

Abstract

O-Methylation modulates the pharmacokinetic and pharmacodynamic (PK/PD) properties of small-molecule natural products, affecting their bioavailability, stability, and binding to targets. Diversity-oriented combinatorial biosynthesis of new chemical entities for drug discovery and optimization of known bioactive scaffolds during drug development both demand efficient O-methyltransferase (OMT) biocatalysts with considerable substrate promiscuity and tunable regioselectivity that can be deployed in a scalable and sustainable manner. Here we demonstrate efficient total biosynthetic and biocatalytic platforms that use a pair of fungal OMTs with orthogonal regiospecificity to produce unnatural O-methylated benzenediol lactone polyketides. We show that rational, structure-guided active-site cavity engineering can reprogram the regioselectivity of these enzymes. We also characterize the interplay of engineered regioselectivity with substrate plasticity. These findings will guide combinatorial biosynthetic tailoring of unnatural products toward the generation of diverse chemical matter for drug discovery and the PK/PD optimization of bioactive scaffolds for drug development.
Introduction
Alkylations, most importantly the methylation of O-, C-, N-, and S-centered nucleophiles, are ubiquitous tailoring reactions during the biosynthesis of all major classes of small-molecule natural products.1 Among these reactions, O-methylation is used by nature and medicinal chemistry for the hydrophobic masking of peripheral hydroxyl or carboxylic acid moieties to adjust the physicochemical and biological properties of bioactive compounds. The resulting “methyl effect” increases the lipophilicity and membrane permeability of small-molecule scaffolds, enhancing their membrane transport, oral bioavailability, absorption, and excretion.2,3 It also modulates the reactivity of small molecules by umpolung (reversal of polarity) of hydroxyl groups,4 affecting the in vivo metabolic stability of drugs and channeling the biosynthesis of natural products through a series of reactive intermediates.5,6O-Methylation may also alter the conformation of natural product scaffolds through stereoelectronic and steric effects and confers weak interactions that contribute to binding to various targets, including receptors.7 As opposed to chemical synthesis, enzymatic O-methylation offers regio- and stereospecific outcomes even for highly complex and reactive scaffolds without resorting to expensive multistep protection/deprotection strategies. It also proceeds with a high yield under mild, environmentally friendly reaction conditions and does not have to contend with the formation of C-methylated side products in the case of phenolic substrates.8,9 In the biotechnology industries, O-methylation is conducted by utilizing in vivo biocatalytic platforms instead of in vitro reconstituted enzymatic reactions because the availability of the methyl donor co-substrate S-adenosyl-methionine (SAM) is limited and its in vitro regeneration is inefficient.1,10
O-Methylation has been engineered in bacterial natural product biosynthetic pathways,7 mostly those of polyketides and non-ribosomal peptides, by deleting the genes encoding native O-methyltransferase (OMT) enzymes or augmenting the pathways with genes for heterologous OMTs despite the often-constrained substrate flexibility of these enzymes. Extensive work has also been conducted on dissecting and altering the chemo- and regioselectivities of the mammalian and bacterial catechol OMTs and those of the plant and bacterial flavonoid and phenylpropanoid OMTs.7,10 Such studies show that regio- and chemospecificity are intimately connected in each enzyme scaffold,11−13 although divorcing these properties is possible to some extent by directed evolution or active-site cavity engineering informed by enzyme–substrate docking studies.14−17 However, little attention has been paid before now to define the substrate promiscuity and modulate the regioselectivity of fungal secondary metabolite OMTs, despite these enzymes constituting the most common tailoring activities during the biosynthesis of such natural products.
OMTs are also crucial to modulate the biological activities of benzenediol lactones (BDLs), a growing group of fungal polyketide natural products with an astonishing range of biological activities.18,19 BDLs are defined by a 1,3-benzenediol moiety fused to a macrocyclic lactone ring, with C2–C7 connectivity as in resorcylic acid lactones (RALs) or with C3–C8 connectivity as in dihydroxyphenylacetic acid lactones (DALs). Natural RALs contain 12- or 14-membered macrocycles (RAL12 or RAL14), while natural DALs most often include 12-membered macrolactones (DAL12). Among RAL12, lasiodiplodin (1a) with a 3-methoxy moiety displays ATP- and prostaglandin synthesis inhibitory activities, while its unmethylated congener, 3-desmethyl-lasiodiplodin (DLD, 1) is devoid of such activities. Instead, DLD 1 is a non-steroidal mineralocorticoid receptor antagonist and pancreatic lipase inhibitor.20 RAL14 compounds with a 5-methoxy group are also important pharmacophores. Thus, hypothemycin is a selective inhibitor of mitogen-associated protein kinases, 7-oxozeaenol inhibits the tumor growth factor β activated kinase 1, and neocosmosin C is an agonist of δ-opioid receptors.18,19 Conversely, BDLs devoid of O-methylation also reveal exciting activities. Thus, RAL14 compounds in the pochonin/monocillin/radicicol family are potent Hsp90 inhibitors, zearalenone is an estrogen agonist, and the DAL12 curvularins display anti-inflammatory and induced nitric oxide synthase inhibitory activities.18,19
The polyketide scaffolds of these and other BDLs are assembled in fungi by two iterative Type I polyketide synthases (iPKSs) that collaborate as BDL synthase (BDLS) subunits, and they are further tailored by oxidations, reductions, halogenations, and O-methylations to afford the mature bioactive molecules.21−23 We have been developing combinatorial biosynthetic platforms that exploit the flexibility of secondary metabolic biosynthetic enzymes to generate structurally complex molecules that may be utilized for drug discovery and agricultural applications. We have generated novel BDL scaffolds in the heterologous host Saccharomyces cerevisiae by BDLS subunit shuffling, combinatorial domain exchanges, and rational engineering of the active sites of domains,24−27 and we have elaborated these biosynthons by exploiting combinatorial tailoring reactions in total biosynthetic or biocatalytic platforms.28 Here, we evaluate the combinatorial biosynthetic potential of two orthologous OMT enzymes that afford orthogonal regioisomeric outcomes when modifying the same BDL substrate. Using protein homology modeling and substrate docking, we show that subtle differences in the active-site cavity architecture of these OMTs force the substrates to approach the conserved active-site base in distinct poses. Rational engineering of these “plasticity residues” then allows us to morph the regioselectivity of one enzyme to that of the other and to create non-selective or iterative OMT variants. These experiments also reveal that regioselectivity is influenced by the substrate; thus, engineering artificial BDL biosynthetic pathways demands OMTs adapted for the chosen scaffold and optimized to yield the desired regioisomer.
