;25(1):42–7. [DOI] [PubMed] [Google Scholar]
- 69.Tan SL, Parker PJ. Emerging and diverse roles of protein kinase C in immune cell signalling. Biochem J 2003;376(Pt 3): 545–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Spitaler M, Cantrell DA. Protein kinase C and beyond. Nat Immunol 2004;5(8):785–90. [DOI] [PubMed] [Google Scholar]
- 71.Trushin SA, Pennington KN, Carmona EM, Asin S, Savoy DN, Billadeau DD, et al. Protein kinase Calpha (PKCalpha) acts upstream of PKCtheta to activate IkappaB kinase and NF-kappaB in T lymphocytes. Mol. Cell. Biol 2003;23(19):7068–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Szamel M, Appel A, Schwinzer R, Resch K. Different protein kinase C isoenzymes regulate IL-2 receptor expression or IL-2 synthesis in human lymphocytes stimulated via the TCR. Journal of immunology 1998;160(5):2207–14. [PubMed] [Google Scholar]
- 73.Guo B, Su TT, Rawlings DJ. Protein kinase C family functions in B-cell activation. CurrOpinImmunol 2004;16(3):367–73. [DOI] [PubMed] [Google Scholar]
- 74.Numaga T, Nishida M, Kiyonaka S, Kato K, Katano M, Mori E, et al. Ca2+ influx and protein scaffolding via TRPC3 sustain PKCbeta and ERK activation in B cells. J. Cell. Sci 2010;123(Pt 6): 927–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Campbell KJ, Perkins ND. Post-translational modification of RelA(p65) NF-kappa B. Biochemical Society transactions 2004;32:1087–9. [DOI] [PubMed] [Google Scholar]
- 76.Harris J, Oliere S, Sharma S, Sun Q, Lin R, Hiscott J, et al. Nuclear accumulation of cRel following C-terminal phosphorylation by TBK1/IKK epsilon. JImmunol 2006;177(4):2527–35. [DOI] [PubMed] [Google Scholar]
- 77.Gerondakis S, Siebenlist U. Roles of the NF-kappa B Pathway in Lymphocyte Development and Function. Cold Spring Harbor Perspectives in Biology 2010;2(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hwang YJ, Lee EW, Song J, Kim HR, Jun YC, Hwang KA. MafK positively regulates NF-kappaB activity by enhancing CBP-mediated p65 acetylation. Sci Rep 2013;3:3242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Geng H, Wittwer T, Dittrich-Breiholz O, Kracht M, Schmitz ML. Phosphorylation of NF-kappaB p65 at Ser468 controls its COMMD1-dependent ubiquitination and target gene-specific proteasomal elimination. EMBO Rep 2009;10(4):381–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Campbell KJ, Perkins ND. Post-translational modification of RelA(p65) NF-kappaB. Biochemical Society transactions 2004;32(Pt 6): 1087–9. [DOI] [PubMed] [Google Scholar]
- 81.Zhong H, May MJ, Jimi E, Ghosh S. The phosphorylation status of nuclear NF-kappa B determines its association with CBP/p300 or HDAC-1. MolCell 2002;9(3):625–36. [DOI] [PubMed] [Google Scholar]
- 82.Fognani C, Rondi R, Romano A, Blasi F. cRel-TD kinase: a serine/threonine kinase binding in vivo and in vitro c-Rel and phosphorylating its transactivation domain. Oncogene 2000;19(18):2224–32. [DOI] [PubMed] [Google Scholar]
- 83.Wang BL, Wei H, Prabhu L, Zhao W, Martin M, Hartley AV, et al. Role of Novel Serine 316 Phosphorylation of the p65 Subunit of NF-kappa B in Differential Gene Regulation. J. Biol. Chem 2015;290(33):20336–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ea CK, Baltimore D. Regulation of NF-kappaB activity through lysine monomethylation of p65. Proc Natl Acad Sci U S A 2009;106(45):18972–7. Epub 2009/10/30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hochrainer K, Racchumi G, Zhang S, Iadecola C, Anrather J. Monoubiquitination of nuclear RelA negatively regulates NF-kappaB activity independent of proteasomal degradation. Cell. Mol. Life Sci 2012;69(12):2057–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Li H, Wittwer T, Weber A, Schneider H, Moreno R, Maine GN, et al. Regulation of NF-kappaB activity by competition between RelA acetylation and ubiquitination. Oncogene 2012;31(5):611–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mattioli I, Geng H, Sebald A, Hodel M, Bucher C, Kracht M, et al. Inducible phosphorylation of NF-kappa B p65 at serine 468 by T cell costimulation is mediated by IKK epsilon. J. Biol. Chem 2006;281(10):6175–83. [DOI] [PubMed] [Google Scholar]
