

Abstract
Amyloid deposits of apolipoprotein A-I (apoA-I) and inflammation are common in atherosclerotic arteries. In this study, we investigated the interplay between oxidation of apoA-I methionine residues (Met(O)-ApoA-I), a known amyloidogenic modification of apoA-I, and the inflammatory response of immune cells. Soluble pre-fibrillar Met(O)-ApoA-I, but not apoA-I, induced intracellular accumulation of pro-interleukin (IL)-1β and secretion of the pro-inflammatory cytokines tumor necrosis factor α (TNFα) and IL-6 in mouse bone marrow–derived macrophages (BMDMs) and human primary monocytes. Additionally, secretion of mature IL-1β was also activated in human monocytes. The pro-inflammatory activity of Met(O)-ApoA-I was Toll-like receptor 4 (TLR4)-dependent and CD36-independent and was solely determined by oxidation of apoA-I methionine residues, in particular Met-86 and Met-148. In contrast, amyloid fibrils or reconstituted high-density lipoproteins (HDLs) generated from Met(O)-ApoA-I did not induce cytokine production in BMDMs. Although lipid-free Met(O)-ApoA-I remained functional in extracting lipids from cells and generating HDL, it gained strong pro-inflammatory properties that may aggravate local inflammation in the arteries and atherosclerosis. Our study indicates that oxidation of apoA-I methionine residues produces a potent danger-associated molecular pattern capable of stimulating pro-inflammatory cytokine secretion at levels similar to those induced by known pathogen-associated molecular patterns, such as lipopolysaccharide.
Keywords: apolipoprotein, amyloid, atherosclerosis, cytokine induction, inflammation, interleukin, macrophage, oxidation-reduction (redox), Toll-like receptor (TLR), methionine oxidation, apolipoprotein A-I, cytokines, DAMP, inflammation, methionine oxidation
Introduction
Conditions such as atherosclerosis, Alzheimer's disease, and type 2 diabetes are often described as sterile inflammation diseases (1–8). In Alzheimer's disease and type 2 diabetes, deposition of amyloid fibrils produced by β-amyloid (Aβ)2 and islet amyloid polypeptide (IAPP), respectively, contributes to the inflammatory process (2, 9). In atherosclerosis, oxidized LDL (10, 11) and cholesterol crystals (12, 13) accumulate in atherosclerotic lesions where they induce inflammasome activation in macrophages and secretion of the potent pro-inflammatory mediator IL-1β (10–15), thus exacerbating the pathological process. Activation of the NLRP3 inflammasome is a highly regulated process that, upon caspase-1–dependent cleavage of the inactive IL-1β precursor (pro-IL-1β), culminates with generation and secretion of mature IL-1β (16, 17). Release of IL-1β requires two signals (3, 18). Signal 1 (priming) up-regulates the synthesis of pro-IL-1β and of some components of the NLRP3 inflammasome following activation of pattern recognition receptors, such as the Toll-like receptors (TLRs) (19). Whereas in infectious inflammation, TLRs are triggered by microbial components (pathogen-associated molecular patterns, PAMPs), sterile inflammation is mediated by endogenous molecules that work as signal 1 either in their native conformation or after redox-mediated changes (20) and that are released by injured cells and tissues (danger-associated molecular patterns (DAMPs)). Signal 2, which drives assembly and activation of the inflammasome, is provided by several PAMPs and DAMPs, as diverse as extracellular ATP, pore-forming toxins, and particulate substances, including amyloids, pathogenic fibers, and crystals (3). Interestingly, soluble precursors of the particulate substances (e.g. oxidized LDL, free cholesterol, Aβ, and IAPP) can also activate the NLRP3 inflammasome through lysosome disruption induced by CD36-dependent intra-lysosomal nucleation of cholesterol crystals or amyloids (2, 21).
With aging, amyloid material accumulates in the media and intima layers of arteries (22–29). Among the amyloidogenic proteins that colocalize in amyloids extracted from atherosclerotic lesions (30), apolipoprotein A-I (apoA-I), the main structural protein of high-density lipoproteins (HDLs), is the most abundant (24–26, 28), suggesting an association between apoA-I amyloid formation and atherosclerosis.
In atherosclerotic lesions, macrophage-secreted myeloperoxidase targets apoA-I locally (31–33). Previously, we demonstrated that myeloperoxidase at levels found in atherosclerotic lesions oxidizes apoA-I methionine residues, thus reducing the structural stability of the protein and affording it amyloidogenic properties (34, 35). In vitro, and presumably in the arteries, lipid binding stabilizes oxidized apoA-I and prevents amyloid formation (34). Although the plasma concentration of lipid-free apoA-I is very low, compared with that of HDL-associated apoA-I (36–39), recent studies detected much higher levels of lipid-free apoA-I in the subendothelial space of the arteries, compared with plasma, and a 100-fold increase in atherosclerotic lesions, compared with normal arteries (40). Notably, this lipid-free apoA-I is heavily oxidized (41–43) and cross-linked (40).
We investigated the ability of soluble and amyloidogenic Met(O)-ApoA-I (an apoA-I with H2O2-oxidized methionines) to induce synthesis and secretion of pro-inflammatory cytokines in monocytes and macrophages, as release of these cytokines can promote sterile inflammation in the arteries. Our results indicate that Met(O)-ApoA-I acts as a powerful DAMP that induces cellular accumulation of pro-IL-1β and secretion of other inflammasome-independent pro-inflammatory cytokines (i.e. IL-6 and TNFα). Notably, in human monocytes, Met(O)-ApoA-I not only induces intracellular accumulation of pro-IL-1β, but also processing and secretion of large amounts of mature IL-1β.
Furthermore, we demonstrated that the pro-inflammatory properties of Met(O)-ApoA-I depend on the activity of the TLR4 receptor. Finally, we found that aggregation into amyloid-like fibrils or association with lipids to form HDL ablated the pro-inflammatory effect of Met(O)-ApoA-I. Further details of the molecular and cellular mechanisms underlying such response were also investigated.
Results
Oxidation of methionine residues imparts pro-inflammatory properties to apoA-I
As a simple and reproducible model for apoA-I methionine oxidation, we used a 1000-fold molar excess of H2O2 to completely oxidize all three apoA-I methionine residues (Met(O)-ApoA-I) (Table 1). Although methionine oxidation destabilizes the tertiary structure and disrupts self-association (quaternary structure) of apoA-I (35), the protein remains completely soluble until a secondary destabilizing stimulus (e.g. lowering the pH or applying sheer force) initiates protein aggregation and amyloid fibril formation (34, 35, 44).
Table 1.