Results and Discussion
Regiospecificity and Substrate Selectivity of LtOMT and HsOMT during Combinatorial Biosynthesis
Previous studies identified LtOMT and HsOMT as two orthologous O-methyltransferases (OMTs) encoded in the biosynthetic gene clusters for two fungal benzenediol lactones (BDLs).21,29 LtOMT from Lasiodiplodia theobromae methylates the C-3 phenolic hydroxyl of the RAL12 desmethyl-lasiodiplodin (DLD, 1) to produce lasiodiplodin (1a, Figure 1).21 HsOMT (Hpm5) from Hypomyces subiculosus methylates the C-5 phenolic hydroxyl of the RAL14trans-14,15-dehydrozearalenol (DHZ, 3) to afford 5-O-methyl-DHZ (3b, Figure 1), an intermediate of hypothemycin.29 To investigate whether these apparently different regiospecificities are intrinsic to the enzymes or dictated by the substrates, we used a S. cerevisiae host to co-express LtOMT in a heterocombination with the DHZ-synthesizing iPKS pair and, separately, HsOMT with the DLD-producing iPKSs. Gratifyingly, both OMTs accepted the BDLs offered by the non-cognate iPKS pairs and efficiently processed each of these compounds to a single O-methylated product. Thus, LtOMT afforded 3-O-methyl-DHZ (3a), while HsOMT yielded 5-O-methyl-DLD (1b); these derivatives are the regioisomers of the cognate OMT products 5-O-methyl-DHZ (3b) and lasiodiplodin (1a), respectively (Figure 1). This suggested that the size, shape, and functionalization of the macrocyclic ring may not affect the regiospecificities of these enzymes. Co-expression of both OMTs in a single chassis with the DLD- or the DHZ-producing iPKS pairs yielded mixtures of the 3-O-methylated, the 5-O-methylated, and the 3,5-O-dimethylated products 1a, 1b, and 1c or 3a, 3b, and 3c, respectively. This indicated that at least one of the OMTs is able to accept the methylated product of the other OMT as an alternative substrate.
Figure 1.
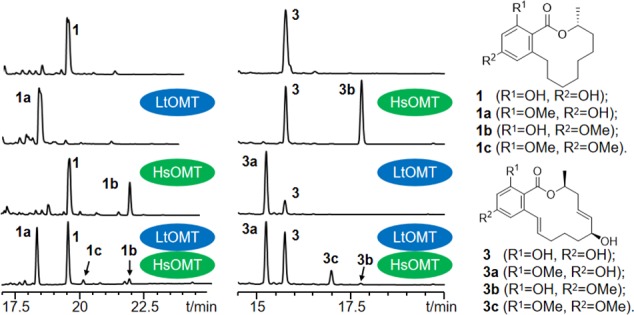
Total biosynthesis of O-methylated BDLs using LtOMT and HsOMT. Product profiles (reversed-phase LC-MS traces recorded at 210 nm with a photodiode array detector) are shown for cultures of S. cerevisiae BJ 5464-NpgA strains co-transformed with the relevant iPKS pairs,21,29 and with LtOMT and/or HsOMT as indicated. The identities of the products were validated by comparison with isolated and structurally characterized compounds.
To further explore the breadth of substrate tolerance and the fidelity of regioselectivity of these two OMTs, we assembled a collection of model substrates that represent the natural and “unnatural” (combinatorial biosynthetic) structural space of BDLs (Figure 2 and Supporting Information, Figure S1), including RAL12 (DLD 1, radiplodin 2, and trans-resorcylide), RAL14 (DHZ 3, zearalenol 4, lasicicol 5, radicicol, monocillin I, and monocillin II), DAL12 (10(11)-dehydrocurvularin and epi-dehydrocurvularin), and DAL14 (lasilarin 6 and radilarin). To explore the requirement for a macrolactone ring, we also tested non-macrocyclic BDL congeners such as isocoumarins (IC15, IC18, and IC28), acyl resorcylic acids (ARA7 7 and ARA8 8), an acyl dihydroxybenzoic acid (ADA5), and a benzaldehyde (pre-asperfuranone). Initial testing of these potential substrates was conducted in a total biosynthetic format by co-expressing the appropriate iPKS pairs with the OMTs. Successful reactions were repeated using a standardized biocatalytic platform whereby the overall rate of O-methylation and the product distribution were quantified after feeding purified substrates to the OMT-expressing yeast host.
Figure 2.

Combinatorial tailoring with LtOMT and HsOMT. Stacked polar bar charts show the product distributions among o-methoxy- (blue), p-methoxy- (green), and o,p-dimethoxy-BDL (red) congeners during biocatalytic transformation of the indicated substrates (bold numbers above the bars) with S. cerevisiae BJ5464-NpgA expressing LtOMT (substrates 1–8, 1b–4b, and 6b–8b), HsOMT (substrates 1–8 and 1a–8a), and both LtOMT and HsOMT (substrates 1–8). Percent values below the compound numbers on these charts show the overall conversion of the indicated substrate into all corresponding methylated products. See Supporting Information, Table S3, for tabulated percent conversion values as the mean ± SD from three independent experiments of three replicates each (n = 9). The structures of substrates 1–8 are shown with the carbonyl “handle” (see text for details) highlighted in yellow, and the sites of O-methylation are identified by blue (LtOMT-type ortho-OMT activity) and green ovals (HsOMT-type para-OMT activity). Scheme: Unmethylated substrates (X) may be converted to the o-methoxy congener (Xa) by the LtOMT and to the p-methoxy congener (Xb) by the HsOMT enzyme. Xa and Xb may also be converted to the o,p-dimethoxy congener (Xc) by the appropriate enzyme (X = 1–8). See Supporting Information, Figure S2, for examples of the resulting biocatalytic grids.
The two OMTs displayed considerable substrate flexibility. Thus, 6 out of the 20 BDL congeners were successfully O-methylated by both LtOMT and HsOMT, with an additional 2 compounds also processed by LtOMT (Figure 2 and Supporting Information, Figure S1). Remarkably, differences in the geometry of the benzenediol lactone ring system (RAL vs DAL), variations in the size (12- or 14-membered) or the functionalization of the macrocycle, and even the absence of the macrocyclic ring were all well tolerated. However, α-pyrone and benzaldehyde analogues were not accepted, nor could the substrate be simplified to a single phenolic ring, as shown by the absence of O-methylation of orsellinic acid, 2,4-dihydroxybenzoic acid, resorcinol, or phenol (Supporting Information, Figure S1). For macrocyclic substrates, the presence of an enone moiety at C9–C11 was found to be incompatible with O-methylation by LtOMT or HsOMT, as shown by radicicol, monocillin I, monocillin II, trans-resorcylide, radilarin, 10(11)-dehydrocurvularin, and epi-dehydrocurvularin (Supporting Information, Figure S1).
HsOMT is apparently more fastidious than LtOMT in accepting non-cognate substrates. First, neither radiplodin (RDN, 2) nor lasicicol (LCL, 5) is O-methylated by this enzyme (Figure 2). For RDN 2, the highly constrained macrocycle may be sterically incompatible with the active-site pocket of the enzyme. For LCL 5, enolization of the C-9 carbonyl allows the resulting hydroxyl group to serve as a proton donor, and the resulting spurious H-bond(s) may restrict the substrate from assuming a productive orientation within the active-site cavity of the enzyme. Although a carbonyl is also present equidistant to the target hydroxyl group in the acceptable substrate lasilarin (LLN 6, the skeletal isomer of LCL 5), that carbonyl at C-1 is part of the lactone and thus would not be subject to enolization. Even for those non-cognate substrates that were accepted by HsOMT, the conversion rates were frequently inferior to those of LtOMT with the same scaffold, as seen with DLD 1 and zearalenone (ZEA, 4), and to a lesser extent with ARA7 7 and LLN 6 (Figure 2 and Supporting Information, Table S3).
