

Abstract
Purpose of review:
Environmental toxicants and psychosocial stressors share many biological substrates and influence overlapping physiological pathways. Increasing evidence indicates stress-induced changes to the maternal milieu may prime rapidly developing physiological systems for disruption by concurrent or subsequent exposure to environmental chemicals. In this review, we highlight putative mechanisms underlying sex-specific susceptibility of the developing neuroendocrine system to the joint effects of stress or stress correlates and environmental toxicants (bisphenol A, alcohol, phthalates, lead, chlorpyrifos and traffic-related air pollution).
Recent findings:
We provide evidence indicating that concurrent or tandem exposure to chemical and non-chemical stressors during windows of rapid development is associated with sex-specific synergistic, potentiated and reversed effects on several neuroendocrine endpoints related to hypothalamic-pituitary-adrenal axis function, sex steroid levels, neurotransmitter circuits and innate immune function. We additionally identify gaps, such as the role that the endocrine-active placenta plays, in our understanding of these complex interactions. Finally, we discuss future research needs, including the investigation of non-hormonal biomarkers of stress.
Summary:
We demonstrate multiple physiologic systems are impacted by joint exposure to chemical and non-chemical stressors differentially among males and females. Collectively, the results highlight the importance of evaluating sex-specific endpoints when investigating the neuroendocrine system and underscore the need to examine exposure to chemical toxicants within the context of the social environment.
Keywords: sexually dimorphic, neurodevelopment, stress, chemical, neurotoxicant, prenatal
Introduction
For decades, the fields of psychology and child development have embraced the concept that an “umbilical transference” [1] occurs during prenatal life, in which the developing fetus is not only susceptible to risks conferred by physical and chemical exposures, but is sensitive to the vicissitudes of maternal psychological state and affect. Indeed, in the 90 years since Freud inferred the importance of the maternal-fetal bond (“[there is] much more continuity between intra-uterine life and earliest infancy than the impressive caesura of birth would have us believe” [2]), maternal emotional state and hormonal fluctuations have been associated with spontaneous abortion, fetal distress, premature labor and other pregnancy complications [3–5].
More recently, the fields of toxicology and environmental epidemiology have begun to adopt these principles and move towards an ‘exposome’ approach, in which an individual’s cumulative “internal chemical environment” is thought to reflect exposure from both exogenous sources (i.e. environmental toxicants) and endogenous processes (i.e. stress-induced hormonal changes) [6].
From a health perspective, embracing the exposome is critical as environmental toxicants and psychosocial stressors share many biological substrates and increasing evidence indicates that stress-induced changes to the maternal milieu may prime rapidly developing physiological systems for disruption by concurrent or subsequent exposure to environmental chemicals and vice versa [7, 8].
In this review, we summarize four putative mechanisms underlying sex-specific susceptibility of the developing neuroendocrine system to the joint effects of psychosocial stress and environmental toxicants (Figure 1). We focus on this system given the sex bias of many neurocognitive and behavioral disorders [9] and evidence demonstrating stress and an array of toxicants independently disrupt neurodevelopmental trajectories and alter programing of the fetal brain, including organization of sexually dimorphic regions [10–12]. We support each mechanism with examples from animal research, and when available, we discuss parallel epidemiologic findings. We draw from studies examining six well-established developmental neurotoxicants, including: bisphenol A (BPA), alcohol, phthalates, lead, chlorpyrifos and traffic-related air pollution. An exhaustive review of sex-specific neurodevelopmental effects associated with isolated exposure to psychosocial stress and these toxicants is beyond the scope of this paper, however, when available we point readers to previously published review articles. Finally, we identify gaps in our current understanding of these complex interactions and discuss future research needs.
Figure 1.
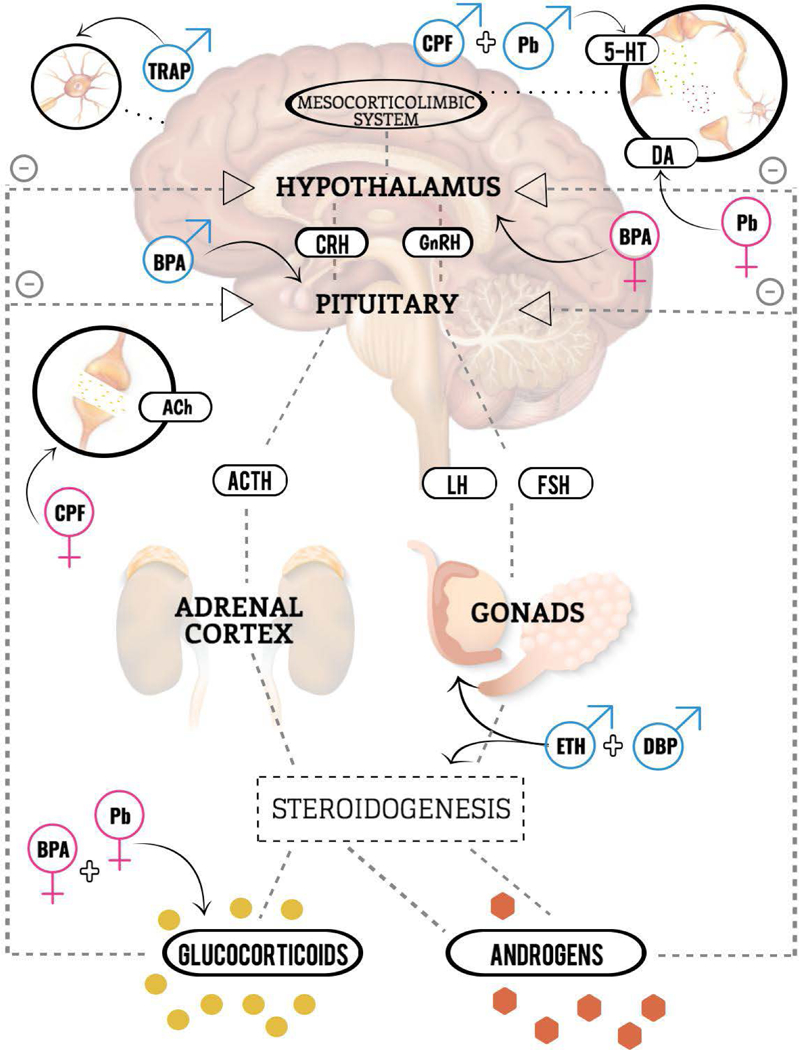
Conceptual model illustrating sex-specific neuroendocrine targets of chemical and non-chemical stressors. All toxicant associations reflect interactions with stress; however, stress is not visually depicted. Abbreviations: 5-HT: serotonin; ACh: acetylcholine; ACTH: adrenocorticotropic hormone; BPA: bisphenol A, CRH: corticotropin releasing hormone; CPF: chlorpyrifos; DA: dopamine; DBP: dibutyl phthalate; ETH: alcohol; FSH: follicular stimulating hormone; GnRH: gonadotropin releasing hormone; LH: luteinizing hormone; Pb: lead; TRAP: traffic related air pollution.
