

Abstract
The choanoflagellate Salpingoeca rosetta contains a chimeric rhodopsin protein composed of an N-terminal rhodopsin (Rh) domain and a C-terminal cyclic nucleotide phosphodiesterase (PDE) domain. The Rh-PDE enzyme light-dependently decreases the concentrations of cyclic nucleotides such as cGMP and cAMP. Photoexcitation of purified full-length Rh-PDE yields an “M” intermediate with a deprotonated Schiff base, and its recovery is much faster than that of the enzyme domain. To gain structural and mechanistic insights into the Rh domain, here we expressed and purified the transmembrane domain of Rh-PDE, Rh-PDE(TMD), and analyzed it with transient absorption, light-induced difference UV-visible, and FTIR spectroscopy methods. These analyses revealed that the “K” intermediate forms within 0.005 ms and converts into the M intermediate with a time constant of 4 ms, with the latter returning to the original state within 4 s. FTIR spectroscopy revealed that all-trans to 13-cis photoisomerization occurs as the primary event during which chromophore distortion is located at the middle of the polyene chain, allowing the Schiff base to form a stronger hydrogen bond. We also noted that the peptide backbone of the α-helix becomes deformed upon M intermediate formation. Results from site-directed mutagenesis suggested that Glu-164 is protonated and that Asp-292 acts as the only Schiff base counterion in Rh-PDE. A strong reduction of enzymatic activity in a D292N variant, but not in an E164Q variant, indicated an important catalytic role of the negative charge at Asp-292. Our findings provide further mechanistic insights into rhodopsin-mediated, light-dependent regulation of second-messenger levels in eukaryotic microbes.
Keywords: phosphodiesterase, spectroscopy, photoreceptor, proton transport, structure-function, rhodopsin, microbial rhodopsin, photoisomerization, proton transfer, structural change, optogenetics, light-activated enzyme, chromophore
Introduction
Rhodopsins are heptahelical membrane proteins that contain retinal as chromophore (1–5). Light-activated enzyme activity is a relatively new function of microbial rhodopsins compared with other functions, such as light-driven cation and anion pumps, light-gated cation and anion channels, positive and negative phototaxis sensors, and photochromic sensors. The first discovered light-activated enzyme was an algal histidine kinase (6), whereas a fungal light-activated guanylyl cyclase (GC)2 was also reported (7–9). These proteins are composed of a membrane-embedded rhodopsin domain and a C-terminal cytoplasmic enzyme domain that are activated when light is absorbed by the all-trans-retinal chromophore.
Light-activated enzyme rhodopsins attract much attention in the field of optogenetics. Whereas the early stages of rhodopsin-based optogenetics required rapid temporal resolution as well as light-gated channels and light-driven ion pumps to excite and silence neurons, respectively (10, 11), optical control of intracellular signaling processes is in high demand for a wider spectrum of biological functions. In particular, there is acute demand for optogenetic control of secondary messengers such as cyclic nucleotides (12, 13). Photoactivated adenylyl cyclase with a FAD chromophore was discovered in Euglena gracilis in 2002 (14), and a microbial rhodopsin containing a GC domain was discovered in Blastocladiella emersonii (7). In contrast to these light-activated proteins that are able to increase cAMP and cGMP concentrations, an efficient native light-dependent enzyme capable of lowering the concentration of cyclic nucleotides was unknown until recently.
In 2017, we reported a novel type of microbial rhodopsin with a C-terminal PDE domain (15). This protein, Rh-PDE, was found in the choanoflagellate Salpingoeca rosetta, a unicellular and colonial single flagellate eukaryote that is considered to be one of the closest living relatives of metazoans. We successfully expressed and purified the full-length Rh-PDE from mammalian cells, and its enzymatic functions were studied in cells, membranes, and detergent, both biochemically and spectroscopically. Rh-PDE exhibits light-dependent hydrolysis activity toward both cGMP and cAMP, which is an unusual promiscuity, as normally the binding sites are very specific for one or the other cyclic nucleotide. A putative molecular mechanism to explain Rh-PDE activation was proposed based on a series of studies that examined the full-length protein. Two recent papers revealed that Rh-PDE possesses eight transmembrane helices, not seven, such that both the N and C terminus face the cytoplasmic side (16, 17). In addition, the crystal structure of the PDE domain was determined (16), and a better construct than the WT for optogenetic application was reported (17). The light-induced mechanism of activation of Rh-PDE enzymatic activity is particularly intriguing, and to assess it, in vivo and in vitro studies have been extensively initiated. On the other hand, detailed structural and mechanistic studies require large amounts of purified sample, which is not easy for full-length proteins, in this case Rh-PDE(full).
In this study, to gain structural and mechanistic information about the Rh domain, we expressed and purified the transmembrane domain of Rh-PDE and Rh-PDE(TMD) and applied various spectroscopic methods such as transient absorption, UV-visible, and FTIR spectroscopy. We observed the K and M intermediates during the Rh-PDE(TMD) photocycle, and light-induced difference FTIR spectroscopy provided information on the structural dynamics of Rh-PDE(TMD). Phylogenetic analysis showed that Rh-PDE is homologous to other enzyme rhodopsins and channelrhodopsins such as cation channelrhodopsin 2 from Chlamydomonas reinhardtii (CrCCR2) (Fig. S1; sequential comparison with other microbial rhodopsins is shown in Fig. S2). In fact, the observed structural dynamics of Rh-PDE(TMD) resemble those of CrCCR2 and a chimera of CrCCR1 and CrCCR2, CrCCR(C1C2). On the other hand, CCR opens the channel, whereas Rh-PDE activates the enzymatic PDE domain. The mechanism of Rh-PDE activation will be discussed based on the present spectroscopic observations.
Results
Transient absorption study of Rh-PDE(TMD)
Fig. S3 shows the absorption spectrum of a purified sample of Rh-PDE(TMD) at pH 8.0. The λmax is located at 497 nm, which is 5-nm red-shifted from that reported previously for Rh-PDE(full) at pH 6.5 (15). We first examined the photoreaction dynamics of Rh-PDE(TMD) by using flash photolysis with nanosecond laser excitation. In the first paper (15), we studied the photoreaction dynamics of Rh-PDE(full) by using a conventional spectrophotometer, whose time resolution took a few seconds in kinetic measurements, and determined the time constant of the M decay (τ1/e) to be 45 s at 4 °C (15). From the linear temperature dependence of the logarithm of the rate constant (ln k) between 4 and 24 °C, the time constant of the M decay (τ1/e) was estimated to be 6.8 s at 27 °C, which was the temperature of the GloSensor assay.
With a higher time resolution, we expected to obtain the time constant of the rise in M as well as the origin of its precursor. Fig. 1A shows the appearance of transient absorption at 600 nm within 5 μs (time resolution), which corresponds to the red-shifted K-like intermediate. The K-like intermediate decayed with a time constant of 4.0 ms. Fig. 1B monitors the transient absorption at 400 nm for the blue-shifted M intermediate, which formed with a time constant of 2.2 ms (24%) and 4.6 ms (76%), and decayed with a time constant of 2.2 s (48%) and 5.4 s (52%). Fig. 1C monitors the transient absorption at 500 nm, corresponding to the bleaching signal of Rh-PDE(TMD), and the kinetics represent a mirror image of the kinetics at 400 nm. In fact, transient absorption at 500 nm decreased with a time constant of 0.6 ms (17%) and 4.0 ms (83%) and increased with a time constant of 3.7 s, which coincided well with the M formation and decay, respectively. Thus, the present transient absorption study revealed that the K-like intermediate converted into the M intermediate with a time constant of 4 ms and that the M intermediate returned to its original state with a time constant of 4 s. Fig. 1A clearly shows the lack of an O intermediate at a late stage of the photocycle. Recovery of the Rh-PDE(TMD) photocycle (4 s) is close to that of Rh-PDE(full) (7 s) (15), indicating little influence of the PDE domain on the photocycle of the Rh domain. As the enzymatic activity remained in a minute time scale, any delay of structural changes to the PDE domain takes place independently in Rh-PDE(full).
Figure 1.