The 14 resulting O-methylated products, including 11 that are new to nature, were isolated, and their structures were elucidated by comparison of their 1H and 13C NMR data with those reported and by careful analysis of their 1D NOESY, 1H–1H COSY, HSQC, and HMBC NMR spectra to ascertain the position of the methoxy moieties (Supporting Information, Figure S6 and Table S2). For all accepted substrates, O-methylation regioselectivity was found to be idiosyncratic for the two OMTs, despite the large structural differences of the BDL congeners (Figure 2). Thus, LtOMT is specific for the phenolic hydroxyl residing at the ortho position to the aromatic carbon bearing the carbonyl (hereafter, ortho-OMT activity). Conversely, HsOMT O-methylates the phenolic hydroxyl that is at the para position to the same (henceforth, para-OMT activity). The carbonyl “handle” for this recognition can be part of a lactone, an ester, or a ketone. No “off-target” O-methylation was observed with either enzyme. This strict regiospecificity is in contrast to the chemical (non-enzymatic) O-methylation of phenolic hydroxyl groups which either affords a mixture of methoxy regioisomers and disubstituted products or requires laborious protection–deprotection reactions to achieve regioselectivity.30
Mono-O-methylated Congeners as Substrates for Subsequent Combinatorial O-Methylation
We also investigated whether pre-existing O-methylation at one phenolic hydroxyl group would influence the suitability of a scaffold toward subsequent O-methylation at the other hydroxyl (Figure 2 and Supporting Information, Table S3). To do this, we compared results from three biocatalytic systems. First, we offered o-methoxy-BDL congeners to the S. cerevisiae strain expressing HsOMT. Second, we provided p-methoxy compounds to the yeast host producing LtOMT. Third, we interrogated O-methylation of unmethylated substrates by yeast strains co-expressing LtOMT and HsOMT in the same chassis. These experiments revealed that seemingly very similar final product distributions can result from very different processes in the coupled biocatalytic production system, where two enzymes are co-expressed in the same host chassis and compete for the same unmethylated substrate, and where the mono-O-methylated product of either enzyme may or may not serve as a second-stage substrate for the other enzyme (Figure 2). The resulting six o,p-dimethoxy-BDL congeners, all of which are new to nature, were isolated, and their structures were elucidated by 1D and 2D NMR spectroscopy (Supporting Information, Table S2 and Figure S6).
In some cases, pre-existing O-methylation severely reduced the ability of both OMTs to accept a BDL scaffold as a substrate, as was seen with O-methylated LLN 6 congeners (Figure 2 and Supporting Information, Figure S2A and Table S3). Consequently, only a small amount of the dimethoxy-LLN analogue 6c was made in the LtOMT–HsOMT co-expression system. In other cases, pre-existing O-methylation had no effect on the ability of either LtOMT or HsOMT to carry out subsequent methylation of the other phenolic hydroxyl, as was observed with methoxy congeners of the DLD 1 scaffold (Figure 2 and Supporting Information, Figure S2B and Table S3). Thus, the dimethoxy-DLD congener 1c could be made either through the 1→1a→1c or the 1→1b→1c route. However, since O-methylation of 1/1a by HsOMT is far less complete than that of 1/1b by LtOMT, lasiodiplodin 1a continued to accumulate in the LtOMT–HsOMT co-expression system, while 5-O-methyl-DLD 1b was mostly converted to the dimethoxy-DLD congener 1c.
Pre-existing O-methylation was often inhibitory to HsOMT but not to LtOMT (Figure 2 and Supporting Information, Figure S2C and Table S3). Thus, ortho-O-methylation (as in 3-O-methyl-DHZ 3a, 3-O-methyl-ZEA 4a, 3-O-methyl-ARA7 7a, and 3-O-methyl-ARA8 8a) prevented further O-methylation by HsOMT, while the regioisomeric congeners (such as 5-O-methyl-DHZ 3b, 5-O-methyl-ZEA 4b, 5-O-methyl-ARA7 7b, and 5-O-methyl-ARA8 8b) remained excellent substrates for LtOMT. As a result, the corresponding o,p-dimethoxy derivatives could only be made via the X→Xb→Xc route in the LtOMT-HsOMT co-expression system (X = 3, 4, 7, or 8). At the same time, most of the available substrates remained trapped as the ortho-O-methyl congeners (as with 3-O-methyl-ZEA 4a, 3-O-methyl-ARA7 7a, and 3-O-methyl-ARA8 8a) since these could not be further converted by HsOMT. A contrary situation was observed for RDN 2 that is not a substrate for HsOMT (Figure 2 and Supporting Information, Figure S2D and Table S3). Here, pre-existing ortho-O-methylation (as in 3-O-methyl-RDN 2a) made this scaffold acceptable, albeit only marginally, to serve as a substrate for HsOMT. Thus, a small amount of the o,p-dimethoxy-RDN derivative 2c was obtained through the 2→2a→2c route in the co-expression system. Since 5-O-methyl-RDN 2b, an otherwise excellent substrate for LtOMT, was not produced by HsOMT, the dimethoxy-RDN congener 2c could not be made by the 2→2b→2c route.
Docking Identifies Distinct Substrate Binding Poses
Despite the mutually orthogonal regiospecificities of LtOMT and HsOMT against multiple and structurally varied substrates, these enzymes are orthologous, with a relatively high sequence similarity (58% identity). This suggests that the target site for O-methylation may be determined by a limited number of key amino acid residues. To identify the structural basis for the programming of these distinct regiospecificities, we constructed homology protein structure models for LtOMT and HsOMT (Figure 3) based on the known crystal structure of the mitomycin 7-O-methyltransferase of Streptomyces lavendulae (MmcR, PDB 3GXO;31 Z-score 43.3, rmsd 0.9, and identity of 26% with LtOMT; and Z-score 41.6, rmsd 2.1, and identity of 24% with HsOMT). Both OMT models feature an N-terminal dimerization domain each (amino acids 1–130, Supporting Information, Figure S3) and a C-terminal OMT Class I, α/β Rossmann-fold catalytic domain with the conserved SAM-binding signature motif DxGGGxG (amino acids 231–237).31−33 Modeling of S-adenosyl-l-homocysteine (SAH) into LtOMT and HsOMT indicated that the key enzyme–co-product interactions are well conserved among LtOMT, HsOMT, MmcR,31 and similar MTs such as the calicheamicin orsellinic acid methyltransferase CalO1 of Micromonospora echinospora(32) (Supporting Information, Figure S4). Thus, the adenine moiety of SAH is predicted to be stabilized by π-stacking with F284, while the sulfur atom of SAH could engage in hydrophobic interactions with F197 and M201. The SAH adenine N-6 amino group forms a hydrogen bond with D283, while the ribose O-2 and O-3 may engage in a hydrogen-bonding network with R262 and E261, with the latter residue also interacting with the SAH adenine N-3. Finally, the terminal carboxylic acid group of SAH may form hydrogen bonds with E203, while the terminal amino group could interact with the main-chain carbonyl oxygen of G233.
Figure 3.

Structure modeling of LtOMT and HsOMT. Cartoon representations of homology models and predicted active-site cavities of LtOMT (A, C) and HsOMT (B, D) with docked substrates DLD (carbon atoms in orange), DHZ (carbons in magenta), and co-product SAH (carbons in green), all represented as sticks. The predicted active site H303 and the residues stabilizing the active site (D331 and E360 for LtOMT, E331 and E360 for HsOMT) are also shown as sticks (carbons in yellow). Residues in the active-site cavity that were found to influence substrate orientation are also shown as sticks (carbons in salmon for LtOMT and gray for HsOMT) and housed in the protein segments (orange on the cartoon diagrams of A and B) that were exchanged in hybrid enzymes H1 and H2 (see Figure 4). Key hydrogen-bonding networks are displayed as yellow dashes.