Background
Measures of Stress
Stress is a complex, multilevel construct characterized by cognitive appraisal of potentially stressful stimuli and consequent physiological reactions at both the cellular and emotional levels. Variation in stress responses reflects features of the stimuli (i.e. context, intensity, chronicity) and individual (i.e. sex, life stage, appraisal, coping capacity), thus requiring a diverse set of instruments and protocols for measurement. In epidemiologic research, measures of physical strain (e.g., malnutrition, sleep deprivation) and socioeconomic correlates of stress (e.g., education, resource accessibility), as well as scales of negative life events, stress perception, and negative affect are often used [13]. In experimental animal studies, common protocols include physical restraint, forced swimming or restricted access to bedding material. Additionally, both animal and human studies frequently measure biomarkers of stress, such as glucocorticoids (i.e. cortisol in humans or corticosterone in rodents), under basal conditions and/or following exposure to stressful stimuli. Similarly, researchers can evaluate the timing and intensity of stress responses by examining physiologic and molecular changes following administration of synthetic glucocorticoids.
Mechanism 1: Disrupted HPA Axis Function
Circulating glucocorticoid levels are maintained by the Hypothalamic-Pituitary-Adrenal Axis (HPA), which regulates physiological responses to potentially stressful stimuli [14]. The HPA axis includes the hypothalamus and pituitary gland located in the brain, and the adrenal glands, which are situated above the kidneys. In response to signals from brain nuclei involved in emotion-regulation, hypothalamic neuroendocrine cells produce corticotropin releasing hormone (CRH), which triggers secretion of adrenocorticotropin hormone (ACTH) from the pituitary gland. ACTH signals the adrenal cortex to synthesize and release cholesterol, which undergoes steroidogenic conversion to glucocorticoids, mineralocorticoids, and to a lesser extent androgens (i.e. testosterone). Glucocorticoid receptors located on the hypothalamus and pituitary detect circulating levels and terminate axis activity though a feedback inhibition mechanism.
Bisphenol-a (BPA)
BPA is an endocrine disrupting chemical associated with sex-specific neurobehavioral problems in children [15]. It has been widely used in food cans, plastic bottles and other consumer products leading to nearly ubiquitous human exposure [15]. In silica research indicates BPA is a glucocorticoid receptor agonist [16], and it has been associated with altered HPA axis function in juvenile trout [17] and pregnant women [18]. During pregnancy, surges in glucocorticoid levels are essential for normal maturation of several organ systems, however, elevated glucocorticoids at incorrect developmental stages have been shown to disrupt fetal programming of the HPA axis [19, 20].
Based on these factors, Pantagiotidou et al investigated sex-specific effects of perinatal exposure to BPA on HPA axis responsiveness during adolescence using a murine model [21]. Among female rats, BPA was associated with altered basal corticosterone (increased) and hypothalamic glucocorticoid receptor (decreased) levels. In response to acute stress (forced swimming), BPA-exposed females exhibited anxious coping behaviors and a dampened corticosterone response with failed downregulation of hypothalamic glucocorticoid receptor expression. In contrast, BPA-exposed males did not show altered basal HPA axis function, however, they failed to upregulate pituitary CRH receptor 1 expression in response to acute-stress (Table 1). Taken together, these findings suggest prenatal BPA exposure may program a hyperactive HPA axis with impaired negative feedback responsiveness to circulating corticosterone levels among females and a dampened stress response among males. Notably, HPA axis hyperactivity has been associated with anxiety and depression-like behaviors in rats [22] and diagnosis of anxiety and major depressive disorder in children and adolescents [23]. These findings suggest sex-specific reprogramming of the HPA axis by in utero exposure to BPA may permanently alter individual responses to stressful stimuli and contribute to life-long neuropsychological problems.
Table 1.
Summary of effects from combined exposure to stress and several developmental toxicants on neuroendocrine and immune endpoints in rodents
| Toxicant | Stressor | Male | Female | Ref |
|---|---|---|---|---|
| BPA (prenatal) | Forced swimming (adolescence) | Failed upregulation of pituitary corticotropin releasing hormone receptor 1 | ↑ anxious coping behavior, ↓ corticosterone response; failed down regulation of hypothalamic glucocorticoid receptor expression | [21] |
| Alcohol (prenatal) | Restraint (prenatal) | Delayed & abbreviated fetal testosterone surge, feminized sexual behavior | Not studied | [28, 29] |
| DBP (prenatal) | Dexamethasone (prenatal) | ↓ fetal intra-testicular testosterone levels, ↓ expression of gonadal steroidogenesis genes, ↑ severity & incidence of reproductive organ malformations | Not studied | [41] |
| Pb (prenatal) | Restraint (prenatal) | Disrupted mesocorticolimbic serotonin function and altered delay discounting behavioral performance (trend only, not significant) | ↓ frontal cortex dopamine levels; ↑ circulating corticosterone levels, ↑ learning deficits; ↑ impulsive choice behavior | [48, 49, 50, 51] |
| Chlorpyrifos (neonatal) | Dexamethasone (prenatal) | ↓ serotonin turnover, ↓ upregulation of serotonin receptor & transport protein expression, ↑ hyperactivity | ↓ choline transport protein binding, ↓ choline acetyltransferase activity, ↓ postsynaptic receptor binding | [65, 66, 67] |
| Diesel exhaust | Nest restriction (prenatal) | ↑ expression of microglial toll-like receptor-4 and caspase-1, ↑ pro-inflammatory bias, ↑ anxiety | No interaction observed | [75] |
BPA: bisphenol a; DBP: dibutyl phthalate; Pb: lead
Mechanism 2: Altered Sex Steroid Levels
Similar to adrenal steroidogenesis, the gonads produce sex steroids under the control of the Hypothalamic-Pituitary-Gonadal (HPG) axis. Precisely timed surges of gonadal and adrenal sex steroids are critical for sexually-dimorphic differentiation, including de-feminization and masculinization of the male brain [24, 25, 9].
Alcohol
Increasing evidence indicates stressful stimuli [26] and environmental toxicants [27] disrupt the fetal HPG axis. Given these findings, Ward et al. conducted a series of rodent studies to examine the effects of combined exposure to prenatal stress (restraint) and alcohol on fetal testosterone patterns and later sexual behavior [28]. Male rats born to dams exposed to alcohol or stress during pregnancy were characterized by a testosterone surge (timing and duration) similar to that of controls, however, at each gestational day studied levels of testosterone were elevated in the alcohol-only group and depressed in the stress-only group [28]. Among males in the combined exposure group, the fetal testosterone surge was significantly delayed and abbreviated (p=0.02) compared to the unexposed and single-exposed groups [28]. As adults, males in the combined exposure group displayed feminized sexual behavior (e.g., lordosis) and reduced incidence of copulation with estrous females (Table 1) [29, 30]. Notably, in typically developing rats, the fetal testosterone surge corresponds with development of the sexually dimorphic nucleus in the hypothalamic preoptic area (SDN-POA), which plays important roles in controlling expression of sexual behaviors [26]. These findings suggest that combined exposure to alcohol and stress may desynchronize the temporal overlap between the fetal testosterone surge and development of the SDN-POA, resulting in feminization of the male brain.
Phthalates
Consistent with the effects of alcohol, exposure to phthalates during the fetal masculinization window has been shown to alter rat gonadal steroidogenesis [31–33] and disrupt development of the male reproductive tract. In humans, prenatal exposure to phthalates has been associated with altered sex steroid levels [34–36] and de-masculinized phenotypes among boys, including reduced anogenital distance [37]. In turn, anogenital distance is considered a sensitive marker of androgen activity in early gestation [38] and correlates with sex-specific neurobehaviors [39]. Dibutyl phthalate (DBP) is a plasticizer that has been used extensively in toys and personal care products [40]. Given nearly ubiquitous human exposure to both DBP and stressful stimuli, Drake et al used a rat model to investigate the effects of concurrent exposure to DBP and the synthetic glucocorticoid dexamethasone [41]. Compared to unexposed controls, male offspring prenatally exposed DBP had lower fetal intra-testicular testosterone levels, reduced expression of key genes (StAR, CYP11a1) involved in gonadal steroidogenesis, and several anatomical malformations, including shortened anogenital distance. Exposure to dexamethasone alone showed no effects on male reproductive endpoints, however, when combined with DBP it enhanced the severity or incidence of most DBP-induced effects. Furthermore, combined exposure revealed effects on anogenital distance at lower doses of DBP (Table 1).