- 88.Xing D, Gong K, Feng W, Nozell SE, Chen YF, Chatham JC, et al. O-GlcNAc modification of NFkappaB p65 inhibits TNF-alpha-induced inflammatory mediator expression in rat aortic smooth muscle cells. PLoS One 2011;6(8):e24021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kim JE, Kim SY, Lim SY, Kieff E, Song YJ. Role of Ca2+/calmodulin-dependent kinase II-IRAK1 interaction in LMP1-induced NF-kappaB activation. Mol Cell Biol 2014;34(3):325–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Jang MK, Goo YH, Sohn YC, Kim YS, Lee SK, Kang H, et al. Ca2+/calmodulin-dependent protein kinase IV stimulates nuclear factor-kappa B transactivation via phosphorylation of the p65 subunit. J. Biol. Chem 2001;276(23):20005–10. [DOI] [PubMed] [Google Scholar]
- 91.Christian F, Smith EL, Carmody RJ. The Regulation of NF-kappaB Subunits by Phosphorylation. Cells 2016;5(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Brownell E, Mathieson B, Young HA, Keller J, Ihle JN, Rice NR. Detection of c-rel-related transcripts in mouse hematopoietic tissues, fractionated lymphocyte populations, and cell lines. Mol. Cell. Biol 1987;7(3):1304–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Carrasco D, Weih F, Bravo R. Developmental expression of the mouse c-rel proto-oncogene in hematopoietic organs. Development 1994;120(10):2991–3004. [DOI] [PubMed] [Google Scholar]
- 94.Nolz JC, Fernandez-Zapico ME, Billadeau DD. TCR/CD28-stimulated actin dynamics are required for NFAT1-mediated transcription of c-rel leading to CD28 response element activation. Journal of immunology 2007;179(2):1104–12. [DOI] [PubMed] [Google Scholar]
- 95.Grumont R, Lock P, Mollinari M, Shannon FM, Moore A, Gerondakis S. The mitogen-induced increase in T cell size involves PKC and NFAT activation of Rel/NF-kappaB-dependent c-myc expression. Immunity 2004;21(1):19–30. [DOI] [PubMed] [Google Scholar]
- 96.Vallabhapurapu S, Karin M. Regulation and Function of NF-kappa B Transcription Factors in the Immune System. Annual Review of Immunology 2009;27:693–733. [DOI] [PubMed] [Google Scholar]
- 97.Berchtold CM, Wu ZH, Huang TT, Miyamoto S. Calcium-dependent regulation of NEMO nuclear export in response to genotoxic stimuli. Molecular and Cellular Biology 2007;27(2):497–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sun SC. The non-canonical NF-kappa B pathway in immunity and inflammation. Nature Reviews Immunology 2017;17(9):545–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Shinkura R, Kitada K, Matsuda F, Tashiro K, Ikuta K, Suzuki M, et al. Alymphoplasia is caused by a point mutation in the mouse gene encoding Nf-kappa b-inducing kinase. Nat. Genet 1999;22(1):74–7. [DOI] [PubMed] [Google Scholar]
