

Abstract
Here, I propose that cancer stem cells (CSCs) would be equivalent to para-embryonic stem cells (p-ESCs), derived from adult cells de-re-programmed to a ground state. p-ESCs would differ from ESCs by the absence of genomic homeostasis. A p-ESC would constitute the cancer cell of origin (i-CSC or CSC0), capable of generating an initial tumor, corresponding to a pre-implantation blastocyst. In a niche with proper signals, it would engraft as a primary tumor, corresponding to a post-implantation blastocyst. i-CSC progeny would form primary pluripotent and slow self-renewing CSCs (CSC1s), blocked in an undifferentiated state, corresponding to epiblast cells; CSC1s would be tumor-initiating cells (TICs). CSC1s would generate secondary CSCs (CSC2s), corresponding to hypoblast cells; CSC2s would be tumor growth cells (TGCs). CSC1s/CSC2s would generate tertiary CSCs (CSC3s), with a mesenchymal phenotype; CSC3s would be tumor migrating cells (TMCs), corresponding to mesodermal precursors at primitive streak. CSC3s with more favorable conditions (normoxia), by asymmetrical division, would differentiate into cancer progenitor cells (CPCs), and these into cancer differentiated cells (CDCs), thus generating a defined cell hierarchy and tumor progression, mimicking somito-histo-organogenesis. CSC3s with less favorable conditions (hypoxia) would delaminate and migrate as quiescent circulating micro-metastases, mimicking mesenchymal cells in gastrula morphogenetic movements. In metastatic niches, these CSC3s would install and remain dormant in the presence of epithelial/mesenchymal transition (EMT) signals and hypoxia. But, in the presence of mesenchymal/epithelial transition (MET) signals and normoxia, they would revert to self-renewing CSC1s, reproducing the same cell hierarchy of the primary tumor as macro-metastases. Further similarities between ontogenesis and oncogenesis involving crucial factors, such as ID, HSP70, HLA-G, CD44, LIF, and STAT3, are strongly evident at molecular, physiological and immunological levels. Much experimental data about these factors led to considering the cancer process as ectopic rudimentary ontogenesis, where CSCs have privileged immunological conditions. These would consent to CSC development in an adverse environment, just like an embryo, which is tolerated, accepted and favored by the maternal organism in spite of its paternal semi-allogeneicity. From all these considerations, novel research directions, potential innovative tumor therapy and prophylaxis strategies might, theoretically, result.
Keywords: HSP70, HLA-G, ESCs, MSCs, CSCs, tumor hierarchy/immunoevasion/therapy/prophylaxis
Introduction
Nearly 30 years ago, I proposed that cancer stem cells (CSCs) would be cells blocked at early steps of their genic program, with reiterated expression of embryonic factors responsible for malignant characters and loss of differentiated factors for terminal genomic homeostasis: thus, CSCs would be equivalent to para-embryonic stem cells (p-ESCs) (Manzo, 1989). The main aspects of stem cells (SCs) are self-renewal, pluripotency (Liu et al., 2007) and the need for a niche (Plaks et al., 2015) with proper stereotrophic factors (space, oxygen) and persistent specific signals (ACTIVIN-A, BMP, WNT, LIF, FGF, TGFb) (Okita and Yamanaka, 2006; Xiao et al., 2006).
ESCs arise from the inner cell mass (ICM) of mammalian pre-implantation blastocyst (Henderson et al., 2002; Ginisa et al., 2004; Figure 1B); they can self-renew symmetrically and indefinitely, maintain the widest pluripotency and generate all cell lineages of the body. This phenomenon requires defined transcription factors (TFs) specifically expressed in SCs, such as OCT4, SOX2, NANOG, STAT3, KLF4, c-MYC et al., that together constitute a pluripotency gene regulatory network (PGRN) (HaKashyap et al., 2009; Do et al., 2013; Festuccia et al., 2013). Human ESCs (hESCs) and human embryos express comparable stage-specific embryonic antigens (Henderson et al., 2002) and can differentiate into the trophectoderm (TE) by BMP4 (Xu et al., 2002; Figure 1B). hESCs are epithelial cells (Ullmann et al., 2006), but during in vitro differentiation they can acquire a mesenchymal phenotype (Eastham et al., 2007).
FIGURE 1.
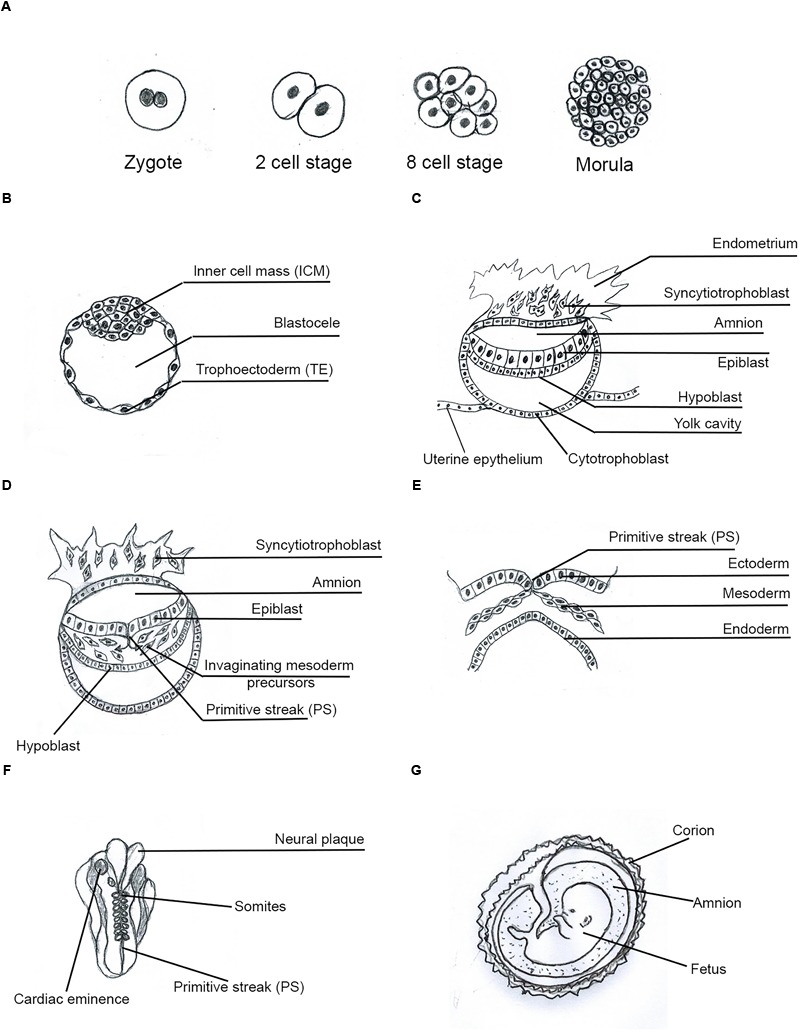
Human Embryo development. Main phases and structures of the embryogenesis process. (A) Zygote to morula transition; (B) pre-implantation blastocyst; (C) implanted blastocyst; (D) early gastrula; (E) late gastrula; (F) somito-histo-organogenesis; (G) fetal growth-differentiation.
MSCs (mesenchymal stem cells) have a mesenchymal phenotype and markers (Ullmann et al., 2006; Eastham et al., 2007; Thiery et al., 2009). MSCs, in Matrigel, grow at the periphery of hESC clusters, have an undifferentiated phenotype and preserve potential expression of pluripotency TFs such as NANOG and OCT4. This indicates that ESCs can undergo epithelial–mesenchymal transition (EMT) without loss of pluripotency, which would be expressed after mesenchymal–epithelial transition (MET) (Ullmann et al., 2006; Thiery et al., 2009). Cells with mesenchymal features largely lie at the primitive streak (PS) in the embryo and in the tumor stroma (Thiery et al., 2009; Nishimura et al., 2012; Figure 1C).
CSCs are tumor cells that are able to generate all the cell types present in the primary tumor and to form metastases, with identical cell types and hierarchy (Marjanovic et al., 2013; Cabrera et al., 2015). CSCs are a small portion of the tumor mass (Collins et al., 2005; Liu et al., 2014) and are often distinct in tumor-initiating cells (TICs) and tumor migrating cells (TMCs) (Hermann et al., 2007; Biddle et al., 2011). TICs have an epithelial phenotype and are able to grow in an anchorage-independent way, to produce spheroids in vitro by self-renewal and to initiate tumor development. TMCs have a mesenchymal phenotype, are free, migrating, invasive and generally quiescent, but are able to generate metastases (Dieter et al., 2011; Brabletz, 2012; Liu et al., 2014).
Therefore, cells with ESC, MSC, and CSC features are at the basis of both embryo development and cancer process (Figure 2).
FIGURE 2.
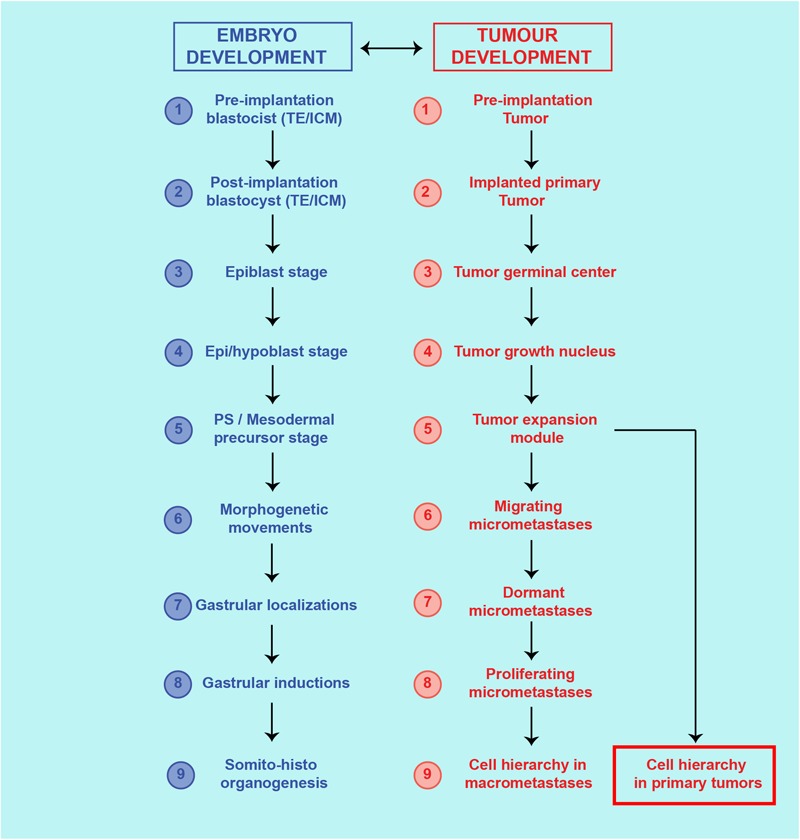
Theoretical similarities between cancer process and ontogenetic development. Correspondence of steps and structures between the cancer process and embryo development.
The Tumor Process as Ectopic Rudimentary Ontogenesis
Cell of Origin (CSC0): Initial Cancer Stem Cell (i-CSC) as a Reprogrammed Para-ESC
Reprogramming would be the main mechanism for genesis and proliferation of the initial i-CSC (CSC0): it has been shown that somatic cell reprogramming requires a MET at its initiation (Li et al., 2010; Samavarchi-Tehrani et al., 2010), with subsequent reversal to a self-renewal and pluripotency state (Silva et al., 2008; Nichols and Smith, 2009; Gillich et al., 2013). Thus, I propose that i-CSCs would be equivalent to ESCs, with one important difference: while an ESC has an integral genic program, an i-CSC would have a program impaired in terminal genome homeostasis (Katoh, 2007; Silva et al., 2008). In this model, an i-CSC would be a p-ESC (Figure 3). At a molecular level, iCSC genesis would presumably require the following steps in the original cell: (a) De-programming (de-differentiation), by impairment of the systems of genomic homeostasis, carried out by growth modulators (p16, p21, p53, p27, E2A, pRb) (Prabhu et al., 1997; Ouyang et al., 2002; Menendez et al., 2010; Tapia and Schöler, 2010) and/or by specific autocrine and paracrine signal pathways (WNT, BMP, LIF, FGF, ERK, TGFb) (Sato et al., 2004; Katoh, 2007; Scheel et al., 2011; Chen et al., 2015), with a consequent differentiation loss and reversal to a previous more primitive (mesenchymal/epithelial/undifferentiated) state (Silva et al., 2008; Schwitalla et al., 2013). (b) Stable PGRN reactivation, by de-regulation of the activity of defined genes (NANOG, OCT4, SOX2, STAT3, ID), with reacquisition of self-renewal and pluripotency (Silva et al., 2008; Do et al., 2013; Yin et al., 2015). (c) Reprogramming, through ID1 gene expression, that prevents i-CSC/CSC0s (p-ESCs) and their direct progeny from normally differentiating (“blocking event”) (Ying et al., 2003; Wang et al., 2012; Nair et al., 2014). ID proteins associate with ubiquitous E proteins, preventing their DNA binding and differentiation activity (Ruzinova and Benezra, 2003; Wang and Baker, 2015). ID1 constitutive expression determines neo-angiogenesis, survival, anti-apoptosis and invasion/migration, both in cancer and embryo development.
FIGURE 3.
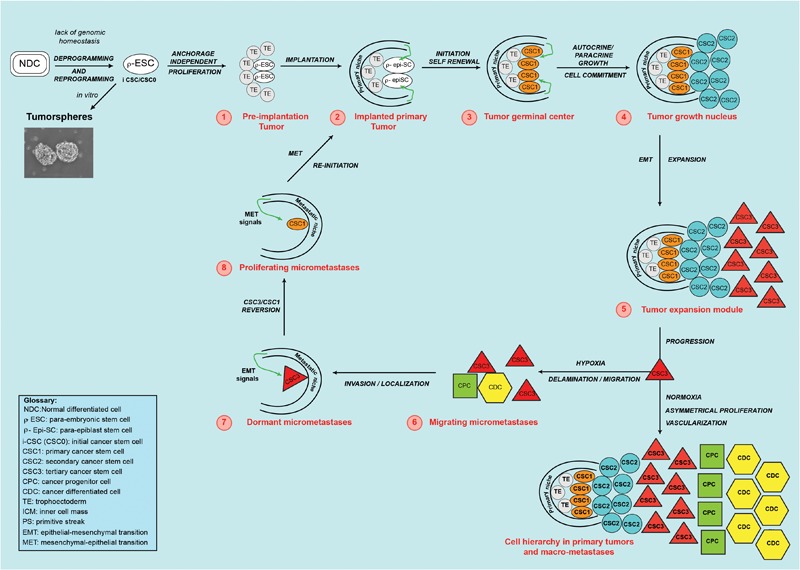
Theoretical cancer process. A normal differentiated cell (NDC), in the absence of genomic homeostasis, would be de-programmed and re-programmed to a para-ESC (p-ESC), constituting the initial cancer stem cell (i-CSC or CSC0). (1) An i-CSC, by anchorage-independent proliferation, would generate p-ESC progeny, forming an initial tumor in vivo (1-red) (or a tumorsphere in vitro), corresponding to a pre-implantation blastocyst (TE/ICM). (2) The initial tumor would install in a niche with proper factors and develop a primary tumor, mimicking an implanted blastocyst in the endometrium. (3) By PGRN activity, primary undifferentiated, pluripotent, slow self-renewing CSCs (CSC1s/TICs) would arise. CSC1s, epigenetically blocked in a ground/primed state, corresponding to the epiblast state, would form a “tumor germinal center,” continuously feeding the tumor. (4) Through autocrine/paracrine growth, from CSC1s, committed non-self-renewing secondary CSCs (CSC2s) would arise. CSC1s and CSC2s, together, would form a “tumor growth nucleus” corresponding to the epiblast/hypoblast state. (5) From CSC1s/CSC2s, tertiary CSCs (CSC3s), with a mesenchymal phenotype (EMT), would be generated. CSC3s would be able to migrate and invade adjoining sites, mimicking delamination of mesodermal precursors at the primitive streak. CSC1s, CSC2s, and CSC3s, together, would form a “tumor expansion module”. In the growing primary tumor, favorable conditions (normoxia) would induce CSC3s to proliferate asymmetrically and generate CPCs and, then, CDCs. This would determine a CSC1-CSC2-CSC3-CPC-CDC cell hierarchy and tumor progression, thus mimicking a partial, rudimentary somito-histo-organogenesis process. (6) In parallel, unfavorable conditions (hypoxia) would induce CSC3s to migrate as circulating micro-metastases, mimicking the gastrula morphogenetic movements. (7) In distant niches with proper EMT signals, CSC3s in the niche would locate as quiescent micro-metastases, mimicking embryonic locations of mesodermal cells at somitogenesis sites. (8) Micro-environmental proper MET signals, would induce mesenchymal CSC3 to revert, into epithelial self-renewing CSC1s mimicking gastrular induction (TICs). Self-renewing CSC1s, as TICs, would form a new tumor germinal center and re-initiate the tumor process in metastatic sites, repeating the same steps and reproducing the same cell types and hierarchy of the primary tumor. Tumorsphere figure was adapted from Bond et al. (2013).