Diethylhexyl phthalate (DEHP) is a second anti-androgenic phthalate that is used to enhance flexibility of plastic-based consumer products [42]. Using data from a multi-center birth cohort, Barrett et al investigated prenatal exposure to stress and DEHP among a sample of infants (n=137 boys, n=136 girls) [43]. Stress was measured using a scale of stressful life events administered to couples during pregnancy and DEHP metabolites were measured in a maternal spot urine sample. Consistent with previous findings [44], stressful life events and DEHP exposure were independently associated with reduced anogenital distance among boys, albeit the association with stressful life events was not statistically significant [43]. Interestingly, when stratified by stress (<4 vs. 4+ stressful life events), DEHP metabolites were associated with reduced anogenital distance only among boys in the low stress group. These findings suggest that among males, stress in combination with DEHP exposure may interact such that their joint exposure protects against the anti-androgenic effects conferred by each in isolation. Among girls, prenatal stress was associated with significantly longer anogenital distance, indicative of a masculinizing phenotype, however, no interaction with DEHP metabolites was observed. Importantly, given the relatively small size of the study sample, it is important that these findings be replicated by future studies.
Collectively, these results provide evidence that in rats exposure to stress during pregnancy may augment the effects of concurrent exposure to environmental chemicals on male reproductive track outcomes and development of sexually-dimorphic brain regions, however, more research is needed to translate these findings to humans.
Mechanism 3: Changes to Neurotransmitter Systems
The brain is organized into complex neural networks that rely on the action of neurotransmitter-specific synapses for communication. Several of these circuits, such as the mesocorticolimbic dopamine circuit, also innervate key regulatory systems, including the HPA axis [45]. The mesocorticolimbic neurotransmitter system projects from the ventral tegmental area to the nucleus accumbens, striatum, cortex and limbic centers; its coordinated function is important in motivation, memory, and positive reinforcement of emotion-related behaviors [46].
Lead (Pb)
Prenatal exposure to Pb and stress have each been shown to independently act on the mesocorticolimbic circuit [47, 48], leading Cory-Slechta et al to investigate the effects of combined exposure in a series of rat studies. The researchers found that among adult female rats prenatally exposed to acute stress (restraint) and Pb, but neither in isolation, frontal cortex dopamine levels were reduced and circulating corticosterone levels were elevated [49]. Females in the combined exposure group also exhibited learning deficits [50] and increased rates of stress-induced food reward responding, an indicator of impulsive choice behavior [48]. Parallel findings were not observed among males. In a subsequent study, the research group found indications of a trend towards disrupted mesocorticolimbic serotonin function and altered delay discounting behavioral performance among males exposed to Pb and stress, however, a statistically significant interaction was not observed [51]. Serotonin is a critical mediator of dopamine function [52], is important to HPA axis programming [53], and has been associated with sex-specific impulsive choice behavior in rats [54] and humans [55]. While substantial evidence indicates that stress and Pb act at the intersection of the mesocorticolimbic dopamine circuit and HPA axis, future research is needed to understand whether serotonin modulates this interaction.
Chlorpyrifos
Chlorpyrifos is a widely used agricultural pesticide [56] that disrupts neurotransmitter circuits by inhibiting acetylcholinesterase, the enzyme responsible for breaking down acetylcholine at neuromuscular junctions. Unsurprisingly, human exposure results in similar effects on acetylcholine systems [57], as well as disruption of serotonin circuits [58, 59]. Among children, prenatal exposure has been associated with tremor [60] and lower scores on measures of cognitive and neurobehavioral development [61, 62]. Glucocorticoids also target cholinergic [63] and serotonergic [64] circuits and recent findings suggest prenatal exposure to glucocorticoids may prime these neurotransmitter systems for enhanced disruption by subsequent chlorpyrifos exposure. Using a murine model, Slotkin et al examined several neurochemical and behavioral endpoints in offspring exposed to the synthetic glucocorticoid dexamethasone and/or chlorpyrifos during the prenatal and neonatal periods, respectively [65–67]. Among males, each exposure was associated with reduced presynaptic acetylcholine activity and combined exposure demonstrated additive effects on this endpoint. Conversely, among females, dexamethasone and chlorpyrifos were each associated with increased presynaptic activity, whereas tandem exposure was associated with significantly reduced presynaptic activity (indexed by decreased choline transport protein binding and reduced choline acetyltransferase activity) and lost postsynaptic reactivity (indexed by decreased postsynaptic receptor binding) [66]. Tandem exposure was also associated with enhanced deficiencies in serotonin turnover, an indicator of pre-synaptic impulse activity, and attenuated upregulation of both serotonin receptor and transport protein expression [65]. Notably, serotonin effects were stronger among male pups, who additionally exhibited significantly greater hyperactivity compared to males in the control and single-exposure groups (Table 1) [67]. These findings suggest prenatal exposure to elevated glucocorticoids may enhance vulnerability of cholinergic (girls) and serotonergic (boys) systems to disruption by postnatal chlorpyrifos exposure.
Mechanism 4: Immune Dysregulation
Exposure to stress during fetal and neonatal life has been shown to alter fetal immune pathways [68, 69, 70], which play important roles in development of sexually dimorphic brain regions. For example, research conducted in male rodents suggests that increased estradiol levels resulting from aromatization of testosterone upregulates several microglial immune response genes resulting in the release of inflammatory signaling molecules (e.g. cytokines, prostaglandins) [71]. In turn, elevated prostaglandin E2 triggers a signaling cascade that ultimately augments dendritic spine density in the medial preoptic area, which play a central role in the ability of males to detect olfactory cues from sexually receptive females (71). This complex molecular pathway has been substantiated by experimental studies demonstrating male copulatory behavior during adulthood is completely suppressed among rats subjected to microglial ablation during the neonatal period (72). Additional pathways through which the immune system influences programming of the developing brain have been reviewed by Bilbo et al. [73].
Traffic-Related Air Pollution
Prenatal exposure to traffic-related air pollution is associated with sex-specific disruption of brain development, neurocognitive and behavioral endpoints, and innate immune function [74]. The effects of combined exposure to stress and diesel exhaust particles on immune markers was recently studied using a murine model. Bolton et al found that male rat pups born to dams exposed to diesel exhaust and subjected to nest restriction during pregnancy had significantly elevated expression of microglial toll-like receptor-4 (TLR4) and its downstream effector caspase-1. TLR4 is an innate immune receptor responsive to both exogenous and endogenous danger-associated molecular patterns [75]. As adults, male pups in the combined exposure group also exhibited a greater pro-inflammatory bias (pro-inflammatory IL-1β /anti-inflammatory IL-10 ratio) in microglial derived cytokine levels compared to females. Subsequent analyses demonstrated these sex-specific molecular changes extended to cognitive and behavioral impairments, with males displaying significant hippocampal-dependent memory deficiency and increased anxiety-like behavior compared to males in the control and single-exposure groups (Table 1) [75]. These findings suggest maternal stress-induced changes in TLR4 signaling may enhance the effects of diesel exhaust exposure through microglia-mediated inflammation pathways within the fetal rat brain.