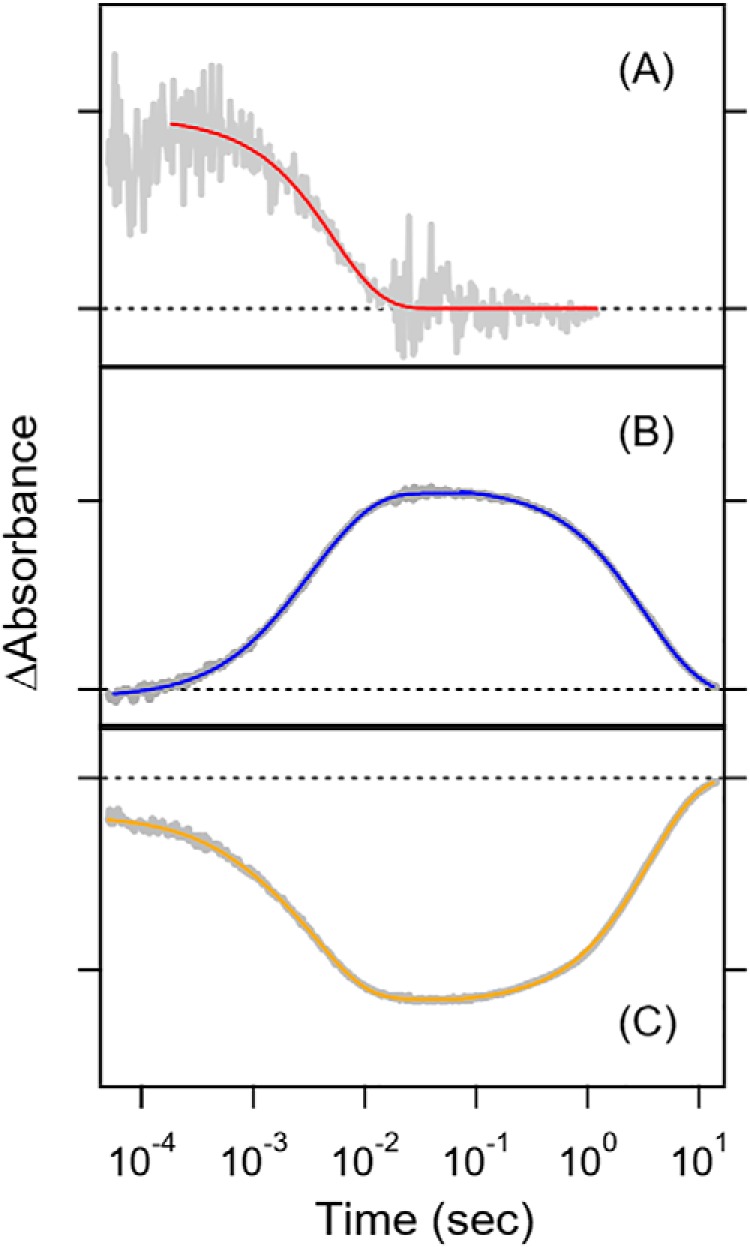
Transient absorption of Rh-PDE(TMD) in detergent (0.03% DDM) at 600 nm (A), 400 nm (B), and 500 nm (C) after excitation at 490 nm. Gray lines represent the experimental data, whereas red (A), blue (B), and yellow lines (C) are the fitting curves. Broken lines show the zero line in each panel. One division of the y axis corresponds to 0.002 (A), 0.03 (B), and 0.03 (C) absorbance units.
Light-induced difference UV-visible spectroscopy of Rh-PDE(TMD)
To gain structural insight into the K and M intermediates, we applied light-induced difference FTIR spectroscopy. Prior to the analysis, we first examined the experimental conditions by light-induced difference UV-visible spectroscopy. Transient absorption was measured for the Rh-PDE(TMD) sample solubilized in 0.03% DDM, whereas FTIR spectroscopy was applied to the hydrated film sample reconstituted into membranes. In this study, the purified Rh-PDE(TMD) sample was reconstituted into POPE/POPG (3:1) liposomes, and the dry film was hydrated by placing a drop of water next to the film.
The black curve in Fig. 2A shows the difference spectra of Rh-PDE(TMD) after-minus-before illumination at 480 nm and at 77 K. The negative and positive peaks at 460/490 and 550 nm correspond to Rh-PDE(TMD) and the red-shifted K intermediate, respectively. Illumination at >570 nm yielded a mirror-image difference spectrum (red curve in Fig. 2A), indicating that the K intermediate had reverted into the original state. The black curve in Fig. 2B, which shows the difference spectrum of Rh-PDE(TMD) after-minus-before illumination at >500 nm and at 295 K, exhibits positive and negative peaks at 406 and 505 nm, respectively. Therefore, the photoproduct is the M intermediate under the present conditions. It should be noted that the difference spectra in Fig. 2B are considerably red-shifted from those reported previously. In the previous study (15), Rh-PDE(full) was solubilized in detergent (0.02% DDM), and these differences in sample conditions presumably affect absorption.
Figure 2.
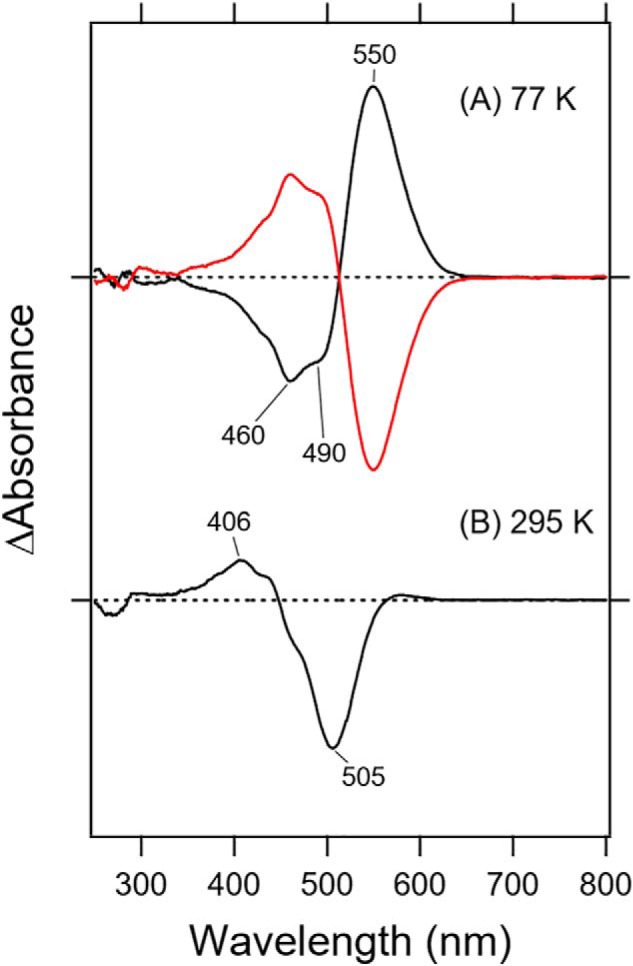
Light-induced difference UV-visible spectra of Rh-PDE(TMD). A, light-minus-dark difference absorption spectra of Rh-PDE(TMD) with 480-nm (black line) and >570-nm (red line) light in the UV-visible region at 77 K. B, light-minus-dark difference absorption spectra of Rh-PDE(TMD) with >500 nm light (black line) at 295 K. Hydrated films of Rh-PDE(TMD) in membranes (POPE/POPG = 3:1) with H2O were used for the measurements, whose sample conditions were identical for FTIR spectroscopy. One division of the y axis corresponds to 0.19 absorbance units.
Light-induced difference FTIR spectroscopy of Rh-PDE(TMD): Chromophore structure
Fig. 3 (A and B) shows the light-induced difference FTIR spectra of Rh-PDE(TMD) in the 1800–900 cm−1 region, which were measured at 77 and 295 K, after hydration with H2O (black lines) and D2O (red lines), respectively. These spectra correspond to the K and M intermediates based on UV-visible absorption. In Fig. 3A, the strongest negative and positive peaks appear at 1553 and 1538 cm−1, respectively, in H2O. The strongest peaks were observed at 1553 and 1536 cm−1 in D2O, which are assignable to the C=C stretching vibrations of the retinal chromophore. The lower frequency shift corresponds to the spectral red shift. Near the positive 1538 cm−1 peak, an additional positive peak was observed at 1527 cm−1, which was shifted to a lower frequency in D2O. There was no positive peak at this frequency region in Fig. 3B, consistent with the formation of the M intermediate in which the Schiff base was deprotonated.
Figure 3.

Light-induced difference FTIR spectra of Rh-PDE(TMD). A, difference FTIR spectra between the K intermediate and unphotolyzed states in the 1800–900 cm−1 region. Black and red lines represent the spectra of samples measured under H2O and D2O hydration, respectively, at 77 K. B, difference FTIR spectra between the M intermediate and unphotolyzed states in the 1800–900 cm−1 region. Black and red lines represent the spectra of samples measured under H2O and D2O hydration, respectively, at 295 K. Hydrated films of Rh-PDE(TMD) in membranes (POPE/POPG = 3:1) were used for the measurements. One division of the y axis corresponds to 0.01 absorbance units. The FTIR spectra of Rh-PDE(TMD) at 77 K was reused in both Fig. 3 and Fig. S4.
In the 1250–1150 cm−1 region, which is the frequency region of the C–C stretch, five negative bands were observed at 1241, 1235, 1210, 1199, and 1183 cm−1, whereas only a sharp positive band was observed at 1191 cm−1. In general, the C–C stretch of the retinal chromophore is insensitive to H/D exchange, and thus only such peaks correspond to the bands at 1241 (−), 1199 (−), and 1191 (+) cm−1. Therefore, these bands are likely to originate from the C–C stretching vibrations of the retinal chromophore. It is important to note that the positive 1191 cm−1 band represents a characteristic frequency of 13-cis-retinal, owing to the C14–C15 stretching vibration (18). This indicates that the K intermediate contains 13-cis chromophore and that the primary photoreaction is the all-trans to 13-cis isomerization of retinal, as well as other microbial rhodopsins (1). No positive peak at this frequency region in Fig. 3B is consistent with formation of the M intermediate.
Hydrogen-out-of-plane (HOOP) vibrations generally appear at 1000–900 cm−1, and the presence of strong HOOP modes represents a distortion of the retinal molecule at the corresponding position. Strong HOOP vibrations are characteristic of the primary K intermediate, and the HOOP modes near the Schiff base are sensitive to H/D exchange. In the case of Rh-PDE(TMD), there were two peaks at 1002 and 957 cm−1, which were insensitive to H/D exchange. The band at 1002 cm−1 is very high, as much as the HOOP vibration, and the frequency is characteristic of C9 and C13 methyl-rock vibrations, as was seen for a light-driven proton pump bacteriorhodopsin (BR) (negative 1009 cm−1 band) and a photochromic sensor Anabaena sensory rhodopsin (ASR) (negative 1006 cm−1 band) (Fig. S4) (19). Therefore, the 1002 cm−1 band originates from a methyl-rock vibration or a HOOP vibration at the middle of the polyene chain of retinal chromophore. A positive peak was observed at 968 cm−1 in D2O, but the corresponding peak in H2O was unclear. A positive band at 1027 cm−1 in H2O shifted to 1019 cm−1 in D2O, but it was too high to assign HOOP vibrations. Fig. S4 clearly shows H/D-exchangeable HOOP vibrations at 1000–900 cm−1 for CrCCR(C1C2), BR, and ASR, which reflect a chromophore distortion near the Schiff base. However, Rh-PDE(TMD) did not exhibit HOOP bands, such as N–H or C15–H, in the K intermediate. Thus, Rh-PDE(TMD) lacks chromophore distortion near the Schiff base. Instead, the distortion is located at the middle of the chromophore. No positive peak at this frequency region in Fig. 3B is consistent with formation of the M intermediate, where the Schiff base is deprotonated.