Thus, I suggest an iCSC/p-ESC model for the tumor initiation.
Primary Cancer Stem Cells (CSC1s) as Epiblast Cells
Once generated, i-CSC/CSC0s would survive and symmetrically proliferate early in an anchorage-independent way. Due to its presumed nature as a p-ESC, an i-CSC/CSC0, like an ESC, could generate all cell lineages of the body and, therefore, an initial structure similar to a pre-implantation blastocyst in vivo (Figure 1B), or a tumorsphere in vitro (Figure 3; Johnson et al., 2013), that is able to implant in a surrounding microenvironment (niche) (Stewart et al., 1992), where ID1 proteins would synchronize stemness and anchorage to the niche (Niola et al., 2012). Here, the p-ESC direct progeny would proliferate in an anchorage-dependent way, with an autocrine symmetric mechanism, generating primary cancer stem cells (CSC1s), presumably corresponding to epiblast cells (Figure 1C). CSC1s would form a “tumor germinal center” that continuously feeds the neo-forming tumor (Figure 2, 3). CSC1s would be blocked in a pluripotent undifferentiated/epithelial state (p-ESCs/p-Epi-SCs) (Nichols and Smith, 2009; Gillich et al., 2013), owing to NANOG, OCT4, SOX2, and STAT3 (PGRN) overexpression (Wen et al., 2010; Miyanari and Torres-Padilla, 2012), or to other genomic conditions inducing ID1 constitutive expression (Ruzinova and Benezra, 2003; Ying et al., 2003; Nair et al., 2014). CSC1s would express self-renewal and pluripotency markers (ALDH1, CD44, CD133) (Wakamatsu et al., 2012). The number of CSC1s would be low, stable and strictly controlled by the “niche contact”, as a limiting factor. ALDH1+ CD44+ Ki67+ CSCs, detected in a central position in mammary tumors, might be hypothetical CSC1s, constituting about 0.084% of the tumor mass (Liu et al., 2014). CSC1s would be TICs, like the identified small population of “slow-cycling melanoma cells,” essential for continuous tumor growth (Roesch et al., 2010), or the “long-term renewing” TICs (LT-TICs), that drive tumor feed and metastasis formation in colon cancer (Dieter et al., 2011).
Thus, I propose that tumor initiation and growth might be continuously sustained by cells (CSC1s) with features (pluripotency and self-renewal) typical of epiblast cells.
Secondary Cancer Stem Cells (CSC2s) as Hypoblast Cells
When niche contact becomes limiting, only one CSC1 daughter could retain maternal place and phenotype, while the other could acquire a new epithelial phenotype (CSC2) (Ezashi et al., 2005; Quail et al., 2012). CSC2s would correspond to hypoblast cells (Figure 1C) and therefore they would not have self-renewal and pluripotency, differently to CSC1s. However, CSC2s might, eventually, revert to CSC1s when a niche contact becomes available (Ezashi et al., 2005; Quail et al., 2012). The CSC2 phenotype could occur via the LIF-STAT3-RAS-MAPK-ERK-MYC-ID2 pathway, by NANOG inhibition (Kunath et al., 2007; Yamanaka et al., 2010), ID1/2 switching, loss of stemness/pluripotency (Itahana et al., 2003; Park et al., 2013) and acquisition of fast paracrine growth (Iavarone et al., 1994). Hypothetical CSC2s might be the CSCs with an ALDH1+ CD44- Ki67+ profile detected in mammary tumors (Liu et al., 2014). The “tumor transient-amplifying cells” (T-TACs) with limited or no self-renewal in human colon cancer, and the “rapidly proliferating main population” surrounding the previously mentioned “slow-cycling melanoma cells” (Roesch et al., 2010; Dieter et al., 2011) also could be hypothetical CSC2s. The CSC2 number would be related to the niche volume, nutriments and oxygen quantities (Ezashi et al., 2005; Mohyeldin et al., 2010; Quail et al., 2012): in mammary carcinomas, ALDH1+ CD44- Ki67+ CSCs (CSC2s) constitute about 5.54% of the tumor mass and lie in a sub-central site, physically distinct from ALDH1+ CD44+ Ki67+ CSCs (CSC1s) (Liu et al., 2014). CSC1s and CSC2s, together, would form a presumable “tumor growth nucleus” responsible for tumor expansion (Figure 3).
Thus, I think that tumor growth might result from an autocrine/paracrine signaling-driven proliferation of cells (CSC2s) with features typical of hypoblast cells.
Tertiary Cancer Stem Cells (CSC3s) as Mesoderm Precursor Cells
When the niche microenvironment becomes limiting for growth, certain stereotrophic factors, such as hypoxia, and particular autocrine or paracrine signals (LIF, STAT3, TGFb, WNT, NOTCH) (Ezashi et al., 2005; Mohyeldin et al., 2010; Scheel et al., 2011) could induce CSC1s/CSC2s to generate a new phenotype (CSC3), that is able to migrate, invade and search for more favorable survival conditions elsewhere (Sahlgren et al., 2008; Conley et al., 2012; Tiwari et al., 2012). This phenotype would have mesenchymal features, that would result as a downstream effect of the STAT3-RAS-MAPK-ERK-MYC pathway, regulating ID3/E47 interactions and promoting tumor cell migration and invasion (Bain et al., 2001) through expression of mesenchymal genes such as MMPs, SNAIL1, TWIST1, and PRRX1. The CSC3 phenotype would be the result of an EMT that recalls the EMT of mesoderm precursors at the PS (Figure 1D,E). STAT3-SNAIL1 would confer delamination, migration and invasive properties to CSC3s, as TMCs (Barrallo-Gimeno and Nieto, 2005; Thiery et al., 2009; Kamran et al., 2013; Yin et al., 2015). TWIST1 would increase their invasive properties and, together with PRRX1, would favor installation as micro-metastases in metastatic niches (Yang et al., 2008; Eckert et al., 2011; Tran et al., 2011; Ocaña et al., 2012), thus presumably mimicking the onset of somitogenesis at gastrulation sites. ALDH1- CD44+ Ki67- CSCs, shown within mammary tumors and located in a peripheral position, at the invasive front, would be presumable CSC3s, constituting about 12.87% of the tumor mass (Liu et al., 2014). CSC1s, CSC2s, and CSC3s, together, would form a “tumor progression module” (Figure 2, 3). I hypothesize that, here, CSC3s with more favorable stereotrophic conditions (normoxia) would become TWIST1+ CSC3s, that is able to install and proliferate asymmetrically via ID3/E47, generating more differentiated oligopotent cancer progenitor cells (CPCs). CPCs, in turn, would yield further differentiated cancer cells (CDCs) (Ezashi et al., 2005; Cakouros et al., 2010; Mohyeldin et al., 2010). The resulting CSC1s/CSC2s/CSC3s/CPCs/CDCs, together, would mimic an ectopic, rudimentary somito-histo-organogenesis process (Figure 1F), and would account for the cell hierarchic heterogeneity in tumor progression (Figure 2), where CSCs would be surrounded and protected by CPCs and CDCs (Marjanovic et al., 2013; Liu et al., 2014; Cabrera et al., 2015; Figure 3). On the contrary, CSC3s with less favorable stereotrophic conditions (hypoxia) would undergo EMT, becoming SNAIL1+ CSC3s, that would be induced to delaminate and migrate as quiescent circulating micro-metastases (Ezashi et al., 2005; Sahlgren et al., 2008; Mohyeldin et al., 2010; Conley et al., 2012; Tiwari et al., 2012; Figure 3).
Thus, I propose that tumor progression might occur by appearance of cells (CSC3s) with features of mesoderm precursor cells, that are able to migrate, invade and colonize new sites.
Micro-Metastases and Macro-Metastases as Localization, Induction, and Growth of Gastrula Migrating Cells
After installation as micro-metastases in defined metastatic sites (Arvelo et al., 2016), by specific niche signals, SNAIL1+ CSC3s would undergo MET (Chaffer et al., 2006; Brabletz, 2012; Ocaña et al., 2012; Teo et al., 2014), reverting to self-renewing CSC1s (TICs), that are able to finally generate macro-metastases (Figure 2, 3). Development of macro-metastases would occur via a hypothetical bidirectional genic system TWIST1/ID3/E47/ID1/PRRX1/CD44/PGRN, balanced by TGFb (Gupta et al., 2007; Bourguignon et al., 2012; Ocaña et al., 2012; Stankic et al., 2013; Teo et al., 2014; Wang and Baker, 2015). This system would form a genic switch point in (re)programming: back, for stemness (MET) via PRRX1-CD44-PGRN-ID1 and, forward, for growth-differentiation via ID3-E47-TWIST1 (Stankic et al., 2013). This would lead, in macro-metastases, simultaneously to self-renewal, angiogenesis and differentiation growth, like in the primary tumor. Presumably, PRRX1 downregulation would suppress the mesenchymal state, restoring the epithelial state (MET) through PGRN reactivation via CD44-ID1. It has been shown that downregulation of PRRX1 expression is necessary for micro-metastases to undergo MET and generate macro-metastases (Brabletz, 2012; Ocaña et al., 2012; Hirata et al., 2015), conferring TIC properties to quiescent CSC3s via an ID3/E47/ID1 switch (O’Brien et al., 2012; Hirata et al., 2015; Wang and Baker, 2015; Figure 3). ID1 and ID3 are necessary for TIC functions in the genesis of both primary tumors and metastases, sustaining proliferation in early stages via p21 (Gupta et al., 2007; O’Brien et al., 2012). Moreover, ID1 and ID3 are required for angiogenesis and vascularization of tumor xenografts (Lyden et al., 1999), necessary for macro-metastasis development. TGFb/ID1 signals promote metastatic colonization via a MET, antagonizing TWIST1 EMT (Gupta et al., 2007; Stankic et al., 2013) in normoxic metastatic sites, but not in hypoxic primary tumor sites (Mohyeldin et al., 2010), where EMT is governed by SNAIL1 (Stankic et al., 2013). I hypothesize that EMT/MET switching could occur through ID3/E47/ID1 balance (Bain et al., 2001; Cubilio et al., 2013; Bohrer et al., 2015; Wang and Baker, 2015) and that it could involve ID3b and ID1b isoforms, generated by alternative splicing of the ID1 and ID3 genes (Deed et al., 1996; Tamura et al., 1998).
Crucial Molecular Factors Common to Embryos and Cancer
Pre-implantation Embryos/Initial Tumors (Initiation)
Pre-implantation embryos lie in unfavorable nutriment and microenvironment conditions. Thus, it is necessary they reach and are installed in a niche that is able to supply proper stereotrophic factors for survival. To this end, molecules such as HSP70, HLA-G, ID, and LIF are crucial both for embryos and cancer.
ID Proteins - ID1, ID2, ID3, and ID4 proteins are highly expressed in normal ontogenetic development, where their function is associated with the primitive proliferative phenotype, and regulate differentiation associating to ubiquitous E proteins (Ruzinova and Benezra, 2003; Wang and Baker, 2015). Numerous studies indicate that ID1, ID2, and ID3 have an oncogenic function, whereas ID4 promotes the survival of adult SCs, differentiation and/or differentiation time (Patel et al., 2015). ID proteins are key regulators of CSCs and tumor aggressiveness (Lasorella et al., 2014). During distinct stages of breast metastases, ID proteins mediate phenotypic switching of CSCs (Stankic et al., 2013) and control CSC niches in an autocrine/paracrine way (Niola et al., 2012; Nair et al., 2014). ID1 has multiple roles in cancer progression, such as implantation in primary and metastatic niches, angiogenesis, CSC survival, chemoresistance, growth, apoptosis inhibition and activation of WNT signaling (Ling et al., 2006; Niola et al., 2012; Nair et al., 2014). In particular, the ID1b isoform has been shown to maintain cell quiescence, confer self-renewal and CSC-like properties, and impair malignancy, inhibiting proliferation and angiogenesis (Tamura et al., 1998; Nguewa et al., 2014; Manrique et al., 2015). Thus, ID1b could be related to slow symmetrical self-renewal, niche anchorage and pluripotency maintenance. The ID1a isoform, on the contrary, could be related to fast asymmetrical self-renewal, angiogenesis and evolutionary growth (Lyden et al., 1999), allowing ID1/ID2 switching, with overcoming of pluripotency and subsequent lineage commitment, like in epi/hypoblast segregation. ID2 enhances cell proliferation by binding pRb, an inhibitor of cell cycle progression, and is directly repressed by p53 (Iavarone et al., 1994; Paolella et al., 2011). A WNT-bCAT signal increases ID2 expression level and the incidence of CSC-like phenotype, mediating the effects of hypoxia on the breast and colorectal CSC hierarchy (Rockman et al., 2001; Dong et al., 2016). I hypothesize that ID2 would be involved in overcoming stemness/pluripotency, thus leading to cell commitment and fast autocrine/paracrine growth, via LIF-STAT3-RAS-MAPK-ERK-MYC (Park et al., 2003; Pankaj et al., 2013), with ERK blocking NANOG-ID1 (Itahana et al., 2003; Kunath et al., 2007; Park et al., 2013). ID1/ID2 switching would occur via ID1-WNT/bCAT-ID2, induced by hypoxia (Rockman et al., 2001), and might be related to the epi/hypoblast and CSC1/CSC2 transitions. Moreover, ID2 expression might lead, via RAS-MAPK-ERK, to the activation of ID3 genes (Bain et al., 2001), with subsequent ID2/ID3 switching and mesenchymal gene expression, namely to EMT. Because the RAS-MAPK-ERK cascade regulates ID3/E2A (E47) interaction (Bain et al., 2001), and ID3/E47 balance would determine stem/precursor differentiation (Bohrer et al., 2015), and because ID3 interacts with ID1 (Wang and Baker, 2015), I hypothesize that ID3/E47/ID1 balance would be related to the potential stemness of mesenchymal phenotypes, such as MSCs and CSC3s (O’Brien et al., 2012; Cubilio et al., 2013; Teo et al., 2014; Bohrer et al., 2015). In more detail, the ID3a isoform could be related to overcoming of the CSC3 SNAIL1+ mesenchymal state and to the stem/progenitor switch via TWIST1 (Cakouros et al., 2010; Tran et al., 2011; Bohrer et al., 2015). ID3b isoforms, on the contrary, would be related to the maintenance of the SNAIL1+ mesenchymal state of dormant CSC3s and, thus, to a potential EMT/MET switch for reversion to the CSC1 (TIC) phenotype. Indeed, ID3b inhibits vascular formations (Deed et al., 1996; Forrest et al., 2004) and, thus, the possibility of macro-metastasis development, maintaining dormancy. ID4 acts as an ID1/ID2/ID3 inhibitor and promotes E47 binding and transcriptional activity in a differentiation direction (Umetani et al., 2004; Yun et al., 2004; Sharma et al., 2015). ID4 is a potential tumor suppressor (Umetani et al., 2004; Carey et al., 2009) and suppresses MMP2-mediated invasion of glioblastoma-derived cells by direct inactivation of TWIST1 (Rahme and Israel, 2015). I hypothesize that ID4 might be related to terminal differentiation growth, via E47, and to global genomic homeostasis (Umetani et al., 2004; Sharma et al., 2015). Thus, ID proteins seem to be crucial factors both in cancer and embryos.