Methionine oxidation in apoA-I samples as measured by LC-MS
| Sample name | Oxidation system | H2O2/apoA-I | Percentage of oxidized methionine (mean ± S.D.) |
||
|---|---|---|---|---|---|
| Met(O)-86 | Met(O)-112 | Met(O)-148 | |||
| mol/mol | % | % | % | ||
| ApoA-I control | None | 0:1 | 11.2 ± 0.0 | 6.2 ± 1.1 | 1.6 ± 0.0 |
| Met(O)-ApoA-I | H2O2 | 200:1 | 57.2 ± 0.6 | 97.5 ± 0.1 | 29.7 ± 0.0 |
| Met(O)-ApoA-I | H2O2 | 500:1 | 94.3 ± 0.2 | 99.7 ± 0.1 | 85.7 ± 0.7 |
| Met(O)-ApoA-I | H2O2 | 1000:1 | 98.9 ± 0.2 | 99.5 ± 0.2 | 99.0 ± 0.1 |
To test the hypothesis that, like other amyloid fibrils (2, 9, 11, 14), Met(O)-ApoA-I amyloid fibrils (particulate) may activate the NLRP3 inflammasome in macrophages, we primed mouse bone marrow-derived macrophages (BMDMs) with LPS (signal 1) for 3.5 h and then incubated the cells for 4 h in the presence of soluble pre-fibrillar Met(O)-ApoA-I or aggregated amyloid fibrils, as potential signals 2. Under these conditions, no secretion of mature IL-1β (17 kDa) was detected (Fig. 1A), indicating that Met(O)-ApoA-I is not active as a signal 2 in mouse macrophages in any of the forms tested (soluble or amyloid fibrils). Prolonged incubation (16–18 h) of LPS-primed BMDMs with Met(O)-ApoA-I samples did not increase the levels of secreted IL-1β (data not shown). Unexpectedly, intracellular levels of pro-IL-1β (34 kDa) were significantly elevated in LPS-primed BMDMs incubated with Met(O)-ApoA-I, compared with cells incubated with buffer (Fig. 1B).
Figure 1.
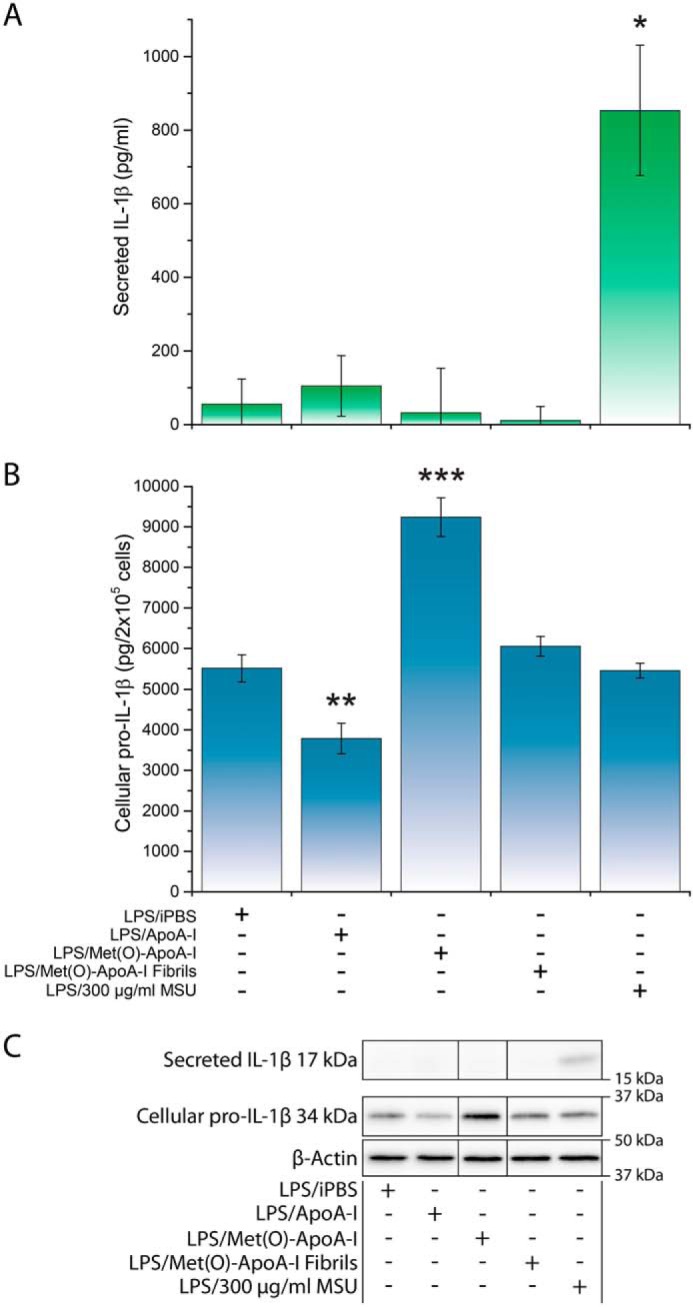
Met(O)-ApoA-I did not activate cleavage of pro-IL-1β (34 kDa) and secretion of mature IL-1β (17 kDa) in murine macrophages. BMDMs were incubated with LPS (0.5 μg/ml) for 3.5 h (priming). The cell culture medium was then replaced with new medium containing LPS (0.5 μg/ml) and (as signal 2) apoA-I samples (1 μm) or monosodium urate crystals (MSU) (300 μg/ml) and incubated for an additional 4 h. MSU was used as a positive control for signal 2 induction (80). Results from a representative experiment (of three) are shown in the figure. Values are means and S.D. (error bars) of three independent determinations. t test significance values versus LPS/iPBS results are reported as p < 0.05 (*), p < 0.01 (**), and p < 0.001 (***). A, the concentration of mature IL-1β in incubation medium was measured by ELISA. B, pro-IL-1β levels were measured in cell extracts by Western blot analysis (see C) and are reported as pg of pro-IL-1β in cell extracts (from 2 × 105 cells). C, representative Western blot analysis of cellular pro-IL-1β and secreted mature IL-1β with β-actin as a loading control. Vertical black lines in the Western blots indicate positions where unnecessary lanes were removed from the original scans.
Prompted by this observation, we investigated the signal 1 activity of Met(O)-ApoA-I on unprimed BMDMs. Nonoxidized apoA-I and Met(O)-ApoA-I amyloid fibrils did not induce any increase in cellular pro-IL-1β levels (Fig. 2). In contrast, BMDMs incubated with Met(O)-ApoA-I, the soluble precursor of the fibrils, accumulated high levels of pro-IL-1β (Fig. 2).
Figure 2.
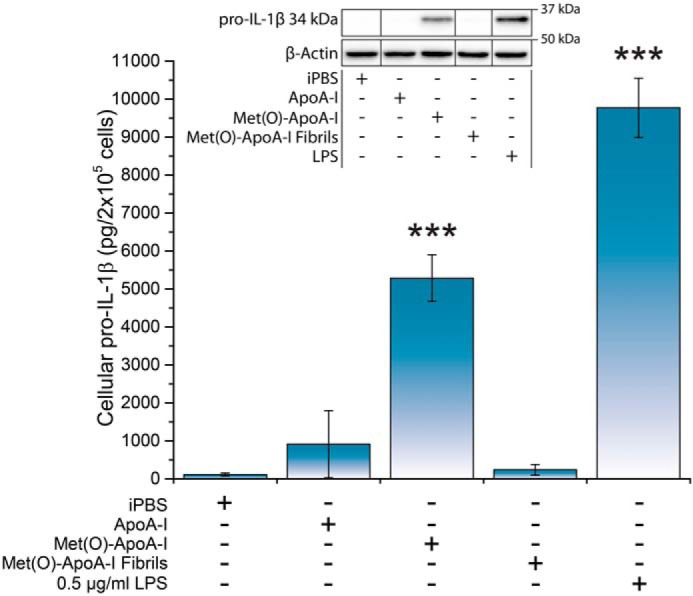
Met(O)-ApoA-I, but not Met(O)-ApoA-I amyloid fibrils, induced accumulation of pro-IL-1β (34 kDa) in mouse BMDMs. Unprimed mouse BMDMs were incubated with either buffer (iPBS), apoA-I samples (1 μm), or LPS (0.5 μg/ml) for 4 h. Pro-IL-1β levels were measured in cell extracts by Western blotting. A representative Western blot analysis with β-actin as a loading control is shown in the inset. Vertical black lines in the Western blots indicate positions where unnecessary lanes were removed from the original scans. No mature IL-1β (17 kDa) or degradation products of pro-IL-1β were detected in the cell extracts. Values are means and S.D. (error bars) of at least eight independent determinations from four independent experiments for all samples but Met(O)-ApoA-I fibrils, for which the result is the mean and S.D. of four independent determinations from two independent experiments. Probabilities that Met(O)-ApoA-I and Met(O)-ApoA-I fibril results are significantly different from ApoA-I results and that LPS results are significantly different from iPBS results were evaluated by the t test. Significance values are reported as p < 0.001 (***).
