

Supplemental Digital Content is available in the text.
Keywords: angiotensin II; apolipoproteins E; fatty acids, volatile; immunology; inflammation; microbiota; Th17 cells; T-lymphocytes, regulatory
Abstract
Background:
Arterial hypertension and its organ sequelae show characteristics of T cell–mediated inflammatory diseases. Experimental anti-inflammatory therapies have been shown to ameliorate hypertensive end-organ damage. Recently, the CANTOS study (Canakinumab Antiinflammatory Thrombosis Outcome Study) targeting interleukin-1β demonstrated that anti-inflammatory therapy reduces cardiovascular risk. The gut microbiome plays a pivotal role in immune homeostasis and cardiovascular health. Short-chain fatty acids (SCFAs) are produced from dietary fiber by gut bacteria and affect host immune homeostasis. Here, we investigated effects of the SCFA propionate in 2 different mouse models of hypertensive cardiovascular damage.
Methods:
To investigate the effect of SCFAs on hypertensive cardiac damage and atherosclerosis, wild-type NMRI or apolipoprotein E knockout–deficient mice received propionate (200 mmol/L) or control in the drinking water. To induce hypertension, wild-type NMRI mice were infused with angiotensin II (1.44 mg·kg–1·d–1 subcutaneous) for 14 days. To accelerate the development of atherosclerosis, apolipoprotein E knockout mice were infused with angiotensin II (0.72 mg·kg–1·d–1 subcutaneous) for 28 days. Cardiac damage and atherosclerosis were assessed using histology, echocardiography, in vivo electrophysiology, immunofluorescence, and flow cytometry. Blood pressure was measured by radiotelemetry. Regulatory T cell depletion using PC61 antibody was used to examine the mode of action of propionate.
Results:
Propionate significantly attenuated cardiac hypertrophy, fibrosis, vascular dysfunction, and hypertension in both models. Susceptibility to cardiac ventricular arrhythmias was significantly reduced in propionate-treated angiotensin II–infused wild-type NMRI mice. Aortic atherosclerotic lesion area was significantly decreased in propionate-treated apolipoprotein E knockout–deficient mice. Systemic inflammation was mitigated by propionate treatment, quantified as a reduction in splenic effector memory T cell frequencies and splenic T helper 17 cells in both models, and a decrease in local cardiac immune cell infiltration in wild-type NMRI mice. Cardioprotective effects of propionate were abrogated in regulatory T cell–depleted angiotensin II–infused mice, suggesting the effect is regulatory T cell–dependent.
Conclusions:
Our data emphasize an immune-modulatory role of SCFAs and their importance for cardiovascular health. The data suggest that lifestyle modifications leading to augmented SCFA production could be a beneficial nonpharmacological preventive strategy for patients with hypertensive cardiovascular disease.
Clinical Perspective.
What Is New?
The present study shows that propionate, a short-chain fatty acid produced by intestinal bacteria, has profound beneficial anti-inflammatory properties limiting cardiovascular disease progression in 2 independent mouse models.
Oral propionate administration beneficially influenced T helper cell homeostasis, thereby reducing cardiac hypertrophy and fibrosis, susceptibility to cardiac arrhythmias, and atherosclerotic lesion burden.
Propionate exhibited antihypertensive effects in both models.
Cardioprotective effects of propionate effects are mainly dependent on regulatory T cells.
What Are the Clinical Implications?
The data suggest increased attention to propionate in hypertensive patients with a high cardiovascular risk and evidence of ongoing chronic inflammation.
Oral propionate supplementation is simple, inexpensive, and regarded safe in humans.
Further studies are needed to characterize the effect of propionate supplementation or interventions increasing propionate production by the gut microbiota in humans.
Hypertension drives cardiovascular disease by causing an array of pathological organ sequelae. Hypertensive tissue damage is largely mediated by different immune cells, which are activated in hypertension.1 Hypertensive stimuli like angiotensin II (AngII) promote the activation of T cells and macrophages,2 which subsequently may infiltrate target organs like the heart and the vasculature to cause tissue damage. Increased generation of effector memory T cells (TEM) is indicative of a chronic inflammatory response in hypertension.3 In particular, T helper cell subtypes Th17 and Th1 may promote hypertension and target organ injury by releasing proinflammatory cytokines like interleukin (IL)–17A and interferon-γ, respectively.4,5 In contrast, regulatory T cells (Treg) counterbalance tissue inflammation by secreting anti-inflammatory IL-10.6 Thus, the extent of hypertensive target organ damage is not solely dependent on the hemodynamic load, but, in addition, depends on the balance of pro- and anti-inflammatory immune cells. In clinical practice, hypertension usually coexists with atherosclerosis and accelerates atherosclerosis progression.7 Atherosclerosis is similarly a chronic inflammatory disease of the vasculature, although the precise nature of the inflammatory response differs from hypertension.8 Inflammation and vascular damage are intensified when both hypertension and atherosclerosis are present.9 Furthermore, experimental and clinical anti-inflammatory approaches, as recently shown in the CANTOS trial (Canakinumab Antiinflammatory Thrombosis Outcome Study),10 ameliorate hypertensive inflammatory tissue damage11 and atherosclerosis.12 In particular, enhancement of Treg by adoptive transfer has been shown to reduce effector T cells and ameliorate cardiac damage,13 hypertension, vascular injury,14 and atherosclerosis.15
The gut microbiota is considered important for cardiovascular health,16 and abnormal bacterial communities have been associated with hypertension17,18 and atherosclerosis.19 Bacterial metabolites mediate interactions with the host. Through resorption and distribution, they can affect intestinal health, and distant functions of the immune system, the vasculature, and the heart, as well. Short-chain fatty acids (SCFAs) are a major class of bacterial metabolites and are mainly produced in the colon by bacterial fermentation of otherwise indigestible polysaccharides (fibers).20 Beyond their importance for intestinal integrity, SCFAs have potent anti-inflammatory effects on immune cell functions. The SCFA propionate (C3) has been shown to induce the differentiation and suppressive capacity of CD25+ Foxp3+ Treg.21,22 Propionate treatment of murine Treg donors significantly enhanced the protective effect of a Treg adoptive transfer in experimental autoimmune encephalomyelitis.23 In vitro, propionate was shown to enhance the differentiation of different murine T cells dependent on the cytokine milieu.24 It is interesting to note that propionate promoted a regulatory phenotype among T cells under Th17 polarization conditions.24 Beneficial effects of SCFAs have been demonstrated in several other disease models, such as colitis, airway disease, metabolic syndrome, or ischemia-reperfusion injury of the kidney.25 Although their mode of action has yet to be elucidated, SCFAs may exert their effects via inhibition of histone deacetylases (HDACs) or via G-protein–coupled receptors (Gpr) 41 and 43 and olfactory receptor 78.25,26 In addition, a direct effect of SCFAs on renin release and vasomotor function leading to blood pressure reduction was recently suggested.26–28
Considering the prominent role of inflammation in both hypertension and atherosclerosis and recent insights about anti-inflammatory and Treg-promoting effects of SCFAs, we hypothesized that the SCFA propionate protects from AngII-induced cardiac and vascular damage. To test our hypothesis, we treated AngII-infused wild-type NMRI (WT) and AngII-infused apolipoprotein E knockout (ApoE–/–) mice with C3 and analyzed inflammatory response, cardiac remodeling, and atherosclerotic lesion burden. Our data underscore the importance of propionate for cardiovascular health.
Methods
The authors declare that all supporting data and analytical methods are available within the article and its online-only Data Supplement. The data, analytical methods, and study materials that support the findings of this study are available from the corresponding author on reasonable request.
Animal Ethics
All experiments were in accordance with the German/European law for animal protection and were approved by the local ethic committees (G0280/13, G250/12, and G301/18). Mice were maintained on a 12:12 hour day:night cycle with constant access to food and water.
AngII-Induced Hypertension in WT Mice
Twelve-week-old male NMRI mice (purchased from Janvier Labs, outbred strain, not genetically altered, referred to as wild-type [WT] in the article), received AngII infusion (1.44 mg·kg–1·d–1, Calbiochem) for 14 days via subcutaneous osmotic minipumps (Alzet). Implantation was performed under isoflurane anesthesia. Mice were fed a purified diet low in fiber (Ssniff, S0087-E050). To study the effects of propionate, mice received sodium propionate (200 mmol/L, Sigma-Aldrich) or sodium chloride as control (200 mmol/L, Sigma-Aldrich) in the drinking water ad libitum. On day 13, echocardiography was performed under isoflurane anesthesia. After 14 days of AngII infusion, the mice were euthanized; blood and organs were collected for further analysis. In a subset of mice, blood pressure was measured using an implantable subcutaneous radiotelemetry transmitter (Data Science International), implanted on day –10 before AngII infusion.
Treg depletion in WT mice was achieved by intraperitoneal injections of anti-CD25 antibody (clone PC61, kindly provided by T. Hünig, University of Würzburg, Germany; 250 µg on days –1, 2, and 5 of AngII infusion). Control mice received the IgG1 isotype (Bio X Cell). Control and Treg-depleted mice received a low-fiber diet, sodium propionate in drinking water, AngII infusion subcutaneously, and implantable radiotelemetry blood pressure transmitters.
All mice were held in 1 room of the animal facility. Animals were age-matched, stratified for their body weight, and randomly assigned to the different treatment groups (n=18 WT Sham, n=36 WT AngII, n=33 WT AngII+C3, n=4 WT AngII+C3+IgG, n=4 WT AngII+C3+PC61). Two mice from the WT AngII+C3 group were excluded because of suture insufficiency at the implantation site of the minipump. Mice assigned to the same treatment group were held in the same cage. Cages were inhabited by 2 to 3 mice. All researchers were blinded during the experiment and analysis. Measured values were excluded if a technical failure of the analysis occurred or by statistical testing described below. Animal numbers for the WT experiment were a priori calculated with G*power software (Heinrich-Heine-Universität Düsseldorf) and an estimated effect size from the literature. For Treg depletion experiments, sample sizes were calculated using G*power and the estimated effect size from the WT experiments. Exact numbers are shown in the respective figures.