HSP70 (heat shock protein 70 kDa) molecules are precociously expressed in ontogenesis, from zygotic gene activation, through blastulation, implantation, gastrulation, and organogenesis to fetal maturation (Figure 1A–G) (Bensaude et al., 1983; Luft and Dix, 1999). In early ontogenesis and oncogenesis, HSP70 could be a first system of protection and survival for pre-implantation embryos, as well as for initial tumors. Protective action of HSP70 for pre-implantation embryos might occur at an endocellular level for preventing apoptosis (Samali and Cotter, 1996); in effect, HSP70 knockdown renders embryonic cells weaker and apoptotic (Neuer et al., 1998). A small amount of HSP70.1-3 is necessary for pre-implantation embryogenesis (Luft and Dix, 1999). HSP70 pre-implantation expression includes a constitutive component (HSC70 and HSP70.1-3) from zygote to four-blastomer morula, and a component (HSP70.1-2) inducible by heat and chemical agents in four- to eight-blastomer morula and in blastocyst (Figure 1A,B) (Luft and Dix, 1999). In adult normal tissues, HSP70s are normally absent, except for transient expression in normal mitosis (Stangl et al., 2011).
HSP70 deregulated overexpression is associated with tumor transformation. In human tumor cells, global profiling of the surface proteome has revealed HSP70.1-2 abundance (Stangl et al., 2011). HSP70s are highly expressed in various tumor cell types, thus rendered resistant to adverse microenvironments and chemotherapy (Shu and Huang, 2008). In human melanoma cell lines, HSP70.1-2 constitutive expression occurs (Dressel et al., 1998). During tumor development, HSP70s can be expressed on the cell surface or exported in the circulation (Shu and Huang, 2008). Human tumor cell lines of colon, breast, lungs and melanomas bind anti-HSP70.1 monoclonal antibodies (mAbs) (Stangl et al., 2011); highly metastatic tumors, but not their primary or poorly malignant counterparts, express membrane HSP70 (mHSP70) (Stangl et al., 2011). mHSP70+ tumors actively release lipidic vesicles (exosomes) with an HSP70+ surface (Stangl et al., 2011). Therefore, I suggest that HSP70s might constitute a first important system of protection and survival in early ontogenesis and oncogenesis.
HLA-G (human leukocyte antigen-G) molecules could be a second important system of protection and survival both for initial embryos and cancer. HLA-Gs appear to be evolutionally and genetically linked to HSP70s: heat shock, a major inductor of HSP70.1-2 expression, and arsenite chemical shock also induce HLA-G expression in tumor cell lines (Ibrahim et al., 2000; Yao et al., 2005). In human pre-implantation embryos, HLA-Gs are expressed as several isoforms, including HLA-G1 and HLA-G5. HLA-G1s are already expressed in two- to eight-blastomer embryos and in all blastocysts as membrane-bound molecules (mHLA-G), whereas, HLA-G5s are expressed only from the blastocyst onwards, as soluble forms (sHLA-G) secreted in biological liquids, or generated by mHLA-G shedding (Yao et al., 2005). HLA-Gs circulating in biological fluids might also be associated with extracellular vesicles (EVs) (Rebmann et al., 2016), containing too antigens, ligands, receptors, cytokines, GFs, mRNAs, and miRNAs (da Silva Nardi et al., 2016). Human pre-implantation embryos express and secrete HLA-Gs, the level of which might be predictive of their implantation capacity (Yao et al., 2005). HLA-Gs orchestrate the early interaction of human trophoblasts with the maternal niche (Gregori et al., 2015). HLA-Gs are expressed early in human ICM and ESCs but, later, mainly in invasive TE, and no more in ICM; after implantation, HLA-Gs are expressed in hypoblast, but no longer in epiblast; yolk-sac mesoderm, endothelial cells of developing vessels, mesenchymal cells and progenitor cells express sHLA-Gs (Hunt et al., 2005; Yao et al., 2005; Verloes et al., 2011) (Figure 1). During pregnancy, sHLA-Gs can be detected in maternal serum. In adult normal tissues, HLA-G constitutive expression is mainly restricted to the fetal extravillous trophoblasts (EVTs), which invade the maternal decidua, rich in NK cells and macrophages (Tilburgs et al., 2015).
Tumor and mesenchymal cells also secrete HLA-Gs in EVs (Yen et al., 2009; Burrello et al., 2016). Tumor cells utilize EVs for dictating a defined phenotype to surrounding cells (Naito et al., 2017). Recent data show that tumor EVs contain molecules for intercellular communications (da Silva Nardi et al., 2016; Rebmann et al., 2016), that act on and impair the recipient immune cells, favoring immune evasion, initiation, tumor development, angiogenesis, invasion, metastasis, CSC and EMT preservation and chemoresistance (Sheu and Shih, 2010; Kosaka, 2016). HLA-G expression has been shown in 22/33 primary tumor tissues of human ovarian carcinoma, but not in normal tissue (Lin et al., 2007), and in 30% of surgically removed melanoma lesions (Yan et al., 2005). In lung cancer, HLA-G1/5 upregulation associates with a high-grade histology, HLA-Ia loss and immunosuppressive IL10 production (Urosevic et al., 2001). Increased HLA-G expression correlates with immune evasion during colorectal cancer progression (Fukushima et al., 1998) and in gastric cancer (Du et al., 2011). These data might, thus, indicate that, in general, in embryos and cancer, HLA-Gs and HSP70s might act not only as protective shields in potentially adverse environments (Sheu and Shih, 2010), but also as means for their invasion and colonization (Yao et al., 2005). More in detail, it seems that HLA-G1 is preferentially expressed precociously and in more peripheral embryonic structures in an essentially protective scope, whereas HLA-G5 is produced later and in invasive embryonic structures and in tumor metastatic cells, in order to favor protection, invasion, installation and development in the host (Rouas-Freiss et al., 2007; Sheu and Shih, 2010). HLA-Gs are induced by hypoxia via HIF-1a (Rebmann et al., 2003) and are upregulated by IL10 with autocatalytic feedback (Urosevic et al., 2001, 2002; Stankic et al., 2013). HLA-Gs induce IL6 production (Urosevic et al., 2002), and thus activation of the gp130-STAT3 pathway, regulating proliferation, invasion, migration and angiogenesis (Kamran et al., 2013). In my opinion, all these data suggest that HLA-G molecules could be key inter-players and shared actors in the initiation and maintenance of both embryogenesis and carcinogenesis. Particularly, HLG together to HSP70, would represent an essential system for protection and survival of both embryo and tumor.
Peri-Implantation Embryos/Primary Tumors (Implantation and Growth)
Beyond a certain stage, a blastocyst might develop further only if a supply of nutrients is possible from the outer environment: this would implicate for the embryo the necessity of implantation in the maternal uterus, and for initial tumors in a niche. Blastocyst implantation occurs thanks to the structure and properties of the syncytiotrophoblast (Figure 1C,D; Park et al., 2003) and to a parallel maternal endometrium condition, suited to accepting implantation. The process requires some crucial interacting factors, such as LIF/LIFr, IL6/IL6r, IL10, IL11, GP130, JAK, and STAT3, and correlated signal pathways, leading to the switch from anchorage-independent to anchorage-dependent growth, both in embryos and cancer.
LIF/LIF-r, IL6/IL6-r, gp130-JAK-STAT3 - In peri-implantation, LIF (leukemia inhibitory factor) is crucial: in LIF knockout mice, blastocyst implantation does not occur; in women, LIF strongly increases in the implantation window (Sherwin et al., 2002; Aghajanova, 2004). Uterine expression of LIF coincides with the onset of blastocyst implantation, and this depends on maternal expression of LIF (Bhatt et al., 1991; Cheng et al., 2001; Sherwin et al., 2002; Yue et al., 2015). LIF is essential for inducing a receptive uterus, but not for embryogenesis (Chen et al., 2000). LIF and LIFr expression in the human endometrium suggests a potential autocrine/paracrine function in regulating embryo implantation (Cullinam et al., 1996). In implantation, LIF carries on its biological functions mainly by activation and regulation of the JAK-STAT3, AKT, ERK1-2, and MAPK signal pathways, inducing expression of integrin α5β1, that realizes implantation, endothelial proliferation and endometrial vascularization (Cheng et al., 2001; Sherwin et al., 2002; Park et al., 2003). LIF-gp130-STAT3 is also linked to HLA-G expression through IL6 and IL10 (Urosevic and Dummer, 2003; Yue et al., 2015). In initial embryos, HLA-G expression is a fundamental prerequisite for obtaining pregnancy: indeed, in in vitro-fertilized human embryos, only those whose culture surnatant contains sHLA-G are able to perform implantation (Fuzzi et al., 2002; Alizadeh et al., 2016).
A LIF signal is also expressed at high levels in a wide spectrum of human cancers, including melanomas, skin, kidney, prostate, pancreas and breast cancer, where cell proliferation is stimulated by paracrine and autocrine pathways, as in embryo implantation (Cullinam et al., 1996; Kellokumpu-Lehtinen et al., 1996). The amount of LIF secreted by a tumor seems to regulate cancerogenesis (Guo et al., 2015); LIF overexpression in breast cancer patients is significantly associated with an unfavorable rate of survival without relapses (Guo et al., 2015). High expression of LIFr identifies very malignant melanocytic lesions at an early stage, and it is a crucial condition for the nevus/implanted melanoma transition (Guo et al., 2015). Analysis of 441 melanomas and 90 nevi showed low LIFr expression for all nevus stages, whereas the presence of this receptor starts to increase in dysplastic nevi, with significantly higher expression in primary melanomas (implantation), and even higher in metastatic melanomas (Guo et al., 2015), suggesting a striking correlation between LIF/LIFr expression and oncogenesis. LIFr knockdown inhibits melanoma cell migration in wound-healing tests (Guo et al., 2015). In general, LIFr activation can promote metastasis and increase the invasion potential of solid tumors (Guo et al., 2015). In human colorectal cancer cells and in solid tumors, hypoxia is an important factor inducing LIF mRNA expression, mediated mainly by HIF-2a (Yue et al., 2015). TGFb also induces expression of LIF mRNA; LIF induction is important to maintain the self-renewal of glioma-initiating cells and prevent their differentiation (Yue et al., 2015). LIF binds to human breast cancer cells and stimulates their proliferation (Estrov et al., 1999). I hypothesize that in the tumor process, LIF/LIFr might be at the basis of: (a) implantation of the primary tumor (CSC1s) via gp130-JAK-STAT3-ID1, with self-renewal and pluripotency (Niola et al., 2012); (b) subsequent paracrine growth (CSC1s/CSC2s) via gp130-JAK-STAT3-RAS-MAPK-ERK-MYC-WNT-ID2 (Cheng et al., 2001; Park et al., 2003); (c) genesis of cells with a mesenchymal phenotype (CSC3s) via CD44-STAT3-RAS-SNAIL1-MMPs-ID3-TWIST1 (Bain et al., 2001; Cheng et al., 2006; Su et al., 2011; Zeilstra et al., 2014; Yan et al., 2015). All these data clearly indicate that LIF/LIFr and related signaling pathways are crucial in cancer initiation, implantation, growth and diffusion, in a way that recalls their function in embryo development.
Post-implantation Embryos/Metastatic Tumors (Progression)
In the post-implantation embryo, at the PS, cells with a mesenchymal phenotype appear, migrating between epiblast and hypoblast cells (Figure 1D). The mesenchymal phenotype also characterizes metastatic TMCs (Sahlgren et al., 2008; Thiery et al., 2009; Ocaña et al., 2012). The main factors that correlate with the embryonic mesenchymal state, tumor metastases and progression include HLA-G, HSP70, hypoxia, STAT3, CD44, SNAIL1, TWIST1, PRRX1, TGFb, WNT/bCAT, ID, and MMPs. Mesenchymal progenitors derived from hESCs (EMPs) express surface HLA-G1 (Yen et al., 2009). Mesenchymal cells and progenitor cells also express HLA-Gs. IL10 selectively induces HLA-G expression in human invasive trophoblasts and monocytes (Moreau et al., 1999). In mesenchymal CSCs of kidney cancer, HLA-Gs and EVs enhance metastasis and progression (Grange et al., 2015). Highly metastatic tumors show the presence of membrane HSP70s (Stangl et al., 2011). In progression of many cancer types, migration is stimulated by the LIF-STAT3 pathway, constitutively activated, that regulates proliferation, migration, angiogenesis and metastasis (Kamran et al., 2013; Guo et al., 2015). The mesenchymal phenotype is highly characterized by the surface marker CD44.
CD44 is a cell-surface glycoprotein constituting a signal platform that regulates the expression of genes related to cell-matrix adhesion, migration, proliferation, survival and differentiation in development (Okamoto et al., 1999; Williams et al., 2013; Yan et al., 2015). The main CD44 receptor is hyaluronic acid (HA), a component of the extracellular matrix (ECM), that envelops tumor cells and bulk, regulating proliferation and motility in cancer progression and metastasis (Chanmee et al., 2015; Avnet and Cortini, 2016). The CD44-STAT3 complex induces Cyclin D1, MMP9, HIF-a2, c-MYC, TWIST1, cytoskeleton remodeling (Su et al., 2011; Yan et al., 2015) and activation of RAS signaling (Cheng et al., 2006). Expression of CD44s (standard) is ubiquitous, while CD44v (variant) isoforms seem restricted to aggressive tumors (Zeilstra et al., 2014). CD44s/CD44v switching is a critical event during EMT (Yan et al., 2015).