Using data from a Boston, MA-based birth cohort, we previously investigated relationships between maternal report of negative life events during pregnancy and prenatal exposure to black carbon, a measure of traffic-related air pollution, on memory and learning domains among 6-year old children (n=145 boys, n=113 girls) [76]. Consistent with Bolton et al, we found that high exposure to black carbon was associated with significant deficits in attention-concentration scores only among boys born to mothers with high negative life event scores. While we did not study immune pathways, previous research in children has demonstrated stress [77] and black carbon [78] are independently associated with significant increases in pro-inflammatory IL-1β levels, supporting the role of the immune system as a potential mediator of our observed findings.
Summary of Mechanisms
Collectively, the studies reviewed here demonstrate that psychosocial stress and chemical toxicants interact to disrupt several physiological systems important to neurodevelopment and that these interactions vary by sex. However, given the limited research on these complex interactions, especially among humans, consistent patterns of disruption across chemicals, stressors and physiological systems remain poorly understood.
Future Research Needs
Role of the Placenta
While the studies summarized here focus directly on the fetal and maternal systems, emerging research indicates the placenta also plays a critical role in shaping sex-specific neurodevelopment [79]. Despite its design as a constitutional barrier between the mother and fetus, the placenta is penetrated by several neurodevelopmental toxicants and recent research indicates it is sensitive to changes in maternal state [79–81]. For example, in mice exposure to stressful stimuli during pregnancy has been shown to downregulate placental expression of O-linked-N-glycosyl transferase (OGT), an x-linked gene involved in regulating epigenetic modification of a global repressive histone mark (H3K27me3) [82, 83]. Notably, OGT escapes x-inactivation, leading to basal levels that are twice as low in males compared to females [82, 83]. The additional stress-induced decrease in OGT may result in an activated state among males via reduced transcriptional repression, ultimately rendering males more sensitive to concurrent or subsequent insults. Moreover, prenatal exposure to stress has been shown to significantly reduce associations between OGT and the 17-beta-hyroxysteroid dehydrogenase-3 (HSD17β3) gene locus in male placentas. This decrease results in a corresponding reduction in placental expression of HSD17β3, which is responsible for converting androstenedione to testosterone [82]. As expected, prenatally stressed male mice present with increased androstenedione and decreased testosterone, as well as a dysmasculanized behavioral phenotype characterized by stress responses, cognitive function, and spatial strategies more similar to control females than control males. Despite these findings, no studies have investigated the sex-specific combined effects of stress and neurotoxicants on placental structure or function. Future research at the intersection of the maternal-placental-fetal unit is needed to more fully understand how gestational perturbations affect placental function, including altered gene expression patterns.
Improved Biomarkers of Stress
The majority of biomarkers currently used as indicators of stress are hormones (i.e. cortisol), which typically fluctuate in response to acute stress [84], may not accurately reflect maternal stress responses during pregnancy due to altered endocrine system homeostasis [85], and are affected by variation in several enzymes, including placental HSD11β2, which converts cortisol to inactive cortisone [86]. Recently, telomeres have been identified as a potential biomarker of cumulative wear and tear on the body [87, 88]. Telomeres are repetitive, non-coding T2AG3 sequences located at terminal chromosome ends. During cell division, chromosomes erode owing to limitations of DNA replication machinery, thus telomeres serve a self-sacrificing role against damage and degradation of protein coding regions [89]. Several recent epidemiological studies have demonstrated associations between early life social disadvantage and shorter telomere length [90–93], however, these studies are largely limited by cross-sectional designs, small sample sizes, and retrospective reporting of childhood experiences. Future research investigating telomere dynamics during pregnancy and throughout early life is needed to understand whether telomeres can serve as a stress-sensitive biomarker during periods of rapid growth and development.
Expanded Research in Humans
As is evident from our focus on animal research, epidemiologic research investigating sex-specific neuroendocrine effects of developmental exposure to chemical and non-chemical stressors is limited. While animal research provides a unique opportunity to experimentally manipulate and control study conditions, substantial anatomical, functional, developmental and behavioral differences between species limit the generalizability of results to humans. For example, rat adrenal glands to not produce androgens, which are important contributors to sexually dimorphic differentiation of the brain in humans [94]. Likewise, the masculinization programming window is only 3–5 days in rats compared to 4–6 weeks in humans, potentially providing a greater time-opportunity for physiological disruption or recovery [41]. More observational epidemiology research investigating stress-chemical-sex interactions is needed to confirm the biological mechanisms elucidated by animal research.
Investigation of Other Systems and Mechanisms
The reviewed studies do not encompass all psychosocial stressors, toxicants, physiological systems or biological mechanisms known to influence brain development, but rather were selected to highlight enhanced disruption of pathways central to sex-specific programming. Moreover, while reviewed separately, these pathways overlap extensively and should be conceptualized as contributing to a single integrated system. For example, the immune system plays an important role in regulating levels of stress-sensitive neurotransmitters [95], in turn, neurotransmitters influence sex steroid levels via modulation of the HPG axis at the level of the hypothalamus [96]. Moreover, the extent to which stress may act to increase exposure at the biological level is poorly understood. For example, rats exposed to stress have increased brain Pb [51] and decreased blood alcohol [97] levels compared to non-stressed animals exposed to the same dose of chemical. These results suggest that rather than directly acting on overlapping biological substrates, stress may alter blood brain barrier permeability, alcohol metabolism, and/or other pathways involved in toxicant distribution and excretion. Expanded research on the integration of these systems, as well as studies on other biological systems (i.e. autonomic) and cellular mechanisms (i.e. oxidative stress) sensitive to stress and chemical toxicants is needed to more fully understand the pathways through which these exposures interact.
Implications for Health Disparities
Members of low-income, minority communities experience more frequent negative life events, such as witnessing violence and suffering discrimination, which in turn may activate stress responses and precipitate feelings of anxiety, exclusion, and anger [98–102]. Likewise, the multiple challenges associated with financial strain, such as inadequate housing conditions, inability to afford food, and reduced access to health care, can tax individual coping strategies and lead to heightened emotional distress [103, 104]. In addition, parents facing multiple adversities are less likely to spend time at home (i.e. due to working multiple jobs with longer commutes and unfavorable shifts [105]) and are more likely to employ controlling, restrictive and punitive parenting strategies, often as an adaptive and protective response to neighborhood crime or other dangerous circumstances [104].
As described extensively by the environmental justice literature, minority and socioeconomically disadvantaged individuals not only experience greater levels of psychosocial stress, but often bear a disproportionate burden of environmental risk [106–108]. For example, nationwide statistics indicate members of racial and ethnic minorities are more likely to live within 150 meters of a major U.S. highway, putting these groups at increased risk for exposure to traffic-related air pollution [109]. Likewise, exposure to Pb has historically been higher among black [110] and low-income [111] children, who disproportionately live in urban neighborhoods with older housing that may contain deteriorating lead-based paint.
The socioeconomic and environmental challenges faced by these populations likely contribute to the persistence of health disparities across ethnic and economic groups in the United States [109]. For example, the rate of premature birth is significantly higher among black (17%) compared to white (11%) infants [109]. Premature birth is estimated to account for one third of all infant deaths and is associated with numerous childhood and later life disorders, including neurocognitive and behavioral problems [112]. Notably, black children (8.4%) and those living below the federal poverty level (11%) are more likely to be diagnosed with a learning disability or attention deficit hyperactivity disorder compared to white children (7.5%) and those living at or above 200% of the poverty level (5.8%), respectively [113]. Likewise, infants born to mothers with less than a high school education, an indicator of socioeconomic status, are nearly twice as likely to die in the first year of life and approximately six times more likely to be rated in poor or fair health during childhood [114]. Similar trends have been documented for other indicators of socioeconomic status (i.e. family structure) and childhood morbidities (i.e. asthma) [113].