The negative band at 1666 cm−1 in H2O appears to have shifted to 1638 cm−1 in D2O (Fig. 3A) and thus originates from the C=N stretching vibrations of the protonated Schiff base. The upshift in frequency of the C=N stretching vibration in H2O is caused by its coupling to the N-H bending vibration of the Schiff base, and the difference in frequency between H2O and D2O is regarded as a measure of the hydrogen-bonding strength of the Schiff base. Such a difference for Rh-PDE(TMD) (28 cm−1) is much larger than that of BR (13 cm−1), suggesting that the hydrogen bond of the Schiff base in Rh-PDE is stronger than in BR. It should be noted, however, that the negative peak at 1666 cm−1 is not necessarily accurate because there is a sharp positive peak at about 1660 cm−1 in D2O (Fig. 3A) that influences the negative peak at 1666 cm−1. In fact, Fig. 3B shows that a strong negative peak at 1658 cm−1 reduces the half-intensity in D2O. Therefore, the C=NH stretch of Rh-PDE(TMD) is probably located at 1658 cm−1. In Fig. 3B, the C=ND stretch at 1638 cm−1 in D2O is canceled by the positive peak at 1633 cm−1 in D2O. In contrast to the negative band, the C=N stretch of the positive band (K intermediate) is complicated, as shown in Fig. 3A. There are two peaks at 1628 and 1618 cm−1 in H2O and three peaks at ∼1625, 1605, and 1589 cm−1 in D2O, and it is not easy to identify the C=N stretch. The C=N stretch is H/D-insensitive for the M intermediate, whereas the corresponding positive peak is unclear in Fig. 3B.
Light-induced difference FTIR spectroscopy of Rh-PDE(TMD): X–D stretching vibrations
Fig. 4, which monitors X–D stretching vibrations, shows the difference spectra at 2800–1800 cm−1 in D2O. We compared the difference spectra of Rh-PDE(TMD) (A) with those of CrCCR(C1C2) (B) (20), BR (C) (21), and ASR (D) (19). A spectral comparison of the samples hydrated with D2O and D218O identified O–D stretching vibrations of water molecules that changed their frequencies upon retinal photoisomerization, which were tagged by green numbers.
Figure 4.

FTIR spectral comparison of X–D stretching vibrations in D2O at 77 K. A–D, the Rh-PDE(TMD)K minus Rh-PDE(TMD) (A), CrCCR(C1C2)K minus CrCCR(C1C2) (B), BRK minus BR (C), and ASRK minus ASR (D) spectra in the 2800–1800 cm−1 region measured at 77 K upon hydration with D2O (red) or D218O (blue). Green-labeled frequencies correspond to those identified as O–D stretching vibrations of water. Purple-labeled and underlined frequencies in C are O–D stretches of Thr-89 and N–D stretches of the Schiff base in BR, respectively. Orange-labeled frequencies in D are S–H stretches of Cys in ASR. One division of the y axis corresponds to 0.001 absorbance units. The data in B–D are reproduced from Refs. 18, 19, and 17, respectively.
Fig. 4A shows that almost all bands at >2400 cm−1 shifted to lower frequencies in 18O water. In contrast, no bands shifted at <2400 cm−1, indicating that these bands originate from N–D or nonwater O–D stretching vibrations. The N–D stretching vibration of the protonated Schiff base appears in this frequency region, and a previous [ζ-15N]lysine labeling study identified the 2171/2123 and 2466 cm−1 bands as the N–D stretches of BR and BRK (Fig. 4C) (22) and the 2163/2125 and 2470 cm−1 bands as the N–D stretches of ASR and ASRK (Fig. 4D) (23), respectively. We inferred the 2022 cm−1 band as the N–D stretch of CrCCR(C1C2) (Fig. 4B) (20), although this was not identified by any isotope. In the case of Rh-PDE(TMD), a broad negative spectral feature at 2250–2100 cm−1 is a candidate of the N–D stretch of the Schiff base, and its frequency is consistent with the results of the analysis of the C=N stretch.
The Schiff base forms a strong hydrogen bond in Rh-PDE(TMD), which is also the case in CrCCR(C1C2), BR, ASR, and many other rhodopsins. On the other hand, a unique spectral feature in Rh-PDE(TMD) was observed for the K intermediate after retinal photoisomerization. Fig. 4A shows a broad positive band at 2050–1900 cm−1, centered at 1960 cm−1, which does not originate from water vibrations. From a similar broad spectral feature of the bands at 2250–2100 cm−1 (−) and at 2050–1900 cm−1 (+), it is likely that these bands correspond to each other, presumably the N–D stretch of the Schiff base. In this case, a lower frequency shift indicates that the hydrogen bond of the Schiff base becomes stronger in the K intermediate. This is unique because photoisomerization weakens the hydrogen bond of the Schiff base, as can be seen in Fig. 4, C and D.
We assigned green-tagged bands in Fig. 4A to the O–D stretching vibrations of water in Rh-PDE(TMD); seven negative bands as the water O–D stretching vibrations of Rh-PDE(TMD) at 2717, 2682, 2672, 2648, 2559, 2494, and 2486 cm−1; and six positive bands as the water O–D stretching vibrations of the K intermediate at 2691, 2636, 2539, 2516, 2503, and 2456 cm−1. The unbalanced number of positive and negative bands suggests that positive peaks contain vibrations of multiple water molecules. It should be noted that isotope shifts of water were observed only between 2720 and 2480 cm−1 for Rh-PDE(TMD), and no water bands were observed at <2480 cm−1.
BR exhibited unusually low-frequency water vibrations at 2323, 2292, and 2171 cm−1 (Fig. 4C), which were identified as the three water molecules involved in the pentagonal cluster in the Schiff base region (24). From comprehensive studies of water vibrations of various rhodopsins, we found that strongly hydrogen-bonded water molecules (O–D stretch at <2400 cm−1) are always present in rhodopsins exhibiting proton-pumping activity (25, 26). For example, green-absorbing proteorhodopsin from marine bacteria (27), Gloeobacter rhodopsin from cyanobacteria (28), and a fungal Leptosphaeria rhodopsin (29) exhibit water O–D stretching vibrations at 2315, 2295, and 2257 cm−1, respectively. This strong correlation between water's hydrogen bond and proton-pumping function allowed us to conclude that a strongly hydrogen-bonded water molecule is the functional determinant of proton pumping. CrCCR(C1C2) contains the water band at 2378 cm−1 (Fig. 4B) (20), falling into the category of strongly hydrogen-bonded water. In contrast, Rh-PDE(TMD) did not contain a strongly hydrogen-bonded water molecule, as was the case for ASR (Fig. 4D) (19).
Although Rh-PDE has no strong hydrogen-bonded water, seven water bands for the resting state is a considerably large number. Comprehensive analysis of protein-bound water molecules revealed that microbial and animal rhodopsins show changes in 2–9 and 6–8 water bands, respectively, upon retinal photoisomerization at 77 K (Table S1). Smaller numbers of altered water molecules in microbial rhodopsins compared with animal rhodopsins may originate from specific differences in each protein's environment or from the structural changes upon all-trans to 13-cis isomerization in microbial rhodopsins being smaller than those upon 11-cis to all-trans isomerization in animal rhodopsins (25). CrCCR(C1C2) contains the largest number of water bands, nine in total (Fig. 4B), and Rh-PDE presumably involves a similar water-containing hydrogen-bonding network.
Light-induced difference FTIR spectroscopy of Rh-PDE(TMD): Protein structure
Fig. 5 enlarges the 1780–1620 cm−1 region of Fig. 3, which contains many protein vibrations in addition to the C=N stretch of the Schiff base of retinal chromophore. Structural changes of the peptide backbone are reflected in the amide-I vibration at 1700–1600 cm−1, which is weakly sensitive to H/D exchange, and a characteristic frequency of the α-helix was observed at 1660–1650 cm−1. The strong negative peak at 1666 cm−1 originates from the C=N stretch in Fig. 5A, as it is shifted to 1638 cm−1 in D2O. Note that the difference spectrum in D2O shows peaks at 1668 (−)/1660 (+)/1650 (−) cm−1, which were assigned as the result of helical structural perturbation upon retinal photoisomerization.
Figure 5.