CD44v (v3, v6, v8) isoforms are CSC markers and have a crucial role in regulation of stemness, self-renewal, tumor initiation, metastasis and chemoresistance (Bourguignon et al., 2012; Chanmee et al., 2015; Yan et al., 2015; Avnet and Cortini, 2016). A positive feedback loop couples RAS activation and CD44v isoform expression (Cheng et al., 2006). CD44/osteopontin (the main component of metastatic niches) interaction activates NANOG-STAT3, OCT4-SOX2-NANOG and c-MYC, namely PGRN, and it is induced by hypoxia via HIF-a1 (Krishnamachary et al., 2012; Pietras et al., 2014). CD44 transcription and cell growth are suppressed by p53 (Godar et al., 2008). Thus, I hypothesize that CD44v could be a sort of molecular trigger that is able to directly activate PGRN in the initial MET for reprogramming (Su et al., 2011; Yan et al., 2015) and, thus, a crucial factor in the generation of signaling for the switch between mesenchymal and epithelial states, usually occurring both in embryos and cancer.
Immunological Aspects Common to Embryos and Cancer: Host Tolerance and Favoring
HSP70s and HLA-Gs are able to strongly influence many important functions of the immune system. Membrane HSP70s affect NK cell cytotoxicity by acting as recognition/activator ligands (Luft and Dix, 1999; Stangl et al., 2008), whereas HLA-Gs are able to inhibit practically all immune system components (NK, T, DC, and B cells) (Hofmeister and Weiss, 2003; Hunt et al., 2005; Gros et al., 2008; Loumagne et al., 2014; Gregori et al., 2015; Tilburgs et al., 2015; Lopatina et al., 2016). In ESCs and pre-implantation embryos, mHLA-G and sHLA-G block all the uterine immune cells, binding the KIR2DL4 inhibitory receptors on NK cells, and ILT2 (LILRB1) and ILT4 (LILRB2) inhibitor receptors on all the leukocytes and macrophages (Hofmeister and Weiss, 2003; Li et al., 2004; Verloes et al., 2011; Tilburgs et al., 2015; Rebmann et al., 2016). Both membrane and soluble HLA-Gs would confer immune-suppressive properties to the cells producing them, in various ways: (a) For HLA-G1, direct interaction with ILT2 and ILT4, determining CD8+ T cell apoptosis, NK cell immobilization, and impairment of monocyte/DC differentiation, with a high production of IL4 and IL10 immunosuppressive cytokines (Rebmann et al., 2003; Urosevic and Dummer, 2003; Gros et al., 2008; Gregori et al., 2015; Rebmann et al., 2016). Mesenchymal progenitors derived from hESCs (EMPs) strongly suppress NK and T cells by HLA-G1 (Yen et al., 2009). (b) For induction of cascade signaling and biological activity in the host cells, thanks to the factors contained in EVs, internalized by trogocytosis or pinocytosis (Rouas-Freiss et al., 2007). CSCs cross-talk with MSCs through EVs containing mRNA and miRNA, identified as the main factors responsible for the phenotypic changes induced in the cells receiving EVs (Lopatina et al., 2016). (c) For amplification of mechanisms and conditions favorable to the cells releasing EVs, with further involvement of other surrounding cells. In human neuroblastomas, tumor cells instruct monocytes to produce and release sHLA-G (Rouas-Freiss et al., 2003). HLA-G transfer from antigen-presenting cells (APCs) to activated T cells has been reported in embryos, with reversion of these cells to a regulator phenotype blocking the allo-immune response (Rebmann et al., 2016). A co-culture of activated decidual NK (dNK) cells or peripheral NK (pNK) cells with EVTs results in mHLA-G initial acquisition within 18 h, and complete acquisition in 36 h (Tilburgs et al., 2015).
In tumors, HLA-Gs transfer quickly from APCs or cancer cells to T and NK cells, and convert these cells into temporarily suppressor HLA-G+ cells (Rebmann et al., 2003). Once acquired by NK cells, HLA-Gs are degraded, and pNK cells revert to their previous cytotoxic phenotype (Tilburgs et al., 2015). Both HLA-G5 and HLA-G1 endocytosed by NK cells lead to NFkB pathway activation and, finally, to transcription of immunosuppressor (IL10) and pro-angiogenetic (IL6) factors (Rebmann et al., 2016). It has been shown that pNK cells might acquire via trogocytosis HLA-Gs from a transfected melanoma M8 cell line, and that, after acquisition, NK cells are no longer cytotoxic and are unable to realize immune synapses (Lesport et al., 2009; Tilburgs et al., 2015). In gastric cancer, HLA-G overexpression associates with immune escape and correlates with a local increase of T regulatory cells (TREGs) (Du et al., 2011). NK cytolysis depends on the amount of HLA-G1 expressed, that in malignant tumors can go from 0 to 100%, with complete NK inhibition, reduced local and systemic immunosurveillance and tumor progression (Rebmann et al., 2003; Chen et al., 2013). HLA-G1s and HLA-G5s have an additive suppressor effect on NK cytolysis dependent on their level, but HLA-G5 is a more potent inhibitor (Zhang et al., 2014). Within a population of HLA-G- tumor cells, few HLA-G+ cells have significant immune inhibitory effects (Lesport et al., 2009). HLA-G upregulation also occurs through factors such as cytokines (IL10), stress and chemotherapeutic demethylating agents (Rouas-Freiss et al., 2003; Yan et al., 2005). IL10 upregulates HLA-G, that induces an immunosuppressive Th2 profile (Urosevic et al., 2002; Rebmann et al., 2003), with further IL10 increase in a vicious circle (Moreau et al., 1999). Interaction of HLA-G+ cells with NK cells also enhances IL6 production, that induces angiogenesis and SC proliferation via gp130-JAK-STAT3-OCT4. The inhibiting effects of HLA-G on NK cells are eliminated by the action of IL15, IL2 and IL12 (Tilburgs et al., 2015). CSC immune evasion also occurs through shedding of MIC-A/B, HSP70 and HLA-G1 (Ames et al., 2015). It has been shown that highly metastatic tumors, but not their primary or poorly malignant counterparts, are mHSP70+, capable of shedding (Stangl et al., 2011). It is presumable that a high number of molecules such as HSP70, HLA-G, and MIC-A/B released in circulation could lead to ectopic activation and subsequent neutralization of immune cells, without these coming into contact with CSC targets (Stangl et al., 2011), thus favoring diffusion of metastasis.
Current Anticancer Therapeutic Approaches in Agreement With the I-CSC/P-ESC Model
Several current partially successful approaches for cancer therapy are in agreement with the i-CSC/p-ESC model, targeting crucial factors common to embryos and tumors, such as HSP70, HLA-G, ID, LIF/LIFr, and CD44. CSCs seem to be preferential targets for NK cells through upregulation of antigens (HSP70, MIC-A/B, FAS, DR5) induced by stress, by which NK cells are capable of targeting (only) quiescent, non-proliferating cells (Ames et al., 2015). Thus, in solid tumors, slow-renewing or quiescent CSCs might be more easily killed by NK cells after depletion of proliferating non-CSCs (CPCs) through anti-proliferative therapies (Ames et al., 2015).
HLA-Gs are a valid target in cancer therapy (Lin and Yan, 2015; Zhang et al., 2018); cytotoxicity studies show that HLA-Gs drastically inhibit lysis of human ovarian carcinoma cells, with subsequent immune evasion, and that lysis can be restored by the conformational anti-HLA-G mAb 87G (Tilburgs et al., 2015). Interestingly, dNK cells in culture with IL2, IL12, and IL15 lose the acquired surface HLA-Gs by internalization and degradation, reacquiring cytolytic activity (Tilburgs et al., 2015). In breast cancer patients treated with NACT (neo-adjuvant chemotherapy), high levels of HLA-Gs/EVs before NACT correlate to tumor progression and the presence of circulating tumor stem-like cells, releasing EVs, while high sHLA-G levels (presumably from tumor lysis) indicate a better clinical outcome (König et al., 2016). HLA-G expression is induced in the melanoma cell line OCM-1A after treatment with 5-aza-2’-deoxycytidine (Yan et al., 2005).
Anticancer therapeutic approaches based on endocellular and exocellular HSP70s have been carried out (Didelot et al., 2007; Shu and Huang, 2008; Jego et al., 2010). It has been reported that mHSP70+ tumors actively release lipid vesicles (exosomes) with an HSP70+ surface, and that they can attract already activated NK cells (but not resting NK cells) (Stangl et al., 2011), causing their ectopic degranulation and neutralization. The TKD peptide (14 amino acids common to all HSP70s), combined with IL2 low doses, has been found to stimulate NK cell migratory and cytolytic activity against HSP70+ tumor cells (Stangl et al., 2011). A murine antibody anti-TKD (cmHsp70.1 mAb) binds all human vital tumor cell lines of colon, breast, lungs and melanomas; thus, HSP70s could be an immune-therapeutic target in a wide spectrum of tumor types (Shu and Huang, 2008; Stangl et al., 2011). In fact, cmHsp70.1 mAb injected into mice with CT26 colon tumors can significantly reduce the bulk of mHSP70+ tumors and increase survival via ADCC induction, that can be further enhanced by NK cells pre-activated with TKD/IL2 (Stangl et al., 2011).
ID genes and proteins are a promising target in cancer therapy for inhibiting tumor cells and their supply in the blood (Benezra, 2001; Fong et al., 2004; Iavarone and Lasorella, 2006). ID protein inhibition by a peptide aptamer induces cell cycle arrest and apoptosis in ovarian cancer cells (Mern et al., 2010a). Suppression of invasion and metastasis in aggressive salivary and breast cancer cells is targeted through inhibition of ID1 expression (Fong et al., 2003; Murase et al., 2016). Inactivation of ID1 genes induces sensitivity of prostate cancer cells to chemotherapeutic drugs (Wong et al., 2008). Co-suppression of ID1 and ID3 results in a significant reduction in the proliferation rate, invasiveness and anchorage-independent growth, reduced angiogenesis and increased apoptosis in small-cell lung cancer (Chen et al., 2014), and significantly reduces the ability of gastric cancer cells to form peritoneal metastases (Tsuchiya et al., 2005). Moreover, ID1 and ID3 knockdown inhibits the metastatic potential of pancreatic cancer, both for proliferation and migration (Shuno et al., 2010). Targeting ID1 and ID3 by a specific peptide aptamer induces E-box promoter activity, cell cycle arrest and apoptosis in breast cancer cells (Mern et al., 2010b). TGFb receptor inhibitors target the CD44high/Id1high glioma-initiating cells in human glioblastoma (Anido et al., 2010). It has been shown that ID2 knockdown by an inhibitor of WNT-bCAT signaling markedly suppresses the formation of CSC spheres in vitro, and metastases in vivo (Rockman et al., 2001), by inhibition of CSC-like phenotypes (Jang et al., 2015).
LIFr expression is a crucial condition for nevus/implanted melanoma transition; LIFr knockdown inhibits migration of melanoma cells in wound-healing tests; thus, LIFr could be a potential target for developing therapies in initial tumor interventions (Guo et al., 2015). Neutralizing antibodies knock down activity or expression of LIF and reduce in vitro the stem cell-like properties of murine slow-growing CSCs (American Association for Cancer Research, 2012).
CD44 is a biomarker and therapeutic target in CSCs (Su et al., 2011; Yan et al., 2015), and CD44v is a promising approach for elimination of CSCs (Jin et al., 2006; Orian-Rousseau and Ponta, 2015; Yan et al., 2015). In breast cancer, cells with an EMT phenotype can be inhibited by Abs specific for CD44 (Yan et al., 2015); knockdown of CD44 induces differentiation of breast CSCs and is a promising differentiation therapy (Pham et al., 2011). CD44 targeting reduces tumor growth and prevents post-chemotherapy relapse of human breast cancer xenografts (Marangoni et al., 2009). Inhibition of CD44v3 and v6 by A5G27 peptide copolymer blocks tumor invasion and metastatic colonization (Zaiden et al., 2017). DNA vaccination with CD44v isoforms reduces mammary tumor local growth and lung metastases (Wallach-Dayan et al., 2008).
Immunological approaches to target CSCs have been carried out, and through CSC vaccination, significant antitumor immunity can be conferred (Ning et al., 2012; Pan et al., 2015). For improving the efficacy of breast cancer treatment, a combination therapy has been used, that permits targeting of both CSC-like and bulk tumor cells (Wang et al., 2016). Thus, it is evident that many current therapeutic strategies are addressed to target crucial factors, common to embryos and cancer, at the basis of this i-CSC/p-ESC model.
Discussion and Conclusion: Proposals for Innovative Cancer Therapy and Prophylaxis Strategies
Considerations for a Proper Antitumor Therapeutic Strategy
From the numerous experimental data reported above, it is evident that embryo and tumor development occurs in very similar physio-pathological conditions of immune tolerance by the host, which accepts and even favors them. Anticancer therapies carried out so far, targeting separately the factors HLA-G, ID, LIF, HSP70, and CD44, have got important positive, but only partial and non-resolutive, results. This limit might be due to several factors: (a) a particular hierarchic tumor structure, immunologically protective for CSCs, surrounded by CPCs and CDCs (Nishimura et al., 2012; Marjanovic et al., 2013; Liu et al., 2014; Ames et al., 2015; Cabrera et al., 2015); (b) interconversion among CSCs (CSC1/CSC2/CSC3/CSC1) via ID1, ID2 and ID3 proteins (Hermann et al., 2007; Biddle et al., 2011; Dieter et al., 2011; Brabletz, 2012; Quail et al., 2012; Do et al., 2013; Cabrera et al., 2015); (c) the presence of HLA-G and HSP70 on the surface of CSCs and CPCs, with impairment of various immune surveillance mechanisms, NK activity in particular (Samali and Cotter, 1996; Rouas-Freiss et al., 2007; Sheu and Shih, 2010; Stangl et al., 2011); (d) anti-proliferative chemotherapies, that only eliminate actively proliferating CCs (CSC2s and CPCs), but spare slow-renewing CSCs (CSC1s) in niches, or quiescent circulating CSCs (CSC3s), and non-proliferating CDCs (Ames et al., 2015; Wang et al., 2016); (e) release into the circulation of a high quantity of HSP70 by shedding, exosomes or chemotherapeutic necrosis (Shu and Huang, 2008; Stangl et al., 2011), attracting ectopically activated NK cells and neutralizing them at a distance from CSC targets (Stangl et al., 2011); (f) release into the circulation, by shedding, EVs or chemotherapeutic necrosis, of high amounts of HLA-G, that is able to block any immune mechanism, or even instruct normal cells to favor the tumor (Hofmeister and Weiss, 2003; Li et al., 2004; Rouas-Freiss et al., 2007; Gros et al., 2008; Loumagne et al., 2014; Lopatina et al., 2016); (g) negative collateral effects of anti-angiogenetic therapies that, by blocking tumor vascularization, might induce hypoxia and a subsequent increase of metastatic CSCs (CSC3s) (Conley et al., 2012).