Conclusions
Health inequalities, including disparities in neurocognitive and behavioral outcomes, persist across ethnic and economic groups in the United States. As demonstrated here, multiple sexually dimorphic biological systems involved in programming the developing brain are susceptible to enhanced disruption by concurrent or consecutive exposure to psychosocial stress and chemical toxicants, which often co-occur among minority and socioeconomically disadvantaged communities. The reviewed studies highlight the importance of examining exposure to chemical toxicants within the context of the social environment, as well as the need to consider the influence of sex when investigating neuroendocrine and immune endpoints. Despite challenges associated with investigating these relationships among humans, such as the large sample size requirements needed to investigate 3-way interactions in observational studies and ethical considerations associated with randomized controlled trials, future research focused on studying these interactions among human populations is critically needed.
Acknowledgments
During her training and preparation of this manuscript, WJC was supported by the National Institute Environmental Health Science grants T32 ES023772 and T32 ES007322 and Environmental Protection Agency STAR Fellowship: FP-91779001.
Footnotes
Disclaimers
This publication was developed under STAR Fellowship Assistance Agreement no. FP-91779001 awarded by the U.S. Environmental Protection Agency (EPA). It has not been formally reviewed by EPA. The views expressed in this publication are solely those of the authors.
Compliance with Ethical Standards
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Conflict of Interest
Whitney J. Cowell and Rosalind J. Wright declare that they have no conflict of interest.
References
Papers of particular interest, published recently, have been highlighted as:
• Of importance
- 1.Culbert-Koehn J Don’t get stuck in the mother: regression in analysis. Journal of Analytical Psychology. 1997;42:99–104. [Google Scholar]
- 2.Freud S Inhibitions, Symptoms and Anxiety. London: Hogarth Press; 1926. [Google Scholar]
- 3.Mitrani JL. Toward an understanding of unmentalized experience. Psychoanal Q 1995;64(1):68–112. [PubMed] [Google Scholar]
- 4.Beydoun H, Saftlas AF. Physical and mental health outcomes of prenatal maternal stress in human and animal studies: a review of recent evidence. Paediatr Perinat Epidemiol. 2008;22(5):438–66. [DOI] [PubMed] [Google Scholar]
- 5.Savitz D, Dunkel-Schetter C. Preterm Birth: Causes, Consequences and Prevention In: Behrman R, AS B, editors. Behavioral and psychosocial contributors to preterm birth. Washington DC: National Academy Press; 2006. p. 87–123. [Google Scholar]
- 6.Rappaport SM. Implications of the exposome for exposure science. J Expo Sci Environ Epidemiol. 2011;21(1):5–9. [DOI] [PubMed] [Google Scholar]
- 7.Weiss B, Bellinger DC. Social ecology of children’s vulnerability to environmental pollutants. Environ Health Perspect. 2006;114(10):1479–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wright RJ. Moving towards making social toxins mainstream in children’s environmental health. Curr Opin Pediatr. 2009;21(2):222–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pfaff DW, Christen Y. Multiple origins of sex differences in brain neuroendocrine functions and their pathologies Research and perspectives in endocrine interactions. Heidelberg; New York: Springer; 2013. [Google Scholar]
- 10.•Antonelli MC, Pallares ME, Ceccatelli S, Spulber S. Long-term consequences of prenatal stress and neurotoxicants exposure on neurodevelopment. Prog Neurobiol. 2016.The authors provide a detailed summary of neurodevelopmental consequences of exposure to gestational stress and environmental toxicants, including sex-specific effects, and the overlapping pathways they each target.
- 11.Graignic-Philippe R, Dayan J, Chokron S, Jacquet AY, Tordjman S. Effects of prenatal stress on fetal and child development: a critical literature review. Neurosci Biobehav Rev. 2014;43:137–62. [DOI] [PubMed] [Google Scholar]
- 12.Jurewicz J, Polanska K, Hanke W. Exposure to widespread environmental toxicants and children’s cognitive development and behavioral problems. Int J Occup Med Environ Health. 2013;26(2):185–204. [DOI] [PubMed] [Google Scholar]
- 13.Cohen S, Kessler R, Underwood Gorden L. Measuring Stress: A guide for health and social scientists. New York: Oxford University Press; 1995. [Google Scholar]
- 14.The Handbook of Stress Science. New York: Springer Publishing Company; 2010. [Google Scholar]
- 15.Mustieles V, Perez-Lobato R, Olea N, Fernandez MF. Bisphenol A: Human exposure and neurobehavior. Neurotoxicology. 2015;49:174–84. [DOI] [PubMed] [Google Scholar]
- 16.Prasanth GK, Divya LM, Sadasivan C. Bisphenol-A can bind to human glucocorticoid receptor as an agonist: an in silico study. J Appl Toxicol. 2010;30(8):769–74. [DOI] [PubMed] [Google Scholar]
- 17.Aluru N, Leatherland JF, Vijayan MM. Bisphenol A in oocytes leads to growth suppression and altered stress performance in juvenile rainbow trout. PLoS One. 2010;5(5):e10741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Giesbrecht GF, Liu J, Ejaredar M, Dewey D, Letourneau N, Campbell T et al. Urinary bisphenol A is associated with dysregulation of HPA-axis function in pregnant women: Findings from the APrON cohort study. Environ Res. 2016;151:689–97. [DOI] [PubMed] [Google Scholar]
- 19.Moisiadis VG, Matthews SG. Glucocorticoids and fetal programming part 2: Mechanisms. Nat Rev Endocrinol. 2014;10(7):403–11. [DOI] [PubMed] [Google Scholar]
- 20.Tegethoff M, Pryce C, Meinlschmidt G. Effects of intrauterine exposure to synthetic glucocorticoids on fetal, newborn, and infant hypothalamic-pituitary-adrenal axis function in humans: a systematic review. Endocr Rev. 2009;30(7):753–89. [DOI] [PubMed] [Google Scholar]
- 21.Panagiotidou E, Zerva S, Mitsiou DJ, Alexis MN, Kitraki E. Perinatal exposure to low-dose bisphenol A affects the neuroendocrine stress response in rats. J Endocrinol. 2014;220(3):207–18. [DOI] [PubMed] [Google Scholar]
- 22.Chen F, Zhou L, Bai Y, Zhou R, Chen L. Hypothalamic-pituitary-adrenal axis hyperactivity accounts for anxiety- and depression-like behaviors in rats perinatally exposed to bisphenol A. J Biomed Res. 2015;29(3):250–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Steingard R, Biederman J, Keenan K, Moore C. Comorbidity in the interpretation of dexamethasone suppression test results in children: a review and report. Biol Psychiatry. 1990;28(3):193–202. [DOI] [PubMed] [Google Scholar]
- 24.Corbier P, Roffi J, Rhoda J. Female sexual behavior in male rats: effect of hour of castration at birth. Physiol Behav. 1983;30(4):613–6. [DOI] [PubMed] [Google Scholar]
- 25.Hoepfner BA, Ward IL. Prenatal and neonatal androgen exposure interact to affect sexual differentiation in female rats. Behav Neurosci. 1988;102(1):61–5. [DOI] [PubMed] [Google Scholar]