As several soluble precursors of particulate substances can induce a pro-inflammatory response (2, 21), we further characterized the effect of Met(O)-ApoA-I on secretion of two non-NLRP3-dependent pro-inflammatory cytokines, IL-6 and TNFα, in mouse macrophages, and of IL-6, TNFα, and mature IL-1β in human monocytes. Soluble Met(O)-ApoA-I promoted secretion of IL-6 and TNFα in both mouse BMDMs (Fig. 3, A and B) and primary human monocytes (Fig. 3, C and D). In experiments with human monocytes, secreted levels of TNFα and IL-6 were determined at 3 and 18 h, respectively, as these incubation times were previously established as optimal for secretion of these cytokines by LPS (45).
Figure 3.

Met(O)-ApoA-I induced secretion of IL-6 and TNFα in mouse BMDMs (A and B) and human monocytes (C and D). In human monocytes, Met(O)-ApoA-I also induced secretion of mature IL-1β (E). A and B, unprimed mouse BMDMs were incubated with either buffer (iPBS), apoA-I samples (1 μm), or LPS (0.5 μg/ml) for 4 h. Secreted IL-6 and TNFα were measured by ELISA (A and B, respectively). C–E, unprimed human monocytes were incubated with apoA-I samples (1 μm) or LPS (0.1 μg/ml) for 3, 6, and 18 h. Levels of IL-6 (18 h) (C), TNFα (3 h) (D), and IL-1β (6 and 18 h) (E), were measured in incubation media by ELISA. Values are means and S.D. (error bars) of at least three independent experiments, with at least two independent determinations for each experiment. Probabilities that Met(O)-ApoA-I and LPS results are significantly different from ApoA-I results were evaluated by the t test. Significance values are reported as p < 0.05 (*), p < 0.01 (**), and p < 0.001 (***), unless otherwise indicated.
In mouse BMDMs, Met(O)-ApoA-I induced cellular accumulation of pro-IL-1β (Fig. 2), but mature IL-1β was secreted by the cells only when an exogenous signal 2 was provided in the form of ATP (Fig. 4), confirming that in murine macrophages, Met(O)-ApoA-I acts exclusively as a signal 1, similarly to the TLR4 agonist LPS. It is known that in human monocytes, LPS provides both signal 1 and 2, triggering synthesis, processing, and secretion of IL-1β (46). Likewise, Met(O)-ApoA-I induced both cellular accumulation of pro-IL-1β (Fig. S1) and secretion of mature IL-1β (Fig. 3E). Of note, the amount of IL-1β secreted upon incubation of human monocytes with 1 μm Met(O)-ApoA-I was similar to the levels induced by 0.1 μg/ml LPS (Fig. 3E) and displayed similar kinetics, rising slowly for at least 18 h (45).
Figure 4.
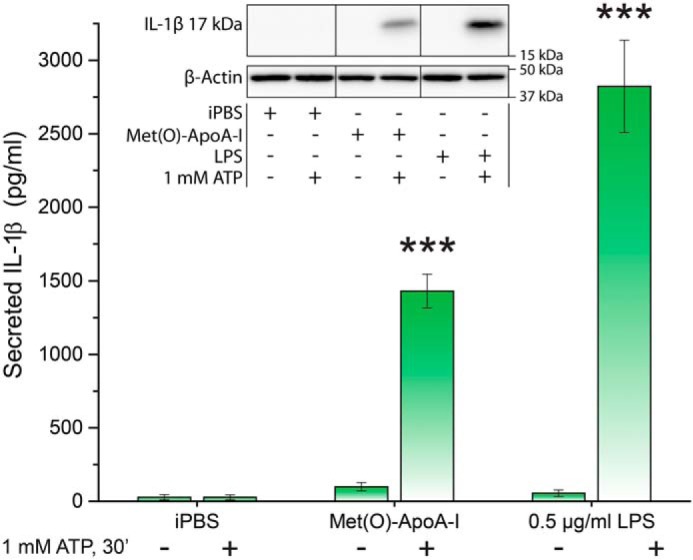
ATP induced secretion of mature IL-1β (17 kDa) from Met(O)-ApoA-I–primed macrophages. BMDMs were primed with iPBS, apoA-I samples (1 μm), or LPS (0.5 μg/ml) for 3.5 h, followed by the addition of ATP (1 mm) (as a signal 2) and 30-min incubation. Levels of secreted IL-1β were quantified in incubation media by Western blot analysis, as described under “Experimental procedures.” A representative Western blot analysis is shown in the inset. Vertical black lines in the Western blots indicate positions where unnecessary lanes were removed from the original scans. β-Actin was extracted from cells of the same well where medium originated. Values are means and S.D. (error bars) of at least four independent determinations from two independent experiments. t test significance values are reported as p < 0.001 (***) for results with ATP versus without ATP.
Met(O)-ApoA-I–induced secretion of IL-1β in human monocytes was caspase-1–dependent, as inhibition of caspase-1 activity by Ac-YVAD-CMK reduced the secreted levels of IL-1β by ∼90% (Fig. S2). This result suggests that interaction of human monocytes with Met(O)-ApoA-I activates the NLRP3 inflammasome and produces active caspase-1, the enzyme that processes pro-IL-1β into mature IL-1β (47). In summary, soluble Met(O)-ApoA-I has pro-inflammatory properties similar to those of other recognized DAMP molecules (48).
The pro-inflammatory properties of Met(O)-ApoA-I depend on oxidation of Met-86 and Met-148
The pro-inflammatory response of mouse BMDMs to Met(O)-ApoA-I, based on cellular pro-IL-1β levels, followed a dose/response trend, with maximal induction achieved for concentrations ≥1 μm (Fig. S3). Furthermore, when intermediate levels of Met(O)-86 and Met(O)-148 were produced by oxidation of apoA-I at lower H2O2 concentrations (the degree of methionine oxidation was measured by LC-MS; Table 1) and unprimed BMDMs were incubated in the presence of these preparations, the extent of cellular pro-IL-1β accumulation correlated with the fraction of oxidized Met-86 and Met-148, but not with Met(O)-112 (Fig. 5). These results indicate that the observed pro-inflammatory properties of Met(O)-ApoA-I depend on the oxidation of two (Met-86 and Met-148) of the three methionine residues of apoA-I. However, we cannot rule out that oxidation of Met-112 is necessary, but not sufficient, to produce the observed pro-inflammatory effect.