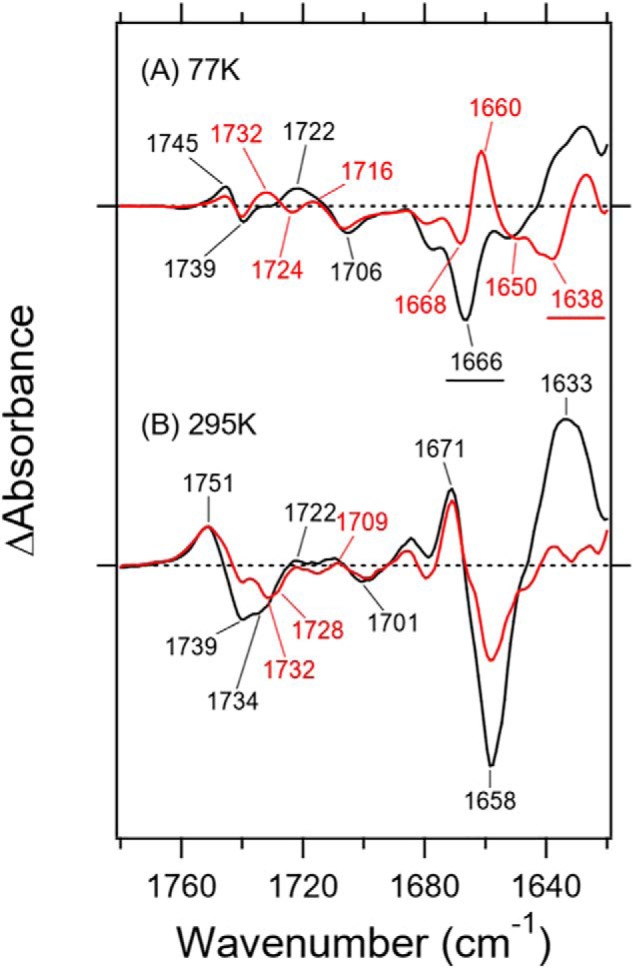
Enlarged difference FTIR spectra of Rh-PDE(TMD) at 77 K (A) and 295 K (B) in the 1780–1620 cm−1 region. Black and red lines represent the spectra of samples measured under H2O and D2O hydration, respectively. One division of the y axis corresponds to 0.008 absorbance units.
Much larger spectral changes were observed in the amide-I region for the M intermediate (Fig. 5B), where peaks appeared at 1671 (+)/1658 (−)/1633 (+) cm−1. Nevertheless, half of the negative peak at 1658 cm−1 in H2O is ascribable to the C=N stretch of the chromophore because of the exchangeable nature of H/D. The positive 1671 cm−1 peak originates exclusively from the amide-I vibration, because there is no change in D2O. In contrast, the positive 1633 cm−1 peak in H2O is considerably diminished in D2O, presumably shifting to 1612 cm−1. Therefore, the strong positive peak at 1633 cm−1 does not originate from amide-I, and its origin is an open question. It should be noted that the C=ND stretch in the unphotolyzed state is located at 1638 cm−1 in Fig. 5A, which also exists in the red spectrum of Fig. 5B. The Schiff base is not protonated in the M intermediate, whose H/D-insensitive C=N stretch appears at the same frequency of the C=ND stretch in D2O. It is thus likely that the positive C=N stretch (M intermediate) and negative C=ND stretch (unphotolyzed state) cancel each other out at 1638 cm−1 in D2O. Consequently, the spectral changes of amide-I in Rh-PDE(TMD) are from 1658 cm−1 in the unphotolyzed state to 1671 cm−1 in the M intermediate, indicating that the hydrogen bond of the α-helix is disrupted or largely weakened. This entire situation is in contrast to the case of CrCCR2, where amide-I largely changes from 1662–1664 to 1648–1649 cm−1 upon channel opening (30–35), where the peptide backbone of the α-helix is strengthened, possibly because of water hydration (35). The spectral feature of amide-I in Rh-PDE(TMD) resembles to some extent the features of CrCCR1 (36) and CrCCR(C1C2) (34). where a spectral upshift was observed. Rh-PDE does not possess an ion-transporting ability unlike channelrhodopsins, and the observed structural perturbation of the α-helix is probably related to its unique structural changes.
A C=O stretching vibration of protonated carboxylic acids, which appears in the 1800–1700 cm−1 region, is a powerful marker of carboxylic acids. Fig. 5A shows bands at 1745 (+)/1739 (−)/1722 (+)/1706 (−) cm−1 in H2O and at 1745 (+)/1739 (−)/1732 (+)/1724 (−)/1716 (+)/1706 (−) cm−1 in D2O for the difference with the K intermediate (77 K), whereas Fig. 5B shows bands at 1751 (+)/1739 (−)/1734 (−)/1722 (+)/1709 (+)/1701 (−) cm−1 in H2O and 1751 (+)/1739 (−)/1732 (−)/1728 (−)/1709 (+)/1701 (−) cm−1 in D2O for the difference with the M intermediate (295 K). From the results at 77 K, it is suggested that three carboxylic acids are protonated, and their frequencies are 1739, 1739, and 1706 cm−1 in the resting state. The measurements in H2O and D2O exhibited one band at 1739 cm−1 (COOH) that was down-shifted to 1724 cm−1 (COOD), whereas another band at 1739 cm−1 did not display any shift in D2O. The spectral feature in D2O indicates that the corresponding band of the negative 1724 cm−1 band is the positive 1732 cm−1 band. Regarding other bands at 1745 (+)/1739 (−) cm−1, the H/D-insensitive carboxylic C=O stretch was similarly reported for channelrhodopsins, Asp-195 in CrCCR1 and CrCCR(C1C2) (20) and Asp-156 in CrCCR2 (37). Furthermore, aspartic acid formed a DC gate with a cysteine in TM-3. Rh-PDE contains the corresponding residue, Asp-194 in TM-4 (Fig. S5), as well as Cys-169 in TM-3 (Fig. S2).
Fig. 5B shows an intense positive band at 1751 cm−1 and intense negative bands at 1739 and 1734 cm−1, together with small bands at 1725–1700 cm−1 in H2O. In D2O, spectra are more complicated with peaks at 1751 (+)/1739 (−)/1732 (−)/1728 (−)/1709 (+)/1701 (−) cm−1. In addition to the three bands at 77 K, a new negative peak appears at 1732 cm−1. Appearance of the M intermediate accompanies deprotonation of the Schiff base, where a specific group accepts the proton. If the acceptor is a carboxylate such as Asp-85 in BR, a new positive signal should appear in the frequency region of protonated carboxylic acids. Nevertheless, the area of the positive band is considerably smaller than that of the negative bands.
In addition to Asp-194, there are three candidate carboxylates near the retinal chromophore, although the structure of Rh-PDE is unknown. Glu-164 and Asp-292 in Rh-PDE correspond to Asp-85 and Asp-212 in BR, respectively, both of which act as Schiff base counterions (Fig. S5). A unique carboxylate of Rh-PDE is Glu-271 in TM-6, which corresponds to Trp-189 of BR located near the β-ionone ring. Rh-GC and Rh-histidine kinase possess tryptophan as well as anion channelrhodopsin, whereas CrCCR2 has phenylalanine. Among the four carboxylates, three are protonated in the resting state, and their hydrogen bonds are altered upon retinal photoisomerization at 77 K. Here we tentatively assigned Asp-194 as the H/D-unexchangeable band at 1739 cm−1. It is reasonable to postulate that Glu-271 is protonated. If so, either Glu-164 or Asp-292 may be protonated, or a distant carboxylic acid may change. Next, we provide the outcome of a mutation study on two key carboxylates near the Schiff base, Glu-164 and Asp-292.
As Rh-PDE contains Asp-194 and Cys-169, we expected spectral changes in the S–H stretching region. Indeed, we observed S–H stretching bands at 2532 (+)/2522 (−) cm−1 at 77 K (Fig. 6A). The stretching frequency of cysteine S–H is in the 2580–2525 cm−1 region, and this low frequency indicates that cysteine formed a very strong hydrogen bond in Rh-PDE(TMD). Fig. 6B shows the spectral changes in the S–D stretching frequency region, and no band indicates that the S-H group was H/D-unexchanged in D2O. This is also the case for CrCCR(C1C2) (20), suggesting the same origin. Fig. 6C shows the results for the M intermediate; two positive peaks were observed at 2557 and 2539 cm−1, as well as a single negative peak at 2526 cm−1. Thus, two cysteines appear to change the environment of the S–H group, where Fig. 6D shows no deuteration.
Figure 6.
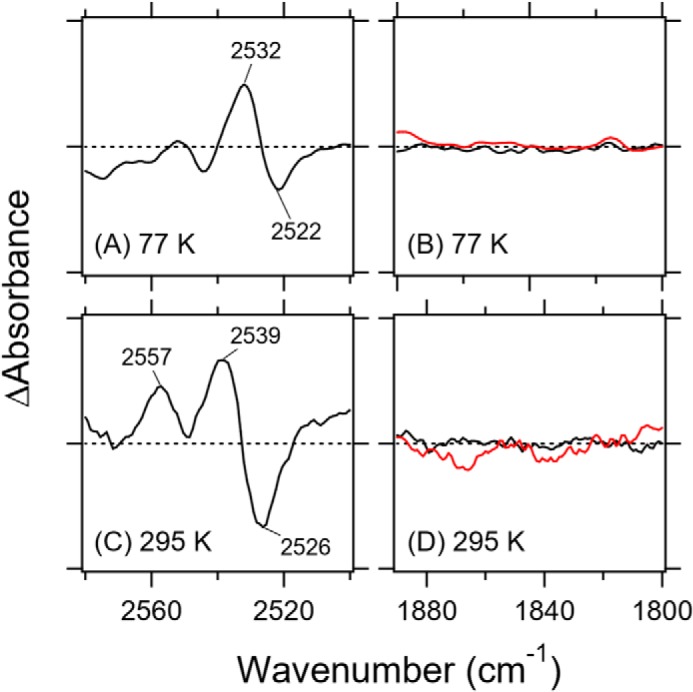
Difference FTIR spectra of Rh-PDE(TMD) in the S–H (A, 77 K; C, 295 K) and S–D (B, 77 K; D, 295 K) stretch region. Black and red lines represent the spectra in H2O and D2O, respectively.