Docking of DLD 1 and DHZ 3 indicated that, similar to other phenolic OMT enzymes,31,32 substrate binding is dominated by hydrophobic and van der Waals interactions with residues such as F52* and W55* from the other subunit of the dimer (for both enzymes), residues F197, M201, A200, and M344 (for LtOMT), and amino acids V136, F197, M201, and I349 (for HsOMT). Importantly, docking also revealed characteristically different binding poses for these substrates in LtOMT vs HsOMT (Figure 3 and Supporting Information, Figure S4). For LtOMT, both DLD and DHZ are predicted to present their OH-3 to the Nε of the deduced active-site base H303,33 while OH-5 may be contacted by the main-chain carboxyl group of D202. Conversely, rotation of both substrates within the active-site cavity may position OH-5 for attack by H303 of HsOMT, while OH-3 and the C-1 carbonyl could interact with the side-chain hydroxyl group of S200. Abstraction of the proton from the substrates by H303 of either enzyme would then lead to a nucleophilic attack of the phenolate anion of the substrate on the reactive methyl group of the co-substrate S-adenosyl-l-methionine (SAM). Meanwhile, the catalytic base may be oriented and stabilized by a hydrogen-bonding network between Nδ of H303 with D331 and E360 (LtOMT) or with D331 and D360 (HsOMT)33 (Figure 3 and Supporting Information, Figure S4).
Remodeling of the Active-Site Cavity Alters Regioselectivity
To identify the residues that may influence the choice of the O-methylation target site in the substrates, we built hybrid enzymes from LtOMT and HsOMT by fusion PCR, expressed these in the S. cerevisiae chassis, and quantified the regioselectivity and the extent of O-methylation after feeding DLD 1 as a model substrate. First, we created chimeras from LtOMT and HsOMT. In chimera H1, amino acids 1–329 of LtOMT were replaced with those of HsOMT. Despite the extensive replacement, the regioselectivity did not change, but the efficiency of H1 declined dramatically (Figure 4). The complementary hybrid H2 (amino acids 330–398 from HsOMT) retained reasonable activity (26.1 ± 3.0% total conversion), but the regioselectivity completely switched to that of HsOMT (i.e., para-OMT). Chimera H3, with a shorter HsOMT-derived segment (amino acids 366–398), produced 1a:1b in a 3:1 ratio at 34.5 ± 0.8% conversion. These chimeras indicated that regioselectivity is determined by the C-terminal end of the enzyme and suggested that extensive replacements are detrimental to enzymatic activity.
Figure 4.

Engineering the O-methylation regioselectivity of LtOMT. Left, schematic representation of chimeric and mutant enzymes created from LtOMT (in blue) and HsOMT (in green), with the positions of amino acid replacements or fragment boundaries indicated. Middle, product profiles (reversed-phase LC-MS traces recorded at 210 nm with a photodiode array detector) of representative in vivo bioconversion reactions (DLD 1 as the substrate fed to cultures of S. cerevisiae BJ5464-NpgA expressing the gene encoding the indicated enzyme). The identities of the products were validated by comparison with isolated and structurally characterized compounds. Right, total conversion (in percent) of DLD 1 into all corresponding methylated product congeners, shown as a bar graph. Percentage distributions of the O-methylated products are represented as pie charts (yields of the individual O-methylated products divided by the total yield of all products, multiplied by 100). Color code: lasiodiplon 1a (blue), 5-O-methyl-DLD 1b (green), and dimethoxy-DLD congener 1c (red). See Supporting Information, Table S4, for percent conversion values (mean ± SD from three independent experiments of three replicates each, n = 9).
Next, we turned to structure-guided site-directed amino acid replacements to examine the influence of residues located within 6 Å of the substrates in the active-site pockets of LtOMT and HsOMT. Replacement of a cluster of four of these amino acids at the C-terminal end of LtOMT with those of HsOMT (Q384K, G386R, W387H, and Q388H) by site-directed mutagenesis provided mutant enzyme M1 that retained the overall high conversion rate of LtOMT (90.0 ± 2.9% of substrate converted to methylated products). M1 preserved most of the ortho-OMT activity of LtOMT (lasiodiplodin 1a, 82.9 ± 0.8% of methylated products) but also gained HsOMT-type para-OMT activity, as shown by the accumulation of products 5-O-methyl-DLD 1b (3.7 ± 0.8%) and dimethoxy-DLD congener 1c (13.4 ± 1.5%). Thus, replacement of these four amino acids is sufficient to accommodate the model substrate within the active-site cavity in orientations that expose not only OH-3 but also OH-5 to the active site. Dissecting the influences of these amino acids by creating all possible 3-point, 2-point, and 1-point replacements (Figure 4 and Supporting Information, Table S4) indicated that W387H has the highest influence toward enabling a para-OMT activity. Q388H (and to a lesser extent Q384K) plays a supplementary role when combined with W387H, while the effect of G386R is context-dependent. Thus, enzyme M4, with only the W387H mutation, shows para-OMT activity in addition to a robust ortho-OMT activity, as evidenced by the production of 7.7 ± 0.3% of dimethoxy-DLD congener 1c at 76.6 ± 20.2% overall substrate conversion. Addition of Q388H (as in M3) and further introduction of Q384K (as in M2) gradually improves the para-OMT activity while preserving a strong ortho-OMT activity (1a:1b:1c = 4:1:1) and continues to support high overall conversion (Figure 4).
Introduction of segment 330–366 of HsOMT into the four-point mutant M1 yielded enzyme HM1 that acted exclusively as a para-OMT, indicating that this short region is decisive in setting regioselectivity. Surprisingly, the chimeric mutant HM1 converted more of the DLD substrate to 5-O-methyl-DLD 1b than HsOMT, the native para-OMT parent (58.3 ± 11.4% vs 41.6 ± 1.2%). We then investigated two residues of this segment that are situated within 6 Å of the substrates. Adding the F330V mutation to those already present in enzyme M1 (as in M5) improved the para-OMT activity, as evidenced by an increase of the 1b yield to 13.3 ± 5.5% (cf. 3.7 ± 0.8% for M1), while the production of the 3,5-di-O-methoxy product 1c and the total conversion rate remained unchanged. Introduction of the T356M mutation into M1 (as in M6) led to a dramatic reversal of the regioselectivity, with 5-O-methyl-DLD 1b representing 74.3 ± 4.2% of the products and lasiodiplodin 1a reduced to 10.0 ± 1.9%, albeit at the expense of a slight penalty in product yield (70.1 ± 5.8% total conversion in M6 vs 90.0 ± 2.9% in M1). Introduction of both changes into M1 afforded enzyme M7 (a six-point mutant) with an almost perfect para-OMT specificity (94.1 ± 1.0% 1b) and high overall conversion (93.7 ± 1.1%). Thus, mutant enzyme M7 far surpassed the parent HsOMT as a DLD para-OMT. Surprisingly, mutant M8, with only a single amino acid replacement (T356M), had an apparently balanced ortho-OMT/para-OMT activity, with the three product congeners present in roughly a 1:1:1 ratio at 80.1 ± 10.2% total conversion. Thus, this residue seems to relax regioselectivity and allows the substrate to assume either of the two binding poses with equal likelihood. Introduction of a second mutation into enzyme M8 (W387H, cf. mutant M4) yielded M9, which afforded the 3,5-di-O-methyl-DLD 1c almost exclusively and with a very high productivity (96.4 ± 0.7% conversion of the substrate).