Figure 5.

Accumulation of pro-IL-1β (34 kDa) in BMDMs was proportional to the Met oxidation levels of Met(O)-ApoA-I. Different levels of Met oxidation were generated by oxidizing apoA-I with increasing concentrations of H2O2 (see Table 1). Unprimed BMDMs were incubated with apoA-I samples (1 μm) for 4 h. A, pro-IL-1β levels were measured in cell extracts by Western blotting and are expressed as a percentage of the pro-IL-1β levels induced by Met(O)-ApoA-I that was completely oxidized with a 1000-fold excess of H2O2 (absolute pro-IL-1β values obtained for this reference sample were 2086 ± 342 pmol of pro-IL-1β/2 × 105 cells (n = 4)). Results from a representative experiment (of two) are shown. Values are means and S.D. (error bars) of two independent determinations. t test significance values are reported as follows: ***, p < 0.001 for Met(O)-ApoA-I H2O2 200:1 versus iPBS, Met(O)-ApoA-I H2O2 500:1 versus Met(O)-ApoA-I H2O2 200:1, and Met(O)-ApoA-I H2O2 1000:1 versus Met(O)-ApoA-I H2O2 500:1. A representative Western blot analysis with β-actin as a loading control is shown in the inset. B, correlation between the extent of oxidation of each Met residue and the pro-IL-1β levels in the cell extracts.
Methionine oxidation solely accounts for the pro-inflammatory effect of Met(O)-ApoA-I
To test whether protein modifications other than methionine oxidation are responsible for the observed pro-inflammatory effect of Met(O)-ApoA-I, we compared cellular pro-IL-1β levels in mouse BMDMs incubated with recombinant WT apoA-I and an apoA-I variant in which all three methionine residues were replaced with leucines (3ML-ApoA-I). Whereas nonoxidized plasma-purified apoA-I did not promote any significant increase in cellular pro-IL-1β levels (Fig. 2), recombinant apoA-I induced some activation of BMDMs (Fig. 6, inset), most likely due to endotoxin contamination retained by the bacterially expressed recombinant proteins even after extensive purification (see “Experimental procedures”). To compare the pro-inflammatory effects, the results are reported as the ratio of the cellular levels of pro-IL-1β induced in BMDMs by the oxidized and the nonoxidized forms of the recombinant apoA-Is (Fig. 6). As expected, the response of macrophages to recombinant Met(O)-WT-ApoA-I was significantly higher (1.6-fold increase) than that induced by recombinant nonoxidized WT-ApoA-I (Fig. 6). In contrast, there was no increase in cellular levels of pro-IL-1β when BMDMs were incubated in the presence of oxidized 3ML-ApoA-I, compared with nonoxidized 3ML-ApoA-I (Fig. 6), indicating that methionine oxidation is responsible for the pro-inflammatory properties of Met(O)-ApoA-I.
Figure 6.
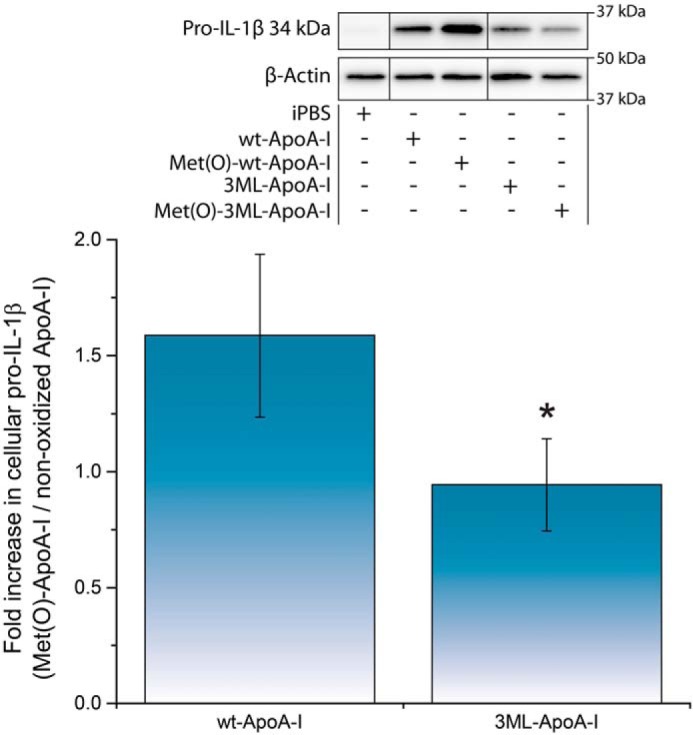
Oxidation of a Met-to-Leu apoA-I mutant (3ML-ApoA-I) failed to induce an increase in cellular pro-IL-1β (34 kDa) levels in mouse macrophages. Unprimed BMDMs were incubated with different preparations of apoA-I (1 μm) for 4 h. Pro-IL-1β levels were measured in cell extracts by Western blotting. A representative Western blot analysis with β-actin as a loading control is shown in the inset. Vertical black lines in the Western blots indicate positions where unnecessary lanes were removed from the original scans. Ratios of pro-IL-1β levels in cells incubated with oxidized apoA-I and nonoxidized apoA-I are reported. Values are means and S.D. (error bars) of at least four independent determinations from two independent experiments. t test significance values are reported as p < 0.05 (3ML-ApoA-I versus WT-ApoA-I) (*).
Evidence of the absence of contaminants in apoA-I samples isolated from human plasma
One source of concern could be that the observed pro-inflammatory response was caused by unknown PAMP/DAMP contaminants in the purified human plasma apoA-I preparations used for this study. Several lines of evidence against this case are available. First, for each oxidation experiment, nonoxidized apoA-I controls were exposed to the same experimental conditions as Met(O)-ApoA-I, except for the presence of the oxidizing agent, yet no significant response to nonoxidized apoA-I was detected in mouse BMDMs or human monocytes. Second, only very low levels of endotoxin contamination (<7 EU/ml) were measured in the human plasma apoA-I samples by the limulus amebocyte lysate kinetic-QCL assay (Fig. S4). Notably, when the same samples were oxidized, no increase in endotoxin score was detected (Fig. S4), indicating that no contaminants were introduced during oxidation and the subsequent steps. Third, comparison of the pro-inflammatory response (based on cellular pro-IL-1β levels) induced by apoA-I samples and the concentration of endotoxins in the samples revealed no correlation between these two variables at these low levels of endotoxins (Fig. S4).
It is also to be noted that the maximal cellular pro-IL-1β level induced by Met(O)-ApoA-I (≥1 μm) was always lower than the maximal level induced by LPS (Fig. S3). Further increasing the concentration of Met(O)-ApoA-I (e.g. 2 μm) did not produce a surge in the cytokine levels (Fig. S3), thus providing additional evidence that the pro-inflammatory effect of Met(O)-ApoA-I was not caused by LPS-like contaminants. Furthermore, the correlation between cellular response to partially oxidized apoA-I and the extent of oxidation of Met-86 and Met-148 (Fig. 5) corroborates the conclusion that the pro-inflammatory effect of Met(O)-ApoA-I depends on the oxidation state of specific methionine residues and not on the presence of contaminants.