AngII-Induced Atherosclerosis in ApoE–/– Mice
To verify the effects of propionate in another AngII-dependent cardiovascular mouse model, we used ApoE–/– mice. In line with the NC3Rs recommendation, we reduced sample sizes by working without a sham group. ApoE–/–mice (Taconic) were backcrossed on a C57BL/6 background at least 10 times. Eight-week-old mice received sodium propionate (200 mmol/L, Sigma-Aldrich) or sodium chloride as control (200 mmol/L, VWR) in the drinking water ad libitum during the whole experimental period, starting 5 days before the implantation of the osmotic minipumps. AngII osmotic minipumps (0.72 mg·kg–1·d–1, Sigma-Aldrich) were implanted subcutaneously as described previously.29 In the fourth week of AngII infusion, systolic blood pressure was measured in conscious mice by tail-cuff plethysmography (BP-98A; Softron). During week 4, 10 measurements per mouse were recorded daily. For habituation during week 3 of AngII minipump implantation, mice were trained daily for 5 consecutive days as follows. Mice were set daily for at least 15 minutes in the mice restrainer; each day at least 20 blood pressure measurements were performed. Systolic blood pressure mean was calculated from all measured days per mouse. After 4 weeks of AngII infusion, mice were euthanized, and blood and organs were collected and used for further analyses.
For ApoE–/– experiments, housing, randomization, blinding, and exclusion were performed as described for the WT experiments. Thirty mice were randomly assigned per group. None of the mice were excluded during the experiment. Sample sizes were calculated using G*power and the estimated effect size from the WT experiments. Exact numbers are shown in the respective figures.
Statistical Analysis
Outliers identified by Grubbs test were excluded, and normality was assessed by Kolmogorov-Smirnov test. In groups with n < 5, normality could not be assessed and nonparametric distribution was assumed. To compare 2 groups, a 1-tailed unpaired t test or 1-tailed Mann-Whitney test was used, as appropriate. To compare >2 groups, a 1-way ANOVA followed by post hoc Tukey test or a Kruskal-Wallis test with post hoc Dunn test was used, as appropriate. Survival of the mice was visualized by Kaplan-Meier curves and statistically compared by using a 1-tailed log-rank test. Statistical analyses were performed using GraphPad Prism 6. Blood pressure time courses measured by telemetry were compared using linear mixed model calculations in R (Version 3.1.1, R Foundation). We calculated a model for each time point by including all time points before. Thereby, we received a specific time point for each longitudinal analysis where the P value fell short of 0.05. To compare the blood pressure area under the curve per week of AngII infusion, we used a 2-way repeated-measures ANOVA followed by a post hoc Sidak test (GraphPad Prism 6). A P value <0.05 was considered statistically significant.
Results
Propionate Prevents AngII-Induced Systemic Inflammatory Response
To investigate the effect of propionate (C3) on immune homeostasis in an established model for hypertensive cardiac damage, WT mice were infused with AngII (1.44 mg·kg–1·d–1) for 2 weeks and concomitantly treated with C3 (sodium propionate, 200 mmol/L) or control (sodium chloride, 200 mmol/L) in the drinking water (Figure 1A). Saline-infused WT controls served as nonhypertensive shams. To specifically investigate the effect of exogenous C3, mice were fed a fiber-depleted purified diet to reduce intestinal SCFA production. C3 administration significantly increased C3 serum levels in AngII-infused WT mice as measured by gas chromatography–mass spectrometry (Figure IA in the online-only Data Supplement). C3 was well tolerated, as indicated by similar body weights in mice treated with C3 or control (Figure IB in the online-only Data Supplement). C3 significantly improved the survival in comparison with control-treated AngII-infused WT mice (Figure 1B). To investigate the role of C3 on systemic inflammation, spleens were harvested after 14 days and analyzed by flow cytometry. In comparison with sham-treated mice, AngII increased splenic CD4+ TEM (CD44+CD62L–) and conversely decreased CD4+ naive T cells (TN; CD44–CD62L+), indicating a significant inflammatory response. The increase in TEM and the decrease in TN were prevented by C3 treatment (Figure 1C). CD4+ central memory T cells (C44+CD62L+) remained unaffected (Figure IC in the online-only Data Supplement). Further analysis of splenic Th17 cells revealed an increase in CD4+IL-17A+IL-10– and CD4+RORγt+Foxp3– frequencies after AngII infusion, which was normalized by C3 treatment (Figure 1D and 1E). Splenic Th1 frequencies, as measured by CD4+Tbet+ (Figure ID in the online-only Data Supplement) and CD4+interferon-γ+ cells (Figure IE in the online-only Data Supplement), were not affected. It is interesting to note that Treg frequencies (CD4+CD25+Foxp3+ and CD4+IL-10+IL-17A–) increased after AngII, signaling a compensatory Treg response to the hypertensive stimulus, which was not observed on C3 treatment (Figure 1C and 1D).
Figure 1.
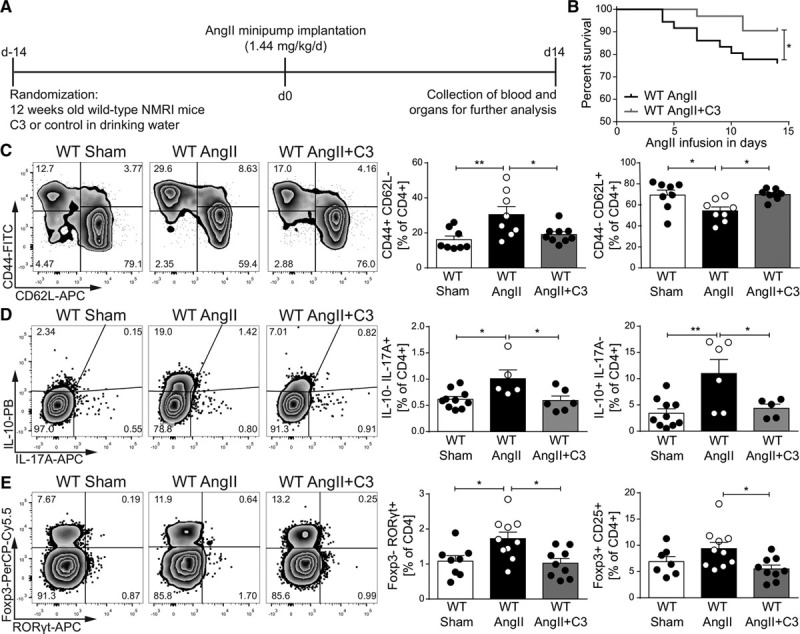
Propionate provides beneficial modulation of effector T cells in AngII-infused wild-type NMRI (WT) mice. A, AngII-infused WT mice were treated with sodium propionate (C3) or sodium chloride as a control, starting 2 weeks before AngII infusion. Saline-infused mice served as nonhypertensive control group (sham). B, Survival curves of AngII-infused WT mice treated with C3 or control. WT AngII n=36, WT AngII+C3 n=31, *P<0.05 by log-rank test. C, After 14 days of AngII, splenocytes were analyzed for CD4+ effector memory (CD44+CD62L–) and naive (CD44– CD62L+) subsets. Left, Representative flow cytometry plots. Right, Quantification in percentage of CD4+ cells. WT Sham n=8, WT AngII n=8, WT AngII+C3 n=9. D, Restimulated splenocytes were analyzed for IL-10 and IL-17A by flow cytometry. Left, Representative flow cytometry plots. Right, Quantification in percentage of CD4+. WT Sham n=10, WT AngII n=5 to 6, WT AngII+C3 n=5 to 6. E, Quantification of FoxP3+CD25+ and RORγt+ in CD4+ splenocytes. Left, Representative flow cytometry plots. Right, Quantification in percentage of CD4+. WT Sham n=7 to 8, WT AngII n=10, WT AngII+C3 n=9. *P<0.05, **P<0.01, 1-way ANOVA and Tukey post hoc for C through E. AngII indicates angiotensin II; APC, Allophycocyanin; FITC, fluorescein isothiocyanate; IL, interleukin; PB, Pacific Blue; and PerCP-Cy5.5, Peridinin-chlorophyll protein cyanine 5.5.
We next tested whether C3 also affects immune homeostasis in hypertensive mice prone to develop atherosclerosis. Therefore, we infused atherosclerosis-prone ApoE–/– mice fed a normal chow with AngII (0.72 mg·kg–1·d–1) for 4 weeks and administered C3 or control (200 mmol/L) via the drinking water (Figure 2A). Similar to WT mice, C3 had no nutritive effect (Figure IF in the online-only Data Supplement). C3 treatment protected AngII-infused ApoE–/– mice from aortic rupture and thereby reduced mortality during the 4 weeks of AngII infusion (Figure 2B). In congruence with our findings in hypertensive mice without atherosclerosis, C3 reduced splenic TEM and increased splenic TN populations in AngII-infused ApoE–/– (Figure 2C), whereas splenic central memory T cells were not significantly regulated (Figure IG in the online-only Data Supplement). In addition, C3 treatment significantly reduced CD4+Foxp3–RORγt+ Th17 cells, whereas CD4+CD25+FoxP3+ Treg were not affected (Figure 2D). Concordant with WT, no significant regulation of Th1 cells was observed (Figure IH in the online-only Data Supplement). Our data suggest that C3 treatment ameliorates systemic inflammation in hypertensive mice with and without atherosclerosis.
Figure 2.