- 100.Matsushima A, Kaisho T, Rennert PD, Nakano H, Kurosawa K, Uchida D, et al. Essential role of nuclear factor (NF)-kappa B-inducing kinase and inhibitor of kappa B (I kappa B) kinase alpha in NF-kappa B activation through lymphotoxin beta receptor, but not through tumor necrosis factor receptor I. Journal of Experimental Medicine 2001;193(5):631–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Yilmaz ZB, Weih DS, Sivakumar V, Weih F. RelB is required for Peyer’s patch development: differential regulation of p52-RelB by lymphotoxin and TNF. EMBO Journal 2003;22(1):121–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Caamano JH, Rizzo CA, Durham SK, Barton DS, Raventos-Suarez C, Snapper CM, et al. Nuclear factor (NF)-kappa B2 (p100/p52) is required for normal splenic microarchitecture and B cell-mediated immune responses. Journal of Experimental Medicine 1998;187(2):185–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Franzoso G, Carlson L, Poljak L, Shores EW, Epstein S, Leonardi A, et al. Mice deficient in nuclear factor (NF)-kappa B/p52 present with defects in humoral responses, germinal center reactions, and splenic microarchitecture. Journal of Experimental Medicine 1998;187(2):147–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Mills DM, Bonizzi G, Karin M, Rickert RC. Regulation of late B cell differentiation by intrinsic IKK alpha-dependent signals. Proceedings of the National Academy of Sciences of the United States of America 2007;104(15):6359–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Claudio E, Brown K, Park S, Wang HS, Siebenlist U. BAFF-induced NEMO-independent processing of NF-kappa B2 in maturing B cells. Nature Immunology 2002;3(10):958–65. [DOI] [PubMed] [Google Scholar]
- 106.Brightbill HD, Jackman JK, Suto E, Kennedy H, Jones C, Chalasani S, et al. Conditional Deletion of NF-kappa B-Inducing Kinase (NIK) in Adult Mice Disrupts Mature B Cell Survival and Activation. Journal of immunology 2015;195(3):953–64. [DOI] [PubMed] [Google Scholar]
- 107.Yu JY, Zhou XF, Nakaya M, Jin W, Cheng XH, Sun SC. T Cell-Intrinsic Function of the Noncanonical NF-kappa B Pathway in the Regulation of GM-CSF Expression and Experimental Autoimmune Encephalomyelitis Pathogenesis. Journal of immunology 2014;193(1):422–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Murray SE, Polesso F, Rowe AM, Basak S, Koguchi Y, Toren KG, et al. NF-kappa B-inducing kinase plays an essential T cell-intrinsic role in graft-versus-host disease and lethal autoimmunity in mice. Journal of Clinical Investigation 2011;121(12):4775–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Li YC, Wang H, Zhou XF, Xie XP, Chen X, Jie ZL, et al. Cell intrinsic role of NF-kappa B-inducing kinase in regulating T cell-mediated immune and autoimmune responses. Sci Rep-Uk 2016;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Rowe AM, Murray SE, Raue HP, Koguchi Y, Slifka MK, Parker DC. A Cell-Intrinsic Requirement for NF-kappa B-Inducing Kinase in CD4 and CD8 T Cell Memory. Journal of immunology 2013;191(7):3663–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Xiao GT, Harhaj EW, Sun SC. NF-kappa B-inducing kinase regulates the processing of NF-kappa B2 p100. Molecular cell 2001;7(2):401–9. [DOI] [PubMed] [Google Scholar]
- 112.Senftleben U, Cao YX, Xiao GT, Greten FR, Krahn G, Bonizzi G, et al. Activation by IKK alpha of a second, evolutionary conserved, NF-kappa B signaling pathway. Science 2001;293(5534):1495–9. [DOI] [PubMed] [Google Scholar]
- 113.Dejardin E, Droin NM, Delhase M, Haas E, Cao YX, Makris C, et al. The lymphotoxin-beta receptor induces different patterns of gene expression via two NF-kappa B pathways. Immunity 2002;17(4):525–35. [DOI] [PubMed] [Google Scholar]
- 114.Xiao GT, Fong A, Sun SC. Induction of p100 processing by NF-kappa B-inducing kinase involves docking I kappa B kinase alpha (IKK alpha) to p100 and IKK alpha-mediated phosphorylation. J Biol Chem 2004;279(29):30099–105. [DOI] [PubMed] [Google Scholar]