The pro-inflammatory response of monocytes and macrophages to Met(O)-ApoA-I requires a functional TLR4, but not a functional CD36
As the cellular responses to Met(O)-ApoA-I and LPS were comparable in magnitude and kinetics (e.g. see Fig. 3), we tested whether TLR4, the receptor responsible for transducing the pro-inflammatory signal of LPS, also senses Met(O)-ApoA-I. In the presence of two different LPS antagonists (LPS-RS and tlrl-mkLPS), Met(O)-ApoA-I–induced secretion of mature IL-1β was reduced in human monocytes by ∼50 and ∼80%, respectively (Fig. 7), suggesting that TLR4 is necessary for signal transduction.
Figure 7.

Inhibition of Met(O)-ApoA-I– and LPS–induced secretion of mature IL-1β in human monocytes in the presence of two different LPS antagonists. Unprimed human monocytes were incubated with Met(O)-ApoA-I (1 μm) or LPS (0.1 μg/ml) for 18 h in the presence or absence of LPS antagonists tlrl-mkLPS and LPS-RS (1 μg/ml). IL-1β levels in incubation medium were measured by ELISA. Values are means and S.D. (error bars) of at least three independent determinations. Probabilities that the results in the presence of the inhibitors are significantly different from those obtained in the absence of the LPS antagonists were evaluated by the t test. Significance values are reported as p < 0.05 (*) and p < 0.01 (**).
To confirm that the pro-inflammatory effect of Met(O)-ApoA-I is TLR-mediated, we used BMDMs from MyD88−/−TRIF−/− (MyD88 × TRIF DKO) mice, in which transduction of the TLR signal is inactivated, and BMDMs from TLR4−/− mice. In both mouse models, when BMDMs were incubated with Met(O)-ApoA-I, no increase in cellular pro-IL-1β levels was detected (Fig. 8 and 9, respectively). Secretion of TNFα and IL-6 was also abolished in TLR4−/− BMDMs (Fig. 9), indicating that the pro-inflammatory signal of Met(O)-ApoA-I is transduced through a TLR4-dependent mechanism.
Figure 8.
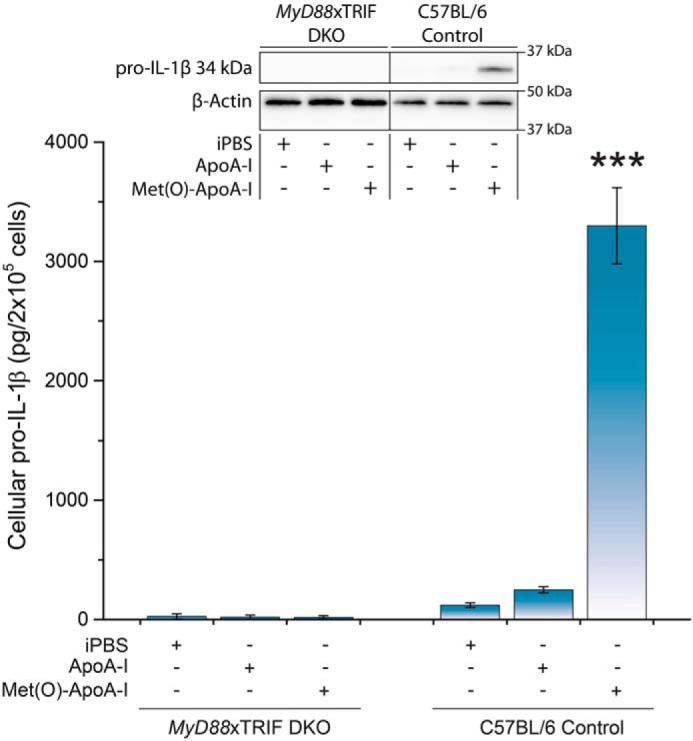
Met(O)-ApoA-I did not increase intracellular pro-IL-1β (34 kDa) levels in MyD88 × TRIF DKO macrophages. Unprimed BMDMs isolated from C57BL/6 and MyD88 × TRIF DKO mice were incubated with apoA-I samples (1 μm) for 4 h. Pro-IL-1β levels were measured in cell extracts by Western blot analysis. A representative Western blot analysis with β-actin as a loading control is shown in the inset. Vertical black lines in the Western blots indicate positions where unnecessary lanes were removed from the original scans. Values are means and S.D. (error bars) of at least three independent determinations for each mouse strain. t test significance values are reported as p < 0.001 versus ApoA-I (***).
Figure 9.
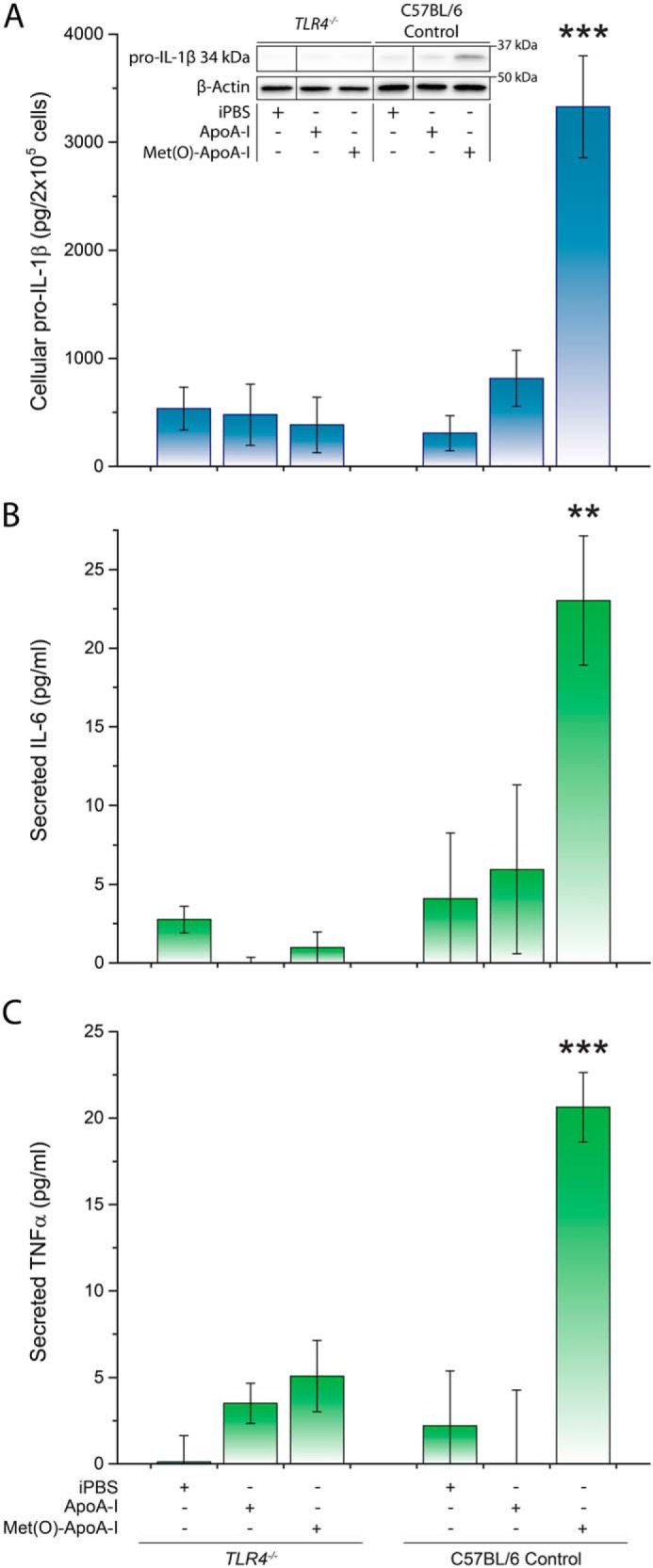
Met(O)-ApoA-I did not induce intracellular accumulation of pro-IL-1β (34 kDa) or secretion of IL-6 or TNFα in TLR4−/− macrophages. Unprimed BMDMs isolated from C57BL/6 and TLR4−/− mice were incubated with apoA-I samples (1 μm) for 4 h. Pro-IL-1β levels were measured in cell extracts by Western blot analysis (A). A representative Western blot analysis with β-actin as a loading control is shown in the inset. Vertical black lines in the Western blots indicate positions where unnecessary lanes were removed from the original scans. Secreted IL-6 and TNFα were measured by ELISA (B and C, respectively). Values are means and S.D. (error bars) of four independent determinations for each mouse strain. t test significance values are reported as p < 0.01 (**) and p < 0.001 (***) versus ApoA-I.