Influence of mutations on color and enzymatic activity
To further gain structural and functional information about the Schiff base region, we prepared E164Q and D292N mutants and examined their molecular properties. Glu-164 and Asp-292 are presumably located near the protonated Schiff base in Rh-PDE, as Asp-85 and Asp-212 act as the Schiff base counterions in BR (1). The Asp → Asn mutations caused a spectral red shift, where the shift was larger for Asp-85 than for Asp-212 (38), and thus Asp-85 and Asp-212 are called the primary and secondary counterion of the Schiff base, respectively. This is also the case for CrCCR2 (39) and CrCCR(C1C2) (20), where the corresponding Asp-85 residue is the primary counterion, as the spectral red shift is larger.
In this experiment, we prepared the full-length protein of Rh-PDE because we wanted to measure the enzymatic activity of E164Q and D292N as well as their absorptions. Whereas the enzymatic activity was measured by an in vivo GloSensor assay, absorption maxima were also examined by hydroxylamine bleach experiments without protein purification. As the sample was not purified, the λmax values were unclear from the absolute absorption spectra, whose absorption is high at shorter wavelengths, because of scattering and contained colored proteins, such as cytochromes. Nevertheless, Fig. 7 successfully depicted absorption spectra of WT, E164Q, and D292N of Rh-PDE(full) in the after-minus-before illumination in the presence of 500 mm hydroxylamine. The obtained λmax were 487, 490, and 495 nm for E164Q, WT, and D292N, respectively. Due to lower expression of the mutants, spectral accuracy was less than that of WT, particularly at 400–500 nm. Thus, the λmax of E164Q at 487 nm is not very accurate. Nevertheless, the wavelength region at >500 nm is relatively accurate, and the spectral red shift for D292N and blue shift for E164Q from WT are clear. D292N exhibited a 5-nm spectral red shift relative to WT, implying that Asp-292 acts as the Schiff base counterion by containing a negative charge at the 292 position. In contrast, E164Q showed a slightly blue-shifted absorption relative to WT. The straightforward interpretation of this observation is that Glu-164 does not act as the Schiff base counterion because of its protonation.
Figure 7.
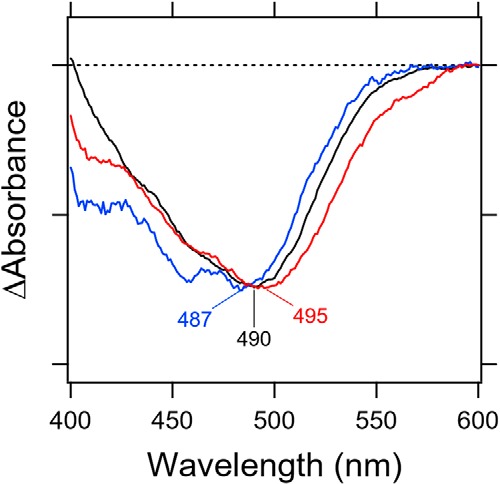
Light-induced difference absorption spectra of WT (black line), E164Q (blue line), and D292N (red line) of Rh-PDE(TMD) in the presence of 500 mm hydroxylamine bleach. Negative signals correspond to each absorption spectrum.
We then measured the enzymatic activity of these proteins as cAMP formation using a GloSensor assay. Fig. 8 compares the activity of E164Q and D292N with that of WT after normalizing the amount of protein estimated by hydroxylamine bleach. E164Q maintained the enzymatic activity of WT, whereas D292N significantly reduced it (<10%). In general, Glu → Gln and Asp → Asn mutations do not largely alter hydrogen-bonding conditions, suggesting little difference if carboxylate is protonated. The results of Figs. 7 and 8 strongly suggest that Asp-292 is negatively charged by deprotonation, whereas Glu-164 is protonated. The present study showed similar structural dynamics between Rh-PDE and channel rhodopsins, which is consistent with the phylogenetic analysis (Fig. S1). The PDE function was proven (15–17), but none of the previous papers reported ion-transport activity of Rh-PDE(TMD). Therefore, we performed electrophysiology experiments by expressing Rh-PDE(full) in mammalian cells (ND7/23). Fig. S6 clearly demonstrates no ion-transport activity of Rh-PDE.
Figure 8.
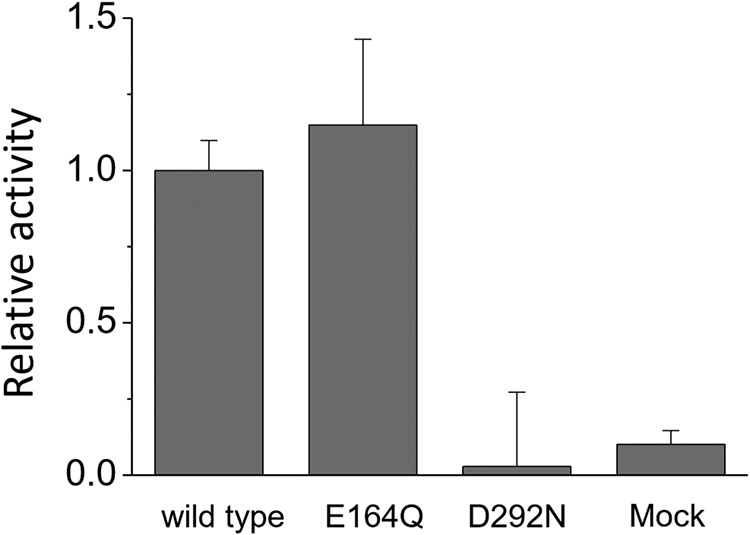
Comparison of enzymatic activity among WT, E164Q, and D292N by an in vivo assay. Error bars, S.D.
Discussion
The discovery of enzyme rhodopsins impacted researchers involved with fundamental and applied studies. In the former category, structural changes of the transmembrane domain lead to changes of the PDE domain, although the underlying mechanism remains unknown. In the latter category, enzyme rhodopsins such as Rh-GC and Rh-PDE can serve as new optogenetic tools to control the concentration of intracellular cyclic nucleotides. Therefore, after the discovery of Rh-PDE, extensive in vivo and in vitro studies began (15–17). In this study, we focused on the mechanism underlying the functioning of the Rh-PDE transmembrane domain. Three spectroscopic methods (transient absorption, light-induced difference UV-visible, and FTIR spectroscopy) were applied to Rh-PDE(TMD). Transient absorption changes were detected at only three wavelengths at a fixed pH (8.0). Nevertheless, the obtained results provided useful information about Rh-PDE(TMD), for which we propose a photoreaction scheme (Fig. 9).
Figure 9.

Schematic drawing of the structural model of the Schiff base region in Rh-PDE and their intermediates during the photocycle.
The unphotolyzed state contains an all-trans chromophore that forms a protonated Schiff base linkage with Lys-296. Glu-164 and Asp-292, which are located near the Schiff base in Rh-PDE, correspond to Asp-85 and Asp-212 in BR, respectively. In BR, both are negatively charged by deprotonation and constitute the counterion complex. Carboxylate deprotonation is normally evidenced by an Asp → Asn or Glu → Gln mutation that causes a spectral red shift. This is because neutralization of the counterion weakens the interaction with the protonated Schiff base, leading to destabilization of the electronic ground state of the retinal chromophore. In BR, Asp-85 and Asp-212 are referred to as principal and secondary counterions, respectively, as the spectral red shift is larger for D85N than for D212N (38). In Rh-PDE(TMD), D292N shows a 5-nm spectral red shift (Fig. 7), which is fully consistent with the function of the counterion. In contrast, E164Q did not display a red shift, and instead a slight blue shift was observed. From these results, we propose that Glu-164 is protonated in Rh-PDE (Fig. 9). This proposal is supported by a functional assay in which E164Q shows similar enzymatic activity as the WT (Fig. 8). This suggests that the negative charge at position 164 is not a prerequisite for activation. In contrast, diminished activity of D292N supports the important role of the negative charge at position 292 in enzymatic activity. In BR and CrCCR(C1C2), water molecule(s) form strong hydrogen bonds with negatively charged carboxylate(s). In Rh-PDE(TMD), the lack of strongly hydrogen-bonded water molecules (Fig. 4) suggests the absence of such water near Asp-292. Thus, the protonated Schiff base forms a direct interaction with Asp-292 in the model (Fig. 9).
The absorption of light causes retinal photoisomerization from the all-trans to 13-cis form, which is common in microbial rhodopsins. The primary K intermediate of Rh-PDE(TMD) lacks chromophore distortion near the Schiff base, and instead, the distortion is located at the middle of the chromophore, as was shown from an analysis of the HOOP region (Fig. 4). In addition, the hydrogen bond of the Schiff base strengthens in the K intermediate, which is a unique phenomenon because photoisomerization weakens the hydrogen bond of the Schiff base in most microbial rhodospins, as can be seen in Fig. 4, C and D. In Fig. 9, we tentatively illustrate the N–H group of the Schiff base interacting with the negatively charged Asp-292. Experimentally, there is little chromophore distortion near the Schiff base (Fig. 4), and thus in our model, isomerization follows the bicycle-pedal model, leading to a syn-like C=N configuration (Fig. 9). Note that the 13-cis, 15-syn configuration is thermally stable, as seen in the dark-adapted BR, and the normal K intermediate contains a 13-cis, 15-anti configuration. In Fig. 9, we propose that the K intermediate in Rh-PDE(TMD) possesses a 13-cis, 15-syn like configuration, with a distorted polyene structure in the middle. Another possibility for the K intermediate is that the N–H dipole points toward the cytoplasmic side (top part of Fig. 9). In this case, the lack of distortion around the Schiff base is consistent with the HOOP signals (Fig. 4), but what remains unclear is the hydrogen-bonding acceptor of the Schiff base that forms a very strong hydrogen bond (Fig. 5). The water molecule may be a possible acceptor, although the very strong hydrogen bond of the Schiff base would still need to be explained.