To further characterize the native and the engineered enzymes, LtOMT, HsOMT, and selected variants of LtOMT (M1, M6–M9) were heterologously expressed in E. coli as His-tagged proteins and purified to substantial homogeneity (Supporting Information, Figure S5). The purified enzymes afforded optimal conversion of the model substrate DLD 1 in the presence of 10 mM MgCl2. While NaCl (0–100 mM), CaCl2 (10 mM), and MnCl2 (10 mM) did not influence the activity, EDTA, CuCl2, and ZnSO4 (each at 10 mM) were strongly inhibitory. The enzymes showed a broad optimum at pH 7.5–9.5 in Tris or phosphate buffers, with the activity disappearing at pH 5.0 and above 10.0. The temperature optimum was 30 °C, with residual activity still measurable at 4 °C and at 65 °C.
The kinetic parameters for LtOMT and its variants with DLD 1 as the substrate are shown in Table 1. HsOMT was found to be kinetically slow, and the reaction did not reach saturation under the conditions tested; thus, Km and Vmax were not determined for this enzyme. Interestingly, the Km values of LtOMT and its five variants were similar. We speculate that the substrate binding cavity of LtOMT is suitably spacious to accommodate the altered amino acid residues without reducing the affinity of the mutant enzymes toward binding the model substrate. In contrast, the turnover rate (kcat) and thus the catalytic efficiency (kcat/Km) of the LtOMT parent enzyme were more than 2 orders of magnitude higher than those of the mutants. This may indicate that the altered substrate poses adopted by the bound substrate decrease the rates of formation of the transition state with the mutant enzymes. Nevertheless, the turnover rates of the mutant enzymes were still similar to those of many wild-type secondary metabolite methyltransferases, such as the GenL gentamicin C1/C2 methyltransferase (kcat = 0.043 s–1)34 or many flavone OMTs from plants (kcat often in the range of 0.06–0.09 s–1).15,16
Table 1. Kinetic Parameters of LtOMT and Its Engineered Variants with DLD (1) as the Substratea.
| Km [mM] | Vmax [nmol s–1 mg–1] | kcat [s–1] | kcat/Km [s–1 mM–1] | |
|---|---|---|---|---|
| LtOMT | 1.58 ± 0.01 | 329.8 ± 102.8 | 14.585 ± 4.547 | 9.209 |
| M1 | 2.03 ± 0.07 | 1.5 ± 0.5 | 0.068 ± 0.023 | 0.033 |
| M6 | 1.15 ± 0.02 | 0.6 ± 0.1 | 0.028 ± 0.002 | 0.024 |
| M7 | 2.49 ± 0.22 | 1.0 ± 0.2 | 0.044 ± 0.008 | 0.018 |
| M8 | 1.40 ± 0.03 | 1.2 ± 0.1 | 0.053 ± 0.005 | 0.038 |
| M9 | 1.52 ± 0.30 | 1.4 ± 0.5 | 0.064 ± 0.022 | 0.042 |
kcat/Km was calculated using the averages of kcat and Km. Values represent the mean ± SD from three independent experiments with three replicates each (n = 9).
Taken together, structure-based site-specific replacements of a few key “plasticity residues” in the active site of LtOMT remodeled this regiospecific ortho-OMT enzyme to afford an almost exclusive DLD para-OMT (M7, 6 amino acids replaced), an apparently unselective DLD ortho-OMT/para-OMT (M8, single amino acid replacement), and a DLD o,p-di-OMT (M9, two amino acids replaced). While homology modeling-based “keyhole surgery” was successful in modulating the regioselectivity of LtOMT, protein crystallographic studies would be necessary to disentangle the exact contributions of the individual amino acid replacements in forcing the substrates to adopt altered binding poses, leading to modulated regioselectivities and catalytic efficiencies. Increasing kinetic efficiency would also require further optimization. This is analogous to natural evolution, where functional innovation in the product and substrate spectrum is often the consequence of a few key amino acid changes, followed by the accumulation of compensatory mutations that optimize the novel activity.35
Regioselectivity of the Engineered Enzymes Is Influenced by the Substrate
The regiospecificities of the wild-type LtOMT and HsOMT enzymes were found to be independent of the substrates used (Figure 2): for any given substrate accepted by the enzyme, only a single O-methylated derivative was obtained, and the targeted phenolic OH was always ortho (LtOMT) or para (HsOMT) to the aromatic carbon bearing the carbonyl “handle”. In contrast, the mutant LtOMT enzymes possess both ortho-OMT and para-OMT activities to various degrees. To discover whether the ratio of these activities is an intrinsic characteristic of these enzymes or if regioselectivity is also influenced by the substrates, we selected five mutants (M1, M6–M9) with characteristically different regioselective outcomes when acting on the DLD model substrate. We conducted biocatalytic reactions with these enzymes using our panel of eight structurally varied BDL substrates and their o-methoxy or p-methoxy congeners (Figure 5 and Supporting Information, Table S5).
Figure 5.

Different substrate scaffolds modulate the apparent regiospecificities of selected engineered OMTs. Stacked polar bar charts show the product distributions among o-methoxy- (blue), p-methoxy- (green), and o,p-dimethoxy-BDL (red) congeners during biocatalytic conversion of the indicated substrates (bold numbers above the bars), with S. cerevisiae BJ5464-NpgA expressing LtOMT and its variants M1 and M6–M9. Percent values below the compound numbers on these charts show the overall conversion percentage of the indicated substrate into all corresponding methylated products. See Supporting Information, Table S5, for tabulated percent conversion values as the mean ± SD from three independent experiments of three replicates each (n = 9). Scheme: Unmethylated substrates (X) may be converted to the o-methoxy congener (Xa) by the ortho-OMT activity and to the p-methoxy congener (Xb) by the para-OMT activity of the enzymes. Either or both of these first-stage products may also be converted to the o,p-dimethoxy congener (Xc) by the enzyme (X = 1–8).