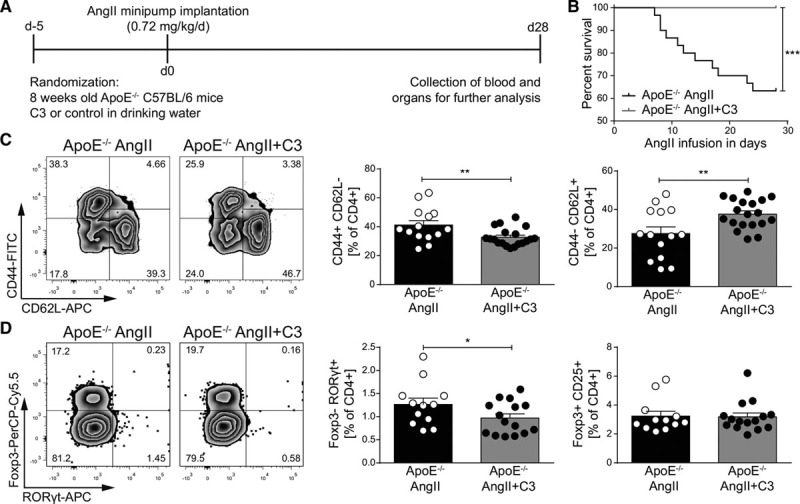
Propionate provides beneficial modulation of effector T cells in AngII-infused ApoE–/– mice. A, AngII-infused ApoE–/– mice were treated with C3 or sodium chloride, starting 5 days before minipump implantation. B, Survival curves of AngII-infused ApoE–/– mice treated with C3 or control. n=30 per group, ***P<0.001 by log-rank test. C, After 28 days of AngII infusion, splenocytes were analyzed for CD4+ effector memory (CD44+CD62L–) and naive (CD44–CD62L+) subsets. Left, Representative flow cytometry plots. Right, Quantification in percentage of CD4+. ApoE–/– AngII n=15, ApoE–/– AngII+C3 n=19. D, Quantification of FoxP3+CD25+ and RORγt+ in CD4+ splenocytes. Left, Representative flow cytometry plots. Right, Quantification in percentage of CD4+. ApoE–/– AngII n=12, ApoE–/– AngII+C3 n=15. *P<0.05, **P<0.01, by 1-tailed t test. AngII indicates angiotensin II; APC, Allophycocyanin; ApoE–/–, apolipoprotein E knockout–deficient; C3, propionate; FITC, fluorescein isothiocyanate; and PerCP-Cy5.5, Peridinin-chlorophyll protein cyanine 5.5.
Propionate Attenuates Vascular Inflammation and Atherosclerosis
Inflammation of the vascular wall is a hallmark of atherosclerosis and is amplified in the presence of elevated AngII levels.9 To investigate if C3 modulates the atherosclerotic inflammatory response, we analyzed aortic immune cells from AngII-infused ApoE–/– by flow cytometry. Aortic CD4+, CD8+ T cell, and F4/80+ macrophage numbers were reduced after C3 treatment (Figure 3A, Figure IIA in the online-only Data Supplement). Similar to splenic immune cells, the frequencies of aortic CD4+ TEM decreased and CD4+ TN increased after C3 treatment, whereas CD4+ central memory T cells remained unaltered (Figure 3B, Figure IIB and IIC in the online-only Data Supplement). Further verification by immunohistochemical staining was obtained in atherosclerotic plaques of the brachiocephalic artery. Fewer CD3+ T cells and F4/80+ macrophages were detected in brachiocephalic artery sections from C3-treated AngII-infused ApoE–/– mice (Figure 3C and 3D). To determine whether these potentially beneficial effects would translate into a reduction in atherosclerotic lesion burden, we performed en face Oil Red O staining of whole aortas. Aortic atherosclerotic lesion burden was significantly reduced in C3-treated mice (Figure 3E). Likewise, considerably less stenosis of the brachiocephalic artery was detected in C3-treated mice (Figure 3F). Because AngII infusion may induce cardiac remodeling in addition to atherosclerosis, we measured heart weight and analyzed cardiac fibrosis in AngII-infused ApoE–/– by Sirius red staining. Hypertrophy index and interstitial fibrosis were significantly attenuated in C3-treated AngII-infused ApoE–/– (Figure 3G through 3H). C3 did not affect serum levels of total cholesterol, triglycerides, high-density lipoprotein, or low-density lipoprotein cholesterol (Table I in the online-only Data Supplement). Taken together, C3 treatment reduced vascular inflammation, atherosclerotic lesion burden, and cardiac remodeling in AngII-infused ApoE–/–independent of blood lipid levels.
Figure 3.
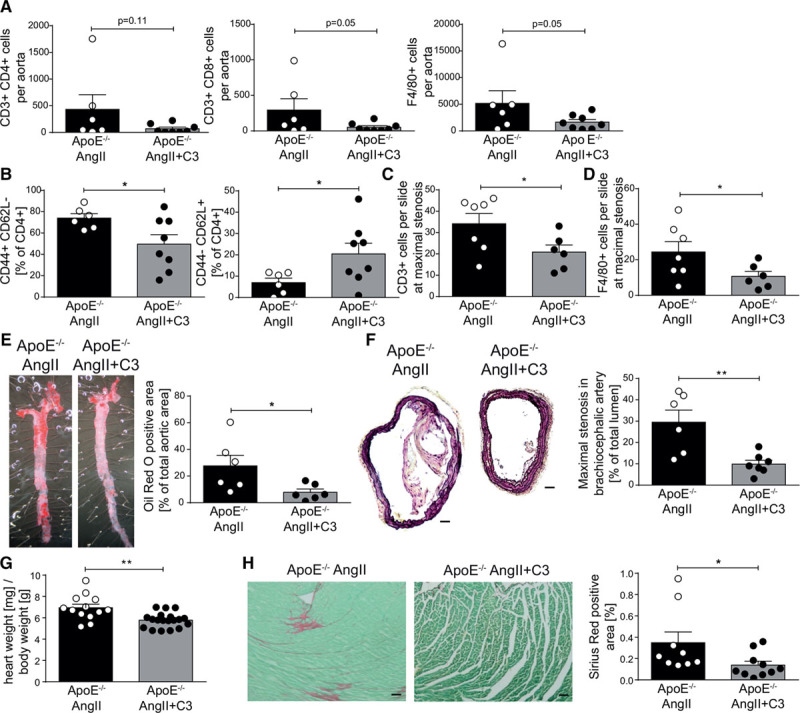
Propionate reduces aortic inflammation and atherosclerotic lesion burden in AngII-infused ApoE–/–. A,Single-cell suspensions from whole aortas of were analyzed for T helper (CD3+CD4+), cytotoxic T cells (CD3+CD8+), and macrophages (F4/80+) by flow cytometry. B, Aortic CD4+ T cells were analyzed for CD4+ effector memory (CD44+CD62L–) and naive (CD44–CD62L+) subsets by flow cytometry. C and D, Quantification of CD3 and F4/80 positive cells in sections of the brachiocephalic artery, respectively. E, En face Oil Red O staining of whole aortas for the quantification of atherosclerotic lesion burden. Left, Representative aortas. Right, Quantification. F, The degree of stenosis in the brachiocephalic artery was determined in Movat-stained cross-sections. Left, Representative sections (scale bar=100 µm). Right, Quantification. Athrough F, ApoE–/– AngII n=6, ApoE–/– AngII+C3 n=8. G, Cardiac hypertrophy index (heart weight [mg]/body weight [g]) of AngII-infused ApoE–/– mice treated with C3 or control, ApoE–/– AngII n=16, ApoE–/– AngII+C3 n=21. H, Left ventricular cardiac fibrosis as analyzed by Sirius red staining. Left, Representative photomicrographs (scale bar=100 µm). Right, Quantification. ApoE–/– AngII n=9, ApoE–/– AngII+C3 n=10. *P<0.05, **P<0.01 by 1-tailed t test or Mann-Whitney test. AngII indicates angiotensin II; ApoE–/–, apolipoprotein E knockout–deficient; and C3, propionate.
Propionate Ameliorates Cardiac Immune Cell Infiltration and Remodeling
Alongside vascular injury, hypertension generates an inflammatory response in the heart, which promotes cardiac remodeling.30 Flow cytometric analysis of heart-infiltrating lymphocytes on day 14 of AngII infusion in WT mice revealed a significant increase in the number of cardiac CD4+ T cells, CD8+ T cells, and F4/80+ macrophages, which was significantly decreased by C3 treatment (Figure 4A through 4E). These results could be confirmed by analysis of CD4 and CD8 immunofluorescence of cardiac cryosections (Figure IIIA and IIIB in the online-only Data Supplement). We further analyzed the proportion of Th17, Treg, and Th1 subsets among infiltrating cardiac T cells. The AngII-induced increase in cardiac CD4+ RORγt+Foxp3– frequencies was prevented by C3 treatment, whereas the fraction of CD4+FoxP3+RORγt– T cells and CD4+T-bet+ T cells was similar between groups (Figure 4F, Figure IIIC in the online-only Data Supplement). IL-10 mRNA levels in cardiac tissue were analyzed by quantitative polymerase chain reaction. In line with IL-10 expression in CD4+ splenocytes, C3 prevented the AngII-induced increase in Il-10 expression (Figure IIID in the online-only Data Supplement).
Figure 4.