The K intermediate is converted to the M intermediate, the active state, with a time constant of 4 ms. The time constant is much slower than that of BR (40 μs) and CrCCR2 (10 μs). Absence of the blue-shifted L and red-shifted O intermediates is characteristic of the Rh-PDE(TMD) photocycle, which is composed exclusively of K and M intermediates (Fig. 1). Relaxation of the chromophore–protein interaction leads to the deprotonation of the Schiff base in the M intermediate. The protonation signal of carboxylates appears at 1780–1700 cm−1, but complicated signals in Fig. 5B make it difficult to affirmatively draw this conclusion. A similar area of positive and negative bands suggests environmental changes rather than protonation. Nevertheless, protonation of a new carboxylate and deprotonation of another carboxylic acid is possible, as was seen for the N intermediate of BR (Asp-85 protonation and Asp-96 deprotonation). Thus, in Fig. 9, we propose that Asp-292 is the proton acceptor from the Schiff base. This proposal is supported by the fact that Asp-292 is a prerequisite for enzymatic activity (Fig. 8). In the M intermediate, deformation of the α-helix takes place (Fig. 5B), and such structural changes are connected to the PDE domain. The M intermediate appears with a time constant of 4 ms and decays with a time constant of 4 s, during which the PDE domain has to be activated.
It is interesting how structural changes of the Rh domain are transduced into the PDE domain. Rh-PDE forms a dimer in which the PDE domain is in an inactive form, presumably because of an unexposed catalytic site. Structural changes of the Rh domain, particularly in helix G (and ensuing changes in the HAMP-like domain), possibly dissociate the PDE dimer, leading to the two catalytic domains being exposed to the aqueous phase. The 10 times longer enzymatic activity than the photocycle of the Rh domain alone suggests weak coupling of structural changes between Rh and PDE domains, which may be advantageous for Rh-PDE to sustain its enzymatic activity. In this study, we reported detailed structural changes of the Rh domain. A structural dynamics study of Rh-PDE(full) will provide further useful information of Rh-PDE in action, which is our future focus.
Experimental procedures
Sample preparation of Rh-PDE(TMD) and Rh-PDE(full) mutants
The Rh-PDE(full) gene (NCBI Gene ID: 16078606) was synthesized after optimization of a human codon (GenScript) as described previously (15). The Rh-PDE(TMD) gene (positions 1–316) was subcloned into the pEG BacMam vector, with a C-terminal GFP-His8 tag and a tobacco etch virus cleavage site, and was expressed in HEK293S GnTI− cells (40). Cells were collected by centrifugation (5000 × g, 10 min, 4 °C) and disrupted by sonication in buffer A (20 mm Tris, pH 8.0, 200 mm NaCl, 20% glycerol) supplemented with 100 μm all-trans-retinal (Sigma). The membrane fraction was collected by ultracentrifugation (185,500 × g, 1 h, 4 °C) and solubilized for 1 h at 4 °C in buffer B (20 mm Tris, pH 8.0, 200 mm NaCl, 10 mm MgCl2, 20% glycerol, 1% n-dodecyl-β-d-maltoside (DDM) (Calbiochem), 0.2% cholesteryl hemisuccinate (CHS) (Sigma). Insoluble materials were removed by ultracentrifugation (185,500 × g, 20 min, 4 °C). The supernatant was incubated with TALON metal affinity resin (Clontech) and incubated for 30 min at 4 °C. The resin was washed with buffer C (20 mm Tris, pH 8.0, 500 mm NaCl, 10 mm MgCl2, 15 mm imidazole, 10% glycerol, 0.03% DDM, 0.006% CHS) and eluted with buffer C containing 200 mm imidazole. The eluate was treated with tobacco etch virus protease and dialyzed against buffer D (20 mm Tris-HCl, pH 8.0, 500 mm NaCl, 10 mm MgCl2, 10% glycerol). The protease and cleaved GFP-His8 tag were removed with TALON resin, and the protein was further purified by size-exclusion chromatography on a HiLoad 16/600 Superdex 200-pg column (GE Healthcare), equilibrated with buffer E (20 mm Tris, pH 8.0, 150 mm NaCl, 10% glycerol, 0.03% DDM, 0.006% CHS). The peak fractions were collected and frozen until the following measurement.
The Rh-PDE(TMD) samples prepared in DDM solution were directly applied to the transient absorption study. For light-induced difference UV-visible and FTIR spectroscopy, Rh-PDE(TMD) samples were reconstituted into a mixture of POPE and POPG (3:1 molar ratio), in which the molar ratio of protein/lipid was 1:20.
Spectroscopic and enzymatic analyses of the mutants were performed for Rh-PDE(full). A full-length gene encoding Rh-PDE (NCBI Gene ID: 16078606) was synthesized as described previously. The plasmids for E164Q and D292N mutants were generated by the QuikChange site-directed mutagenesis kit (Agilent Technologies). Each construct was verified by DNA sequencing.
Flash photolysis
The transient absorption changes after photoexcitation of Rh-PDE(TMD) in detergent (0.03% DDM) were monitored by flash photolysis measurements at 25 °C, as described previously (41, 42). The sample was illuminated with a 490-nm beam produced from an OPO system (LT-2214, LOTIS TII, Minsk, Belarus) excited by the third harmonics of a nanosecond-pulsed Nd3+-YAG laser (λ = 355 nm, LS-2134UTF, LOTIS TII). The change in absorption at each wavelength after laser excitation was probed by monochromated light from the output of a xenon arc lamp (L9289-01, Hamamatsu Photonics, Shizuoka, Japan), and the change in intensity of the probe light passed through the sample was monitored by a photomultiplier tube (R10699, Hamamatsu Photonics). The signal from the photomultiplier tube was averaged and stored in a digital storage oscilloscope (DPO7104, Tektronix, Japan). The obtained kinetics data were fitted by single- or double-exponential curves individually.
UV-visible spectroscopy
The UV-visible spectra were measured by a UV-visible spectrometer (V-550, JASCO) equipped with a cryostat (OptistatDN, Oxford Instruments) (43). The Rh-PDE(TMD) sample in POPE/POPG liposomes was washed repeatedly with a buffer containing 10 mm NaCl and 2 mm sodium phosphate (pH 7.0) and dried on a BaF2 window. The dry film was hydrated before measurements by placing the hydrated film at 77 and 295 K. Illumination with 480-nm light through an interference filter at 77 K converted Rh-PDE(TMD) into the K intermediate, which reverted to the original state upon illumination with >570-nm light (O-59 filter), as evidenced by a mirror image of the difference spectra. For measurement of the M intermediate, the sample was illuminated with >500 nm (Y-52 filter).
The absorption maxima of WT, E164Q, and D292N were determined by hydroxylamine bleach measurements, but without purification (44). Each rhodopsin molecule expressed in HEK293 cells was suspended in 2.0% DDM (500 mm hydroxylamine, 100 mm NaCl, Na2HPO4 buffer pH 7.0) and illuminated for 1 min with a 1-kW tungsten–halogen projector lamp (Master HILUX-HR, Rikagaku, Kawasaki, Japan) through a glass filter (Y-52, AGC Techno Glass, Yoshida, Japan) at wavelengths > 500 nm at room temperature.
FTIR spectroscopy
Low-temperature difference FTIR spectroscopy at 77 K was performed as described previously (20, 24). Rh-PDE(TMD) films were hydrated with H2O, D2O, or D218O before measurements. The sample was then placed in the cell of a cryostat (Optistat, Oxford Instruments) mounted in an FTIR spectrometer (Carry670, Agilent Technologies) and cooled to 77 K. Each difference spectrum was calculated from two spectra constructed from 128 interferograms with 2 cm−1 resolution. Illumination lights were the same as those used for low-temperature UV-visible spectroscopy: 480-nm light (1 min) for the conversion of Rh-PDE(TMD) into the K intermediate and >570-nm light (1 min) for the reversion of the K intermediate into the original state. An average of 18, 68, or 50 experiments were conducted for the spectra upon hydration of H2O, D2O, or D218O, respectively.
Difference FTIR spectroscopy at 295 K was performed as described previously (45). Rh-PDE(TMD) films were hydrated with H2O or with D2O before measurements. The sample was then placed in the cell of a cryostat (Optistat, Oxford Instruments) mounted in an FTIR spectrometer (FTS-7000I, Agilent Technologies), and temperature was maintained at 295 K. Each difference spectrum was calculated from two spectra constructed from 128 interferograms with 2 cm−1 resolution. Illumination light was the same as for low-temperature UV-visible spectroscopy, and >500-nm light (10 min) was used for the conversion of Rh-PDE(TMD) into the M intermediate. An average of three experiments were used for the spectra upon hydration of H2O or D2O.
Assay of the enzymatic activity of Rh-PDE in mammalian cells
Enzymatic activity was evaluated by the GloSensor assay as described previously (15). HEK293 cells were purchased from the JCRB Cell Bank and cultured in Eagle's minimal essential medium with l-glutamine and phenol red (Wako) containing 10% (v/v) FBS and penicillin/streptomycin. The cells were co-transfected with the plasmid of the WT, E164Q, and D292N Rh-PDE(TMD) and the pGloSensor-22F cAMP vector (Promega) by using Lipofectamine 2000 (Invitrogen). Changes in the intracellular cAMP concentration of HEK293 cells were measured by the GloSensor assay (Promega). Transfected cells were incubated with or without 0.5 μm all-trans-retinal (Toronto Research Chemicals). Before measurements, the culture medium was replaced with a CO2-independent medium containing 10% (v/v) FBS and 2% (v/v) GloSensor cAMP stock solution (Promega). Cells were then incubated for 2 h at room temperature in the dark. Intracellular cAMP level was observed by monitoring luminescence by using a microplate reader (Corona Electric) at 27 °C. The cells were treated with 3.5 μm forskolin (Wako), a direct activator of adenylyl cyclase, to elevate intracellular cAMP level. The cells were illuminated with 510-nm light from a xenon lamp (LAX-103, Asahi Spectra Co., Ltd., Tokyo, Japan) through an interference filter. Light intensity was adjusted to desired values by a variable ND filter mounted in the LAX-103. Three independent experiments were averaged.
