

Treatment of stage IV cancer, when metastases are detectable by conventional imaging methods, is commonly the only strategy we have to defend against this lethal stage of disease. However, the seeds of those lesions, the disseminated cancer cells (DCCs), can reside in a patient’s organs for long periods before their outgrowth (1). DCCs are proposed to be able to persist for years because they interpret homeostatic signals from the host microenvironment and respond by entering a long-lasting dormant state, with occasional cell divisions (1, 2). In response to signals not yet fully identified, dormant DCCs reawaken and grow into larger lesions. So why not intervene earlier when DCCs are dormant? This has presented a challenge because the biology of DCC dormancy was mechanistically unclear and there was no way to determine whether DCCs were dormant. However, two de cades of mechanistic research (1, 2) and recent work by Pommier et al. (3) and on page XXX of this issue by Albrengues et al. (4) have provided important insights into the micro-environmental control of DCC dormancy and reawakening that leads to metastasis, which could have therapeutic implications.
“…disseminated cancer cells… can reside in a patients’s organs for long periods”
The dormancy of DCCs has been mostly explained by the ability of these cancer cells to enter quiescence, a reversible and programmed growth arrest (1). Clinical evidence and experimental models support this mechanistic view, which helps to explain why in some cancers (for example, estrogen receptor–positive breast cancer and prostate cancer), patients can be free of disease for more than a dozen years. The implications of this biology are profound; it offers a window of opportunity to treat metastasis early and to understand how host and epigenetic factors override the driver genetics that supposedly can only fuel cancer growth. This new theory of metastasis progression (1) has led to innovative questions such as those addressed by Albrengues et al. and Pommier et al.
Pommier et al. investigated how DCCs evade the immune system. They found that mice immunized against pancreatic cancer cells produced an immune response that killed most of the DCCs when they were subsequently engrafted. But some solitary DCCs persisted, which suggests that CD8+ T cells did not recognize them. Careful analysis confirmed that DCCs are quiescent and lack expression of the epithelial cell marker E-cadherin (epithelial cadherin), but do not undergo an obvious epithelial-mesenchymal transition, which has been linked to increased cancer cell adaptability to changing microenvironments and initiation of metastasis. Importantly, they found that dormant DCCs activate components of the unfolded protein response (UPR), a proteotoxicity survival pathway (5). The UPR was previously found to promote the survival of dormant cancer cells implanted in immunodeficient mice [patient-derived xenograft (PDX)], and it has been linked to how cancer cells respond both to internal (metabolic) stress and external stress (adaptation to foreign extracellular matrix composition) (6, 7). Pommier et al. found that the UPR caused posttranscriptional down-regulation of major histocompatibility complex (MHC) class I molecules, which present antigens to CD8+ T cells. Thus, the UPR also controls how dormant DCCs are interacting with the immune microenvironment. This decrease in MHC class I expression was associated with activated UPR and rendered dormant DCCs undetectable by CD8+ T cells; restoration of UPR activity caused DCCs to reexpress E-cadherin and MHC class I, reversing the immune-evasive phenotype so CD8+ T cells could detect them in vivo.
Different cancer types exhibit predictable metastatic distribution, but the timing of relapse is variable and appears to be random. This has prompted the suggestion that both DCC-intrinsic and microenvironmental factors influence the rate of relapse (1, 2). Albrengues et al. investigated what reactivates dormant DCCs. A prevalent theory is that inflammation may activate immune or other host cells to produce signals that break dormancy (8). Albrengues et al. showed that bacterial-derived lipopolysaccharide (LPS), a trigger of inflammation, or cigarette smoke (which may carry LPS as a contaminant) can activate neutrophils to release their DNA content into the lung parenchyma to form neutrophil extracellular traps (NETs) that usually capture microorganisms. NETs are also associated with neutrophil degranulation and release of proteases such as neutrophil elastase (NE) and matrix metalloprotease 9 (MMP9). The NETs work as scaffolds, and with the proteases they enhance the processing of laminin-111, a basement membrane component, which consequently binds α3β1 integrin and activates focal adhesion kinase (FAK) signaling in dormant DCCs. This FAK activation induced proliferation of DCCs in several mouse models of cancer, confirming previous findings from various laboratories (1). An α3β1 integrin-blocking antibody prevented reactivation of dormant DCCs in NET-rich lungs. These findings might explain why heavy smoking, before and after treatment of primary breast tumors, increases the risk of recurrence. However, Albrengues et al. also found that proliferative breast cancer DCCs could induce NETs, which suggests that signals other than smoking-induced inflammation may also reawaken dormant DCCs (patients who do not smoke also relapse). Perhaps NETs that are induced by active DCCs promote their growth and/or induce an immune-suppressive microenvironment.