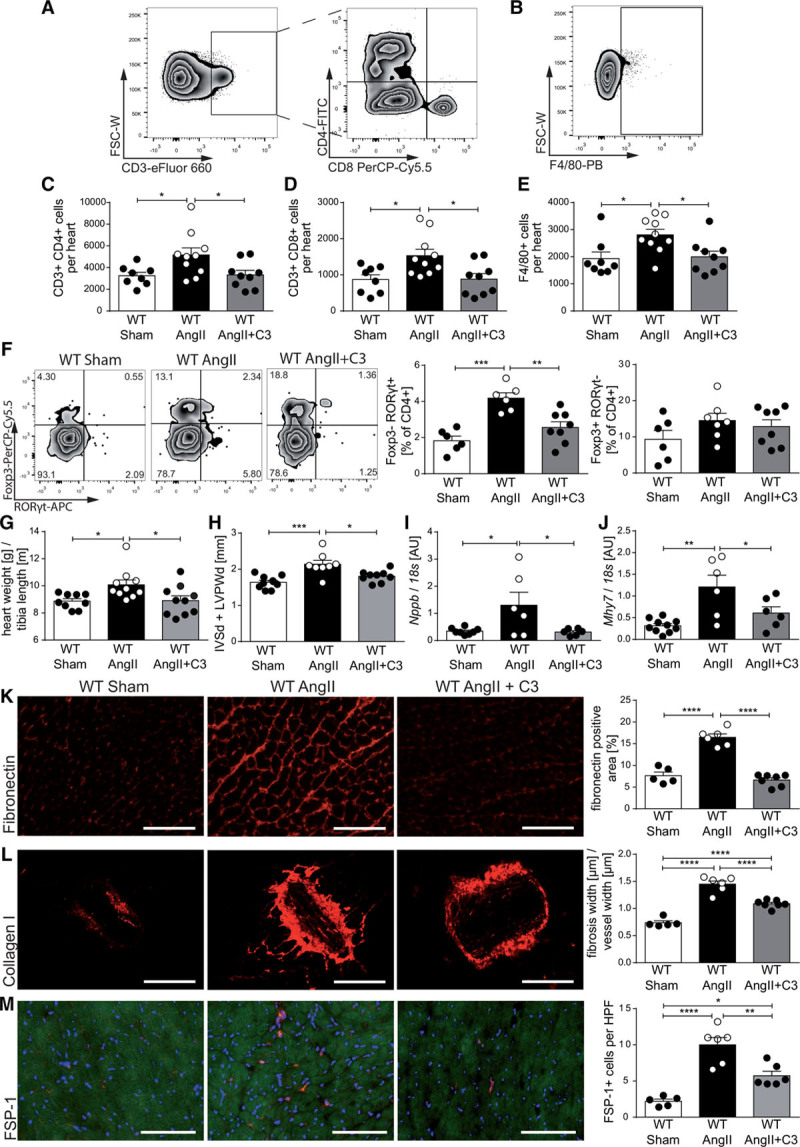
Propionate attenuates hypertensive cardiac damage in AngII-infused wild-type NMRI (WT) mice. A through E, Single cells were isolated from hearts of sham-infused or AngII-infused WT mice treated with C3 or control and analyzed by flow cytometry for T helper cells (CD3+CD4+), cytotoxic T cells (CD3+CD8+), and macrophages (F4/80+), as well. A and B, Representative ratings. C through E, The respective quantifications. WT Sham n=8, WT AngII n=10, WT AngII+C3 n=9. F, Analysis of CD4+FoxP3+ and CD4+RORγt+ cells in heart single-cell suspensions. Left, Representative flow cytometry plots. Right, Quantifications. WT Sham n=6, WT AngII n=6 to 7, WT AngII+C3 n=8. G, Cardiac hypertrophy index (heart weight [g]/tibia length [m]), (WT Sham n=9, WT AngII n=10, WT AngII+C3 n=10). H, Left ventricular wall thickness (sum of IVSd and LVPWd) as measured by echocardiography (WT Sham n=9, WT AngII n=8, WT AngII+C3 n=9). Cardiac Nppb (I) and Mhy7 (J) expression as measured by qPCR at the end of the treatment (WT Sham n=10, WT AngII n=6, WT AngII+C3 n=6). K through M, Immunofluorescence analysis of cardiac left ventricular fibrosis using fibronectin (K), collagen I (L), and FSP-1 (M) antibodies (WT Sham n=5, WT AngII n=6, WT AngII+C3 n=7). Left, Representative photomicrographs (scale bar=100 µm). Right, Quantifications. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001 by 1-way ANOVA and Tukey post hoc. AngII indicates angiotensin II; APC, Allophycocyanin; AU, arbitrary unit; C3, propionate; FITC, fluorescein isothiocyanate; FSC-W, forward scatter width; FSP-1, fibroblast-specific protein 1; HPF, high-power field; IVSd, interventricular septal thickness at diastole; LVPWd, left ventricular posterior wall end diastole; PB, Pacific Blue; PerCP-Cy5.5, Peridinin-chlorophyll protein cyanine 5.5; and qPCR, quantitative polymerase chain reaction.
As expected, AngII increased the cardiac hypertrophy index after 14 days of infusion, an effect that was prevented by C3 treatment (Figure 4G). We confirmed this finding using echocardiography, which revealed an increased left ventricular wall thickness after AngII infusion and a significant reduction on C3 treatment (Figure 4H). Accordingly, the AngII-induced increase in cardiac brain natriuretic peptide (Nppb) and β-myosin heavy chain (Mhy7) mRNA expression was prevented by C3 treatment as measured by quantitative polymerase chain reaction (Figure 4I and 4J). C3 treatment also prevented the AngII-induced increase in interstitial and perivascular cardiac fibrosis as measured by fibronectin and collagen I immunofluorescence, respectively (Figure 4K and 4L). Consistently, the number of cardiac fibroblasts analyzed using fibroblast-specific protein 1 immunofluorescence was similarly regulated (Figure 4M). Cardiac mRNA expression of connective tissue growth factor (Ctgf) and neutrophil gelatinase–associated lipocalin (Ngal) also confirmed the antifibrotic effect (Figure IIIE and IIIF in the online-only Data Supplement).
HDAC inhibitory properties have been attributed to SCFAs,31 and HDAC inhibition is known to inhibit AngII-induced cardiac hypertrophy and fibrosis.32 To address this potential mechanism of action, we cultured rat neonatal cardiomyocytes in vitro in the presence or absence of AngII and tested the effect of C3 on atrial natriuretic peptide (Nppa) mRNA expression as a sensitive hypertrophy marker in comparison with the known class I and II HDAC inhibitor trichostatin A. In contrast to trichostatin A, C3 did not reduce Nppa mRNA expression (Figure IV in the online-only Data Supplement), suggesting that the effect of C3 on AngII-induced cardiac hypertrophy is HDAC-independent.
Effect of Propionate Depends on Treg
C3 has been shown to promote Treg generation and function.22 We hypothesized that the observed beneficial effects of C3 in AngII-infused hypertensive WT mice are Treg-dependent. To test this hypothesis, we depleted Tregs in C3-treated AngII-infused WT mice by injecting anti-CD25 antibody (intraperitoneal injections of antibody clone PC61 on days –1, 2, and 5 of AngII infusion). We assessed inflammation and cardiac fibrosis in comparison with nondepleted C3-treated AngII-infused WT mice receiving IgG control antibodies intraperitoneally; Treg depletion was well tolerated and had no effect on body weight (Figure VA in the online-only Data Supplement). On day 14 of AngII infusion, splenic Tregs were still significantly depleted in comparison with the IgG control group (Figure 5A). Treg-depleted AngII-infused WT mice treated with C3 displayed a significant increase in splenic CD4+IL-17A+ cell frequencies in comparison with nondepleted mice (Figure 5B), reinforced by a similar trend in splenic CD4+RORγt+ Foxp3– frequencies (Figure VB in the online-only Data Supplement). Splenic Th1 cells, as measured by interferon-γ and T-bet expression in CD4+ cells, were not regulated (Figure VC and VD in the online-only Data Supplement). The inhibitory effect of C3 on splenic CD4+ TEM frequencies was abrogated in Treg-depleted mice (Figure 5C), without altering central memory T cells or TN populations (Figure VE and VF in the online-only Data Supplement). Significantly more CD4+ and CD8+ lymphocytes could be detected in heart sections of Treg-depleted AngII-infused mice treated with C3 (Figure 5D and 5E). Cardiac hypertrophy measured by echocardiography and hypertrophy index was only nonsignificantly increased in Treg-depleted C3-treated AngII-infused mice in comparison with mice injected with IgG (Figure 5F, Figure VG in the online-only Data Supplement). However, hearts from Treg-depleted mice displayed a significantly increased interstitial and perivascular fibrosis, as well as increased numbers of fibroblast-specific protein 1+ cells (Figure 5G through 5I). These findings suggest that Treg may partially mediate the cardioprotective effects of C3.
Figure 5.
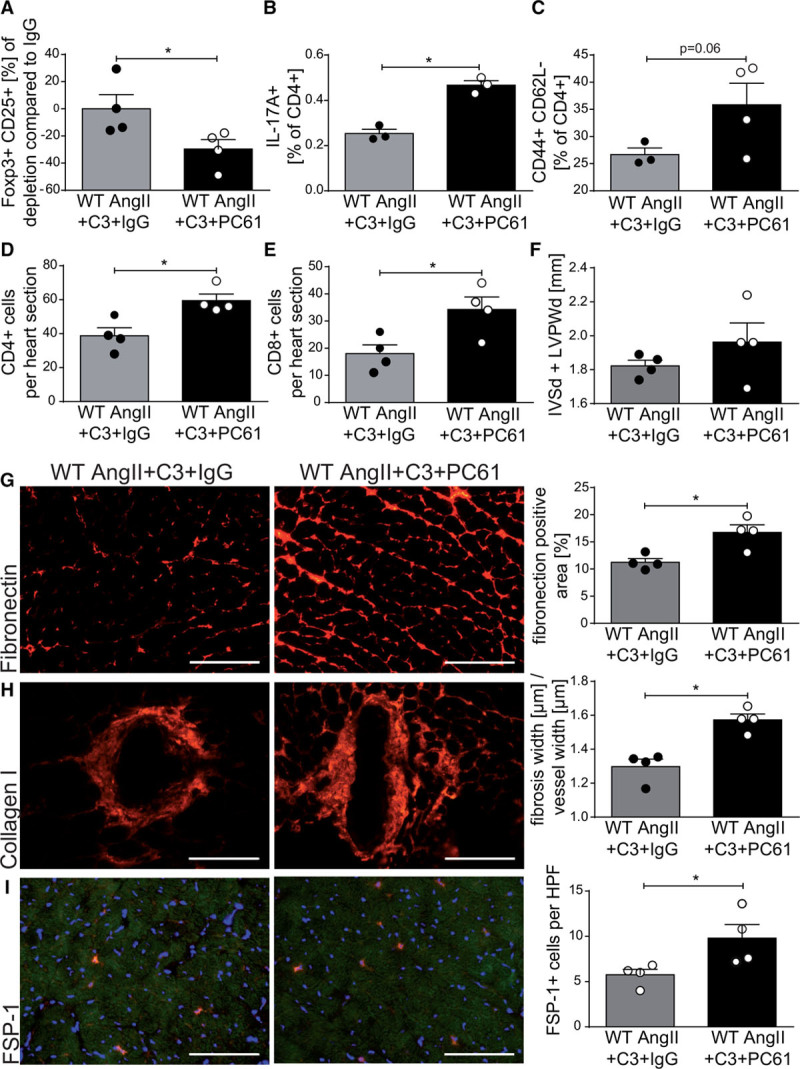
Depletion of regulatory T cells abrogates the effect of propionate in AngII-infused wild-type NMRI (WT) mice. AngII-infused WT mice received propionate treatment with intraperitoneal injections of anti-CD25 (PC61) or IgG control. A, Relative reduction in splenic CD4+CD25+Foxp3+ regulatory T cells at day 14 of AngII infusion in comparison with IgG control. B, IL-17A production in CD4+ restimulated splenocytes measured by flow cytometry. C, Splenic effector memory T cell (CD4+CD44+CD62L–) frequencies. D and E, Analysis of CD4+ (D) and CD8+ (E) lymphocytes in heart sections using immunofluorescence. F, Left ventricular wall thickness (sum of IVSd and LVPWd) as measured by echocardiography. G through I, Immunofluorescence analysis of left ventricular fibrosis using fibronectin (G), collagen I (H), and FSP-1 (I) antibodies. Left, Representative photomicrographs (scale bars=100 µm). Right, quantification. A, D through I, WT AngII+C3+IgG n=4, WT AngII+C3+PC61 n=4; B and C, WT AngII+C3+IgG n=3, WT AngII+C3+PC61 n=4, *P<0.05 by 1-tailed Mann-Whitney test. AngII indicates angiotensin II; C3, propionate; FSP-1, fibroblast-specific protein 1; IgG, immunoglobulin G; IL, interleukin; IVSd, interventricular septal thickness at diastole; and LVPWd, left ventricular posterior wall end diastole.