- 115.Vallabhapurapu S, Matsuzawa A, Zhang WZ, Tseng PH, Keats JJ, Wang HP, et al. Nonredundant and complementary functions of TRAF2 and TRAF3 in a ubiquitination cascade that activates NIK-dependent alternative NF-kappa B signaling. Nature Immunology 2008;9(12):1364–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Shinzawa M, Konno H, Qin J, Akiyama N, Miyauchi M, Ohashi H, et al. Catalytic subunits of the phosphatase calcineurin interact with NF-κB-inducing kinase (NIK) and attenuate NIK-dependent gene expression. Sci Rep 2015;5:10758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Wang XY, Huang Y, Li L, Wei Q. TRAF3 negatively regulates calcineurin-NFAT pathway by targeting calcineurin B subunit for degradation. IUBMB Life 2012;64(9):748–56. [DOI] [PubMed] [Google Scholar]
- 118.Stadanlick JE, Kaileh M, Karnell FG, Scholz JL, Miller JP, Quinn WJ, 3rd, et al. Tonic B cell antigen receptor signals supply an NF-kappaB substrate for prosurvival BLyS signaling. Nat. Immunol 2008;9(12):1379–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Gascoigne NRJ, Rybakin V, Acuto O, Brzostek J. TCR Signal Strength and T Cell Development. Annual Review of Cell and Developmental Biology 2016;32(1):327–48. [DOI] [PubMed] [Google Scholar]
- 120.Stritesky GL, Jameson SC, Hogquist KA. Selection of self-reactive T cells in the thymus. Annu Rev Immunol 2012;30:95–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Caton AJ, Cozzo C, Larkin J, III, Lerman MA, Boesteanu A, Jordan MS. CD4(+) CD25(+) regulatory T cell selection. Ann. NY Acad. Sci 2004;1029:101–14. [DOI] [PubMed] [Google Scholar]
- 122.Bettini ML, Vignali DAA. Development of Thymically-Derived Natural Regulatory T Cells. Annals of the New York Academy of Sciences 2010;1183:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Schuster M, Plaza-Sirvent C, Matthies AM, Heise U, Jeron A, Bruder D, et al. c-REL and IkBNS Govern Common and Independent Steps of Regulatory T Cell Development from Novel CD122-Expressing Pre-Precursors. Journal of immunology 2017;199(3):920–30. [DOI] [PubMed] [Google Scholar]
- 124.Daley SR, Hu DY, Goodnow CC. Helios marks strongly autoreactive CD4(+) T cells in two major waves of thymic deletion distinguished by induction of PD-1 or NF-kappa B. Journal of Experimental Medicine 2013;210(2):269–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Grigoriadis G, Vasanthakumar A, Banerjee A, Grumont R, Overall S, Gleeson P, et al. c-Rel Controls Multiple Discrete Steps in the Thymic Development of Foxp3(+) CD4 Regulatory T Cells. Plos One 2011;6(10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Setoguchi R, Hori S, Takahashi T, Sakaguchi S. Homeostatic maintenance of natural Foxp3+CD25+CD4+ regulatory T cells by interleukin (IL)-2 and induction of autoimmune disease by IL-2 neutralization. The Journal of experimental medicine 2005;201(5):723–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Lio CW, Hsieh CS. A two-step process for thymic regulatory T cell development. Immunity 2008;28(1):100–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Wu Y, Borde M, Heissmeyer V, Feuerer M, Lapan AD, Stroud JC, et al. FOXP3 controls regulatory T cell function through cooperation with NFAT. Cell 2006;126(2):375–87. [DOI] [PubMed] [Google Scholar]
- 129.Tone Y, Furuuchi K, Kojima Y, Tykocinski ML, Greene MI, Tone M. Smad3 and NFAT cooperate to induce Foxp3 expression through its enhancer. Nat. Immunol 2008;9(2):194–202. [DOI] [PubMed] [Google Scholar]
- 130.Bopp T NFATc2 and NFATc3 transcription factors play a crucial role in suppression of CD4(+) T lymphocytes by CD4(+) CD25(+) regulatory T cells 2005;201(2):181–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Schmidt-Supprian M, Courtois G, Tian J, Coyle AJ, Israel A, Rajewsky K, et al. Mature T cells depend on signaling through the IKK complex. Immunity 2003;19(3):377–89. [DOI] [PubMed] [Google Scholar]