Heterologous expression in ND7/23 cells and electrophysiology
ND7/23 cells was purchased from DS Pharma Biomedical (Osaka, Japan) and cultured in high-glucose Dulbecco's modified Eagle's medium (Wako) in a 37 °C, 5% CO2 incubator. Plasmid DNA carrying a full-length Rh-PDE with enhanced GFP at the C terminus was described previously (15). Transfection of ND7/23 cells was performed by Lipofectamine 2000 (Invitrogen). Cells were supplemented with 1 μm all-trans-retinal (Sigma) after transfection. Whole-cell patch clamp recordings on ND7/23 cells were performed as described previously (46). Continuous light was illuminated by OSG L12194-00-39070 (Hamamatsu Photonics) via a light guide into an inverted microscope, IMT-2 (Olympus, Tokyo Japan). Illumination was controlled by a mechanical shutter LS6S with an opening time of 0.7 ms and an closing time of 0.8 ms (Vincent Associates, Rochester, NY). Glass pipettes were fabricated by a micropipette puller, P-97 (Sutter Instruments, Novato, CA) and fire-polished by using a microforge, MF-830 (Narishige, Tokyo, Japan). The pipette resistance was between 1.5 and 2.5 megaohms. The pipette electrode was controlled by a micromanipulator, PCS-5000 (Burleigh Instruments, Fishers, NY). Current traces were recorded at 10 kHz and filtered to 2 kHz by an internal circuit of the amplifier, Axopatch 200B (Molecular Devices, Sunnyvale, CA). Data acquisition and shutter triggering were performed by pClamp version 10 software via a Digidata 1550 system (Molecular Devices). Data were analyzed by Clampfit and Origin software.
The standard external solution contained 140 mm NaCl, 2 mm MgCl2, 2 mm CaCl2, 2 mm KCl, 10 mm Hepes-NaOH (pH 7.2). The standard internal solution contained 110 mm NaCl, 2 mm MgCl2, 1 mm CaCl2, 5 mm KCL, 10 mm EGTA, 10 mm Hepes-NaOH (pH 7.2). Osmolality of the solutions was adjusted to 300 mosm by adding an appropriate amount of sucrose.
Author contributions
T. I., W. S., and O. N. prepared the Rh-PDE(TMD) sample, and M. W. and D. Y. reconstituted the sample into membranes. K. Y. measured transient absorption. M. W. and D. Y. measured light-induced UV-visible and FTIR spectra. K. Y. and S. P. T. conducted hydroxylamine bleach and enzymatic activity measurements. H. K. wrote the paper and directed all of the research. All authors discussed and commented on the manuscript.
Supplementary Material
Acknowledgments
We thank Drs. Keiichi Inoue and Leonid S. Brown for helpful discussions.
This work was supported by Japanese Ministry of Education, Culture, Sports, Science, and Technology Grants 17J30010 and 18K14634 (to W. S.), 18K06109 (to S. P. T.) and 16H06294 (to O. N.), and 25104009, 15H02391, and 18H03986 (to H. K.); by CREST, Japan Science and Technology Agency, Grant JPMJCR1753 (to H. K.); and by PRESTO, Japan Science and Technology Agency, Grant JPMJPR1688 (to S. P. T.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Table S1 and Figs. S1–S6.
- GC
- guanylyl cyclase
- Rh
- rhodopsin
- PDE
- phosphodiesterase
- Rh-PDE(TMD)
- transmembrane domain of Rh-PDE
- Rh-PDE(full)
- full-length protein of Rh-PDE
- CrCCR2
- cation channelrhodopsin 2 from C. reinhardtii
- CrCCR1
- cation channelrhodopsin 1 from C. reinhardtii
- CrCCR(C1C2)
- a chimera of CrCCR1 and CrCCR2
- HOOP
- hydrogen-out-of-plane
- BR
- bacteriorhodopsin
- ASR
- Anabaena sensory rhodopsin
- DDM
- n-dodecyl-β-d-maltoside
- POPE
- phosphatidylethanolamine
- POPG
- phosphatidylglycerol
- H/D
- hydrogen/deuterium
- CHS
- cholesteryl hemisuccinate.
References
- 1. Ernst O. P., Lodowski D. T., Elstner M., Hegemann P., Brown L. S., and Kandori H. (2014) Microbial and animal rhodopsins: structures, functions, and molecular mechanisms. Chem. Rev. 114, 126–163 10.1021/cr4003769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Grote M., Engelhard M., and Hegemann P. (2014) Of ion pumps, sensors and channels—perspectives on microbial rhodopsins between science and history. Biochim. Biophys. Acta 1837, 533–545 10.1016/j.bbabio.2013.08.006 [DOI] [PubMed] [Google Scholar]
- 3. Brown L. S. (2014) Eubacterial rhodopsins—unique photosensors and diverse ion pumps. Biochim. Biophys. Acta 1837, 553–561 10.1016/j.bbabio.2013.05.006 [DOI] [PubMed] [Google Scholar]
- 4. Inoue K., Kato Y., and Kandori H. (2015) Light-driven ion-translocating rhodopsins in marine bacteria. Trends Microbiol. 23, 91–98 10.1016/j.tim.2014.10.009 [DOI] [PubMed] [Google Scholar]
- 5. Govorunova E. G., Sineshchekov O. A., Li H., and Spudich J. L. (2017) Microbial rhodopsins: diversity, mechanisms, and optogenetic applications. Annu. Rev. Biochem. 86, 845–872 10.1146/annurev-biochem-101910-144233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Luck M., Mathes T., Bruun S., Fudim R., Hagedorn R., Tran Nguyen T. M., Kateriya S., Kennis J. T. M., Hildebrandt P., and Hegemann P. (2012) A photochromic histidine kinase rhodopsin (HKR1) that is bimodally switched by ultraviolet and blue light. J. Biol. Chem. 287, 40083–40090 10.1074/jbc.M112.401604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Avelar G. M., Schumacher R. I., Zaini P. A., Leonard G., Richards T. A., and Gomes S. L. (2014) A rhodopsin-guanylyl cyclase gene fusion functions in visual perception in a fungus. Curr. Biol. 24, 1234–1240 10.1016/j.cub.2014.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Scheib U., Stehfest K., Gee C. E., Körschen H. G., Fudim R., Oertner T. G., and Hegemann P. (2015) The rhodopsin-guanylyl cyclase of the aquatic fungus Blastocladiella emersonii enables fast optical control of cGMP signaling. Sci. Signal. 8, rs8 10.1126/scisignal.aab0611 [DOI] [PubMed] [Google Scholar]
- 9. Gao S., Nagpal J., Schneider M. W., Kozjak-Pavlovic V., Nagel G., and Gottschalk A. (2015) Optogenetic manipulation of cGMP in cells and animals by the tightly light-regulated guanylyl-cyclase opsin CyclOp. Nat. Commun. 6, 8046 10.1038/ncomms9046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Boyden E. S., Zhang F., Bamberg E., Nagel G., and Deisseroth K. (2005) Millisecond-timescale, genetically targeted optical control of neural activity. Nat. Neurosci. 8, 1263–1268 10.1038/nn1525 [DOI] [PubMed] [Google Scholar]
- 11. Zhang F., Wang L.-P., Brauner M., Liewald J. F., Kay K., Watzke N., Wood P. G., Bamberg E., Nagel G., Gottschalk A., and Deisseroth K. (2007) Multimodal fast optical interrogation of neural circuitry. Nature 446, 633–639 10.1038/nature05744 [DOI] [PubMed] [Google Scholar]
- 12. Ryu M. H., Moskvin O. V., Siltberg-Liberles J., and Gomelsky M. (2010) Natural and engineered photoactivated nucleotidyl cyclases for optogenetic applications. J. Biol. Chem. 285, 41501–41508 10.1074/jbc.M110.177600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Stierl M., Stumpf P., Udwari D., Gueta R., Hagedorn R., Losi A., Gärtner W., Petereit L., Efetova M., Schwarzel M., Oertner T. G., Nagel G., and Hegemann P. (2011) Light modulation of celler cAMP by a small bacterial photoactivated adenylyl cyclase, bPAC, of the soil bacterium Beggiatoa. J. Biol. Chem. 286, 1181–1188 10.1074/jbc.M110.185496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Iseki M., Matsunaga S., Murakami A., Ohno K., Shiga K., Yoshida K., Sugai M., Takahashi T., Hori T., and Watanabe M. (2002) A blue-light-activated adenylyl cyclase mediates photoavoidance in Euglena gracilis. Nature 415, 1047–1051 10.1038/4151047a [DOI] [PubMed] [Google Scholar]
- 15. Yoshida K., Tsunoda S. P., Brown L. S., and Kandori H. (2017) A unique choanoflagellate enzyme rhodopsin exhibits light-dependent cyclic nucleotide phosphodiesterase activity. J. Biol. Chem. 292, 7531–7541 10.1074/jbc.M117.775569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lamarche L. B., Kumar R. P., Trieu M. M., Devine E. L., Cohen-Abeles L. E., Theobald D. L., and Oprian D. D. (2017) Purification and characterization of RhoPDE, a retinylidene/phosphodiesterase fusion protein and potential optogenetic tool from the choanoflagellate Salpingoeca rosetta. Biochemistry 56, 5812–5822 10.1021/acs.biochem.7b00519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tian Y., Gao S., Yang S., and Nagel G. (2018) A novel rhodopsin phosphodiesterase from Salpingoeca rosetta shows light-enhanced substrate affinity. Biochem. J. 475, 1121–1128 10.1042/BCJ20180010 [DOI] [PubMed] [Google Scholar]