As the TLR4–TLR6–CD36 membrane complex has been implicated in priming and activation of the NLRP3 inflammasome by soluble species that are precursors to particulate matter (2, 11, 21), we tested whether this heterotrimer participates in the transduction of the pro-inflammatory signal of Met(O)-ApoA-I by using BMDMs from CD36−/− mice. In these cells, Met(O)-ApoA-I induced a 3.5-fold increase in cellular pro-IL-1β levels, compared with those accumulated upon incubation with nonoxidized apoA-I (Fig. S5), indicating that a functional TLR4–TLR6–CD36 complex is not necessary for the TLR4-dependent transduction of the pro-inflammatory signal of Met(O)-ApoA-I.
Loss of the pro-inflammatory properties of Met(O)-ApoA-I upon incorporation into HDL particles
To test whether lipid binding affects the pro-inflammatory activity of Met(O)-ApoA-I, we reconstituted HDL particles composed of POPC and free cholesterol (FC) with either Met(O)-ApoA-I or nonoxidized apoA-I and incubated the reconstituted HDL samples with unprimed mouse BMDMs using our routine protocol. Under these conditions, the pro-inflammatory effect of Met(O)-ApoA-I was ablated (Fig. 10), suggesting that lipidation prevents interaction of Met(O)-ApoA-I with TLR4.
Figure 10.
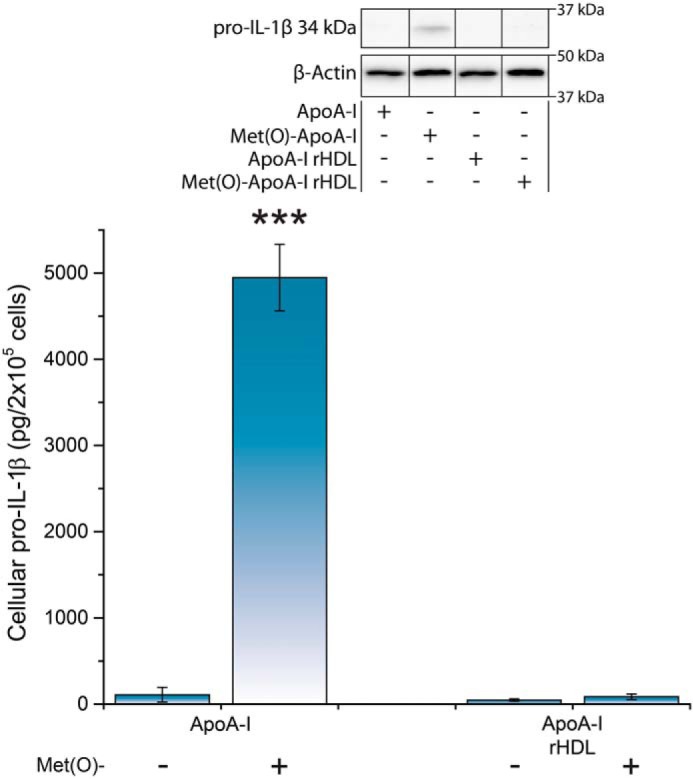
No pro-inflammatory response was detected when Met(O)-ApoA-I was provided in the form of reconstituted HDL. Unprimed BMDMs were incubated with apoA-I and apoA-I/rHDL preparations (1 μm) for 4 h. Pro-IL-1β levels were measured in cell extracts by Western blot analysis. A representative Western blot analysis with β-actin as a loading control is shown in the inset. Vertical black lines in the Western blots indicate positions where unnecessary lanes were removed from the original scans. Values are means and S.D. (error bars) of four independent determinations from two independent experiments. t test significance values are reported as p < 0.001 versus nonoxidized apoA-I (***).
Discussion
Inflammation is a leading cause of atherosclerosis (3, 49). The potent pro-inflammatory cytokine IL-1β mediates sterile inflammation in atherosclerosis through various mechanisms, including enhanced expression of adhesion and chemotactic molecules (50, 51) that in turn, drive the recruitment of monocytes/macrophages to early atherosclerotic lesions. HDL exerts an anti-inflammatory function through several mechanisms (52), one being the ability to inhibit IL-1β production in monocytes/macrophages activated by PAMP signals (53). This anti-inflammatory property might be mediated directly by apoA-I, rather than the whole HDL particle, as lipid-free apoA-I strongly inhibits release of IL-1β from monocytes/macrophages activated by contact with stimulated T lymphocytes (53). Our data are consistent with this hypothesis, as nonoxidized apoA-I was able to reduce the cellular levels of pro-IL-1β in LPS-primed BMDMs (Fig. 1B). Taken together, these observations indicate that HDL and apoA-I act as anti-inflammatory factors in both acute and chronic inflammation conditions (54, 55). However, HDL oxidation by inflammation-induced factors, such as macrophage-produced myeloperoxidase (55, 56), may impact the anti-inflammatory function of HDL and even transform HDL or its oxidized components into pro-inflammatory molecules (56–60). The Hazen and Smith laboratories (58) for example, reported a significant increase in endothelial cell surface expression of the cell adhesion molecule VCAM-1 upon incubation in the presence of myeloperoxidase-oxidized HDL particles. Other DAMPs also acquire pro-inflammatory properties upon oxidation. For instance, extracellular high mobility group protein B1 (HMGB1) recruits leukocytes and induces them to release pro-inflammatory mediators by switching among multiple oxidation states (61).
In this study, we investigated how oxidation of methionine to methionine sulfoxide (Met(O)) imparts pro-inflammatory properties to lipid-free apoA-I. For our experiments, we used a reproducible and well-characterized oxidized human plasma apoA-I (Met(O)-ApoA-I), in which all three methionine residues are oxidized to Met(O) by an excess of H2O2 (34, 35, 44).
When mouse BMDMs and human monocytes were incubated in the presence of Met(O)-ApoA-I, intracellular accumulation of pro-IL-1β and secretion of IL-6 and TNFα were strongly induced. Moreover, in human monocytes, Met(O)-ApoA-I also promoted secretion of mature IL-1β at levels as high as those produced by the bacterial endotoxin LPS at 0.1 μg/ml. Notably, all three cytokines (IL-1β, IL-6, and TNFα) are found in human atherosclerotic plaques and are believed to be directly implicated in atherogenesis (62, 63).