Moderate Blood Pressure–Lowering Effect of Propionate
Recent studies have shown that C3 may directly influence vasomotor function.26 To achieve C3-induced vasorelaxation in isolated perfused kidneys, very high, supraphysiological concentrations (3–100 mmol/L) were needed (data not shown). In addition, atomic force microscopy–based nanoindentation measurements in ApoE–/– mice revealed that C3 treatment softens endothelial cells in comparison with control treatment (data not shown). Next, we examined whether chronic oral C3 treatment influences blood pressure in AngII-infused WT mice and performed continuous radiotelemetric blood pressure measurements. Systolic and diastolic blood pressures in C3-treated WT mice were not affected in the initial phase, but were lowered toward the second week of AngII infusion, reaching statistical significance from day 11 and day 12 on (Figure 6A and 6C). Calculating the area under the curve for both weeks of AngII infusion separately, a significant difference in systolic and diastolic blood pressure was seen only for the second week (Figure 6B and 6D). This pattern was confirmed in the ApoE–/– model, because tail-cuff measurements of the systolic blood pressure were significantly lower in C3-treated mice during the last week of AngII infusion (Figure VIA in the online-only Data Supplement). In parallel with the reduced blood pressure, endothelial dysfunction in both mouse models was significantly ameliorated by C3 treatment as shown by ex vivo analysis of endothelium-dependent relaxation (Figure VIB and VIC in the online-only Data Supplement). To analyze whether C3 affects endothelial dysfunction in an immune cell–free setting, we incubated isolated mesenteric rings from untreated healthy mice with IL-17A and AngII for 24 hours in vitro (Figure VID in the online-only Data Supplement). Coincubation with C3 under cell culture conditions did not prevent endothelial dysfunction, making a direct endothelium-mediated effect of C3 less likely (Figure VID in the online-only Data Supplement).
Figure 6.
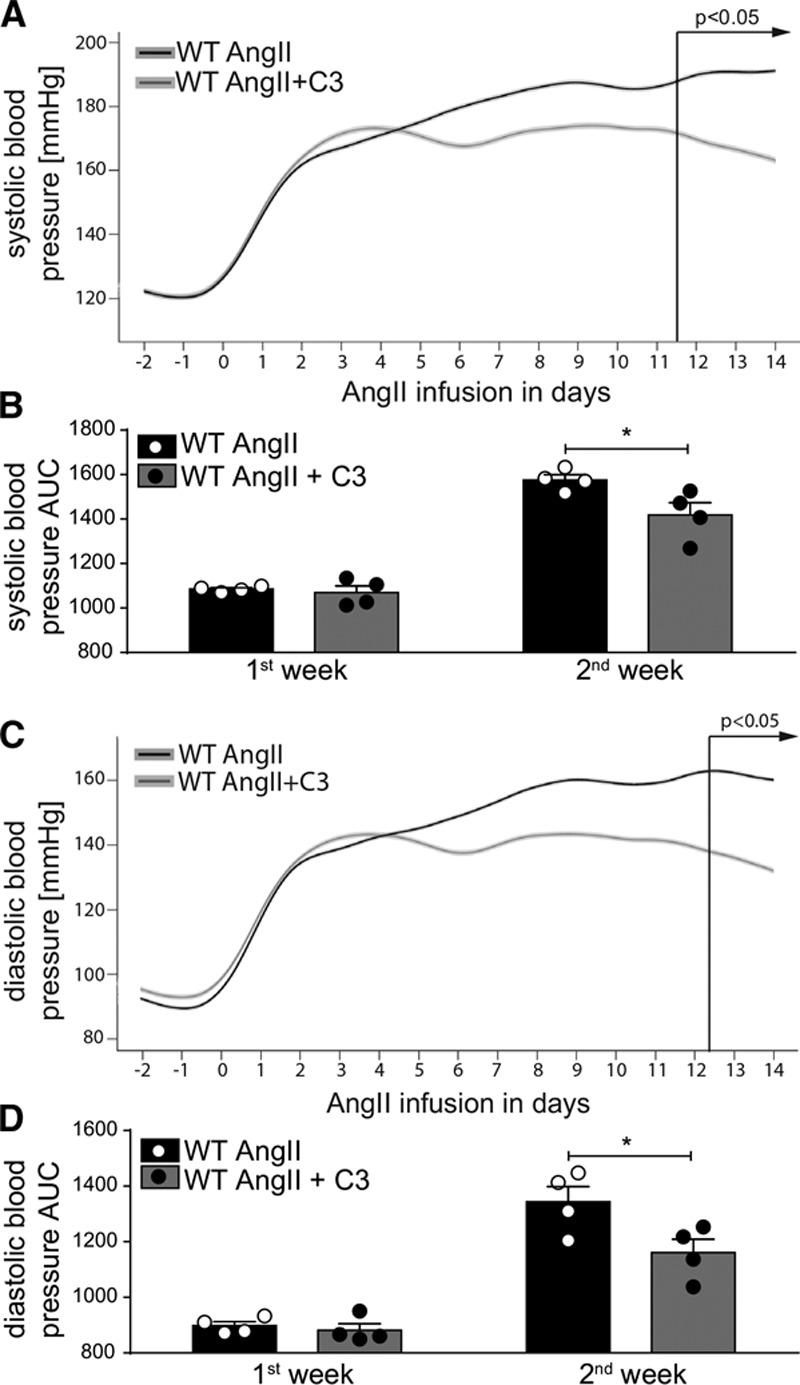
Propionate treatment shows a blood pressure–lowering effect confined to the second week of AngII infusion. A through D, Systolic and diastolic blood pressure were measured continuously by radiotelemetry in AngII-infused WT mice treated with C3 or control. A and C, Shown are smoothened curves over time for systolic and diastolic blood pressure, respectively. P values by linear mixed model. B and D, Shown are systolic and diastolic pressures calculated as AUC in week 1 and week 2 of AngII infusion, respectively. n=4 per group. *P<0.05 by 2-way repeated-measurement ANOVA and Sidak post hoc. AngII indicates angiotensin II; AUC, area under the curve; C3, propionate; and WT, wild-type NMRI.
To elucidate if the blood pressure–lowering effect of C3 is influenced directly by Treg, we measured blood pressure by radiotelemetry in C3-treated AngII-infused WT mice receiving the Treg-depleting anti-CD25 antibody or IgG control. However, systolic and diastolic blood pressures were similar in the initial and late phases of AngII infusion. (Figure VIE and VIF in the online-only Data Supplement). Therefore, the blood pressure–lowering effect of chronic C3 treatment cannot be ascribed to a single mechanism.
Propionate Reduces Susceptibility to Ventricular Arrhythmias
To further explore if the beneficial effects of C3 treatment on AngII-induced cardiac remodeling led to an improved functional outcome, we assessed the susceptibility of AngII-infused WT mice treated with C3 or control to ventricular arrhythmias by in vivo cardiac electrophysiological studies. Ventricular tachyarrhythmias are prognostically relevant in hypertensive heart disease.33 Susceptibility to ventricular tachyarrhythmias was significantly lower in C3-treated animals (5 of 7 C3-treated mice could not be triggered at all), whereas sustained tachyarrhythmias could be triggered in 85% of control-treated mice (Figure 7A and 7B). Connexin 43, a major gap junction protein required for electric integrity, was relocated from the intercalated disk to the lateral border of the cardiomyocytes on AngII infusion as shown by immunofluorescence (Figure 7C). Consequently, the degree of connexin 43 colocalization with N-cadherin (localized at the intercalated disc) was reduced on AngII infusion in comparison with sham-infused mice and maintained by concomitant C3 treatment (Figure 7C). These data show that C3 improves cardiac electric remodeling.
Figure 7.
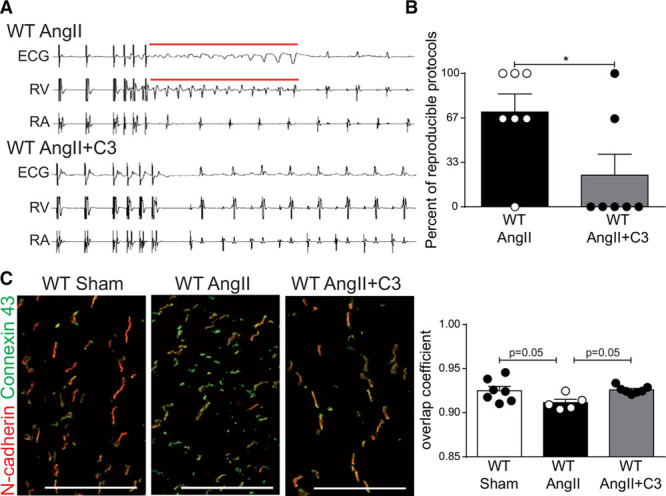
Propionate reduces susceptibility to ventricular arrhythmias in AngII-infused wild-type NMRI (WT) mice. In vivo programmed electric ventricular stimulations were performed in AngII-infused WT mice treated with C3 or control. A, Representative original tracings showing the induction of ventricular tachyarrhythmia. Surface ECG, right ventricular (RV), and right atrial (RA) recordings are shown. B, Quantification of ventricular arrhythmias susceptibility. n=7 per group, *P<0.05 by Mann-Whitney test. C, Immunofluorescent costaining of connexin 43 (green) and N-cadherin (red) in cardiac cryosections from sham-infused or AngII-infused WT mice treated with C3 or control. WT Sham n=7, WT AngII n=5, WT AngII+C3 n=7. Representative photomicrographs (scale bar=100 µm) and quantification of colocalization. P values by 1-way ANOVA and Tukey post hoc. AngII indicates angiotensin II; and C3, propionate.