- 132.Gupta S, Manicassamy S, Vasu C, Kumar A, Shang W, Sun Z. Differential requirement of PKC-theta in the development and function of natural regulatory T cells. Molecular Immunology 2008;46(2):213–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Long M, Park SG, Strickland I, Hayden MS, Ghosh S. Nuclear factor-kappaB modulates regulatory T cell development by directly regulating expression of Foxp3 transcription factor. Immunity 2009;31(6):921–31. [DOI] [PubMed] [Google Scholar]
- 134.Oh H, Ghosh S. NF-kappaB: roles and regulation in different CD4(+) T-cell subsets. Immunol. Rev 2013;252(1):41–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Wan YSY, Chi H, Xie M, Schneider MD, Flavell RA. The kinase TAK1 integrates antigen and cytokine receptor signaling for T cell development, survival and function. Nature Immunology 2006;7(8):851–8. [DOI] [PubMed] [Google Scholar]
- 136.Isomura I, Palmer S, Grumont RJ, Bunting K, Hoyne G, Wilkinson N, et al. c-Rel is required for the development of thymic Foxp3(+) CD4 regulatory T cells (vol 206, pg 3001, 2009). Journal of Experimental Medicine 2010;207(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Ruan QG, Kameswaran V, Tone Y, Li L, Liou HC, Greene MI, et al. Development of Foxp3(+) Regulatory T Cells Is Driven by the c-Rel Enhanceosome. Immunity 2009;31(6):932–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Sivakumar V, Hammond KJ, Howells N, Pfeffer K, Weih F. Differential requirement for Rel/nuclear factor kappa B family members in natural killer T cell development. The Journal of Experimental Medicine 2003;197(12):1613–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Kontgen F, Grumont RJ, Strasser A, Metcalf D, Li R, Tarlinton D, et al. Mice lacking the c-rel proto-oncogene exhibit defects in lymphocyte proliferation, humoral immunity, and interleukin-2 expression. Genes Dev 1995;9(16):1965–77. [DOI] [PubMed] [Google Scholar]
- 140.Venkataraman L, Burakoff SJ, Sen R. FK506 inhibits antigen receptor-mediated induction of c-rel in B and T lymphoid cells. The Journal of Experimental Medicine 1995;181(3):1091–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Gilmore TD, Gerondakis S. The c-Rel Transcription Factor in Development and Disease. Genes & Cancer 2011;2(7):695–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Vaeth M, Schliesser U, Müller G, Reissig S, Satoh K, Tuettenberg A, et al. Dependence on nuclear factor of activated T-cells (NFAT) levels discriminates conventional T cells from Foxp3(+) regulatory T cells. Proc Natl Acad Sci U S A 2012;109(40):16258–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Oh H, Grinberg-Bleyer Y, Liao W, Maloney D, Wang PZ, Wu ZK, et al. An NF-kappa B Transcription-Factor-Dependent Lineage-Specific Transcriptional Program Promotes Regulatory T Cell Identity and Function. Immunity 2017;47(3):450- [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Mosmann TR, Cherwinski H, Bond MW, Giedlin MA, Coffman RL. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. Journal of immunology 1986;136(7):2348–57. [PubMed] [Google Scholar]
- 145.Killar L, MacDonald G, West J, Woods A, Bottomly K. Cloned, Ia-restricted T cells that do not produce interleukin 4(IL 4)/B cell stimulatory factor 1(BSF-1) fail to help antigen-specific B cells. Journal of immunology 1987;138(6):1674–9. [PubMed] [Google Scholar]
- 146.Zhu J, Yamane H, Paul WE. Differentiation of Effector CD4 T Cell Populations. Annu Rev Immunol 2010;28:445–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Hsieh CS, Macatonia SE, Tripp CS, Wolf SF, O’Garra A, Murphy KM. Development of TH1 CD4+ T cells through IL-12 produced by Listeria-induced macrophages. Science 1993;260(5107):547–9. [DOI] [PubMed] [Google Scholar]