The LtOMT variant enzymes processed DLD 1 and RDN 2, the two RAL12 scaffolds of the panel, in a similar manner. The T356M mutation in LtOMT variant M8 created a previously non-existent para-OMT activity against both the unmethylated compounds DLD 1 and RDN 2 and their o-methoxy congeners lasiodiplodin 1a and 3-O-methyl-RDN 2a. The ortho-OMT activity against the unmethylated and the p-methoxy congeners remained operational; thus, the o,p-dimethoxy congeners were produced through both the X→Xa→Xc and the X→Xb→Xc routes (X = 1 or 2). Adding the W387H mutation (as in M9) caused these o,p-dimethoxy congeners to become the main products with substrates DLD 1 and RDN 2. This was the result of improvements in the para-OMT activity toward both lasiodiplodin 1a and 3-O-methyl-RDN 2a and an increase in the ortho-OMT activity toward 5-O-methyl-DLD 1b and 5-O-methyl-RDN 2b. Supplementing the T356M and the W387H mutations with the Q384K, G386R, and Q388H changes (as in M6) reduced activity toward the monomethoxy congeners but improved the para-OMT activity toward the unmethylated substrates DLD 1 and RDN 2. As a result, the amounts of the dimethoxy congeners were reduced, but the p-methoxy congeners 1b and 2b accumulated. Removing the T356M mutation (as in M1) did not affect the ortho-OMT activity but eliminated the para-OMT activity against lasiodiplodin 1a and 3-O-methyl-RDN 2a and limited it against DLD 1 and RDN 2. Introducing the F330V mutation into M6 (as in M7) did not seem to affect the ortho-OMT activity against 1/1b and 2/2b. However, it improved the para-OMT activity against DLD 1 and RDN 2 but not against the o-methoxy congeners 1a and 2a. As a result, 5-O-methyl-DLD 1b became the main product for the DLD scaffold, while 5-O-methyl-RDN 2b production reached its highest level for the RDN scaffold with this enzyme variant.
The unmethylated RAL14 congeners were found to be less amenable than the RAL12 compounds toward para-O-methylation, perhaps as a consequence of the increased steric bulk of the larger macrocycles. In particular, LCL 5 (and its o-methoxy congener 5a) remained completely impervious to para-O-methylation, just as with the other parent enzyme HsOMT, indicating that the suggested inhibitory role of the enol tautomer of the C-9 carbonyl remains a problem even in the context of an LtOMT scaffold. Thus, molding the active-site cavity modulated only the ortho-OMT activity against LCL 5. For the DHZ 3 and ZEA 4 scaffolds, no mutants were able to accommodate the o-methoxy congeners 3a and 4a, perhaps because the increased bulk of the 3-methoxy group obstructs access to the active site in the conformation that is conducive to the para-OMT activity. Consequently, the X→Xa→Xc route (X = 3 or 4) toward the dimethoxy analogues remained unavailable for all enzyme variants. In contrast, the p-methoxy congeners 3b and 4b remained excellent substrates for all LtOMT mutants, indicating that the methoxy group in the ortho-OMT binding pose does not encounter similar steric restrictions. Consequently, the first-stage products 5-O-methyl-DHZ 3b and 5-O-methyl-ZEA 4b did not accumulate but were converted to the dimethoxy congeners through the X→Xb→Xc route (X = 3 or 4).
The only DAL scaffold recognized as a substrate by the two parental enzymes was the 14-membered BDL, LLN 6, although the conversion rate was only moderate for both enzymes (37.7 ± 1.4% for LtOMT and 30.7 ± 3.1% for HsOMT, Figure 2). The introduced mutations allowed the production of 7-O-methyl-LLN 6a, 5-O-methyl-LLN 6b, and the dimethoxy-LLN congener 6c by all engineered enzymes, with the monomethoxy congeners 6a and 6b accepted as substrates for a second round of O-methylation by all mutants except M1. However, overall conversion declined precipitously to 2–5% with M1 and M6–M8, recovering to 15.8 ± 1.4% only with M9.
Just as with the RAL14 scaffolds, the p-methoxy congeners of the two ARAs (5-O-methyl-ARA7, 7b and 5-O-methyl-ARA8, 8b) could be methylated by LtOMT and all its mutants, while the o-methoxy congeners (3-O-methyl ARA7, 7a and 3-O-methyl ARA8, 8a) remained recalcitrant toward subsequent methylation. Presumably, the bulky o-methoxy group prohibits the substrate from binding in the active-site cavity in a pose that could offer the para-phenolic hydroxyl to the active-site base H303, but it can be accommodated in the pose that leads to ortho-O-methylation.
Taking these together, it is striking that the apparent ortho-OMT/para-OMT preference of a given enzyme variant is not an intrinsic and immutable property of the enzyme but emerges from an intimate interplay with the various substrate scaffolds. Similar phenomena were also noted for bacterial and mammalian catechol OMTs.11−13 Product distribution among o-methoxy, p-methoxy, and o,p-dimethoxy derivatives is determined by two major factors: (1) how well the active site accommodates the unmethylated substrate in the two poses that expose one or the other phenolic hydroxyl to the active site and (2) whether the o-methoxy and the p-methoxy first-stage reaction products can be re-accommodated in the complementary pose that offers the free OH to the active site. It is thus perhaps not surprising that mutant enzymes selected by using a single model substrate (in this case, DLD 1) would reveal very different product profiles when offered a different substrate. Analogous phenomena are often encountered in directed enzyme evolution projects, prompting the formulation of the “first law of directed evolution”: you get what you screen for.36
Conclusions
O-Methylation of reactive phenolic hydroxyl groups by regiospecific OMT enzymes is used by BDL-producing fungi in nature to modulate the biological activities of these ecologically important small-molecule natural products. For combinatorial biosynthesis of drug-like BDL scaffolds in the laboratory, appropriate tailoring enzymes need to be employed to process unnatural congeners produced by engineered polyketide biosynthetic pathways. Among these tailoring enzymes, OMTs need to display significant promiscuity to accept structurally varied BDL scaffolds as substrates, while these enzymes must also offer appropriate regioselectivity to deliver the desired methoxy regioisomer. Our work shows that two orthologous OMT enzymes from different BDL biosynthetic pathways possess considerable substrate flexibility but display strict orthogonal regiospecificity. Protein homology modeling and substrate docking reveal that nature uses subtle alterations of the amino acids lining the active-site cavity to force the substrates to adopt different binding poses. In turn, this exposes distinct phenolic hydroxyls to the conserved catalytic base, accounting for the observed orthogonal regiospecificity. This apparent plasticity of the active-site cavity architecture then allows us to re-program the regioselectivity of BDL OMT enzymes by adjusting just a few amino acids in a keyhole surgery-like manner. Doing so, we can transpose the strict regiospecificity of a native OMT to generate a mutant enzyme that produces the orthogonal p-methoxy derivative of the model substrate almost exclusively. We can also engineer a mutant enzyme that apparently eschews regioselectivity toward the model substrate to afford o-methoxy, p-methoxy, and o,p-dimethoxy congeners in equal proportions, or another one that specializes in recursive methylation to yield mostly the o,p-dimethoxy derivative. We also show that apparently similar product distributions may result from different scenarios involving dissimilar recognition and processing of the unmethylated, the o-methoxy, and the p-methoxy substrates by the ortho-OMT and the para-OMT activities engineered into the same enzyme chassis. As their active-site cavity is being remodeled, the enzymes may acquire or lose one or the other activity and come to accept or reject certain substrates. In accord with these dynamic changes, the regiospecificities and the resulting product ratios of the mutant OMT enzymes turn out to be context-dependent, as they are modulated by the complex interactions of the structurally varied substrates with the enzymes. Thus, engineering an OMT to generate a desired methoxy regioisomer of a given “unnatural product” during combinatorial biosynthesis is analogous to enzyme evolution in nature, where catalytic innovations emerge in the context of a particular molecule that provides fitness advantage to the producer, accomplished by a few key mutations in “plasticity residues” followed by the acquisition of supplementary mutations to optimize the novel activity. Recapturing this process in synthetic biology by directed enzyme evolution toward improved catalytic efficiency, coupled with chassis metabolic engineering and fermentation process optimization would doubtless be also necessary to deploy engineered OMTs for practical, industrial-scale applications, as was seen with the recent development of a commercial process for vanillin production that also employs an OMT.40−42 Even so, extension of the scalable, sustainable, and tunable enzymatic approach described here toward the regioselective O-methylation of a wider variety of phenolic polyketide natural products, such as the isocoumarins, isochromanones, anthraquinoids, and flavonoids,7,10,43,44 offers advantages over the well-known chemical methods that require high temperature/pressure and longer reaction times, often leading to undesired C-methylated byproducts.8,9
Acknowledgments
This article is dedicated to Peter F. Leadlay, FRS, on his retirement. This work was supported by the National Natural Science Foundation of China (31570093 and 31870076 to Y.X. and 21807110 to C.W.), the National Program of China for Transgenic Research (No. 2016ZX08009003-002 to M.L.), the National Basic Research Program of China (2015CB755700 to Y.X. and M.L.), China Postdoctoral Science Foundation (2018M640208 to C.W.), the Joint Genomics Institute of the U.S. Department of Energy (WIP ID 1349 to I.M.), and the National Institutes of Health (NIGMS R01 GM114418 to I.M.). We thank the Beijing Center for Physical and Chemical Analysis for providing access to their NMR device.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.8b12967.