Discussion
Metabolites released by the gut microbiota exert an important influence on the cardiovascular health of their host. SCFAs, end products of bacterial metabolism in the intestine, which are primarily derived from dietary fibers, have been attributed health-promoting properties in several diseases, in particular because of their potent action on immune cells.25 Epidemiological studies suggest that sufficient fiber intake may be beneficial in hypertension,34 but we lack a comprehensive understanding of the underlying mechanisms. The current study demonstrates that the SCFA propionate prevents target organ damage in hypertensive mice with and without atherosclerosis by maintaining immune homeostasis.
Hypertension stands out among health risk factors because it promotes several cardiovascular diseases, including hypertensive heart disease and atherosclerosis. Beyond blood pressure control, the need to address the inflammatory response to hypertensive stimuli has been recognized.35 In particular, effector T cells and macrophages are activated in hypertension and mediate damage to the heart and the vasculature. Experimental evidence suggests that immunosuppression11 or adoptive transfer of Treg13,14 limits hypertensive target organ damage, although potential side effects prevent further translation of these interventions.
Propionate improved the survival in both WT and ApoE–/– mice and attenuated the systemic T cell response to AngII as indicated by reduced splenic TEM frequencies together with less proinflammatory Th17 cells. It is more important that this anti-inflammatory effect was also detectable in the respective target organs, because fewer T cells and macrophages infiltrated the heart and the aorta, respectively. AngII-induced cardiac hypertrophy and fibrosis were attenuated by propionate in both mouse models. In ApoE–/– mice, propionate treatment led to a reduced atherosclerotic lesion burden despite unaltered blood lipid levels.
Our data expand on recent observations demonstrating the cardioprotective and renoprotective effects of a high-fiber diet in uninephrectomized deoxycorticosterone acetate–treated hypertensive mice.28 The authors ascribe the observed cardiorenal protection to acetate, another SCFA, and demonstrate a beneficial gene regulation in hearts and kidneys from uninephrectomized deoxycorticosterone acetate mice fed a high-fiber diet or supplemented with acetate. The authors also describe decreased blood pressures at the end of the treatment.28 Our current study expands on the cardiovascular protective effects of SCFAs, pointing to an important contribution of immune homeostasis in the SCFA-mediated effect.
Activation of T cells can be observed in response to hypertensive stimuli, indicated by increased TEM.36 The balance of Treg and effector T cells is critically important in hypertension and hypertensive end-organ damage. Tregs are known to limit target organ damage in hypertension, because adoptive transfer of Treg has been shown to dampen AngII-induced cardiac13 and vascular14 damage. Moreover, depletion of Treg accelerates atherosclerosis in hypercholesterolemic mice.15 Increased proportions of TEM are associated with the development of atherosclerosis in humans and mice.37,38 In addition to the significant effect on TEM, our data suggest that the propionate effect is also Treg-dependent, because Treg depletion abrogated the propionate effect on systemic and cardiac inflammation, and on cardiac fibrosis, as well. It is interesting to note that we observed an increase in splenic Treg frequencies in response to AngII along with an increase in Th17 cells, suggesting that Tregs counterbalance the AngII-elicited effector Th17 response. This observation is supported by a study showing increased plasma levels of the anti-inflammatory cytokine IL-10 in AngII-infused mice.14 Likewise, serum IL-10 is increased in hypercholesterolemic mice,39 and human atherosclerotic lesions show substantial IL-10 expression.40 Enrichment of Treg in nonlymphoid tissues in response to inflammatory stimuli or injury can be observed in various contexts,41 likely as a compensatory response. It is most important that propionate treatment in our study preserved the balance of Th17 and Treg in AngII-infused mice. Furthermore, Treg depletion abrogated the anti-inflammatory effect of propionate, indicated by increased Th17 and TEM frequencies.
The current picture of SCFA signaling to host cells is complex, because both the interaction with Gpr and olfactory receptor 78,26 and HDAC inhibitory properties, as well, have been described. Propionate is known to bind to Gpr41 and Gpr43, and independent studies have shown that either Gpr4142 or Gpr43,21 but also HDAC inhibition,22 may account for the propionate effect on immune cells. Direct propionate signaling to nonimmune cells such as cardiomyocytes may also play a role, because the inhibition of HDAC activity is known to inhibit cardiomyocyte hypertrophy.32 To distinguish effects mediated by immune cells from direct effects on cardiomyocytes, we tested the ability of propionate to inhibit AngII-induced hypertrophy of rat primary neonatal cardiomyocytes in comparison with the established HDAC inhibitor trichostatin A. However, propionate failed to inhibit cardiomyocyte hypertrophy. These data, together with the abrogation of the propionate effects by Treg depletion, suggest that T cells substantially contribute to the protective effect of propionate. Further studies are warranted to dissect other potentially protective pleiotropic effects of propionate.
Our data demonstrate that, in addition to beneficial immunomodulatory effects, propionate moderately reduced blood pressure in both models. Endothelial dysfunction is associated with the development of essential hypertension.43 In isolated mesenteric arteries of AngII-infused WT mice and in isolated perfused kidneys of AngII-infused ApoE–/– mice, we could show that chronic C3 treatment improved endothelium-dependent vasodilation. Earlier reports described acute vasodilation in response to propionate and other SCFAs.26 Similarly, we observed a C3-induced vasodilation in isolated perfused kidneys, albeit only at supraphysiological concentrations. Propionate was previously shown to activate Gpr41 located in the vascular endothelium, mediating the vasodilating effect of propionate, although the downstream signaling cascades are less clear.27 In isolated mesenteric rings, C3 failed to improve endothelial function in a short-term experiment. Nevertheless, we cannot exclude that chronic Gpr41-mediated antihypertensive effects of propionate on the endothelium may have contributed to target organ protection. However, blood pressure reduction was observed only toward the late phase of AngII infusion, suggesting rather a chronic effect of propionate on hypertension, thereby arguing against a direct endothelium-dependent effect.
It is important to note that an increased occurrence of ventricular tachycardia can be observed in hypertensive patients with left ventricular hypertrophy, which is prognostically relevant.33 Spatial redistribution of gap junction proteins is characteristic of pathological electric remodeling and has been described in the human hypertrophic myocardium.44 We show that propionate improves electric remodeling with a reduced susceptibility to programmed ventricular tachycardia in vivo. Our observation is confirmed by attenuated cardiac gap junction remodeling in propionate-treated mice, as indicated by a reduced lateralization of connexin 43 in cardiomyocytes. Similarly, beneficial effects were observed earlier in AngII-infused mice after adoptive Treg transfer.13
In conclusion, propionate treatment protects from cardiac damage and reduces atherosclerosis in experimental hypertension. Target organ protection by propionate is at least partially dependent on Treg, although other pleiotropic effects likely contribute to this result. Our data provide further experimental evidence for the importance of microbiota-derived SCFAs in promoting host cardiovascular health. Hypertension and atherosclerosis account for a substantial proportion of worldwide cardiovascular morbidity and mortality. Propionate could be important in improving cardiovascular health, because both atherosclerosis and hypertensive cardiac remodeling were significantly reduced on propionate treatment in our study. It is interesting to note that several subsets of gut bacteria are capable of producing propionate,45 some of which were shown to be less abundant in experimental hypertension46 and hypertensive patients.17 Consequently, oral supplementation with propionate or its precursors may be beneficial in hypertensive individuals to prevent damage to target organs. Current hypertension guidelines recommend lifestyle modifications before the initiation of any pharmacological antihypertensive treatment.47 Dietary augmentation of propionate is an affordable intervention, and our observations suggest that this could be a novel approach to prevent hypertensive damage to target organs.
Acknowledgments
We thank Gabriele N’diaye, Ilona Kramer, May-Britt Köhler, Jana Czychi, Martin Taube, Dr Sabine Bartel, and Christina Schwandt for technical assistance, and Thomas Hünig for the provision of the PC61 antibody. Drs Wilck, Müller, and Stegbauer and H. Bartolomaeus designed the study. H. Bartolomaeus and Drs Wilck, Balogh, Markó, Avery, Haase, and Kräker performed experiments in wild-type NMRI mice, and analyzed and interpreted the data. Drs Yakoub, Homann, Höges, Hering, Maase, Kusche-Vihrog, Grandoch, and Stegbauer performed experiments in ApoE–/– mice, and analyzed and interpreted the data. Drs A. Kushugulova, S. Kozhakhmetov, Zhumadilov, Dechend, Eckardt, Rump, and Forslund helped with data analysis and interpretation. Drs Wundersitz and Fielitz performed and analyzed neonatal cardiomyocyte experiments. S. Kempa performed and analyzed gas chromatography–mass spectrometry measurements. Dr Krannich performed statistical analyses. Dr Wilck, H. Bartolomaeus, and Drs Stegbauer and Müller wrote the manuscript with input from all authors.
Sources of Funding
Dr Wilck is participant in the Clinician Scientist Program funded by the Berlin Institute of Health. Drs Yakoub, Grandoch, Rump, S. Homann, and Stegbauer were supported by the DFG (German Research Foundation) (IRTG 1902). Dr Fielitz received funding from the DZHK (German Center for Cardiovascular Research) (81Z5400153) and the DFG (German Research Foundation) (FI 965/5-2).