General Suggestions for a Potential Multi-Target Multistep Cancer Therapy
On the basis of the previous considerations, I suggest some general indications for a multistep multi-target therapeutic strategy. To this end, I think that it would be necessary to make use of “micro-antibodies” (mi-Abs), which are artificial, chemically synthetized short chains of amino acids, copied from fully functional natural antibodies (Heap et al., 2005). Their small size allows them to leave the circulation quickly and reach specific target sites in the tissues, normally unapproachable for mAbs. mi-Abs are poorly immunogenic and do not stimulate an immune response versus the host. mi-Abs show neutralizing properties versus viruses, such as HIV, infecting cells in vitro (Fujii, 2009). All these mi-Ab properties might consent the following antitumor therapeutic strategy, leading to progressive back-dismantling of the tumor hierarchic organization, selectively targeting the diverse tumor cell populations in a defined multistep sequence: (a) co-culture of autologous peripheral blood mononuclear cells (PBMCs) with IL15, IL12 and IL2, to restore them from the effects of HLA-G (Tilburgs et al., 2015) and dispose of HLA-G-free PBMCs, to be used in next steps; (b) tumor biopsy for the selection of the various populations: differentiated non-proliferating cells (CDCs), non- or slow-proliferating SCs (CSC1s and CSC3s), actively proliferating cells (CSC2s and CPCs); (c) co-culture of HLA-G-free PBMCs with each selected tumor population, and “in toto” tumor populations, previously treated with anti-HLA-G and anti-HSP70 mi-Abs, allowing in vitro PBMC sensitization without conditioning; (d) anti-HLA-G and anti-HSP70 prophylaxis of the subject by specific mi-Abs; (e) reinfusion of anti-CDC-sensitized PBMCs and/or chemotherapeutic treatment, to reduce the tumor bulk and expose the internal cell populations (Ames et al., 2015; Wang et al., 2016), thus making them more approachable in the next steps; (f) chemotherapeutic anti-proliferative treatment of the subject to eliminate fast-proliferating CSC2s and CPCs, upon protection with anti-HLA-G and anti-HSP70 neutralizing mi-Abs; (g) treatment of the subject with anti-CD44v and anti-ID1/ID3 mi-Abs to neutralize circulating CSC3s from an eventual CSC3/CSC1 transition (MET), and CSC1s in niches from anchorage and self-renewal (Marangoni et al., 2009; Chen et al., 2014; Zaiden et al., 2017); (h) treatment of the subject with anti-LIF/LIFr and anti-ID1/2 mi-Abs to prevent or block CSC1 implantation in niches and angiogenesis, CSC migration by activation of the LIF-STAT3 pathway, and a possible CSC1/CSC2/CSC3 transition (EMT) (Rockman et al., 2001; American Association for Cancer Research, 2012; Guo et al., 2015; Jang et al., 2015); (i) treatment of the subject with anti-HLA-G, anti-HSP70, anti-CD44, anti-ID1/ID2/ID3 and anti-LIF/LIFr mi-Abs for general prophylaxis; (ii) final treatment with “in toto” sensitized HLA-G-free PBMCs to restore natural immunological and physiological conditions in the host.
Considerations and Suggestions for Potential Cancer Prophylaxis
On the basis of the i-CSC/p-ESC model, it might be logically and biologically possible to use anti-HSP70/HLA-G (Qa-2 in mouse)/ID/LIF-LIFr/CD44 mi-Abs for antitumor prophylaxis. Periodical treatments with mi-Abs at defined times and in defined ways could protect healthy subjects from arising tumors (i-CSC/CSC0s) or implanting initial tumors (CSC1s). The validity of this hypothesis might be tested in appropriate animal models, inoculating before, together and after mi-Ab treatments: (a) CSCs of various human tumor types, in nude mice; (b) CSCs of various mouse tumor types in normal autologous mice. Verifying tumor development, an eventual absence of tumors with the treatment before or together with CSC inoculation would indicate a preventive effect of the mi-Abs while, with the subsequent treatment, it could also indicate a drainage effect of the mi-Abs versus initial tumors. The use of mi-Abs for periodical prophylaxis could be evaluated in relation to the subject age, in general. In particular, in women, it could be considered in relation to an eventual pregnancy, because all the mi-Ab tumor targets are crucial embryonic factors.
In conclusion, I hope that this work might make a valid contribution for a better vision of the cancer, and that it might stimulate the interest of others to debate and verify some validity of the ideas expressed, as well as to test the therapeutic potential of the suggested proposals.
Author Contributions
The author confirms being the sole contributor of this work and has approved it for publication.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
I would like to remember Giustino Manzo, who died of cancer on 20 February 1979, whose sacrifice has incited and supported this research constantly. I thank all who have believed in the ideas of this work and have contributed to it scientifically, technically and emotionally. In particular, I thank my son Stefano Manzo for his precious contribution to the discussion and solution to several problems which have arisen in this study.
References
- Aghajanova L. (2004). Leukemia inhibitory factor and human embryo implantation. Ann. N. Y. Acad. Sci. 1034 176–183. 10.1196/annals.1335.020 [DOI] [PubMed] [Google Scholar]
- Alizadeh N., Mosaferi E., Farzadi L., Majidi J., Monfaredan A., Yousefi B., et al. (2016). Frequency of null allele of human leukocyte antigen-G (HLA-G) locus in subjects to recurrent miscarriage. Int. J. Reprod. Biomed. 14 459–464. 10.29252/ijrm.14.7.4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- American Association for Cancer Research (2012). Leukemia Inhibitory Factor May be a Promising Target Against Pancreatic Cancer. Public Release. Available at: https://www.eurekalert.org/pub_releases/2012-06/aafc-lif061412.php [Google Scholar]
- Ames E., Canter R. J., Grossenbacher S. K., Mac S., Chen M., Smith R. C., et al. (2015). NK cells preferentially target tumor cells with a cancer stem cell phenotype. J. Immunol. 195 4010–4019. 10.4049/jimmunol.1500447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anido J., Saez-Borderias A., Gonzalez-Junca A., Rodon L., Folch G., Carmona M. A., et al. (2010). TGF-beta receptor inhibitors target the CD44(high)/Id1(high) glioma-initiating cell population in human glioblastoma. Cancer Cell 18 655–668. 10.1016/j.ccr.2010.10.023 [DOI] [PubMed] [Google Scholar]
- Arvelo F., Sojo F., Cotte C. (2016). Tumor progression and metastasis. Ecancermedicalscience 10:617 10.3332/ecancer.2016.617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avnet S., Cortini M. (2016). Role of pericellular matrix in the regulation of cancer stemness. Stem Cell Rev. 12 464–475. 10.1007/s12015-016-9660-x [DOI] [PubMed] [Google Scholar]
- Bain G., Cravatt C. B., Loomans C., Alberola-Ila J., Hedrick S. M., Murre C. (2001). Regulation of the helix-loop-helix proteins, E2A and Id3, by the Ras-ERK MAPK cascade. Nat. Immunol. 2 165–171. 10.1038/84273 [DOI] [PubMed] [Google Scholar]
- Barrallo-Gimeno A., Nieto M. A. (2005). The Snail genes as inducers of cell movement and survival: implications in development and cancer. Development 132 3151–3161. 10.1242/dev.01907 [DOI] [PubMed] [Google Scholar]
- Benezra R. (2001). The Id proteins: targets for inhibiting tumor cells and their blood supply. Biochim. Biophys. Acta 1551 F39–F47. 10.1016/S0304-419X(01)00028-2 [DOI] [PubMed] [Google Scholar]
- Bensaude O., Babinet C., Morange M., Jacob F. (1983). Heat shock proteins, first major products of zygotic gene activity in mouse embryo. Nature 305 331–333. 10.1038/305331a0 [DOI] [PubMed] [Google Scholar]
- Bhatt H., Brunet L. J., Stewart C. L. (1991). Uterine expression of leukemia inhibitory factor coincides with the onset of blastocyst implantation. Proc. Natl. Acad. Sci. U.S.A. 88 11408–11412. 10.1073/pnas.88.24.11408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biddle A., Liang X., Gammon L., Fazil B., Harper L. J., Emich H., et al. (2011). Cancer stem cells in squamous cell carcinoma switch between two distinct phenotypes that are preferentially migratory or proliferative. Cancer Res. 71 5317–5326. 10.1158/0008-5472.CAN-11-1059 [DOI] [PubMed] [Google Scholar]
- Bohrer C., Pfurr S., Mammadzada K., Schildge S., Plappert L., Hils M., et al. (2015). The balance of Id3 and E47 determines neural stem/precursor cell differentiation into astrocytes. EMBO J. 34 2804–2819. 10.15252/embj.201591118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bond W. S., Akinfenwa P. Y., Perlaky L., Hurwitz M. Y., Hurwitz R. L., Chévez-Barrios P. (2013). Tumorspheres but not adherent cells derived from retinoblastoma tumors are of malignant origin. PLoS ONE 8:e63519. 10.1371/journal.pone.0063519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourguignon L. Y., Wong G., Earle C., Chen L. (2012). Hyaluronan-CD44v3 interaction with Oct4-Sox2-Nanog promotes mir-302 expression leading to self-renewal, clonal formation, and cisplatin resistance in cancer stem cells from head and neck squamous cell carcinoma. J. Biol. Chem. 287 32800–32824. 10.1074/jbc.M111.308528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brabletz T. (2012). EMT and MET in metastasis: where are the cancer stem cells? Cancer Cell 22 699–701. 10.1016/j.ccr.2012.11.009 [DOI] [PubMed] [Google Scholar]
- Burrello J., Monticone S., Gai C., Gomez Y., Kholia S., Camussi G. (2016). Stem cell-derived extracellular vesicles and immune-modulation. Front. Cell. Dev. Biol. 4:83 10.3389/fcell.2016.00083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabrera M. C., Hollingsworth R. E., Hurt E. M. (2015). Cancer stem cell plasticity and tumor hierarchy. World J. Stem Cells 7 27–36. 10.4252/wjsc.v7.i1.27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cakouros D., Raices R. M., Gronthos S., Glackin C. A. (2010). Twist-ing cell fate: mechanistic insights into the role of twist in lineage specification/differentiation and tumorigenesis. J. Cell. Biochem. 110 1288–1298. 10.1002/jcb.22651 [DOI] [PubMed] [Google Scholar]
- Carey J. P., Asirvatham A. J., Galm O., Ghogomu T. A., Chaudhary J. (2009). Inhibitor of differentiation 4 (Id4) is a potential tumor suppressor in prostate cancer. BMC Cancer 9:173. 10.1186/1471-2407-9-173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaffer C. L., Brennan J. P., Slavin J. L., Blick T., Thompson E. W., Williams E. D. (2006). Mesenchymal-to-epithelial transition facilitates bladder cancer metastasis: role of fibroblast growth factor receptor-2. Cancer Res. 66 11271–11278. 10.1158/0008-5472.CAN-06-2044 [DOI] [PubMed] [Google Scholar]
- Chanmee T., Ontong P., Kimata K., Itano N. (2015). Key roles of hyaluronan and its CD44 receptor in the stemness and survival of cancer stem cells. Front. Oncol. 5:180. 10.3389/fonc.2015.00180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B. G., Xu D. P., Lin A., Yan W. H. (2013). NK cytolysis is dependent on the proportion of HLA-G expression. Hum. Immunol. 74 286–289. 10.1016/j.humimm.2012.12.005 [DOI] [PubMed] [Google Scholar]
- Chen D., Forootan S. S., Gosney J. R., Forootan F. S., Ke Y. (2014). Increased expression of ID1 and ID3 promotes tumorigenicity by enhancing angiogenesis and suppressing apoptosis in small cell lung cancer. Genes Cancer 5 212–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H., Guo R., Zhang Q., Guo H., Yang M., Wu Z., et al. (2015). Erk signaling is indispensable for genomic stability and self-renewal of mouse embryonic stem cells. Proc. Natl. Acad. Sci. U.S.A. 112 E5936–E5943. 10.1073/pnas.1516319112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J. R., Cheng J. G., Shatzer T., Sewell L., Hernandez L., Stewart C. L. (2000). Leukemia inhibitory factor can substitute for nidatory estrogen and is essential for inducing a receptive uterus for implantation but is not essential for subsequent embryogenesis. Endocrinology 141 4365–4372. 10.1210/endo.141.12.7855 [DOI] [PubMed] [Google Scholar]
- Cheng C., Yaffe M. B., Sharp P. A. (2006). A positive feedback loop couples Ras activation and CD44 alternative splicing. Genes Dev. 20 1715–1720. 10.1101/gad.1430906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng J. G., Chen J. R., Hernandez L., Alvord W. G., Stewart C. L. (2001). Dual control of LIF expression and LIF receptor function regulate Stat3 activation at the onset of uterine receptivity and embryo implantation. Proc. Natl. Acad. Sci. U.S.A. 98 8680–8685. 10.1073/pnas.151180898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins A. T., Berry P. A., Hyde C., Stower M. J., Maitland N. J. (2005). Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res. 65 10946–10951. 10.1158/0008-5472.CAN-05-2018 [DOI] [PubMed] [Google Scholar]
- Conley S. J., Gheordunescu E., Kakarala P., Newman B., Korkaya H., Heath A. N., et al. (2012). Antiangiogenic agents increase breast cancer stem cells via the generation of tumor hypoxia. Proc. Natl. Acad. Sci. U.S.A. 109 2784–2789. 10.1073/pnas.1018866109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cubilio E., Diaz-Lopez A., Cuevas E. P., Moreno-Bueno G., Peinado H., Montes A., et al. (2013). E47 and ID1 interplay in epithelial-mesenchymal transition. PLoS One 8:e59948. 10.1371/journal.pone.00948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullinam E. B., Abbondanzo S. J., Anderson P. S., Pollard J. W., Lessey B. A., Stewart C. L. (1996). Leukemia inhibitory factor (LIF) and LIF receptor expression in human endometrium suggests a potential autocrine/paracrine function in regulating embryo implantation. Proc. Natl. Acad. Sci. U.S.A. 93 3115–3120. 10.1073/pnas.93.7.3115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Silva Nardi F., König L., Wagner B., Giebel B., Santos Manvailer L. F., Rebmann V. (2016). Soluble monomers, dimers and HLA-G-expressing extracellular vesicles: the three dimensions of structural complexity to use HLA-G as a clinical biomarker. HLA 88 77–86. 10.1111/tan.12844 [DOI] [PubMed] [Google Scholar]