These two elegant studies reveal how dormant DCCs may evade adaptive immunity and can be reawakened by innate immune cells (neutrophils) responding to nontumor inflammation (see the figure). The ability of DCCs to evade adaptive immunity has previously been shown for the survival of dormant leukemic cells in a mouse model of acute my elogenous leukemia (AML) (9). The dormant AML cells up-regulated the immune checkpoint proteins PD-L1 (programmed cell death protein 1 ligand 1) and the CTLA-4 (cytotoxic T lymphocyte–associated antigen 4) ligand B7.1, which prevent T cell activation. It would be interesting to determine whether PD-L1 and CTLA-4 signaling are also involved in immune evasion of dormant DCCs with UPR activation because of the availability of immune checkpoint inhibitors and the development of drugs targeting the UPR (10).
Dormant DCC immune evasion and activation to form metastasis.
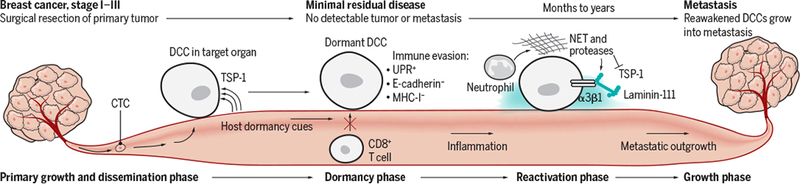
This simplified model shows that microenvironmental cues, such as TSP-1, induce and maintain dormancy. Dormant DCCs down-regulate E-cadherin expression and up- regulate UPR activity, which results in MHC-I down-regulation and immune evasion from CD8+ T cells. Eventually, unpredictable changes in the host cause inflammation and other tissue-wide changes. Inflammation can recruit neutrophils to produce NETs and the associated proteases cause proteolytic processing of laminin-111 and TSP1 degradation. This causes α3β1 integ sin-mediated activation of dormant DCCs that proliferate to form metastasis.
What is the source of UPR activation? Is it simply a response to enhanced translational output from oncogenic signaling (11)? Or is it induced by the microenvironment? Quiescent hair follicle stem cells (HFSCs) reside in niches, specialized microenvironments that control their quiescence, and in this state, HFSCs down-regulate MHC class I and evade CD8+ T cell immune recognition (12). HFSCs also engage specific pathways to repress translation initiation (a function of the UPR) for proper stem cell function during entry into and exit from quiescence (13), which suggests that attenuation of translation is not only a response but also a morphogenetic program. Niche-driven quiescence and translational attenuation are also linked to dormancy of DCCs in various models (even immune-deficient models) (1, 2), raising the possibility that MHC class I down-regulation in DCCs may recapitulate an adult stem cell program that maintains quiescence.
The findings of Albrengues et al. suggest that antibodies that block the binding of α3β1 integrin to laminin-111 (rather than blocking all α3β1 integrin binding events) could prevent reactivation of DCCs. Detection of the laminin-111 epitope, exposed by proteolytic processing, by the antibody Ab28 in patients who have undergone surgery and have minimal residual disease may be used as an early marker for relapse, allowing for prompt intervention. Furthermore, the proteolytic degradation of thrombospondin-1 (TSP-1), a protein linked to dormancy and metastasis suppression (14), also has a role in NET-induced reawakening of DCCs. Albrengues et al. found that laminin-111 processing could not induce DCC activation unless TSP-1 was degraded. Therefore, TSP-1 activity may need to be overcome for inflammation or other signals to reawaken dormant DCCs.
Together, the innovative studies by Pommier et al. and Albrengues et al. open exciting avenues to explore opportunities for monitoring and targeting dormant DCCs during minimal residual disease stages, to explore how DCC dormancy is affected by inflammation, and to identify possible targets in dormant DCCs that may impair the success of stage IV cancer treatments. This work also offers an opportunity to study how the mechanisms found in dormant DCCs are related to organism development and tissue homeostasis. Moreover, how oncogenic driver alterations affect DCCs and how the function of such driver mutations is influenced by dormancy mechanisms can be investigated. This could help to better define safer therapies that specifically target dormant or reactivating DCCs to prevent metastasis and relapse.
REFERENCES
- 1.Sosa MS et al. , Nat. Rev. Cancer 14, 611 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Giancotti FG, Cell 155, 750 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pommier A et al. , Science 360, eaao4908 (2018).29773669 [Google Scholar]
- 4.Albrengues J et al. , Science 361, eaao4227 (2018).30262472 [Google Scholar]
- 5.Hetz C, Papa FR, Mol. Cell 69, 169 (2018). [DOI] [PubMed] [Google Scholar]
- 6.Schewe D, Aguirre-Ghiso JA, Proc. Natl. Acad. Sci. U.S.A 105, 10519 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ranganathan AC et al. , Cancer Res. 66, 1702 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.El Rayes T et al. , Proc. Natl. Acad. Sci. U.S.A 112, 16000 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Saudemont A, Quesnel B, Blood 104, 2124 (2004). [DOI] [PubMed] [Google Scholar]
- 10.Pytel D et al. , PLOS Genet. 12, e1006518 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hart LS et al. , J. Clin. Invest 122, 4621 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Agudo J et al. , Immunity 48, 271 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Blanco S et al. , Nature 534, 335 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ghajar CM et al. , Nat. Cell. Biol 15, 807 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]