- 148.Le Gros G, Ben-Sasson SZ, Seder R, Finkelman FD, Paul WE. Generation of interleukin 4 (IL-4)-producing cells in vivo and in vitro: IL-2 and IL-4 are required for in vitro generation of IL-4-producing cells. The Journal of Experimental medicine 1990;172(3):921–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Swain SL, Weinberg AD, English M, Huston G. IL-4 directs the development of Th2-like helper effectors. Journal of immunology 1990;145(11):3796–806. [PubMed] [Google Scholar]
- 150.Vaeth M, Eckstein M, Shaw PJ, Kozhaya L, Yang J, Berberich-Siebelt F, et al. Store-Operated Ca2+ Entry in Follicular T Cells Controls Humoral Immune Responses and Autoimmunity. Immunity 2016;44(6):1350–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Mason NJ, Liou HC, Hunter CA. T cell-intrinsic expression of c-Rel regulates Th1 cell responses essential for resistance to Toxoplasma gondii. Journal of immunology 2004;172(6):3704–11. [DOI] [PubMed] [Google Scholar]
- 152.Kiani A, Garcia-Cozar FJ, Habermann I, Laforsch S, Aebischer T, Ehninger G, et al. Regulation of interferon-gamma gene expression by nuclear factor of activated T cells. Blood 2001;98(5):1480–8. [DOI] [PubMed] [Google Scholar]
- 153.Das J, Chen CH, Yang L, Cohn L, Ray P, Ray A. A critical role for NF-kappa B in GATA3 expression and TH2 differentiation in allergic airway inflammation. Nat. Immunol 2001;2(1):45–50. [DOI] [PubMed] [Google Scholar]
- 154.Yoshida H, Nishina H, Takimoto H, Marengere LE, Wakeham AC, Bouchard D, et al. The transcription factor NF-ATc1 regulates lymphocyte proliferation and Th2 cytokine production. Immunity 1998;8(1):115–24. [DOI] [PubMed] [Google Scholar]
- 155.Ranger AM, Hodge MR, Gravallese EM, Oukka M, Davidson L, Alt FW, et al. Delayed lymphoid repopulation with defects in IL-4-driven responses produced by inactivation of NF-ATc. Immunity 1998;8(1):125–34. [DOI] [PubMed] [Google Scholar]
- 156.Feske S Immunodeficiency due to defects in store-operated calcium entry. AnnNYAcadSci 2011;1238:74–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Linterman MA, Pierson W, Lee SK, Kallies A, Kawamoto S, Rayner TF, et al. Foxp3+ follicular regulatory T cells control the germinal center response. Nature Medicine 2011;17(8):975–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Johnston RJ, Poholek AC, DiToro D, Yusuf I, Eto D, Barnett B, et al. Bcl6 and Blimp-1 are reciprocal and antagonistic regulators of T follicular helper cell differentiation. Science 2009;325(5943):1006–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Vaeth M, Muller G, Stauss D, Dietz L, Klein-Hessling S, Serfling E, et al. Follicular regulatory T cells control humoral autoimmunity via NFAT2-regulated CXCR5 expression. The Journal of Experimental Medicine 2014;211(3):545–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Kim HP, Korn LL, Gamero AM, Leonard WJ. Calcium-dependent activation of interleukin-21 gene expression in T cells. J Biol Chem 2005;280(26):25291–7. [DOI] [PubMed] [Google Scholar]
- 161.Chen G, Hardy K, Bunting K, Daley S, Ma L, Shannon MF. Regulation of the IL-21 gene by the NF-kappaB transcription factor c-Rel. Journal of immunology 2010;185(4):2350–9. [DOI] [PubMed] [Google Scholar]