- Deed R. W., Jasiok M., Norton J. D. (1996). Attenuated function of a variant form of the helix-loop-helix protein, Id-3, generated by an alternative splicing mechanism. FEBS Lett. 393 113–116. 10.1016/0014-5793(96)00868-X [DOI] [PubMed] [Google Scholar]
- Didelot C., Lanneau D., Brunet M., Joly A. L., De Thonel A., Chiosis G., et al. (2007). Anti-cancer therapeutic approaches based on intracellular and extracellular heat shock proteins. Curr. Med. Chem. 14 2839–2847. 10.2174/092986707782360079 [DOI] [PubMed] [Google Scholar]
- Dieter S. M., Ball C. R., Hoffmann C. M., Nowrouzi A., Herbst F., Zavidij O., et al. (2011). Distinct types of tumor-initiating cells form human colon cancer tumors and metastases. Cell Stem Cell 9 357–365. 10.1016/j.stem.2011.08.010 [DOI] [PubMed] [Google Scholar]
- Do D. V., Ueda J., Messerschmidt D. M., Lorthongpanich C., Zhou Y., Feng B., et al. (2013). A genetic and developmental pathway from STAT3 to the OCT4-NANOG circuit is essential for maintenance of ICM lineages in vivo. Genes Dev. 27 1378–1390. 10.1101/gad.221176.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong H. J., Jang G. B., Lee H. Y., Park S. R., Kim J. Y., Nam J. S., et al. (2016). The Wnt/β-catenin signaling/Id2 cascade mediates the effects of hypoxia on the hierarchy of colorectal-cancer stem cells. Sci. Rep. 6:22966. 10.1038/srep22966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dressel R., Johnson J. P., Günther E. (1998). Heterogeneous patterns of constitutive and heat shock induced expression of HLA-linked HSP70–1 and HSP70–2 heat shock genes in human melanoma cell lines. Melanoma Res. 8 482–492. 10.1097/00008390-199812000-00002 [DOI] [PubMed] [Google Scholar]
- Du L., Xiao X., Wang C., Zhang X., Zheng N., Wang L., et al. (2011). Human leukocyte antigen-G is closely associated with tumor immune escape in gastric cancer by increasing local regulatory T cells. Cancer Sci. 102 1272–1280. 10.1111/j.1349-7006.2011.01951.x [DOI] [PubMed] [Google Scholar]
- Eastham A. M., Spencer H., Soncin F., Ritson S., Merry C. L., Stern P. L. (2007). Epithelial-mesenchymal transition events during human embryonic stem cell differentiation. Cancer Res. 67 11254–11262. 10.1158/0008-5472.CAN-07-2253 [DOI] [PubMed] [Google Scholar]
- Eckert M. A., Lwin T. M., Chang A. T., Kim J., Danis E., Ohno-Machado L., et al. (2011). Twist1-induced invadopodia formation promotes tumor metastasis. Cancer Cell 19 372–386. 10.1016/j.ccr.2011.01.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estrov Z., Samal B., Lapushin R., Kellokumpu-Lehtinen P., Sahin A. A., Kurzrock R., et al. (1999). Leukemia inhibitory factor binds to human breast cancer cells and stimulates their proliferation. J. Interferon Cytokine Res. 15 905–913. 10.1089/jir.1995.15.905 [DOI] [PubMed] [Google Scholar]
- Ezashi T., Das P., Roberts R. M. (2005). Low O2 tensions and the prevention of differentiation of hES cells. Proc. Natl. Acad. Sci. U.S.A. 102 4783–4788. 10.1073/pnas.0501283102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Festuccia N., Osorno R., Wilson V., Chambers I. (2013). The role of pluripotency gene regulatory network components in mediating transitions between pluripotent cell states. Curr. Opin. Genet. Dev. 23 504–511. 10.1016/j.gde.2013.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong S., Debs R. J., Desprez P. Y. (2004). Id genes and proteins as promising targets in cancer therapy. Trends Mol. Med. 10 387–392. 10.1016/j.molmed.2004.06.008 [DOI] [PubMed] [Google Scholar]
- Fong S., Itahana Y., Sumida T., Singh J., Coppe J. P., Liu Y., et al. (2003). Id-1 as a molecular target in therapy for breast cancer cell invasion and metastasis. Proc. Natl. Acad. Sci. U.S.A. 100 13543–13548. 10.1073/pnas.2230238100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forrest S. T., Barringhaus K. G., Perlegas D., Hammarskjold M. L., McNamara C. A. (2004). Intron retention generates a novel Id3 isoform that inhibits vascular lesion formation. J. Biol. Chem. 279 32897–32903. 10.1074/jbc.M404882200 [DOI] [PubMed] [Google Scholar]
- Fujii I. (2009). Beyond antibodies: generation of conformationally constrained peptides for molecular-targeting therapy. Yakugaku Zasshi 129 1303–1309. 10.1248/yakushi.129.1303 [DOI] [PubMed] [Google Scholar]
- Fukushima Y., Oshika Y., Nakamura M., Tokunaga T., Hatanaka H., Abe Y., et al. (1998). Increased expression of human histocompatibility leukocyte antigen-G in colorectal cancer cells. Int. J. Mol. Med. 2 349–351. 10.3892/ijmm.2.3.349 [DOI] [PubMed] [Google Scholar]
- Fuzzi B., Rizzo R., Criscuoli L., Noci I., Melchiorri L., Scarselli B., et al. (2002). HLA-G expression in early embryos is a fundamental prerequisite for the obtainment of pregnancy. Eur. J. Immunol. 32 311–315. 10.1002/1521-4141(200202)32:2<311::AID-IMMU311>3.0.CO;2-8 [DOI] [PubMed] [Google Scholar]
- Gillich A., Bao S., Surani M. A. (2013). Reversion of mouse postimplantation epiblast stem cells to a naïve pluripotent state by modulation of signalling pathways. Methods Mol. Biol. 1074 15–29. 10.1007/978-1-62703-628-3-2 [DOI] [PubMed] [Google Scholar]
- Ginisa I., Luoa Y., Miuraa T., Thiesb S., Brandenbergerb R., Gerecht-Nirc S., et al. (2004). Differences between human and mouse embryonic stem cells. Dev. Biol. 269 360–380. 10.1016/j.ydbio.2003.12.034 [DOI] [PubMed] [Google Scholar]
- Godar S., Ince T. A., Bell G. W., Feldser D., Donaher J. L., Bergh G., et al. (2008). Growth-inhibitory and tumor-suppressive functions of p53 depend on its repression of CD44 expression. Cell 134 62–73. 10.1016/j.cell.2008.06.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grange C., Tapparo M., Tritta S., Deregibus M. C., Battaglia A., Gontero P., et al. (2015). Role of HLA-G and extracellular vesicles in renal cancer stem cell-induced inhibition of dendritic cell differentiation. BMC Cancer 15:1009. 10.1186/s12885-015-2025-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregori S., Amodio G., Quattrone F., Panina-Bordignon P. (2015). HLA-G orchestrates the early interaction of human trophoblasts with the maternal niche. Front. Immunol. 6:128. 10.3389/fimmu.2015.00128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gros F., Cabillic F., Toutirais O., Maux A. L., Sebti Y., Amiot L. (2008). Soluble HLA-G molecules impair natural killer/dendritic cell crosstalk via inhibition of dendritic cells. Eur. J. Immunol. 38 742–749. 10.1002/eji.200736918 [DOI] [PubMed] [Google Scholar]
- Guo H., Cheng Y., Martinka M., McElwee K. (2015). High LIFr expression stimulates melanoma cell migration and is associated with unfavorable prognosis in melanoma. Oncotarget 6 25484–25498. 10.18632/oncotarget.4688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta G. P., Perk J., Acharyya S., de Candia P., Vivek Mittal V., Todorova-Manova K., et al. (2007). ID genes mediate tumor reinitiation during breast cancer lung metastasis. Proc. Natl. Acad. Sci. U.S.A. 194 19506–19511. 10.1073/pnas.0709185104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- HaKashyap V., Rezende N. C., Scotland K. B., Shaffer S. M., Persson J. L., Gudas L. J., et al. (2009). Regulation of stem cell pluripotency and differentiation involves a mutual regulatory circuit of the NANOG, OCT4, and SOX2 pluripotency transcription factors with polycomb repressive complexes and stem cell micro-RNAs. Stem Cells Dev. 18 1093–1108. 10.1089/scd.2009.0113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heap C. J., Wang Y., Pinheiro T. J., Reading S. A., Jennings K. R., Dimmock N. J. (2005). Analysis of a 17-amino acid residue, virus-neutralizing microantibody. J. Gen. Virol. 86 1791–1800. 10.1099/vir.0.80812-0 [DOI] [PubMed] [Google Scholar]
- Henderson J. K., Draper J. S., Baillie H. S., Fishel S., Thomson J. A., Moore H., et al. (2002). Preimplantation human embryos and embryonic stem cells show comparable expression of stage-specific embryonic antigens. Stem Cells 20 329–337. 10.1634/stemcells.20-4-329 [DOI] [PubMed] [Google Scholar]
- Hermann P. C., Huber S. L., Herrler T., Aicher A., Ellwart J. W., Guba M., et al. (2007). Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell 1 313–323. 10.1016/j.stem.2007.06.002 [DOI] [PubMed] [Google Scholar]
- Hirata H., Sugimachi K., Takahashi Y., Ueda M., Sakimura S., Uchi R., et al. (2015). Downregulation of PRRX1 confers cancer stem cell-like properties and predicts poor prognosis in hepatocellular carcinoma. Ann. Surg. Oncol. 22(Suppl. 3) S1402–S1409. 10.1245/s10434-014-4242-0 [DOI] [PubMed] [Google Scholar]
- Hofmeister V., Weiss E. H. (2003). HLA-G modulates immune responses by diverse receptor interactions. Semin. Cancer Biol. 13 317–323. 10.1016/S1044-579X(03)00022-1 [DOI] [PubMed] [Google Scholar]
- Hunt J. S., Petroff M. G., McIntire R. H., Ober C. (2005). HLA-G and immune tolerance in pregnancy. FASEB J. 19 681–693. 10.1096/fj.04-2078rev [DOI] [PubMed] [Google Scholar]
- Iavarone A., Garg P., Lasorella A., Hsu J., Israel M. A. (1994). The helix-loop-helix protein Id-2 enhances cell proliferation and binds to the retinoblastoma protein. Genes Dev. 8 1270–1284. 10.1101/gad.8.11.1270 [DOI] [PubMed] [Google Scholar]
- Iavarone A., Lasorella A. (2006). ID proteins as targets in cancer and tools in neurobiology. Trends Mol. Med. 12 588–594. 10.1016/j.molmed.2006.10.007 [DOI] [PubMed] [Google Scholar]
- Ibrahim E. C., Morange M., Dausset J., Carosella E. D., Paul P. (2000). Heat shock and arsenite induce expression of the nonclassical class I histocompatibility HLA-G gene in tumor cell lines. Cell Stress Chaperones 5 207–218. 10.1379/1466-1268(2000)005<0207:HSAAIE>2.0.CO;2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itahana Y., Singh J., Sumida T., Coppe J. P., Parrinello S., Bennington J. L., et al. (2003). Role of Id-2 in the maintenance of a differentiated and noninvasive phenotype in breast cancer cells. Cancer Res. 63 7098–7105. [PubMed] [Google Scholar]
- Jang G. B., Kim J. Y., Cho S. D., Park K. S., Jung J. Y., Lee H. Y., et al. (2015). Blockade of Wnt/β-catenin signaling suppresses breast cancer metastasis by inhibiting CSC-like phenotype. Sci. Rep. 5:12465. 10.1038/srep12465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jego G., Hazoumè A., Seigneuric R., Garrido C. (2010). Targeting heat shock proteins in cancer. Cancer Lett 332 275–285. 10.1016/j.canlet.2010.10.014 [DOI] [PubMed] [Google Scholar]
- Jin L., Hope K. J., Zhai Q., Smadja-Joffe F., Dick J. E. (2006). Targeting of CD44 eradicates human acute myeloid leukemic stem cells. Nat. Med. 12 1167–1174. 10.1038/nm1483 [DOI] [PubMed] [Google Scholar]
- Johnson S., Chen H., Lo P. K. (2013). In vitro tumor-sphere formation assays. Bio.Protoc. 3:e325 10.21769/BioProtoc.325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamran M. Z., Patil P., Gude R. P. (2013). Role of STAT3 in cancer metastasis and advances. Biomed. Res. Int. 2013:421821. 10.1155/2013/421821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh M. (2007). Networking of WNT, FGF, Notch, BMP, and Hedgehog signaling pathways during carcinogenesis. Stem Cell Rev. 3 30–38. 10.1007/s12015-007-0006-6 [DOI] [PubMed] [Google Scholar]
- Kellokumpu-Lehtinen P., Talpaz M., Harris D., Van Q., Kurzrock R., Estrov Z. (1996). Leukemia inhibitory factor stimulates breast, kidney and prostate cancer cell proliferation by paracrine and autocrine pathways. Int. J. Cancer 66 515–519. 10.1002/(SICI)1097-0215(19960516)66:4<515::AID-IJC15>3.0.CO;2-6 [DOI] [PubMed] [Google Scholar]
- König L., Kasimir-Bauer S., Hoffmann O., Bittner A. K., Wagner B., Manvailer L. F., et al. (2016). The prognostic impact of soluble and vesicular HLA-G and its relationship to circulating tumor cells in neoadjuvant treated breast cancer patients. Hum. Immunol. 77 791–799. 10.1016/j.humimm.2016.01.002 [DOI] [PubMed] [Google Scholar]
- Kosaka N. (2016). Decoding the secret of cancer by means of extracellular vesicles. J. Clin. Med. 5:E22. 10.3390/jcm5020022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnamachary B., Penet M. F., Nimmagadda S., Mironchik Y., Raman V., Solaiyappan M., et al. (2012). Hypoxia regulates CD44 and its variant isoforms through HIF-1a in triple negative breast cancer. PLoS One 7:e44078. 10.1371/journal.pone.0044078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunath T., Saba-El-Leil M. K., Almousailleakh M., Wray J., Meloche S., Smith A. (2007). FGF stimulation of the Erk1/2 signalling cascade triggers transition of pluripotent embryonic stem cells from self-renewal to lineage commitment. Development 134 2895–2902. 10.1242/dev.02880 [DOI] [PubMed] [Google Scholar]
- Lasorella A., Benezra R., Iavarone A. (2014). The ID proteins: master regulators of cancer stem cells and tumour aggressiveness. Nat. Rev. Cancer 14 77–91. 10.1038/nrc3638 [DOI] [PubMed] [Google Scholar]
- Lesport E., Baudhuin J., LeMaoult J., Sousa S., Doliger C., Carosella E. D., et al. (2009). Human melanoma cells secreting human leukocyte antigen-G5 inhibit natural killer cell cytotoxicity by impairing lytic granules polarization toward target cell. Hum. Immunol. 70 1000–1005. 10.1016/j.humimm.2009.07.019 [DOI] [PubMed] [Google Scholar]
- Li L., Baroja M. L., Majumdar A., Chadwick K., Rouleau A., Gallacher L., et al. (2004). Human embryonic stem cells possess immune-privileged properties. Stem Cells 22 448–456. 10.1634/stemcells.22-4-448 [DOI] [PubMed] [Google Scholar]