Experimental and bioinformatics procedures, descriptions of the isolation and structure elucidation of compounds, spectroscopic data, tabulated biocatalytic conversion rates, supplementary figures including additional structures, dissection of demethylation routes, and protein topology and docking models (PDF)
Author Contributions
# X.W., C.W., and L.D. contributed equally to this work.
The authors declare the following competing financial interest(s): I. M. has disclosed financial interests in TEVA Pharmaceuticals Hungary and DSM Nutritional Products, LLC, which are unrelated to the subject of the research presented here. All other authors declare no competing financial interests.
Supplementary Material
References
- Wessjohann L. A.; Keim J.; Weigel B.; Dippe M. Alkylating enzymes. Curr. Opin. Chem. Biol. 2013, 17, 229–235. 10.1016/j.cbpa.2013.02.016. [DOI] [PubMed] [Google Scholar]
- Koirala N.; Thuan N. H.; Ghimire G. P.; Thang D. V.; Sohng J. K. Methylation of flavonoids: Chemical structures, bioactivities, progress and perspectives for biotechnological production. Enzyme Microb. Technol. 2016, 86, 103–116. 10.1016/j.enzmictec.2016.02.003. [DOI] [PubMed] [Google Scholar]
- Liscombe D. K.; Louie G. V.; Noel J. P. Architectures, mechanisms and molecular evolution of natural product methyltransferases. Nat. Prod. Rep. 2012, 29, 1238–1250. 10.1039/c2np20029e. [DOI] [PubMed] [Google Scholar]
- Wessjohann L. A.; Dippe M.; Tengg M.; Gruber-Khadjawi M.. Methyltransferases in biocatalysis. Cascade biocatalysis: integrating stereoselective and environmentally friendly reactions, 1st ed.; Wiley: Somerset, NJ, 2014; pp 393–425. [Google Scholar]
- Henry K. M.; Townsend C. A. Ordering the reductive and cytochrome P450 oxidative steps in demethylsterigmatocystin formation yields general insights into the biosynthesis of aflatoxin and related fungal metabolites. J. Am. Chem. Soc. 2005, 127, 3724–3733. 10.1021/ja0455188. [DOI] [PubMed] [Google Scholar]
- Du Y. L.; Ding T.; Ryan K. S. Biosynthetic O-methylation protects cladoniamides from self-destruction. Org. Lett. 2013, 15, 2538–2541. 10.1021/ol401036f. [DOI] [PubMed] [Google Scholar]
- Struck A. W.; Thompson M. L.; Wong L. S.; Micklefield J. S-Adenosyl-methionine-dependent methyltransferases: Highly versatile enzymes in biocatalysis, biosynthesis and other biotechnological applications. ChemBioChem 2012, 13, 2642–2655. 10.1002/cbic.201200556. [DOI] [PubMed] [Google Scholar]
- Shen Z. L.; Jiang X. Z.; Mo W. M.; Hu B. X.; Sun N. Catalytic O-methylation of phenols with dimethyl carbonate to aryl methyl ethers using BMIm Cl. Green Chem. 2005, 7, 97–99. 10.1039/b411201f. [DOI] [Google Scholar]
- Basak A.; Nayak M. K.; Chakraborti A. K. Chemoselective O-methylation of phenols under non-aqueous condition. Tetrahedron Lett. 1998, 39, 4883–4886. 10.1016/S0040-4039(98)00885-5. [DOI] [Google Scholar]
- Bennett M. R.; Shepherd S. A.; Cronin V. A.; Micklefield J. Recent advances in methyltransferase biocatalysis. Curr. Opin. Chem. Biol. 2017, 37, 97–106. 10.1016/j.cbpa.2017.01.020. [DOI] [PubMed] [Google Scholar]
- Wang J. H.; Pichersky E. Identification of specific residues involved in substrate discrimination in two plant O-methyltransferases. Arch. Biochem. Biophys. 1999, 368, 172–180. 10.1006/abbi.1999.1304. [DOI] [PubMed] [Google Scholar]
- Schmidt A.; Li C.; Shi F.; Jones A. D.; Pichersky E. Polymethylated myricetin in trichomes of the wild tomato species Solanum habrochaites and characterization of trichome-specific 3′/5′- and 7/4’-myricetin O-methyltransferases. Plant Physiol. 2011, 155, 1999–2009. 10.1104/pp.110.169961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee A.; Park E. B.; Lee J.; Choi B. S.; Kang S. J. The N terminus of cGAS de-oligomerizes the cGAS: DNA complex and lifts the DNA size restriction of core-cGAS activity. FEBS Lett. 2017, 591, 954–961. 10.1002/1873-3468.12598. [DOI] [PubMed] [Google Scholar]
- Law B. J.; Bennett M. R.; Thompson M. L.; Levy C.; Shepherd S. A.; Leys D.; Micklefield J. Effects of active-site modification and quaternary structure on the regioselectivity of catechol-O-methyltransferase. Angew. Chem., Int. Ed. 2016, 55, 2683–2687. 10.1002/anie.201508287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berim A.; Hyatt D. C.; Gang D. R. A set of regioselective O-methyltransferases gives rise to the complex pattern of methoxylated flavones in sweet basil. Plant Physiol. 2012, 160, 1052–1069. 10.1104/pp.112.204164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joe E. J.; Kim B. G.; An B. C.; Chong Y.; Ahn J. H. Engineering of flavonoid O-methyltransferase for a novel regioselectivity. Mol. Cells 2010, 30, 137–141. 10.1007/s10059-010-0098-8. [DOI] [PubMed] [Google Scholar]
- Wils C. R.; Brandt W.; Manke K.; Vogt T. A single amino acid determines position specificity of an Arabidopsis thaliana CCoAOMT-like O-methyltransferase. FEBS Lett. 2013, 587, 683–689. 10.1016/j.febslet.2013.01.040. [DOI] [PubMed] [Google Scholar]
- Shen W.; Mao H.; Huang Q.; Dong J. Benzenediol lactones: A class of fungal metabolites with diverse structural features and biological activities. Eur. J. Med. Chem. 2015, 97, 747–777. 10.1016/j.ejmech.2014.11.067. [DOI] [PubMed] [Google Scholar]
- Patocka J.; Soukup O.; Kuca K. Resorcylic acid lactones as the protein kinase inhibitors, naturally occuring toxins. Mini-Rev. Med. Chem. 2013, 13, 1873–1878. 10.2174/13895575113136660096. [DOI] [PubMed] [Google Scholar]