- 26.Shansky RM. Sex differences in the central nervous system Neuroscience Net Reference Book Series Book 4. Boston: Elsevier Academic Press; 2016. [Google Scholar]
- 27.Hampl R, Kubatova J, Starka L. Steroids and endocrine disruptors--History, recent state of art and open questions. J Steroid Biochem Mol Biol. 2016;155(Pt B):217–23. [DOI] [PubMed] [Google Scholar]
- 28.Ward IL, Ward OB, Affuso JD, Long WD 3rd, French JA, Hendricks SE. Fetal testosterone surge: specific modulations induced in male rats by maternal stress and/or alcohol consumption. Horm Behav. 2003;43(5):531–9. [DOI] [PubMed] [Google Scholar]
- 29.Ward OB, Ward IL, Denning JH, Hendricks SE, French JA. Hormonal mechanisms underlying aberrant sexual differentiation in male rats prenatally exposed to alcohol, stress, or both. Arch Sex Behav. 2002;31(1):9–16. [DOI] [PubMed] [Google Scholar]
- 30.Ward IL, Bennett AL, Ward OB, Hendricks SE, French JA. Androgen threshold to activate copulation differs in male rats prenatally exposed to alcohol, stress, or both factors. Horm Behav. 1999;36(2):129–40. [DOI] [PubMed] [Google Scholar]
- 31.Supornsilchai V, Soder O, Svechnikov K. Stimulation of the pituitary-adrenal axis and of adrenocortical steroidogenesis ex vivo by administration of di-2-ethylhexyl phthalate to prepubertal male rats. J Endocrinol. 2007;192(1):33–9. [DOI] [PubMed] [Google Scholar]
- 32.Sekaran S, Jagadeesan A. In utero exposure to phthalate downregulates critical genes in Leydig cells of F1 male progeny. J Cell Biochem. 2015;116(7):1466–77. [DOI] [PubMed] [Google Scholar]
- 33.Akingbemi BT, Youker RT, Sottas CM, Ge R, Katz E, Klinefelter GR et al. Modulation of rat Leydig cell steroidogenic function by di(2-ethylhexyl)phthalate. Biol Reprod. 2001;65(4):1252–9. [DOI] [PubMed] [Google Scholar]
- 34.Araki A, Mitsui T, Goudarzi H, Nakajima T, Miyashita C, Itoh S et al. Prenatal di(2-ethylhexyl) phthalate exposure and disruption of adrenal androgens and glucocorticoids levels in cord blood: The Hokkaido Study. Sci Total Environ. 2017;581–582:297–304. [DOI] [PubMed] [Google Scholar]
- 35.Ferguson KK, Peterson KE, Lee JM, Mercado-Garcia A, Blank-Goldenberg C, Tellez-Rojo MM et al. Prenatal and peripubertal phthalates and bisphenol A in relation to sex hormones and puberty in boys. Reprod Toxicol. 2014;47:70–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Watkins DJ, Tellez-Rojo MM, Ferguson KK, Lee JM, Solano-Gonzalez M, Blank-Goldenberg C et al. In utero and peripubertal exposure to phthalates and BPA in relation to female sexual maturation. Environ Res. 2014;134:233–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Swan SH, Sathyanarayana S, Barrett ES, Janssen S, Liu F, Nguyen RH et al. First trimester phthalate exposure and anogenital distance in newborns. Hum Reprod. 2015;30(4):963–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Thankamony A, Pasterski V, Ong KK, Acerini CL, Hughes IA. Anogenital distance as a marker of androgen exposure in humans. Andrology. 2016;4(4):616–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pasterski V, Acerini CL, Dunger DB, Ong KK, Hughes IA, Thankamony A et al. Postnatal penile growth concurrent with mini-puberty predicts later sex-typed play behavior: Evidence for neurobehavioral effects of the postnatal androgen surge in typically developing boys. Horm Behav. 2015;69:98–105. [DOI] [PubMed] [Google Scholar]
- 40.Schettler T Human exposure to phthalates via consumer products. Int J Androl. 2006;29(1):134–9. [DOI] [PubMed] [Google Scholar]
- 41.Drake AJ, van den Driesche S, Scott HM, Hutchison GR, Seckl JR, Sharpe RM. Glucocorticoids amplify dibutyl phthalate-induced disruption of testosterone production and male reproductive development. Endocrinology. 2009;150(11):5055–64. [DOI] [PubMed] [Google Scholar]
- 42.Zarean M, Keikha M, Poursafa P, Khalighinejad P, Amin M, Kelishadi R. A systematic review on the adverse health effects of di-2-ethylhexyl phthalate. Environ Sci Pollut Res Int. 2016;23(24):24642–93. [DOI] [PubMed] [Google Scholar]
- 43.Barrett ES, Parlett LE, Sathyanarayana S, Redmon JB, Nguyen RH, Swan SH. Prenatal Stress as a Modifier of Associations between Phthalate Exposure and Reproductive Development: results from a Multicentre Pregnancy Cohort Study. Paediatr Perinat Epidemiol. 2016;30(2):105–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Barrett ES, Parlett LE, Sathyanarayana S, Liu F, Redmon JB, Wang C et al. Prenatal exposure to stressful life events is associated with masculinized anogenital distance (AGD) in female infants. Physiol Behav. 2013;114–115:14–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sullivan RM, Dufresne MM. Mesocortical dopamine and HPA axis regulation: role of laterality and early environment. Brain Res. 2006;1076(1):49–59 [DOI] [PubMed] [Google Scholar]
- 46.Strominger N, Demarest R, Laemle L. Noback’s Human Nervous System. New York: Springer Science+Business Media; 2012. [Google Scholar]
- 47.Rossi-George A, Virgolini MB, Weston D, Thiruchelvam M, Cory-Slechta DA. Interactions of lifetime lead exposure and stress: behavioral, neurochemical and HPA axis effects. Neurotoxicology. 2011;32(1):83–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Virgolini MB, Rossi-George A, Lisek R, Weston DD, Thiruchelvam M, Cory-Slechta DA. CNS effects of developmental Pb exposure are enhanced by combined maternal and offspring stress. Neurotoxicology. 2008;29(5):812–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cory-Slechta DA, Virgolini MB, Thiruchelvam M, Weston DD, Bauter MR. Maternal stress modulates the effects of developmental lead exposure. Environ Health Perspect. 2004;112(6):717–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cory-Slechta DA, Stern S, Weston D, Allen JL, Liu S. Enhanced learning deficits in female rats following lifetime pb exposure combined with prenatal stress. Toxicol Sci. 2010;117(2):427–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Weston HI, Weston DD, Allen JL, Cory-Slechta DA. Sex-dependent impacts of low-level lead exposure and prenatal stress on impulsive choice behavior and associated biochemical and neurochemical manifestations. Neurotoxicology. 2014;44:169–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hensler JG, Artigas F, Bortolozzi A, Daws LC, De Deurwaerdere P, Milan L et al. Catecholamine/Serotonin interactions: systems thinking for brain function and disease. Adv Pharmacol. 2013;68:167–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kapoor A, Dunn E, Kostaki A, Andrews MH, Matthews SG. Fetal programming of hypothalamo-pituitary-adrenal function: prenatal stress and glucocorticoids. J Physiol. 2006;572(Pt 1):31–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Baarendse PJ, Vanderschuren LJ. Dissociable effects of monoamine reuptake inhibitors on distinct forms of impulsive behavior in rats. Psychopharmacology (Berl). 2012;219(2):313–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Walderhaug E, Magnusson A, Neumeister A, Lappalainen J, Lunde H, Refsum H et al. Interactive effects of sex and 5-HTTLPR on mood and impulsivity during tryptophan depletion in healthy people. Biol Psychiatry. 2007;62(6):593–9. [DOI] [PubMed] [Google Scholar]