- 162.Bollig N, Brustle A, Kellner K, Ackermann W, Abass E, Raifer H, et al. Transcription factor IRF4 determines germinal center formation through follicular T-helper cell differentiation. Proc Natl Acad Sci U S A 2012;109(22):8664–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Vaeth M, Maus M, Klein-Hessling S, Freinkman E, Yang J, Eckstein M, et al. Store-Operated Ca(2+) Entry Controls Clonal Expansion of T Cells through Metabolic Reprogramming. Immunity 2017;47(4):664–79.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Grumont RJ, Strasser A, Gerondakis S. B cell growth is controlled by phosphatidylinosotol 3-kinase-dependent induction of Rel/NF-kappaB regulated c-myc transcription. Molecular Cell 2002;10(6):1283–94. [DOI] [PubMed] [Google Scholar]
- 165.Kaileh M, Sen R. NF-kappaB function in B lymphocytes. Immunological reviews 2012;246(1):254–71. [DOI] [PubMed] [Google Scholar]
- 166.Allman D, Pillai S. Peripheral B cell subsets. Current opinion in immunology 2008;20(2):149–57. Epub 2008/04/25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Mao X, Zhang J, Han Y, Luan C, Hu Y, Hao Z, et al. Deficient for endoplasmic reticulum calcium sensors Stim1 and Stim2 affects aberrant antibody affinity maturation in B cells. Oncotarget 2016;7(38):60885–95. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 168.Enders A, Bouillet P, Puthalakath H, Xu Y, Tarlinton DM, Strasser A. Loss of the Pro-Apoptotic BH3-only Bcl-2 Family Member Bim Inhibits BCR Stimulation– induced Apoptosis and Deletion of Autoreactive B Cells. The Journal of Experimental Medicine 2003;198(7):1119–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Grossmann M, O’Reilly LA, Gugasyan R, Strasser A, Adams JM, Gerondakis S. The anti-apoptotic activities of Rel and RelA required during B-cell maturation involve the regulation of Bcl-2 expression. EMBO J 2000;19(23):6351–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Grossmann M, Metcalf D, Merryfull J, Beg A, Baltimore D, Gerondakis S. The combined absence of the transcription factors Rel and RelA leads to multiple hemopoietic cell defects. Proc Natl Acad Sci U S A 1999;96(21):11848–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Derudder E, Cadera EJ, Vahl JC, Wang J, Fox CJ, Zha S, et al. Development of immunoglobulin lambda-chain-positive B cells, but not editing of immunoglobulin kappa-chain, depends on NF-kappaB signals. Nat. Immunol 2009;10(6):647–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Sen R Control of B lymphocyte apoptosis by the transcription factor NF-kappaB. Immunity 2006;25(6):871–83. [DOI] [PubMed] [Google Scholar]
- 173.Banerjee A, Grumont R, Gugasyan R, White C, Strasser A, Gerondakis S. NF-κB1 and c-Rel cooperate to promote the survival of TLR4-activated B cells by neutralizing Bim via distinct mechanisms. Blood 2008;112(13):5063–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Chen C, Edelstein LC, Gelinas C. The Rel/NF-kappaB family directly activates expression of the apoptosis inhibitor Bcl-x(L). Mol Cell Biol 2000;20(8):2687–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Fillatreau S, Sweenie CH, McGeachy MJ, Gray D, Anderton SM. B cells regulate autoimmunity by provision of IL-10. Nat. Immunol 2002;3(10):944–50. [DOI] [PubMed] [Google Scholar]
- 176.Saraiva M, Christensen JR, Tsytsykova AV, Goldfeld AE, Ley SC, Kioussis D, et al. Identification of a Macrophage-Specific Chromatin Signature in the IL-10 Locus. The Journal of Immunology 2005;175(2):1041–6. [DOI] [PubMed] [Google Scholar]
- 177.Tumang JR, Hsia CY, Tian W, Bromberg JF, Liou HC. IL-6 rescues the hyporesponsiveness of c-Rel deficient B cells independent of Bcl-xL, Mcl-1, and Bcl-2. Cell Immunol 2002;217(1–2):47–57. [DOI] [PubMed] [Google Scholar]
- 178.Wessells J, Baer M, Young HA, Claudio E, Brown K, Siebenlist U, et al. BCL-3 and NF-kappaB p50 attenuate lipopolysaccharide-induced inflammatory responses in macrophages. J Biol Chem 2004;279(48):49995–50003. [DOI] [PubMed] [Google Scholar]