- Li R., Liang J., Ni S., Zhou T., Qing X., Li H., et al. (2010). A mesenchymal-to-epithelial transition initiates and is required for the nuclear reprogramming of mouse fibroblasts. Cell Stem Cell 7 51–63. 10.1016/j.stem.2010.04.014 [DOI] [PubMed] [Google Scholar]
- Lin A., Yan W. H. (2015). Human leukocyte antigen-G (HLA-G) expression in cancers: roles in immune evasion, metastasis and target for therapy. Mol. Med. 21 782–791. 10.2119/molmed.2015.00083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin A., Yan W. H., Xu H. H., Gan M. F., Cai J. F., Zhu M., et al. (2007). HLA-G expression in human ovarian carcinoma counteracts NK cell function. Ann. Oncol. 18 1804–1809. 10.1093/annonc/mdm356 [DOI] [PubMed] [Google Scholar]
- Ling M. T., Wang X., Zhang X., Wong Y. C. (2006). The multiple roles of Id-1 in cancer progression. Differentiation 74 481–487. 10.1111/j.1432-0436.2006.00083.x [DOI] [PubMed] [Google Scholar]
- Liu N., Lu M., Tian X., Han Z. J. (2007). Molecular mechanisms involved in self-renewal and pluripotency of embryonic stem cells. Cell Physiol. 211 279–286. 10.1002/jcp.20978 [DOI] [PubMed] [Google Scholar]
- Liu S., Cong Y., Wang D., Sun Y., Deng L., Liu Y., et al. (2014). Breast cancer stem cells transition between epithelial and mesenchymal states reflective of their normal counterparts. Stem Cell Rep. 2 78–91. 10.1016/j.stemcr.2013.11.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopatina T., Gai C., Deregibus M. C., Kholia S., Camussi G. (2016). Cross talk between cancer and mesenchymal stem cells through extracellular vesicles carrying nucleic acids. Front. Oncol. 6:125. 10.3389/fonc.2016.00125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loumagne L., Baudhuin J., Favier B., Montespan F., Carosella E. D., Rouas-Freiss N. (2014). In vivo evidence that secretion of HLA-G by immunogenic tumor cells allows their evasion from immunosurveillance. Int. J. Cancer 135 2107–2117. 10.1002/ijc.28845 [DOI] [PubMed] [Google Scholar]
- Luft J. C., Dix D. J. (1999). HSP70 expression and function during embryogenesis. Cell Stress Chaperones 4 162–170. 10.1379/1466-1268(1999)004<0162:HEAFDE>2.3.CO;2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyden D., Young A. Z., Zagzag D., Yan W., Gerald W., O’Reilly R., et al. (1999). Id1 and Id3 are required for neurogenesis, angiogenesis and vascularization of tumor xenografts. Nature 401 670–677. 10.1038/44334 [DOI] [PubMed] [Google Scholar]
- Manrique I., Nguewa P., Bleau A. M., Nistal-Villan E., Lopez I., Villalba M., et al. (2015). The inhibitor of differentiation isoform Id1b, generated by alternative splicing, maintains cell quiescence and confers self-renewal and cancer stem cell-like properties. Cancer Lett. 356 899–909. 10.1016/j.canlet.2014.10.035 [DOI] [PubMed] [Google Scholar]
- Manzo G. (1989). Phylogenesis–ontogenesis–oncogenesis. Med. Hypotheses 30 245–257. 10.1016/0306-9877(89)90033-9 [DOI] [PubMed] [Google Scholar]
- Marangoni E., Lecomte N., Durand L., de Pinieux G., Decaudin D., Chomienne C., et al. (2009). CD44 targeting reduces tumour growth and prevents post-chemotherapy relapse of human breast cancers xenografts. Br. J. Cancer 100 918–992. 10.1038/sj.bjc.6604953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marjanovic N. D., Weinberg R. A., Chaffer C. L. (2013). Cell plasticity and heterogeneity in cancer. Clin. Chem. 59 168–179. 10.1373/clinchem.2012.184655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menendez S., Camus S., Izpisua Belmonte J. C. (2010). p53: guardian of reprogramming. Cell Cycle 9 3887–3891. 10.4161/cc.9.19.13301 [DOI] [PubMed] [Google Scholar]
- Mern D. S., Hasskarl J., Burwinkel B. (2010a). Inhibition of Id proteins by a peptide aptamer induces cell-cycle arrest and apoptosis in ovarian cancer cells. Br. J. Cancer 103 1237–1244. 10.1038/sj.bjc.6605897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mern D. S., Hoppe-Seyler K., Hoppe-Seyler F., Hasskarl J., Burwinkel B. (2010b). Targeting Id1 and Id3 by a specific peptide aptamer induces E-box promoter activity, cell cycle arrest, and apoptosis in breast cancer cells. Breast Cancer Res. Treat. 124 623–633. 10.1007/s10549-010-0810-6 [DOI] [PubMed] [Google Scholar]
- Miyanari Y., Torres-Padilla M. E. (2012). Control of ground-state pluripotency by allelic regulation of Nanog. Nature 483 470–473. 10.1038/nature10807 [DOI] [PubMed] [Google Scholar]
- Mohyeldin A., Garzon-Muvdi T., Quinones-Hinojosa A. (2010). Oxygen in stem cell biology: a critical component of the stem cell niche. Cell Stem Cell 7 150–161. 10.1016/j.stem.2010.07.007 [DOI] [PubMed] [Google Scholar]
- Moreau P., Adrian-Cabestre F., Menier C., Guiard V., Gourand L., Dausset J., et al. (1999). IL-10 selectively induces HLA-G expression in human trophoblasts and monocytes. Int. Immunol. 11 803–811. 10.1093/intimm/11.5.803 [DOI] [PubMed] [Google Scholar]
- Murase R., Sumida T., Kawamura R., Onishi-Ishikawa A., Hamakawa H., McAllister S. D., et al. (2016). Suppression of invasion and metastasis in aggressive salivary cancer cells through targeted inhibition of ID1 gene expression. Cancer Lett. 377 11–16. 10.1016/j.canlet.2016.04.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair R., Teo W. S., Mittal V., Swarbrik A. (2014). ID proteins regulate diverse aspects of cancer progression and provide novel therapeutic opportunities. Mol. Ther. 22 1407–1415. 10.1038/mt.2014.83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naito Y., Yoshioka Y., Yamamoto Y., Ochiya T. (2017). How cancer cells dictate their microenvironment: present roles of extracellular vesicles. Cell. Mol. Life Sci. 74 697–713. 10.1007/s00018-016-2346-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuer A., Mele C., Liu H. C., Rosenwaks Z., Witkin S. S. (1998). Monoclonal antibodies to mammalian heat shock proteins impair mouse embryo development in vitro. Hum. Reprod. 3 987–990. 10.1093/humrep/13.4.987 [DOI] [PubMed] [Google Scholar]
- Nguewa P., Manrique I., Diaz R., Redrado M., Parrondo R., Perez-Stable C., et al. (2014). Id-1B, an alternatively spliced isoform of the inhibitor of differentiation-1, impairs cancer cell malignancy through inhibition of proliferation and angiogenesis. Curr. Mol. Med. 14 151–162. 10.2174/1566524013666131203100643 [DOI] [PubMed] [Google Scholar]
- Nichols J., Smith A. (2009). Naive and primed pluripotent states. Cell Stem Cell 4 487–492. 10.1016/j.stem.2009.05.015 [DOI] [PubMed] [Google Scholar]
- Ning N., Pan Q., Zheng F., Teitz-Tennenbaum S., Egenti M., Yet J., et al. (2012). Cancer stem cell vaccination confers significant antitumor immunity. Cancer Res. 72 1853–1864. 10.1158/0008-5472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niola F., Zhao X., Singh D., Castano A., Sullivan R., Lauria M., et al. (2012). Id proteins synchronize stemness and anchorage to the niche of neural stem cells. Nat. Cell Biol. 14 477–487. 10.1038/ncb2490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura K., Semba S., Aoyagi K., Sasaki H., Yokozaki H. (2012). Mesenchymal stem cells provide an advantageous tumor microenvironment for the restoration of cancer stem cells. Pathobiology 79 290–306. 10.1159/000337296 [DOI] [PubMed] [Google Scholar]
- O’Brien C. A., Kreso A., Ryan P., Hermans K. G., Gibson L., Wang Y., et al. (2012). ID1 and ID3 regulate the self-renewal capacity of human colon cancer-initiating cells through p21. Cancer Cell 21 777–792. 10.1016/j.ccr.2012.04.036 [DOI] [PubMed] [Google Scholar]
- Ocaña O. H., Córcoles R., Fabra A., Moreno-Bueno G., Acloque H., Vega S., et al. (2012). Metastatic colonization requires the repression of the epithelial-mesenchymal transition inducer PRRX1. Cancer Cell 22 709–724. 10.1016/j.ccr.2012.10.012 [DOI] [PubMed] [Google Scholar]
- Okamoto I., Kawano Y., Tsuiki H., Sasaki J., Nakao M., Matsumoto M., et al. (1999). CD44 cleavage induced by a membrane-associated metalloprotease plays a critical role in tumor cell migration. Oncogene 18 1435–1446. 10.1038/sj.onc.1202447 [DOI] [PubMed] [Google Scholar]
- Okita K., Yamanaka S. (2006). Intracellular signaling pathways regulating pluripotency of embryonic stem cells. Curr. Stem Cell Res. Ther. 1 103–111. 10.2174/157488806775269061 [DOI] [PubMed] [Google Scholar]
- Orian-Rousseau V., Ponta H. (2015). Perspectives of CD44 targeting therapies. Arch. Toxicol. 89 3–14. 10.1007/s00204-014-1424-2 [DOI] [PubMed] [Google Scholar]
- Ouyang X. S., Wang X., Ling M. T., Wong H. L., Tsao S. W., Wong Y. C. (2002). Id-1 stimulates serum independent prostate cancer cell proliferation through inactivation of p16(INK4a)/pRB pathway. Carcinogenesis 23 721–725. 10.1093/carcin/23.5.721 [DOI] [PubMed] [Google Scholar]
- Pan Q., Li Q., Liu S., Ning N., Zhang X., Xu Y., et al. (2015). Concise review: targeting cancer stem cells using immunological approaches. Stem Cells 33 2085–2092. 10.1002/stem.2039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pankaj S., Sudha Saryu M., Satish Kumar G. (2013). LIF-STAT signaling and trophoblast biology. JAKSTAT 2:e25155. 10.4161/jkst.25155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paolella B. R., Havrda M. C., Mantani A., Wray C. M., Zhang Z., Israel M. A. (2011). p53 directly represses Id2 to inhibit the proliferation of neural progenitor cells. Stem Cells 29 1090–1101. 10.1002/stem.660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H. J., Hong M., Bronson R. T., Israel M. A., Frankel W. N., Yun K. (2013). Elevated Id2 expression results in precocious neural stem cell depletion and abnormal brain development. Stem Cells 31 1010–1021. 10.1002/stem.1351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J. I., Strock C. J., Ball D. W., Nelkin B. D. (2003). The Ras/Raf/MEK/extracellular signal-regulated kinase pathway induces autocrine-paracrine growth inhibition via the leukemia inhibitory factor/JAK/STAT pathway. Mol. Cell Biol. 23 543–554. 10.1128/MCB.23.2.543-554.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel D., Morton D. J., Carey J., Havrda M. C., Chaudhary J. (2015). Inhibitor of differentiation 4 (ID4): from development to cancer. Biochim. Biophys. Acta 1855 92–103. 10.1016/j.bbcan.2014.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham P. V., Phan N. L., Nguyen N. T., Truong N. H., Duong T. T., Le D. V., et al. (2011). Differentiation of breast cancer stem cells by knockdown of CD44: promising differentiation therapy. Transl. Med. 9:209. 10.1186/1479-5876-9-209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietras A., Katz A. M., Ekstrom E. J., Wee B., Halliday J. J., Pitter K. L., et al. (2014). Osteopontin-CD44 signaling in the glioma perivascular niche enhances cancer stem cell phenotypes and promotes aggressive tumor growth. Cell Stem Cell 14 357–369. 10.1016/j.stem.2014.01.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plaks V., Kong N., Werb Z. (2015). The cancer stem cell niche: how essential is the niche in regulating stemness of tumor cells? Cell Stem Cell 16 225–238. 10.1016/j.stem.2015.02.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prabhu S., Ignatova A., Park S. T., Sun X. H. (1997). Regulation of the expression of cyclin-dependent kinase inhibitor p21 by E2A and ID proteins. Mol. Cell Biol. 17 5888–5896. 10.1128/MCB.17.10.5888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quail D. F., Taylor M. J., Postovit L. M. (2012). Microenvironmental regulation of cancer stem cell phenotypes. Curr. Stem Cell Res. Ther. 7 197–216. 10.2174/157488812799859838 [DOI] [PubMed] [Google Scholar]
- Rahme G. J., Israel M. A. (2015). Id4 suppresses MMP2-mediated invasion of glioblastoma-derived cells by direct inactivation of Twist1 function. Oncogene 34 53–62. 10.1038/onc.2013.531 [DOI] [PubMed] [Google Scholar]
- Rebmann V., König L., Nardi Fda S., Wagner B., Manvailer L. F., Horn P. A. (2016). The potential of HLA-G-bearing extracellular vesicles as a future element in HLA-G immune biology. Front. Immunol. 7:173. 10.3389/fimmu.2016.00173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebmann V., Regel J., Stolke D., Grosse-Wilde H. (2003). Secretion of sHLA-G molecules in malignancies. Semin. Cancer Biol. 13 371–377. 10.1016/S1044-579X(03)00028-2 [DOI] [PubMed] [Google Scholar]
- Rockman S. P., Currie S. A., Ciavarella M., Vincan E., Dow C., Thomas R. J., et al. (2001). Id2 is a target of the beta-catenin/T cell factor pathway in colon carcinoma. J. Biol. Chem. 276 45113–45119. 10.1074/jbc.M107742200 [DOI] [PubMed] [Google Scholar]
- Roesch A., Fukunaga-Kalabis M., Schmidt E. C., Zabierowski S. E., Brafford P. A., Vultur A., et al. (2010). A temporarily distinct subpopulation of slow-cycling melanoma cells is required for continuous tumor growth. Cell 141 583–594. 10.1016/j.cell.2010.04.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouas-Freiss N., Moreau P., Menier C., Carosella E. D. (2003). HLA-G in cancer: a way to turn off the immune system. Semin. Cancer Biol. 13 325–336. 10.1016/S1044-579X(03)00023-3 [DOI] [PubMed] [Google Scholar]
- Rouas-Freiss N., Moreau P., Menier C., LeMaoult J., Carosella E. D. (2007). Expression of tolerogenic HLA-G molecules in cancer prevents antitumor responses. Semin. Cancer Biol. 17 413–421. 10.1016/j.semcancer.2007.07.003 [DOI] [PubMed] [Google Scholar]
- Ruzinova M. B., Benezra R. (2003). Id proteins in development, cell cycle and cancer. Trends Cell Biol. 13 410–418. 10.1016/S0962-8924(03)00147-8 [DOI] [PubMed] [Google Scholar]
- Sahlgren C., Gustafsson M. V., Jin S., Poellinger L., Lendahl U. (2008). Notch signaling mediates hypoxia-induced tumor cell migration and invasion. Proc. Natl. Acad. Sci. U.S.A. 105 6392–6397. 10.1073/pnas.0802047105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samali A., Cotter T. G. (1996). Heat shock proteins increase resistance to apoptosis. Exp. Cell Res. 223 163–170. 10.1006/excr.1996.0070 [DOI] [PubMed] [Google Scholar]