- 56.Chlorpyrifos. U.S. Environmental Protection Agency. Available: https://www.epa.gov/ingredients-used-pesticide-products/chlorpyrifos
- 57.Prueitt RL, Goodman JE, Bailey LA, Rhomberg LR. Hypothesis-based weight-of-evidence evaluation of the neurodevelopmental effects of chlorpyrifos. Crit Rev Toxicol. 2011;41(10):822–903. [DOI] [PubMed] [Google Scholar]
- 58.Slotkin TA, Seidler FJ. The alterations in CNS serotonergic mechanisms caused by neonatal chlorpyrifos exposure are permanent. Brain Res Dev Brain Res. 2005;158(1–2):115–9. [DOI] [PubMed] [Google Scholar]
- 59.Aldridge JE, Seidler FJ, Meyer A, Thillai I, Slotkin TA. Serotonergic systems targeted by developmental exposure to chlorpyrifos: effects during different critical periods. Environ Health Perspect. 2003;111(14):1736–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rauh VA, Garcia WE, Whyatt RM, Horton MK, Barr DB, Louis ED. Prenatal exposure to the organophosphate pesticide chlorpyrifos and childhood tremor. Neurotoxicology. 2015;51:80–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rauh VA, Garfinkel R, Perera FP, Andrews HF, Hoepner L, Barr DB et al. Impact of prenatal chlorpyrifos exposure on neurodevelopment in the first 3 years of life among inner-city children. Pediatrics. 2006;118(6):e1845–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bouchard MF, Chevrier J, Harley KG, Kogut K, Vedar M, Calderon N et al. Prenatal exposure to organophosphate pesticides and IQ in 7-year-old children. Environ Health Perspect. 2011;119(8):1189–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Emgard M, Paradisi M, Pirondi S, Fernandez M, Giardino L, Calza L. Prenatal glucocorticoid exposure affects learning and vulnerability of cholinergic neurons. Neurobiol Aging. 2007;28(1):112–21. [DOI] [PubMed] [Google Scholar]
- 64.St-Pierre J, Laurent L, King S, Vaillancourt C. Effects of prenatal maternal stress on serotonin and fetal development. Placenta. 2016;48 Suppl 1:S66–S71. [DOI] [PubMed] [Google Scholar]
- 65.Slotkin TA, Card J, Seidler FJ. Prenatal dexamethasone, as used in preterm labor, worsens the impact of postnatal chlorpyrifos exposure on serotonergic pathways. Brain Res Bull. 2014;100:44–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Slotkin TA, Card J, Infante A, Seidler FJ. Prenatal dexamethasone augments the sex-selective developmental neurotoxicity of chlorpyrifos: implications for vulnerability after pharmacotherapy for preterm labor. Neurotoxicol Teratol. 2013;37:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Levin ED, Cauley M, Johnson JE, Cooper EM, Stapleton HM, Ferguson PL et al. Prenatal dexamethasone augments the neurobehavioral teratology of chlorpyrifos: significance for maternal stress and preterm labor. Neurotoxicol Teratol. 2014;41:35–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Coussons-Read ME, Okun ML, Nettles CD. Psychosocial stress increases inflammatory markers and alters cytokine production across pregnancy. Brain Behav Immun. 2007;21(3):343–50. [DOI] [PubMed] [Google Scholar]
- 69.Cryan JF, Dinan TG. Unraveling the longstanding scars of early neurodevelopmental stress. Biol Psychiatry. 2013;74(11):788–9. [DOI] [PubMed] [Google Scholar]
- 70.Johnson FK, Kaffman A Early life stress perturbs the function of microglia in the developing rodent brain: new insights and future directions. Brain, Behav and Immun. 2017; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.McCarthy M, Nugent BM, Lenz KM. Neuroimmunology and neuroepigenetics in the establishment of sex differences in the brain. Nature Reviews Neuroscience. 2017;18:471–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.VanRyzin JW, Yu SJ, Perez-Pouchoulen M, McCarthy MM. Temporary depletion of microglia during the early postnatal period induces lasting sex-dependent and sex-independent effects on behavior in rats. eNeuro. 2016;3(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bilbo SD, Schwarz JM. The immune system and developmental programming of brain and behavior. Front Neuroendocrinol. 2012;33(3):267–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.•Costa LG, Cole TB, Coburn J, Chang YC, Dao K, Roque PJ. Neurotoxicity of traffic-related air pollution. Neurotoxicology. 2017;59:133–9.This recent review summarizes results from toxicologic and epidemiologic research investigating neurodevelopmental and neurodegeneraive effects of exposure to air pollution. The paper highlights the need for future research investigating sex differences and gene-environment interactions.
- 75.Bolton JL, Huff NC, Smith SH, Mason SN, Foster WM, Auten RL et al. Maternal stress and effects of prenatal air pollution on offspring mental health outcomes in mice. Environ Health Perspect. 2013;121(9):1075–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cowell WJ, Bellinger DC, Coull BA, Gennings C, Wright RO, Wright RJ. Associations between Prenatal Exposure to Black Carbon and Memory Domains in Urban Children: Modification by Sex and Prenatal Stress. PLoS One. 2015;10(11):e0142492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tyrka AR, Parade SH, Valentine TR, Eslinger NM, Seifer R. Adversity in preschool-aged children: Effects on salivary interleukin-1beta. Dev Psychopathol. 2015;27(2):567–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.De Prins S, Dons E, Van Poppel M, Int Panis L, Van de Mieroop E, Nelen V et al. Airway oxidative stress and inflammation markers in exhaled breath from children are linked with exposure to black carbon. Environ Int. 2014;73:440–6. [DOI] [PubMed] [Google Scholar]
- 79.Bale TL. Sex differences in prenatal epigenetic programming of stress pathways. Stress. 2011;14(4):348–56. [DOI] [PubMed] [Google Scholar]
- 80.Howerton CL, Bale TL. Prenatal programing: at the intersection of maternal stress and immune activation. Horm Behav. 2012;62(3):237–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Murphy VE, Smith R, Giles WB, Clifton VL. Endocrine regulation of human fetal growth: the role of the mother, placenta, and fetus. Endocr Rev. 2006;27(2):141–69. [DOI] [PubMed] [Google Scholar]
- 82.•Howerton CL, Bale TL. Targeted placental deletion of OGT recapitulates the prenatal stress phenotype including hypothalamic mitochondrial dysfunction. Proc Natl Acad Sci U S A. 2014;111(26):9639–44.This important paper demonstrates that knocking out a specific placental gene results in mice pups characterized by stress respones similar to those of mice exposed to stress during gestation. The paper provides convincing evidence that in mice, levels of this protein (OGT) may serve as a placental biomarker of stress.