Despite the rule dictating that signals 1 are required for priming, whereas signals 2 induce inflammasome activation and IL-1β secretion, distinctions among signals 1 and 2 are not always so definite, as some DAMPs, such as oxidized LDL (21, 64) and electronegative LDL (65) (a minor LDL subfraction), can induce IL-1β synthesis, processing, and secretion. It is worth noting that both forms of LDL are involved in the pathophysiology of atherosclerosis. Furthermore, various PAMPs have been shown to trigger synthesis, processing, and secretion of IL-1β in human monocytes independently of the presence of signals 2 (46, 66). Our results indicate that Met(O)-ApoA-I is a very potent DAMP that can induce synthesis, processing, and secretion of IL-1β, IL-6, and TNFα in human monocytes and could then promote sterile inflammation relevant to atherosclerosis.
Endotoxin analysis (Fig. S4) and other lines of evidence presented under “Results” indicate that the pro-inflammatory effect of Met(O)-ApoA-I is not due to endotoxin contamination of the samples used in this study. Further corroborating this evidence, the pro-inflammatory effect was dependent exclusively on oxidation of methionine residues, as illustrated by the lack of response to an oxidized apoA-I variant in which all methionines were substituted with leucines (Fig. 6). Moreover, the pro-inflammatory response was proportional to the degree of oxidation of Met(O)-86 and Met(O)-148 (Fig. 5). Previously, we found that oxidation of Met-86 and Met-148 was also primarily responsible for the transformation of apoA-I into an amyloidogenic peptide (34). Thus, the overall structural changes promoted by Met-86 and Met-148 oxidation that are responsible for protein destabilization and amyloid formation (34, 35) could also facilitate the interaction of apoA-I with macrophages (35) and induce their pro-inflammatory response, as shown in the present study.
Our results also demonstrate that the response of macrophages to Met(O)-ApoA-I depends on the activity of TLR4 (Figs. 7–9) but does not require functional CD36 (Fig. S5), the scavenger receptor implicated in the activation of the NLRP3 inflammasome upon interaction of mouse macrophages with other amyloid precursor peptides, such as Aβ and IAPP (2, 21). The TLR4-dependent pro-inflammatory effect of Met(O)-ApoA-I is therefore distinct from the mechanism of action of other soluble amyloid precursor peptides, wherein activation of the NLRP3 inflammasome requires CD36-dependent cellular uptake and lysosomal damage (21). Notably, when Met(O)-ApoA-I was incorporated into HDL, its pro-inflammatory effect was completely suppressed (Fig. 10).
Although apoA-I is predominantly associated with HDL in circulation, high levels of lipid-free apoA-I have been detected in atherosclerotic lesions (40–43). The methionine residues of this lipid-free apoA-I could be heavily oxidized by the local action of macrophage-secreted myeloperoxidase (31). Additionally, HDL-associated apoA-I can be selectively oxidized at methionine residues by reactive oxygen species (67) and released from HDL in the exchange reaction between HDL and lipid-free apoA-I (68), providing another possible source of apoA-I oxidized at methionine residues. We hypothesize that local production of this powerful DAMP at the level of the arterial wall would promote further inflammation, thus exacerbating atherosclerosis progression.
Unexpectedly, when Met(O)-ApoA-I was incorporated into amyloid fibrils, its pro-inflammatory effect on mouse BMDMs was also drastically reduced (Figs. 1 and 2). Similar results were obtained with human monocytes (data not shown). Particulate substances in the form of amyloids are often believed to have deleterious cellular effects. However, transformation of pro-inflammatory soluble Met(O)-ApoA-I into more inert amyloid material may reduce, rather than increase, the pro-inflammatory burden. This is reminiscent of Alzheimer's disease, in which pre-fibrillar soluble Aβ species, rather than mature amyloid fibrils, are the most cytotoxic and pro-inflammatory molecules (21, 69–71). Our results suggest the intriguing hypothesis that amyloid formation by Met(O)-ApoA-I could actually represent a safety mechanism that eliminates dangerous inflammatory species (i.e. soluble apoA-I with oxidized methionines) by inactivating them in the form of solid aggregates. Such aggregates would accumulate in the atherosclerotic lesions and eventually enlarge the mass of the atheroma, with potential long-term deleterious consequences, but in the short term, this process could reduce the immediate danger represented by high concentrations of active pro-inflammatory oxidized apoA-I in the artery walls (40).
In conclusion, our results indicate that oxidation of the methionine residues of apoA-I produces a very potent DAMP. To the best of our knowledge, this is the first report of a DAMP capable of inducing secretion of IL-1β at levels similar to those induced by the PAMP of reference (i.e. LPS). Of note, this pro-inflammatory activity is mediated by the same receptor, TLR4, that is largely responsible for the cellular response to LPS (72, 73) and a series of DAMPs (48, 74). The pro-inflammatory activity of Met(O)-ApoA-I is effectively abrogated upon amyloid formation. In the arteries, however, amyloid formation by apoA-I with oxidized methionines could be a slow process, and soluble oxidized apoA-I may reside in the subendothelial space long enough to interact with cell types implicated in inflammation and in the development of atherosclerosis.
Arterial deposition of oxidized apoA-I–rich amyloids may manifest within two, not necessarily adverse, scenarios. Atherosclerosis-associated amyloids could be a marker of severe inflammation promoted by high levels of pro-inflammatory pre-fibrillar apoA-I with oxidized methionine residues. Alternatively, atherosclerosis-associated amyloids could be a sign of reduced inflammation in conditions that favor amyloid formation rather than the sustained presence of pro-inflammatory oxidized apoA-I. As these species could unleash their pro-inflammatory potential in the subendothelial space of the arteries, more research aimed at improving our understanding of their pro-atherogenic effect in vivo is warranted.
Experimental procedures
Plasma apoA-I
Plasma of de-identified healthy donors was obtained from the Clinical Laboratory of UCSF Benioff Children's Hospital Oakland. HDL was isolated from EDTA-treated plasma, and apoA-I was isolated and purified from HDL as described (34, 75). Purified protein samples were stored frozen in 6 m guanidine chloride buffer until use. Before experiments, stored proteins were refolded by extensive dialysis against the appropriate experiment-specific buffer.
Production and purification of recombinant proteins used for the experiments reported in Fig. 6
WT recombinant apoA-I (WT-ApoA-I) and the Met-to-Leu mutant (3ML-ApoA-I) were produced in Escherichia coli, purified by affinity chromatography, and refolded by extensive dialysis against isotonic PBS (iPBS; 10 mm Na2HPO4, 2 mm KH2PO4, 137 mm NaCl, 2.7 mm KCl, pH 7.4) as described previously (34, 35, 75). To reduce residual endotoxins, recombinant protein preparations were further purified by SEC on a SuperdexTM 200 Increase 10/300 GL column (GE Healthcare) controlled by an AKTA Pure FPLC system (GE Healthcare). The flow rate was 0.9 ml/min, and the elution buffer was iPBS. Pooled fractions were dialyzed against a total of 6 liters of final buffer with two buffer exchanges. After purification, endotoxin contamination in recombinant protein preparations (as measured by the limulus amebocyte lysate kinetic-QCL assay) was reduced by 4 orders of magnitude from levels as high as 80,000,000 EU/ml before purification to less than 25,000 EU/ml. Purified protein samples were stored frozen in 6 m guanidine chloride buffer and refolded by extensive dialysis against iPBS before use.