Disclosures
None.
Supplementary Material
Footnotes
Drs Müller, Stegbauer, and Wilck jointly supervised this work.
Sources of Funding, see page 1419
The online-only Data Supplement is available with this article at https://www.ahajournals.org/doi/suppl/10.1161/CIRCULATIONAHA.118.036652.
References
- 1.Wenzel U, Turner JE, Krebs C, Kurts C, Harrison DG, Ehmke H. Immune mechanisms in arterial hypertension. J Am Soc Nephrol. 2016;27:677–686. doi: 10.1681/ASN.2015050562. doi: 10.1681/ASN.2015050562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rudemiller NP, Crowley SD. Interactions between the immune and the renin-angiotensin systems in hypertension. Hypertension. 2016;68:289–296. doi: 10.1161/HYPERTENSIONAHA.116.06591. doi: 10.1161/HYPERTENSIONAHA.116.06591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Itani HA, Harrison DG. Memories that last in hypertension. Am J Physiol Renal Physiol. 2015;308:F1197–F1199. doi: 10.1152/ajprenal.00633.2014. doi: 10.1152/ajprenal.00633.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Markó L, Kvakan H, Park JK, Qadri F, Spallek B, Binger KJ, Bowman EP, Kleinewietfeld M, Fokuhl V, Dechend R, Müller DN. Interferon-γ signaling inhibition ameliorates angiotensin II-induced cardiac damage. Hypertension. 2012;60:1430–1436. doi: 10.1161/HYPERTENSIONAHA.112.199265. doi: 10.1161/HYPERTENSIONAHA.112.199265. [DOI] [PubMed] [Google Scholar]
- 5.Madhur MS, Lob HE, McCann LA, Iwakura Y, Blinder Y, Guzik TJ, Harrison DG. Interleukin 17 promotes angiotensin II-induced hypertension and vascular dysfunction. Hypertension. 2010;55:500–507. doi: 10.1161/HYPERTENSIONAHA.109.145094. doi: 10.1161/HYPERTENSIONAHA.109.145094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Didion SP, Kinzenbaw DA, Schrader LI, Chu Y, Faraci FM. Endogenous interleukin-10 inhibits angiotensin II-induced vascular dysfunction. Hypertension. 2009;54:619–624. doi: 10.1161/HYPERTENSIONAHA.109.137158. doi: 10.1161/HYPERTENSIONAHA.109.137158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rosendorff C, Lackland DT, Allison M, Aronow WS, Black HR, Blumenthal RS, Cannon CP, de Lemos JA, Elliott WJ, Findeiss L, Gersh BJ, Gore JM, Levy D, Long JB, O’Connor CM, O’Gara PT, Ogedegbe O, Oparil S, White WB American Heart Association; American College of Cardiology; American Society of Hypertension. Treatment of hypertension in patients with coronary artery disease: a scientific statement from the American Heart Association, American College of Cardiology, and American Society of Hypertension. J Am Coll Cardiol. 2015;65:1998–2038. doi: 10.1016/j.jacc.2015.02.038. doi: 10.1016/j.jacc.2015.02.038. [DOI] [PubMed] [Google Scholar]
- 8.Hansson GK, Hermansson A. The immune system in atherosclerosis. Nat Immunol. 2011;12:204–212. doi: 10.1038/ni.2001. doi: 10.1038/ni.2001. [DOI] [PubMed] [Google Scholar]
- 9.Mazzolai L, Duchosal MA, Korber M, Bouzourene K, Aubert JF, Hao H, Vallet V, Brunner HR, Nussberger J, Gabbiani G, Hayoz D. Endogenous angiotensin II induces atherosclerotic plaque vulnerability and elicits a Th1 response in ApoE-/- mice. Hypertension. 2004;44:277–282. doi: 10.1161/01.HYP.0000140269.55873.7b. doi: 10.1161/01.HYP.0000140269.55873.7b. [DOI] [PubMed] [Google Scholar]
- 10.Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, Fonseca F, Nicolau J, Koenig W, Anker SD, Kastelein JJP, Cornel JH, Pais P, Pella D, Genest J, Cifkova R, Lorenzatti A, Forster T, Kobalava Z, Vida-Simiti L, Flather M, Shimokawa H, Ogawa H, Dellborg M, Rossi PRF, Troquay RPT, Libby P, Glynn RJ CANTOS Trial Group. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. 2017;377:1119–1131. doi: 10.1056/NEJMoa1707914. doi: 10.1056/NEJMoa1707914. [DOI] [PubMed] [Google Scholar]
- 11.Muller DN, Shagdarsuren E, Park JK, Dechend R, Mervaala E, Hampich F, Fiebeler A, Ju X, Finckenberg P, Theuer J, Viedt C, Kreuzer J, Heidecke H, Haller H, Zenke M, Luft FC. Immunosuppressive treatment protects against angiotensin II-induced renal damage. Am J Pathol. 2002;161:1679–1693. doi: 10.1016/S0002-9440(10)64445-8. doi: 10.1016/S0002-9440(10)64445-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bäck M, Hansson GK. Anti-inflammatory therapies for atherosclerosis. Nat Rev Cardiol. 2015;12:199–211. doi: 10.1038/nrcardio.2015.5. doi: 10.1038/nrcardio.2015.5. [DOI] [PubMed] [Google Scholar]
- 13.Kvakan H, Kleinewietfeld M, Qadri F, Park JK, Fischer R, Schwarz I, Rahn HP, Plehm R, Wellner M, Elitok S, Gratze P, Dechend R, Luft FC, Muller DN. Regulatory T cells ameliorate angiotensin II-induced cardiac damage. Circulation. 2009;119:2904–2912. doi: 10.1161/CIRCULATIONAHA.108.832782. doi: 10.1161/CIRCULATIONAHA.108.832782. [DOI] [PubMed] [Google Scholar]
- 14.Barhoumi T, Kasal DA, Li MW, Shbat L, Laurant P, Neves MF, Paradis P, Schiffrin EL. T regulatory lymphocytes prevent angiotensin II-induced hypertension and vascular injury. Hypertension. 2011;57:469–476. doi: 10.1161/HYPERTENSIONAHA.110.162941. doi: 10.1161/HYPERTENSIONAHA.110.162941. [DOI] [PubMed] [Google Scholar]
- 15.Ait-Oufella H, Salomon BL, Potteaux S, Robertson AK, Gourdy P, Zoll J, Merval R, Esposito B, Cohen JL, Fisson S, Flavell RA, Hansson GK, Klatzmann D, Tedgui A, Mallat Z. Natural regulatory T cells control the development of atherosclerosis in mice. Nat Med. 2006;12:178–180. doi: 10.1038/nm1343. doi: 10.1038/nm1343. [DOI] [PubMed] [Google Scholar]
- 16.Tang WH, Hazen SL. The contributory role of gut microbiota in cardiovascular disease. J Clin Invest. 2014;124:4204–4211. doi: 10.1172/JCI72331. doi: 10.1172/JCI72331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li J, Zhao F, Wang Y, Chen J, Tao J, Tian G, Wu S, Liu W, Cui Q, Geng B, Zhang W, Weldon R, Auguste K, Yang L, Liu X, Chen L, Yang X, Zhu B, Cai J. Gut microbiota dysbiosis contributes to the development of hypertension. Microbiome. 2017;5:14. doi: 10.1186/s40168-016-0222-x. doi: 10.1186/s40168-016-0222-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wilck N, Matus MG, Kearney SM, Olesen SW, Forslund K, Bartolomaeus H, Haase S, Mähler A, Balogh A, Markó L, Vvedenskaya O, Kleiner FH, Tsvetkov D, Klug L, Costea PI, Sunagawa S, Maier L, Rakova N, Schatz V, Neubert P, Frätzer C, Krannich A, Gollasch M, Grohme DA, Côrte-Real BF, Gerlach RG, Basic M, Typas A, Wu C, Titze JM, Jantsch J, Boschmann M, Dechend R, Kleinewietfeld M, Kempa S, Bork P, Linker RA, Alm EJ, Müller DN. Salt-responsive gut commensal modulates TH17 axis and disease. Nature. 2017;551:585–589. doi: 10.1038/nature24628. doi: 10.1038/nature24628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Karlsson FH, Fåk F, Nookaew I, Tremaroli V, Fagerberg B, Petranovic D, Bäckhed F, Nielsen J. Symptomatic atherosclerosis is associated with an altered gut metagenome. Nat Commun. 2012;3:1245. doi: 10.1038/ncomms2266. doi: 10.1038/ncomms2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cummings JH, Pomare EW, Branch WJ, Naylor CP, Macfarlane GT. Short chain fatty acids in human large intestine, portal, hepatic and venous blood. Gut. 1987;28:1221–1227. doi: 10.1136/gut.28.10.1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly-Y M, Glickman JN, Garrett WS. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science. 2013;341:569–573. doi: 10.1126/science.1241165. doi: 10.1126/science.1241165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Arpaia N, Campbell C, Fan X, Dikiy S, van der Veeken J, deRoos P, Liu H, Cross JR, Pfeffer K, Coffer PJ, Rudensky AY. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature. 2013;504:451–455. doi: 10.1038/nature12726. doi: 10.1038/nature12726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Haghikia A, Jörg S, Duscha A, Berg J, Manzel A, Waschbisch A, Hammer A, Lee DH, May C, Wilck N, Balogh A, Ostermann AI, Schebb NH, Akkad DA, Grohme DA, Kleinewietfeld M, Kempa S, Thöne J, Demir S, Müller DN, Gold R, Linker RA. Dietary fatty acids directly impact central nervous system autoimmunity via the small intestine. Immunity. 2015;43:817–829. doi: 10.1016/j.immuni.2015.09.007. doi: 10.1016/j.immuni.2015.09.007. [DOI] [PubMed] [Google Scholar]