- 83.Howerton CL, Morgan CP, Fischer DB, Bale TL. O-GlcNAc transferase (OGT) as a placental biomarker of maternal stress and reprogramming of CNS gene transcription in development. Proc Natl Acad Sci U S A. 2013;110(13):5169–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Smith MN, Griffith WC, Beresford SA, Vredevoogd M, Vigoren EM, Faustman EM. Using a biokinetic model to quantify and optimize cortisol measurements for acute and chronic environmental stress exposure during pregnancy. J Expo Sci Environ Epidemiol. 2014;24(5):510–6. [DOI] [PubMed] [Google Scholar]
- 85.Reynolds RM. Glucocorticoid excess and the developmental origins of disease: two decades of testing the hypothesis−−2012 Curt Richter Award Winner. Psychoneuroendocrinology. 2013;38(1):1–11. [DOI] [PubMed] [Google Scholar]
- 86.Benediktsson R, Calder AA, Edwards CR, Seckl JR. Placental 11 beta-hydroxysteroid dehydrogenase: a key regulator of fetal glucocorticoid exposure. Clin Endocrinol (Oxf). 1997;46(2):161–6. [DOI] [PubMed] [Google Scholar]
- 87.Mitchell C, Hobcraft J, McLanahan SS, Siegel SR, Berg A, Brooks-Gunn J et al. Social disadvantage, genetic sensitivity, and children’s telomere length. Proc Natl Acad Sci U S A. 2014;111(16):5944–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.•Shalev I. Early life stress and telomere length: investigating the connection and possible mechanisms: a critical survey of the evidence base, research methodology and basic biology. BioEssays : news and reviews in molecular, cellular and developmental biology. 2012;34(11):943–52.This thourough review paper summarizes the evidence linking early life stress with telomere dynamics and also provides a clear and concise review of methodoligical issues and limitations relating to telomere measurement (i.e. relating to assay choice, cell type, measurement error).
- 89.Bonetti D, Martina M, Falcettoni M, Longhese MP. Telomere-end processing: mechanisms and regulation. Chromosoma. 2013. [DOI] [PubMed] [Google Scholar]
- 90.Entringer S, Epel ES, Kumsta R, Lin J, Hellhammer DH, Blackburn EH et al. Stress exposure in intrauterine life is associated with shorter telomere length in young adulthood. Proc Natl Acad Sci U S A. 2011;108(33):E513–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Entringer S, Epel ES, Lin J, Buss C, Shahbaba B, Blackburn EH et al. Maternal psychosocial stress during pregnancy is associated with newborn leukocyte telomere length. Am J Obstet Gynecol. 2013;208(2):134 e1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kiecolt-Glaser JK, Gouin JP, Weng NP, Malarkey WB, Beversdorf DQ, Glaser R. Childhood adversity heightens the impact of later-life caregiving stress on telomere length and inflammation. Psychosom Med. 2011;73(1):16–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Tyrka AR, Price LH, Kao HT, Porton B, Marsella SA, Carpenter LL. Childhood maltreatment and telomere shortening: preliminary support for an effect of early stress on cellular aging. Biol Psychiatry. 2010;67(6):531–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Moisiadis VG, Matthews SG. Glucocorticoids and fetal programming part 1: Outcomes. Nat Rev Endocrinol. 2014;10(7):391–402. [DOI] [PubMed] [Google Scholar]
- 95.Dantzer R, O’Connor JC, Freund GG, Johnson RW, Kelley KW. From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci. 2008;9(1):46–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Watanabe M, Fukuda A, Nabekura J. The role of GABA in the regulation of GnRH neurons. Front Neurosci. 2014;8:387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ward IL, Ward OB, French JA, Hendricks SE, Mehan D, Winn RJ. Prenatal alcohol and stress interact to attenuate ejaculatory behavior, but not serum testosterone or LH in adult male rats. Behav Neurosci. 1996;110(6):1469–77. [DOI] [PubMed] [Google Scholar]
- 98.King K, Ogle C. Negative life events vary by neighborhood and mediate the relation between neighborhood context and psychological well-being. PLoS One. 2014;9(4):e93539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Myers HF. Ethnicity- and socio-economic status-related stresses in context: an integrative review and conceptual model. J Behav Med. 2009;32(1):9–19. [DOI] [PubMed] [Google Scholar]
- 100.Wade R, Jr., Cronholm PF, Fein JA, Forke CM, Davis MB, M Harkins-Schwarz et al. Household and community-level Adverse Childhood Experiences and adult health outcomes in a diverse urban population. Child Abuse Negl. 2016;52:135–45. [DOI] [PubMed] [Google Scholar]
- 101.Williams DR, Neighbors HW, Jackson JS. Racial/ethnic discrimination and health: findings from community studies. Am J Public Health. 2003;93(2):200–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wilson WC, Rosenthal BS. The relationship between exposure to community violence and psychological distress among adolescents: a meta-analysis. Violence Vict. 2003;18(3):335–52. [DOI] [PubMed] [Google Scholar]
- 103.Becerra BJ, Sis-Medina RC, Reyes A, Becerra MB. Association Between Food Insecurity and Serious Psychological Distress Among Hispanic Adults Living in Poverty. Prev Chronic Dis. 2015;12:E206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Chen E, Miller GE. Socioeconomic status and health: mediating and moderating factors. Annu Rev Clin Psychol. 2013;9:723–49. doi: 10.1146/annurev-clinpsy-050212-185634. [DOI] [PubMed] [Google Scholar]
- 105.Bradley RH, Corwyn RF, McAdoo HP, Coll CG. The home environments of children in the United States part I: variations by age, ethnicity, and poverty status. Child Dev. 2001;72(6):1844–67. [DOI] [PubMed] [Google Scholar]
- 106.•Corburn J. Concepts for Studying Urban Environmental Justice. Curr Environ Health Rep. 2017;4(1):61–7.This review paper summarizes conceptual thinking and current research frameworks related to environmental justice in the United States with a focus on urban neighborhoods.
- 107.Cushing L, Morello-Frosch R, Wander M, Pastor M. The haves, the have-nots, and the health of everyone: the relationship between social inequality and environmental quality. Annu Rev Public Health. 2015;36:193–209. [DOI] [PubMed] [Google Scholar]
- 108.Vrijheid M, Martinez D, Aguilera I, Ballester F, Basterrechea M, Esplugues A et al. Socioeconomic status and exposure to multiple environmental pollutants during pregnancy: evidence for environmental inequity? Journal of epidemiology and community health. 2012;66(2):106–13. [DOI] [PubMed] [Google Scholar]
- 109.CDC. Centers for Disease Control and Prevention, Health Disparities & Ineqalities Report - United States. Morbidity & Mortality Weekly Report (MMWR) Supplement. 2013;62:1–187. [Google Scholar]
- 110.White BM, Bonilha HS, Ellis C, Jr. Racial/Ethnic Differences in Childhood Blood Lead Levels Among Children <72 Months of Age in the United States: a Systematic Review of the Literature. J Racial Ethn Health Disparities. 2016;3(1):145–53. [DOI] [PubMed] [Google Scholar]
- 111.Moody HA, Darden JT, Pigozzi BW. The Relationship of Neighborhood Socioeconomic Differences and Racial Residential Segregation to Childhood Blood Lead Levels in Metropolitan Detroit. J Urban Health. 2016;93(5):820–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.McCormick MC, Litt JS, Smith VC, Zupancic JA. Prematurity: an overview and public health implications. Annu Rev Public Health. 2011;32:367–79. [DOI] [PubMed] [Google Scholar]
- 113.CDC. Centers for Disease Control and Prevention. National Center for Health Statistics, National Health Interview Survey: Summary Health Statistics. 2015. Available: https://www.cdc.gov/nchs/nhis/shs/tables.htm
- 114.Braveman PA, Egerter SA, Mockenhaupt RE. Broadening the focus: the need to address the social determinants of health. Am J Prev Med. 2011;40:S4–18. [DOI] [PubMed] [Google Scholar]