- Samavarchi-Tehrani P., Golipour A., David L., Sung H. K., Beyer T. A., Datti A., et al. (2010). Functional genomics reveals a BMP-driven mesenchymal-to-epithelial transition in the initiation of somatic cell reprogramming. Cell Stem Cell 7 64–77. 10.1016/j.stem.2010.04.015 [DOI] [PubMed] [Google Scholar]
- Sato N., Meijer L., Skaltsounis L., Greengard P., Brivanlou A. H. (2004). Maintenance of pluripotency in human and mouse embryonic stem cells through activation of Wnt signaling by a pharmacological GSK-3-specific inhibitor. Nat. Med. 10 55–63. 10.1038/nm979 [DOI] [PubMed] [Google Scholar]
- Scheel C., Eaton N. E., Li S. H. J., Chaffer C. L., Reinhardt F., Kah K. J., et al. (2011). Paracrine and autocrine signals induce and maintain mesenchymal and stem cell states in the breast. Cell 145 926–940. 10.1016/j.cell.2011.04.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwitalla S., Fingerle A. A., Cammareri P., Nebelsiek T., Goktuna S. I., Ziegler P. K., et al. (2013). Intestinal tumorigenesis initiated by dedifferentiation and acquisition of stem-cell-like properties. Cell 152 25–38. 10.1016/j.cell.2012.12.012 [DOI] [PubMed] [Google Scholar]
- Sharma P., Chinaranagari S., Chaudhary J. (2015). Inhibitor of differentiation 4 (ID4) acts as an inhibitor of ID-1, -2 and -3 and promotes basic helix loop helix (bHLH) E47 DNA binding and transcriptional activity. Biochimie 112 139–150. 10.1016/j.bochi.2015.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherwin J. R. A., Smith S. K., Wilson A., Sharkey A. M. (2002). Soluble gp130 is up-regulated in the implantation window and shows altered secretion in patients with primary unexplained infertility. J. Clin. Endocrinol. Metab. 87 3953–3960. 10.1210/jcem.87.8.8766 [DOI] [PubMed] [Google Scholar]
- Sheu J., Shih I. E. (2010). HLA-G and immune evasion in cancer cells. Med. Assoc. 109 248–257. 10.1016/S0929-6646(10)60050-2 [DOI] [PubMed] [Google Scholar]
- Shu C. W., Huang C. M. (2008). HSP70s: from tumor transformation to cancer therapy. Clin. Med. Oncol. 2 335–345. 10.4137/CMO.S475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuno Y., Tsuno N. H., Okaji Y., Tsuchiya T., Sakurai D., Nishikawa T., et al. (2010). ID1/ID3 knockdown inhibits metastatic potential of pancreatic cancer. J. Surg. Res. 161 76–82. 10.1016/j.jss.2008.10.o31 [DOI] [PubMed] [Google Scholar]
- Silva J., Barrandon O., Nichols J., Kawaguchi J., Theunissen T. W., Smith A., et al. (2008). Promotion of reprogramming to ground state pluripotency by signal inhibition. PLoS Biol. 6:e253. 10.1371/journal.pbio.0060253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stangl S., Gehrmann M., Riegger J., Kuhs K., Riederer I., Sievert W., et al. (2011). Targeting membrane heat-shock protein 70 (HSP70) on tumors by cmHSP70.1 antibody. Proc. Natl. Acad. Sci. U.S.A. 108 733–738. 10.1073/pnas.1016065108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stangl S., Gross C., Pokley A. G., Asea A. A., Multhoff G. (2008). Influence of HSP70 and HLA-E on the killing of leukemic blasts by cytokine/HSP70 peptide activated human natural killer (NK) cells. Cell Stress Chaperones 13 221–230. 10.1007/s12192-007-0008-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stankic M., Pavlovic S., Chin Y., Brogi E., Padua D., Norton L., et al. (2013). TGF-β-Id1 signaling opposes Twist1 and promotes metastatic colonization via a mesenchymal-to-epithelial transition. Cell Rep. 5 1228–1242. 10.1016/j.celrep.2013.11.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart C. L., Kaspar P., Brunet L. J., Bhatt H., Gadi I., Kontgen F., et al. (1992). Blastocyst implantation depends on maternal expression of leukemia inhibitory factor. Nature 359 76–79. 10.1038/359076a0 [DOI] [PubMed] [Google Scholar]
- Su Y. J., Lai H. M., Chang Y. W., Chen J. Y., Lee J. L. (2011). Direct reprogramming of stem cell properties in colon cancer cells by CD44. EMBO J. 30 3186–3199. 10.1038/emboj.2011.211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura Y., Sugimoto M., Ohnishi K., Sakai T., Hara E. (1998). Differential activity of a variant form of the human Id-1 protein generated by alternative splicing. FEBS Lett. 436 169–173. 10.1016/S0014-5793(98)01117-X [DOI] [PubMed] [Google Scholar]
- Tapia N., Schöler H. R. (2010). P53 connects tumorigenesis and reprogramming to pluripotency. J. Exp. Med. 207 2045–2048. 10.1084/jem.20101866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teo W. S., Nair R., Swarbrick A. (2014). New insights into the role of ID proteins in breast cancer metastasis: a MET affair. Breast Cancer Res. 16 305. 10.1186/bcr3654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiery J. P., Acloque H., Huang R. J. Y., Nieto M. A. (2009). Epithelial-mesenchymal transitions in development and disease. Cell 139 871–890. 10.1016/j.cell.2009.11.007 [DOI] [PubMed] [Google Scholar]
- Tilburgs T., Evans J. H., Crespo A. C., Strominger J. L. (2015). The HLA-G cycle provides for both NK tolerance and immunity at the maternal-fetal interface. Proc. Natl. Acad. Sci. U.S.A. 112 13312–13317. 10.1073/pnas.1517724112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiwari N., Gheldof A., Tatari M., Christofori G. (2012). EMT as the ultimate survival mechanism of cancer cells. Semin. Cancer Biol. 22 194–207. 10.1016/j.semcancer.2012.02.013 [DOI] [PubMed] [Google Scholar]
- Tran D. D., Corsa C. A., Biswas H., Aft R. L., Longmore G. D. (2011). Temporal and spatial cooperation of snail1 and twist1 during epithelial-mesenchymal transition predicts for human breast cancer. Mol. Cancer Res. 9 1644–1657. 10.1158/1541-7786.MCR-11-0371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuchiya T., Okaji Y., Tsuno N. H., Sakurai D., Tsuchiya N., Kawai K., et al. (2005). Targeting ID1 and ID3 inhibits peritoneal metastasis of gastric cancer. Cancer Sci. 96 784–790. 10.1111/j.1349-7006.2005.00113.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullmann U., In’t Veld P., Gilles C., Sermon K., De Rycke M., Van de Velde H., et al. (2006). Epithelial-mesenchymal transition process in human embryonic stem cells cultured in feeder-free conditions. Mol. Hum. Reprod. 13 21–32. 10.1093/molehr/gal091 [DOI] [PubMed] [Google Scholar]
- Umetani N., Takeuchi H., Fujimoto A., Shinozaki M., Bilchik A. J., Hoon D. S. B. (2004). Epigenetic inactivation of ID4 in colorectal carcinomas correlates with poor differentiation and unfavorable prognosis. Clin. Cancer Res. 10 7475–7483. 10.1158/1078-0432.CCR-04-0689 [DOI] [PubMed] [Google Scholar]
- Urosevic M., Dummer R. (2003). HLA-G and IL-10 expression in human cancer–different stories with the same message. Semin. Cancer Biol. 13 337–342. 10.1016/S1044-579X(03)00024-5 [DOI] [PubMed] [Google Scholar]
- Urosevic M., Kurrer M. O., Kamarashev J., Mueller B., Weder W., Burg G., et al. (2001). Human leukocyte antigen G up-regulation in lung cancer associates with high-grade histology, human leukocyte antigen class I loss and interleukin-10 production. Am. J. Pathol. 159 817–824. 10.1016/S0002-9440(10)61756-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urosevic M., Willers J., Mueller B., Kempf W., Burg G., Dummer R. (2002). HLA-G protein up-regulation in primary cutaneous lymphomas is associated with interleukin-10 expression in large cell T-cell lymphomas and indolent B-cell lymphomas. Blood 99 609–617. 10.1182/blood.V99.2.609 [DOI] [PubMed] [Google Scholar]
- Verloes A., Van de Velde H., LeMaoult J., Mateizel I., Cauffmann G., Horn P. A., et al. (2011). HLA-G expression in human embryonic stem cells and preimplantation embryos. J. Immunol. 186 2663–2671. 10.4049/jimmunol.1001081 [DOI] [PubMed] [Google Scholar]
- Wakamatsu Y., Sakamoto N., Oo H. Z., Naito Y., Uraoka N., Anami K., et al. (2012). Expression of cancer stem cell markers ALDH1, CD44 and CD133 in primary tumor and lymph node metastasis of gastric cancer. Pathol. Int. 62 112–119. 10.1111/j.1440-1827.2011.02760.x [DOI] [PubMed] [Google Scholar]
- Wallach-Dayan S. B., Rubinstein A. M., Hand C., Breuer R., Naor D. (2008). DNA vaccination with CD44 variant isoform reduces mammary tumor local growth and lung metastasis. Mol. Cancer Ther. 7 1615–1623. 10.1158/1535-7163.MCT-07-2383 [DOI] [PubMed] [Google Scholar]
- Wang L. H., Baker N. E. (2015). E proteins and ID proteins: helix-loop-helix partners in development and disease. Dev. Cell 35 269–280. 10.1016/j.devcel.2015.10.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T., Narayanaswamy R., Ren H., Torchilin V. P. (2016). Combination therapy targeting both cancer stem-like cells and bulk tumor cells for improved efficacy of breast cancer treatment. Cancer Biol. Ther. 17 698–707. 10.1080/15384047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z., Oron E., Nelson B., Razis S., Ivanova N. (2012). Distinct lineage specification roles for NANOG, OCT4, and SOX2 in human embryonic stem cells. Cell 10 440–454. 10.1016/j.stem.2012.02.016 [DOI] [PubMed] [Google Scholar]
- Wen J., Park J. Y., Park K. H., Chung H. W., Bang S., Park S. W., et al. (2010). Oct4 and Nanog expression is associated with early stages of pancreatic carcinogenesis. Pancreas 39 622–626. 10.1097/MPA.0b013e3181c75f5e [DOI] [PubMed] [Google Scholar]
- Williams K., Motiani K., Giridhar P. V., Kasper S. (2013). CD44 integrates signaling in normal stem cell, cancer stem cell and (pre)metastatic niches. Exp. Biol. Med. 238 324–338. 10.1177/1535370213480714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong Y. C., Zhang X. M., Ling M. T., Wang X. H. (2008). Inactivation of ID-1 gene induces sensitivity of prostate cancer cells to chemotherapeutic drugs. Adv. Exp. Med. Biol. 617 565–572. 10.1007/978-0-387-69080-3_58 [DOI] [PubMed] [Google Scholar]
- Xiao L., Yuan X., Sharkis S. J. (2006). Activin A maintains self-renewal and regulates fibroblast growth factor, Wnt, and bone morphogenic protein pathways in human embryonic stem cells. Stem Cells 24 1476–1486. 10.1634/stemcells.2005-0299 [DOI] [PubMed] [Google Scholar]
- Xu R. H., Chen X., Li D. S., Li R., Addicks G. C., Glennon C., et al. (2002). BMP4 initiates human embryonic stem cell differentiation to trophoblast. Nat. Biotechnol. 20 1261–1264. 10.1038/nbt761 [DOI] [PubMed] [Google Scholar]
- Yamanaka Y., Lanner F., Rossant J. (2010). FGF signal-dependent segregation of primitive endoderm and epiblast in the mouse blastocyst. Development 137 715–724. 10.1242/dev.043471 [DOI] [PubMed] [Google Scholar]
- Yan W. H., Lin A. F., Chang C. C., Ferrone S. (2005). Induction of HLA-G expression in a melanoma cell line OCM-1A following the treatment with 5-aza-2’-deoxycytidine. Cell Res. 15 523–531. 10.1038/sj.cr.7290376 [DOI] [PubMed] [Google Scholar]
- Yan Y., Zuo X., Wei D. (2015). Concise review: emerging role of CD44 in cancer stem cells: a promising biomarker and therapeutic target. Stem Cells Transl. Med. 4 1033–1043. 10.5966/sctm.0048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M. H., Wu M. Z., Chiou S. H., Chen P. M., Chang S. Y., Liu C. J., et al. (2008). Direct regulation of TWIST by HIF-1alpha promotes metastasis. Nat. Cell Biol. 10 295–305. 10.1038/ncb1691 [DOI] [PubMed] [Google Scholar]
- Yao Y. Q., Barlow D. H., Sargent I. L. (2005). Differential expression of alternatively spliced transcripts of HLA-G in human preimplantation embryos and inner cell masses. J. Immunol. 175 8379–8385. 10.4049/jimmunol.175.12.8379 [DOI] [PubMed] [Google Scholar]
- Yen B. L., Chang C. J., Liu K. J., Chen Y. C., Hu H. I., Bai C. H., et al. (2009). Human embryonic stem cell-derived mesenchymal progenitors possess strong immunosuppressive effects toward natural killer cells as well as T lymphocytes. Stem Cells 27 451–456. 10.1634/stemcells.2008-0390 [DOI] [PubMed] [Google Scholar]
- Yin X., Zhang B. H., Zheng S. S., Gao D. M., Qiu S. J., Wu W. Z., et al. (2015). Coexpression of gene Oct4 and Nanog initiates stem cell characteristics in hepatocellular carcinoma and promotes epithelial-mesenchymal transition through activation of Stat3/Snail signaling. J. Hematol. Oncol. 8:23. 10.1186/s13045-015-0119-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying Q. L., Nichols J., Chambers I., Smith A. (2003). BMP induction of Id proteins suppresses differentiation and sustains embryonic stem cell self-renewal in collaboration with STAT3. Cell 115 281–292. 10.1016/S0092-8674(03)00847-X [DOI] [PubMed] [Google Scholar]
- Yue X., Wu L., Hu W. (2015). The regulation of leukemia inhibitory factor. Cancer Cell Microenviron. 2:e877. 10.14800/ccm.877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun K., Mantani A., Garel S., Rubenstein J., Israel M. A. (2004). Id4 regulates neural progenitor proliferation and differentiation in vivo. Development 131 5441–5448. 10.1242/dev.01430 [DOI] [PubMed] [Google Scholar]
- Zaiden M., Feinshtein V., David A. (2017). Inhibition of CD44v3 and CD44v6 function blocks tumor invasion and metastatic colonization. J. Control Release 257 10–20. 10.1016/j.conrel.2017.01.021 [DOI] [PubMed] [Google Scholar]
- Zeilstra J., Joosten S. P., van Andel H., Tolg C., Berns A., Snoek M., et al. (2014). Stem cell CD44v isoforms promote intestinal cancer formation in Apc (min) mice downstream of WNT signaling. Oncogene 33 665–670. 10.1038/onc.2012.611 [DOI] [PubMed] [Google Scholar]
- Zhang W. Q., Xu D. P., Liu D., Li Y. Y., Ruan Y. Y., Lin A., et al. (2014). HLA-G1 and HLA-G5 isoforms have an additive effect on NK cytolysis. Hum. Immunol. 75 182–189. 10.1016/j.humimm.2013.11.001 [DOI] [PubMed] [Google Scholar]
- Zhang Y., Yu S., Han Y., Wang Y., Sun Y. (2018). Human leukocyte antigen-G expression and polymorphisms promote cancer development and guide cancer diagnosis/treatment. Oncol. Lett. 15 699–709. 10.3892/ol.2017.7407 [DOI] [PMC free article] [PubMed] [Google Scholar]