- 24.Park J, Kim M, Kang SG, Jannasch AH, Cooper B, Patterson J, Kim CH. Short-chain fatty acids induce both effector and regulatory T cells by suppression of histone deacetylases and regulation of the mTOR-S6K pathway. Mucosal Immunol. 2015;8:80–93. doi: 10.1038/mi.2014.44. doi: 10.1038/mi.2014.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Koh A, De Vadder F, Kovatcheva-Datchary P, Bäckhed F. From dietary fiber to host physiology: short-chain fatty acids as key bacterial metabolites. Cell. 2016;165:1332–1345. doi: 10.1016/j.cell.2016.05.041. doi: 10.1016/j.cell.2016.05.041. [DOI] [PubMed] [Google Scholar]
- 26.Pluznick JL, Protzko RJ, Gevorgyan H, Peterlin Z, Sipos A, Han J, Brunet I, Wan LX, Rey F, Wang T, Firestein SJ, Yanagisawa M, Gordon JI, Eichmann A, Peti-Peterdi J, Caplan MJ. Olfactory receptor responding to gut microbiota-derived signals plays a role in renin secretion and blood pressure regulation. Proc Natl Acad Sci U S A. 2013;110:4410–4415. doi: 10.1073/pnas.1215927110. doi: 10.1073/pnas.1215927110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Natarajan N, Hori D, Flavahan S, Steppan J, Flavahan NA, Berkowitz DE, Pluznick JL. Microbial short chain fatty acid metabolites lower blood pressure via endothelial G protein-coupled receptor 41. Physiol Genomics. 2016;48:826–834. doi: 10.1152/physiolgenomics.00089.2016. doi: 10.1152/physiolgenomics.00089.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marques FZ, Nelson E, Chu PY, Horlock D, Fiedler A, Ziemann M, Tan JK, Kuruppu S, Rajapakse NW, El-Osta A, Mackay CR, Kaye DM. High-fiber diet and acetate supplementation change the gut microbiota and prevent the development of hypertension and heart failure in hypertensive mice. Circulation. 2017;135:964–977. doi: 10.1161/CIRCULATIONAHA.116.024545. doi: 10.1161/CIRCULATIONAHA.116.024545. [DOI] [PubMed] [Google Scholar]
- 29.Stegbauer J, Chen D, Herrera M, Sparks MA, Yang T, Königshausen E, Gurley SB, Coffman TM. Resistance to hypertension mediated by intercalated cells of the collecting duct. JCI Insight. 2017;2:e92720. doi: 10.1172/jci.insight.92720. doi: 10.1172/jci.insight.92720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nicoletti A, Heudes D, Mandet C, Hinglais N, Bariety J, Michel JB. Inflammatory cells and myocardial fibrosis: spatial and temporal distribution in renovascular hypertensive rats. Cardiovasc Res. 1996;32:1096–1107. doi: 10.1016/s0008-6363(96)00158-7. [DOI] [PubMed] [Google Scholar]
- 31.Waldecker M, Kautenburger T, Daumann H, Busch C, Schrenk D. Inhibition of histone-deacetylase activity by short-chain fatty acids and some polyphenol metabolites formed in the colon. J Nutr Biochem. 2008;19:587–593. doi: 10.1016/j.jnutbio.2007.08.002. doi: 10.1016/j.jnutbio.2007.08.002. [DOI] [PubMed] [Google Scholar]
- 32.Kee HJ, Sohn IS, Nam KI, Park JE, Qian YR, Yin Z, Ahn Y, Jeong MH, Bang YJ, Kim N, Kim JK, Kim KK, Epstein JA, Kook H. Inhibition of histone deacetylation blocks cardiac hypertrophy induced by angiotensin II infusion and aortic banding. Circulation. 2006;113:51–59. doi: 10.1161/CIRCULATIONAHA.105.559724. doi: 10.1161/CIRCULATIONAHA.105.559724. [DOI] [PubMed] [Google Scholar]
- 33.McLenachan JM, Henderson E, Morris KI, Dargie HJ. Ventricular arrhythmias in patients with hypertensive left ventricular hypertrophy. N Engl J Med. 1987;317:787–792. doi: 10.1056/NEJM198709243171302. doi: 10.1056/NEJM198709243171302. [DOI] [PubMed] [Google Scholar]
- 34.Streppel MT, Arends LR, van’t Veer P, Grobbee DE, Geleijnse JM. Dietary fiber and blood pressure: a meta-analysis of randomized placebo-controlled trials. Arch Intern Med. 2005;165:150–156. doi: 10.1001/archinte.165.2.150. doi: 10.1001/archinte.165.2.150. [DOI] [PubMed] [Google Scholar]
- 35.Caillon A, Schiffrin EL. Role of inflammation and immunity in hypertension: recent epidemiological, laboratory, and clinical evidence. Curr Hypertens Rep. 2016;18:21. doi: 10.1007/s11906-016-0628-7. doi: 10.1007/s11906-016-0628-7. [DOI] [PubMed] [Google Scholar]
- 36.Itani HA, Xiao L, Saleh MA, Wu J, Pilkinton MA, Dale BL, Barbaro NR, Foss JD, Kirabo A, Montaniel KR, Norlander AE, Chen W, Sato R, Navar LG, Mallal SA, Madhur MS, Bernstein KE, Harrison DG. CD70 exacerbates blood pressure elevation and renal damage in response to repeated hypertensive stimuli. Circ Res. 2016;118:1233–1243. doi: 10.1161/CIRCRESAHA.115.308111. doi: 10.1161/CIRCRESAHA.115.308111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ammirati E, Cianflone D, Vecchio V, Banfi M, Vermi AC, De Metrio M, Grigore L, Pellegatta F, Pirillo A, Garlaschelli K, Manfredi AA, Catapano AL, Maseri A, Palini AG, Norata GD. Effector memory T cells are associated with atherosclerosis in humans and animal models. J Am Heart Assoc. 2012;1:27–41. doi: 10.1161/JAHA.111.000125. doi: 10.1161/JAHA.111.000125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Klingenberg R, Gerdes N, Badeau RM, Gisterå A, Strodthoff D, Ketelhuth DF, Lundberg AM, Rudling M, Nilsson SK, Olivecrona G, Zoller S, Lohmann C, Lüscher TF, Jauhiainen M, Sparwasser T, Hansson GK. Depletion of FOXP3+ regulatory T cells promotes hypercholesterolemia and atherosclerosis. J Clin Invest. 2013;123:1323–1334. doi: 10.1172/JCI63891. doi: 10.1172/JCI63891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xie JJ, Wang J, Tang TT, Chen J, Gao XL, Yuan J, Zhou ZH, Liao MY, Yao R, Yu X, Wang D, Cheng Y, Liao YH, Cheng X. The Th17/Treg functional imbalance during atherogenesis in ApoE(-/-) mice. Cytokine. 2010;49:185–193. doi: 10.1016/j.cyto.2009.09.007. doi: 10.1016/j.cyto.2009.09.007. [DOI] [PubMed] [Google Scholar]
- 40.Mallat Z, Heymes C, Ohan J, Faggin E, Lesèche G, Tedgui A. Expression of interleukin-10 in advanced human atherosclerotic plaques: relation to inducible nitric oxide synthase expression and cell death. Arterioscler Thromb Vasc Biol. 1999;19:611–616. doi: 10.1161/01.atv.19.3.611. [DOI] [PubMed] [Google Scholar]
- 41.Burzyn D, Benoist C, Mathis D. Regulatory T cells in nonlymphoid tissues. Nat Immunol. 2013;14:1007–1013. doi: 10.1038/ni.2683. doi: 10.1038/ni.2683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Trompette A, Gollwitzer ES, Yadava K, Sichelstiel AK, Sprenger N, Ngom-Bru C, Blanchard C, Junt T, Nicod LP, Harris NL, Marsland BJ. Gut microbiota metabolism of dietary fiber influences allergic airway disease and hematopoiesis. Nat Med. 2014;20:159–166. doi: 10.1038/nm.3444. doi: 10.1038/nm.3444. [DOI] [PubMed] [Google Scholar]
- 43.Yang G, Istas G, Höges S, Yakoub M, Hendgen-Cotta U, Rassaf T, Rodriguez-Mateos A, Hering L, Grandoch M, Mergia E, Rump LC, Stegbauer J. Angiotensin-(1-7)-induced Mas receptor activation attenuates atherosclerosis through a nitric oxide-dependent mechanism in apolipoproteinE-KO mice. Pflugers Arch. 2018;470:661–667. doi: 10.1007/s00424-018-2108-1. doi: 10.1007/s00424-018-2108-1. [DOI] [PubMed] [Google Scholar]
- 44.Kostin S, Dammer S, Hein S, Klovekorn WP, Bauer EP, Schaper J. Connexin 43 expression and distribution in compensated and decompensated cardiac hypertrophy in patients with aortic stenosis. Cardiovasc Res. 2004;62:426–436. doi: 10.1016/j.cardiores.2003.12.010. doi: 10.1016/j.cardiores.2003.12.010. [DOI] [PubMed] [Google Scholar]
- 45.Louis P, Flint HJ. Formation of propionate and butyrate by the human colonic microbiota. Environ Microbiol. 2017;19:29–41. doi: 10.1111/1462-2920.13589. doi: 10.1111/1462-2920.13589. [DOI] [PubMed] [Google Scholar]
- 46.Yang T, Santisteban MM, Rodriguez V, Li E, Ahmari N, Carvajal JM, Zadeh M, Gong M, Qi Y, Zubcevic J, Sahay B, Pepine CJ, Raizada MK, Mohamadzadeh M. Gut dysbiosis is linked to hypertension. Hypertension. 2015;65:1331–1340. doi: 10.1161/HYPERTENSIONAHA.115.05315. doi: 10.1161/HYPERTENSIONAHA.115.05315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Whelton PK, Carey RM, Aronow WS, Casey DE, Jr, Collins KJ, Dennison Himmelfarb C, DePalma SM, Gidding S, Jamerson KA, Jones DW, MacLaughlin EJ, Muntner P, Ovbiagele B, Smith SC, Jr, Spencer CC, Stafford RS, Taler SJ, Thomas RJ, Williams KA, Sr, Williamson JD, Wright JT., Jr 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA Guideline for the Prevention, Detection, Evaluation, and Management of High Blood Pressure in Adults: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation. 2018;138:e484–e594. doi: 10.1161/CIR.0000000000000596. doi: 10.1161/CIR.0000000000000596. [DOI] [PubMed] [Google Scholar]