- Liu Z.; Zheng X.; Wang Y.; Tang M.; Chen S.; Zhang F.; Li L.; Zhang C.; Sun Y. Lignans and isoflavonoids from the stems of Pisonia umbellifera. RSC Adv. 2018, 8, 16383–16391. 10.1039/C8RA02240B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y.; Zhou T.; Espinosa-Artiles P.; Tang Y.; Zhan J.; Molnár I. Insights into the biosynthesis of 12-membered resorcylic acid lactones from heterologous production in Saccharomyces cerevisiae. ACS Chem. Biol. 2014, 9, 1119–1127. 10.1021/cb500043g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y.; Espinosa-Artiles P.; Schubert V.; Xu Y.-M.; Zhang W.; Lin M.; Gunatilaka A. L.; Süssmuth R.; Molnár I. Characterization of the biosynthetic genes for 10,11-dehydrocurvularin, a heat shock response-modulating anticancer fungal polyketide from Aspergillus terreus. Appl. Environ. Microbiol. 2013, 79, 2038–2047. 10.1128/AEM.03334-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S.; Xu Y.; Maine E. A.; Wijeratne E. K.; Espinosa-Artiles P.; Gunatilaka A. L.; Molnár I. Functional characterization of the biosynthesis of radicicol, an Hsp90 inhibitor resorcylic acid lactone from Chaetomium chiversii. Chem. Biol. 2008, 15, 1328–1338. 10.1016/j.chembiol.2008.10.006. [DOI] [PubMed] [Google Scholar]
- Xu Y.; Zhou T.; Zhang S.; Xuan L. J.; Zhan J.; Molnár I. Thioesterase domains of fungal nonreducing polyketide synthases act as decision gates during combinatorial biosynthesis. J. Am. Chem. Soc. 2013, 135, 10783–10791. 10.1021/ja4041362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai J.; Lu Y.; Xu Y.-M.; Zhang W.; Chen M.; Lin M.; Gunatilaka A. L.; Xu Y.; Molnár I. Diversity-oriented combinatorial biosynthesis of hybrid polyketide scaffolds from azaphilone and benzenediol lactone biosynthons. Org. Lett. 2016, 18, 1262–1265. 10.1021/acs.orglett.6b00110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y.; Zhou T.; Zhang S.; Espinosa-Artiles P.; Wang L.; Zhang W.; Lin M.; Gunatilaka A. L.; Zhan J.; Molnár I. Diversity-oriented combinatorial biosynthesis of benzenediol lactone scaffolds by subunit shuffling of fungal polyketide synthases. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 12354–12359. 10.1073/pnas.1406999111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y.; Zhou T.; Zhou Z.; Su S.; Roberts S. A.; Montfort W. R.; Zeng J.; Chen M.; Zhang W.; Lin M.; Zhan J.; Molnár I. Rational reprogramming of fungal polyketide first-ring cyclization. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 5398–5403. 10.1073/pnas.1301201110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie L.; Zhang L.; Wang C.; Wang X.; Xu Y.-M.; Yu H.; Wu P.; Li S.; Han L.; Gunatilaka A. L.; Wei X.; Lin M.; Molnár I.; Xu Y. Methylglucosylation of aromatic amino and phenolic moieties of drug-like biosynthons by combinatorial biosynthesis. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, E4980–E4989. 10.1073/pnas.1716046115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeves C. D.; Hu Z.; Reid R.; Kealey J. T. Genes for biosynthesis of the fungal polyketides hypothemycin from Hypomyces subiculosus and radicicol from Pochonia chlamydosporia. Appl. Environ. Microbiol. 2008, 74, 5121–5129. 10.1128/AEM.00478-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai Q.; Jiang Y.; Yu J. T.; Cheng J. Peroxide: A novel methylating reagent. Synthesis 2016, 48, 329–339. 10.1055/s-0035-1560536. [DOI] [Google Scholar]
- Singh S.; Chang A.; Goff R. D.; Bingman C. A.; Grüschow S.; Sherman D. H.; Phillips G. N. Jr.; Thorson J. S. Structural characterization of the mitomycin 7-O-methyltransferase. Proteins: Struct., Funct., Genet. 2011, 79, 2181–2188. 10.1002/prot.23040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang A.; Singh S.; Bingman C. A.; Thorson J. S.; Phillips G. N. Jr. Structural characterization of CalO1: a putative orsellinic acid methyltransferase in the calicheamicin-biosynthetic pathway. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2011, 67, 197–203. 10.1107/S090744491100360X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zubieta C.; He X. Z.; Dixon R. A.; Noel J. P. Structures of two natural product methyltransferases reveal the basis for substrate specificity in plant O-methyltransferases. Nat. Struct. Biol. 2001, 8, 271–279. 10.1038/85029. [DOI] [PubMed] [Google Scholar]
- Li S.; Guo J.; Reva A.; Huang F.; Xiong B.; Liu Y.; Deng Z.; Leadlay P. F.; Sun Y. Methyltransferases of gentamicin biosynthesis. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, 1340–1345. 10.1073/pnas.1711603115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton M. S.; Arcus V. L.; Gerth M. L.; Patrick W. M. Enzyme evolution: innovation is easy, optimization is complicated. Curr. Opin. Struct. Biol. 2018, 48, 110–116. 10.1016/j.sbi.2017.11.007. [DOI] [PubMed] [Google Scholar]
- Hibbert E. G.; Dalby P. A. Directed evolution strategies for improved enzymatic performance. Microb. Cell Fact. 2005, 4, 29. 10.1186/1475-2859-4-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen E. H.; Moller B. L.; Kock G. R.; Bunner C. M.; Kristensen C.; Jensen O. R.; Okkels F. T.; Olsen C. E.; Motawia M. S.; Hansen J. De novo biosynthesis of vanillin in fission yeast (Schizosaccharomyces pombe) and baker’s yeast (Saccharomyces cerevisiae). Appl. Environ. Microbiol. 2009, 75, 2765–2774. 10.1128/AEM.02681-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brochado A. R.; Matos C.; Moller B. L.; Hansen J.; Mortensen U. H.; Patil K. R. Improved vanillin production in baker’s yeast through in silico design. Microb. Cell Fact. 2010, 9, 84. 10.1186/1475-2859-9-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayden E. C. Synthetic-biology firms shift focus. Nature 2014, 505, 598. 10.1038/505598a. [DOI] [PubMed] [Google Scholar]
- Chandler I. M.; McIntyre C. R.; Simpson T. J. Structural revision and synthesis of LL-D253α and related chromanone fungal metabolites. J. Chem. Soc., Perkin Trans. 1 1992, 2271–2284. 10.1039/p19920002271. [DOI] [Google Scholar]
- Chen Z.-G.; Fujii I.; Ebizuka Y.; Sankawa U. Emodin O-methyltransferase from Aspergillus terreus. Arch. Microbiol. 1992, 158, 29–34. 10.1007/BF00249062. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.