- 18. Mizuide N., Shibata M., Friedman N., Sheves M., Belenky M., Herzfeld J., and Kandori H. (2006) Structural changes in bacteriorhodopsin following retinal photoisomerization from the 13-cis form. Biochemistry 45, 10674–10681 10.1021/bi060958s [DOI] [PubMed] [Google Scholar]
- 19. Furutani Y., Kawanabe A., Jung K.-H., and Kandori H. (2005) FTIR spectroscopy of the all-trans form of Anabaena sensory rhodopsin at 77 K: hydrogen bond of a water between the Schiff base and Asp75. Biochemistry 44, 12287–12296 10.1021/bi050841o [DOI] [PubMed] [Google Scholar]
- 20. Ito S., Kato H. E., Taniguchi R., Iwata T., Nureki O., and Kandori H. (2014) Water-containing hydrogen-bonding network in the active center of channelrhodopsin. J. Am. Chem. Soc. 136, 3475–3482 10.1021/ja410836g [DOI] [PubMed] [Google Scholar]
- 21. Tanimoto T., Furutani Y., and Kandori H. (2003) Structural changes of water in the Schiff base region of bacteriorhodopsin: proposal of a hydration switch model. Biochemistry 42, 2300–2306 10.1021/bi026990d [DOI] [PubMed] [Google Scholar]
- 22. Kandori H., Belenky M., and Herzfeld J. (2002) Vibrational frequency and dipolar orientation of the protonated Schiff base in bacteriorhodopsin before and after photoisomerization. Biochemistry 41, 6026–6031 10.1021/bi025585j [DOI] [PubMed] [Google Scholar]
- 23. Kawanabe A., Furutani Y., Jung K. H., and Kandori H. (2006) FTIR study of the photoisomerization processes in the 13-cis and all-trans forms of Anabaena sensory rhodopsin at 77 K. Biochemistry 45, 4362–4370 10.1021/bi052324b [DOI] [PubMed] [Google Scholar]
- 24. Shibata M., and Kandori H. (2005) FTIR studies of internal water molecules in the Schiff base region of bacteriorhodopsin. Biochemistry 44, 7406–7413 10.1021/bi050122+ [DOI] [PubMed] [Google Scholar]
- 25. Kandori H. (2011) in Supramolecular Photochemistry: Controlling Photochemical Processes (Ramamurthy V., and Inoue Y., eds) pp. 571–596, John Wiley & Sons, Inc., Hoboken, NJ [Google Scholar]
- 26. Muroda K., Nakashima K., Shibata M., Demura M., and Kandori H. (2012) Protein-bound water as the determinant of asymmetric functional conversion between light-driven proton and chloride pumps. Biochemistry 51, 4677–4684 10.1021/bi300485r [DOI] [PubMed] [Google Scholar]
- 27. Ikeda D., Furutani Y., and Kandori H. (2007) FTIR study of the retinal Schiff base and internal water molecules of proteorhodopsin. Biochemistry 46, 5365–5373 10.1021/bi700143g [DOI] [PubMed] [Google Scholar]
- 28. Hashimoto K., Choi A. R., Furutani Y., Jung K.-H., and Kandori H. (2010) Low-temperature FTIR study of Gloeobacter rhodopsin: presence of strongly hydrogen-bonded water and long-range structural protein perturbation upon retinal photoisomerization. Biochemistry 49, 3343–3350 10.1021/bi100184k [DOI] [PubMed] [Google Scholar]
- 29. Sumii M., Furutani Y., Waschuk S. A., Brown L. S., and Kandori H. (2005) Strongly hydrogen-bonded water molecule present near the retinal chromophore of Leptosphaeria rhodopsin, the bacteriorhodopsin-like proton pump from a eukaryote. Biochemistry 44, 15159–15166 10.1021/bi0513498 [DOI] [PubMed] [Google Scholar]
- 30. Ritter E., Stehfest K., Berndt A., Hegemann P., and Bartl F. J. (2008) Monitoring light-induced structural changes of Channelrhodopsin-2 by UV-visible and Fourier transform infrared spectroscopy. J. Biol. Chem. 283, 35033–35041 10.1074/jbc.M806353200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Radu I., Bamann C., Nack M., Nagel G., Bamberg E., and Heberle J. (2009) Conformational changes of channelrhodopsin-2. J. Am. Chem. Soc. 131, 7313–7319 10.1021/ja8084274 [DOI] [PubMed] [Google Scholar]
- 32. Eisenhauer K., Kuhne J., Ritter E., Berndt A., Wolf S., Freier E., Bartl F., Hegemann P., and Gerwert K. (2012) In channelrhodopsin-2 Glu-90 is crucial for ion selectivity and is deprotonated during the photocycle. J. Biol. Chem. 287, 6904–6911 10.1074/jbc.M111.327700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lórenz-Fonfría V. A., Resler T., Krause N., Nack M., Gossing M., Fischer von Mollard G., Bamann C., Bamberg E., Schlesinger R., and Heberle J. (2013) Transient protonation changes in channelrhodopsin-2 and their relevance to channel gating. Proc. Natl. Acad. Sci. U.S.A. 110, E1273–E1281 10.1073/pnas.1219502110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Inaguma A., Tsukamoto H., Kato H. E., Kimura T., Ishizuka T., Oishi S., Yawo H., Nureki O., and Furutani Y. (2015) Chimeras of channelrhodopsin-1 and -2 from Chlamydomonas reinhardtii exhibit distinctive light-induced structural changes from channelrhodopsin-2. J. Biol. Chem. 290, 11623–11634 10.1074/jbc.M115.642256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lórenz-Fonfría V. A., Bamann C., Resler T., Schlesinger R., Bamberg E., and Heberle J. (2015) Temporal evolution of helix hydration in a light-gated ion channel correlates with ion conductance. Proc. Natl. Acad. Sci. U.S.A. 112, E5796–E5804 10.1073/pnas.1511462112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lórenz-Fonfría V. A., Muders V., Schlesinger R., and Heberle J. (2014) Changes in the hydrogen-bonding strength of internal water molecules and cysteine residues in the conductive state of channelrhodopsin-1. J. Chem. Phys. 141, 22D507 10.1063/1.4895796 [DOI] [PubMed] [Google Scholar]
- 37. Nack M., Radu I., Gossing M., Bamann C., Bamberg E., von Mollard G. F., and Heberle J. (2010) The DC gate in channelrhodopsin-2: crucial hydrogen bonding interaction between C128 and D156. Photochem. Photobiol. Sci. 9, 194–198 10.1039/b9pp00157c [DOI] [PubMed] [Google Scholar]
- 38. Moltke S., Krebs M. P., Mollaaghababa R., Khorana H. G., and Heyn M. P. (1995) Intramolecular charge transfer in the bacteriorhodopsin mutants Asp85 → Asn and Asp212 → Asn: effects of pH and anions. Biophys. J. 69, 2074–2083 10.1016/S0006-3495(95)80078-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yizhar O., Fenno L. E., Davidson T. J., Mogri M., and Deisseroth K. (2011) Optogenetics in neural systems. Neuron 71, 9–34 10.1016/j.neuron.2011.06.004 [DOI] [PubMed] [Google Scholar]
- 40. Goehring A., Lee C. H., Wang K. H., Michel J. C., Claxton D. P., Baconguis I., Althoff T., Fischer S., Garcia K. C., and Gouaux E. (2014) Screening and large-scale expression of membrane proteins in mammalian cells for structural studies. Nat. Protoc. 9, 2574–2585 10.1038/nprot.2014.173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Inoue K., Reissig L., Sakai M., Kobayashi S., Homma M., Fujii M., Kandori H., and Sudo Y. (2012) Absorption spectra and photochemical reactions in a unique photoactive protein, middle rhodopsin MR. J. Phys. Chem. B 116, 5888–5899 10.1021/jp302357m [DOI] [PubMed] [Google Scholar]
- 42. Inoue K., Ono H., Abe-Yoshizumi R., Yoshizawa S., Ito H., Kogure K., and Kandori H. (2013) A light-driven sodium ion pump in marine bacteria. Nat. Commun. 4, 1678 10.1038/ncomms2689 [DOI] [PubMed] [Google Scholar]
- 43. Kawanabe A., Furutani Y., Jung K. H., and Kandori H. (2007) Photochromism of Anabaena sensory rhodopsin. J. Am. Chem. Soc. 129, 8644–8649 10.1021/ja072085a [DOI] [PubMed] [Google Scholar]
- 44. Singh M., Inoue K., Pushkarev A., Béjà O., and Kandori H. (2018) Mutation study of heliorhodopsin 48C12. Biochemistry 57, 5041–5049 10.1021/acs.biochem.8b00637 [DOI] [PubMed] [Google Scholar]
- 45. Furutani Y., Iwamoto M., Shimono K., Wada A., Ito M., Kamo N., and Kandori H. (2004) FTIR spectroscopy of the O photointermediate in pharaonis phoborhodopsin. Biochemistry 43, 5204–5212 10.1021/bi036316b [DOI] [PubMed] [Google Scholar]
- 46. Tsunoda S. P., Prigge M., Abe-Yoshizumi R., Inoue K., Kozaki Y., Ishizuka T., Yawo H., Yizhar O., and Kandori H. (2017) Functional characterization of sodium-pumping rhodopsins with different pumping properties. PLoS One 12, e0179232 10.1371/journal.pone.0179232 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.