Endotoxin test
Endotoxin levels in all protein preparations (plasma-purified and recombinant) were evaluated by the limulus amebocyte lysate kinetic-QCL assay (Lonza), according to the manufacturer's instructions. Kinetic measurements at 405 nm were performed on a BioTek Synergy H1 Hybrid Multi-Mode Reader thermostated at 37 °C. Note that 1 EU = 0.1–0.2 ng of endotoxin/ml of solution, depending on the reference standard used (76).
Protein oxidation
ApoA-I samples (1.5–2.0 mg/ml) were oxidized with a molar excess of H2O2 (i.e. 200:1, 500:1, and 1000:1 H2O2/protein) as described previously (34). After oxidation, excess H2O2 was eliminated by extensive dialysis (2 days with two buffer exchanges) against iPBS (34). The iPBS buffer at the termination of the last dialysis was stored for use as iPBS in the presented experiments. Samples labeled as ApoA-I were incubated in the same oxidation conditions, but in the absence of H2O2, and dialyzed.
BMDM preparation and incubation with apoA-I samples
All mouse experiments were approved by the UCSF Benioff Children's Hospital Oakland Institutional Animal Care and Use Committee. Bone marrow cells were isolated from 3–6-month-old mice and differentiated according to the procedure described previously (35). For experiments involving MyD88−/−TRIF−/− (77), TLR4−/− (77), and CD36−/− (78) knockout mice in the C57BL/6 background, WT control cells were derived from C57BL/6 mice. For all other experiments, BMDMs were obtained from mice of the FVB/N strain. Replated BMDMs were washed twice with Dulbecco's PBS and treated with 0.1 ml/well (96-well plate) of tested reagents diluted in Hanks' buffered salt solution (HBSS) without phenol red and containing 10 mm HEPES, 100 units/ml penicillin, and 0.1 mg/ml streptomycin. Before each experiment, the HBSS-HEPES medium was preincubated in the CO2 incubator overnight, its pH was adjusted to 7.3, and it was sterilized by filtration. After incubation, the plate was centrifuged at 600 × g for 5 min at 4 °C, and incubation media were transferred to 0.6-ml tubes containing protease inhibitor cocktail (Bimake), fast-frozen, and stored at −80 °C until analyzed. The medium-free wells were washed with PBS and centrifuged, and the PBS was discarded. After three PBS washes, cells were solubilized in radioimmune precipitation assay buffer (Boston BioProducts) (0.1 ml/well) containing protease inhibitor cocktail. The plate was centrifuged at 1000 × g for 10 min, and cell extracts were transferred to microtubes and fast-frozen.
Human monocyte preparation and incubation with apoA-I samples
Peripheral blood mononuclear cells (PBMCs) were isolated from freshly drawn heparinized blood by Ficoll–Paque (Sigma-Aldrich) gradients. PBMCs were plated on 96-well plates at a density of 5 × 105 cells/well. The monocyte population was enriched by allowing PBMCs to adhere to the plate for 45 min. Adherent cells were washed twice with warm Dulbecco's PBS and incubated either with buffer (iPBS), LPS (100 ng/ml), apoA-I (1 μm), or Met(O)-ApoA-I (1 μm) in HBSS without phenol red and containing 10 mm HEPES, 100 units/ml penicillin, and 0.1 mg/ml streptomycin. When indicated, the following inhibitors were added to the cell cultures 30 min before treatment: caspase-1 inhibitor Ac-YVAD-CMK (Cayman Chemical; 20 μm,) and LPS antagonists tlrl-mkLPS and LPS-RS (Invivogen; 1 μg/ml). At 3, 6, and 18 h, supernatants were recovered for ELISA analysis.
Western blot analysis
Thawed media and cell extracts were centrifuged at 1000 × g for 5 min and 14,000 × g for 10 min, respectively, and the supernatants were analyzed by Western blotting after separation on 1.5-mm 13% SDS-PAGE (25 and 10 μl/well, respectively). After transfer (Trans-Blot Turbo Transfer System, Bio-Rad) and blocking (5% nonfat milk in Tris-buffered saline, 0.1% Tween 20 (TBS-T)), polyvinylidene difluoride membranes were incubated with goat polyclonal anti-mouse IL-1β antibody (R&D Systems; 0.1 μg/ml) followed by HRP-conjugated bovine anti-goat IgG (H+L) (AffiniPure, Jackson ImmunoResearch Laboratories; 1:3000 dilution) or with 3ZD anti-human IL-1β mAb (IgG1; obtained from the NCI, National Institutes of Health, Biological Resources Branch; 1 μg/ml) followed by HRP-conjugated rabbit anti-mouse IgG (Dako; 1:10000 dilution). The antibodies were diluted with Can-Get-Signal (Toyobo) for medium analysis and with 5 mg/ml BSA in TBS-T (BSA-TBS-T) for analysis of cellular extracts. ECL (BMDMs, Advansta Western Bright; PBMCs, Clarity Western ECL Substrate; Bio-Rad) generated light was detected on a FluorChem Q instrument (Alpha Innotech) (BMDM) or an Alliance Imaging system (Uvitec) (PBMCs). Partially saturated images were quantified with ImageJ version 1.51a software (National Institutes of Health). One, two, or four IL-1β standards (Cell Signaling Technology) were utilized to quantify the cytokine. Standard curves were practically linear in the assayed range (Can-Get-Signal, 0–20 pg; BSA-TBS-T, 0–1 ng; Fig. S6). Pro-IL-1β values were obtained by multiplying IL-1β standard–derived numbers by 1.778 (ratio of masses of pro-IL-1β (34 kDa) and IL-1β (17 kDa)). After triple washing with TBS-T, the same blot was stained for β-actin (mouse monoclonal (Sigma); 1:10,000 dilution) followed by HRP-conjugated goat anti-mouse IgG (H+L) (Immuno-Pure, Pierce; 1:7500 dilution) (BMDM) or for human α-tubulin mAb (Sigma-Aldrich; 0.5 μg/ml) followed by HRP-conjugated rabbit anti-mouse IgG (Dako; 1:10,000 dilution) (PBMCs).
ELISA
Secreted IL-6, TNFα, and mature IL-1β were measured with the corresponding DuoSet® ELISA Development Kits (R&D Systems) according to the manufacturer's instructions.
Quantification of Met oxidation by LC-MS
ApoA-I preparations were digested with trypsin, and the digests were analyzed by LC-MS using an AdvanceBio Peptide Map column (100 × 2.1 mm; Agilent Technologies) on a 1290 Infinity LC system (Agilent Technologies) interfaced with an Agilent 6490 triple quadrupole mass spectrometer (Agilent Technologies), as described previously (35).
Met(O)-ApoA-I amyloid fibril formation
Met(O)-ApoA-I obtained by oxidation with a 1000-fold molar excess of H2O2 was diluted to 1.0 mg/ml and incubated in fibrillation buffer (10 mm sodium phosphate, pH 6.0) at 37 °C with continuous vortexing for 72 h, as described before (34, 35). The resulting sample, containing aggregated protein, was used for cellular experiments upon appropriate dilution with cell incubation buffer.
HDL reconstitution
ApoA-I and Met(O)-ApoA-I were used to reconstitute HDL particles by the sodium cholate dialysis method, as described previously (68, 79). To reconstitute predominantly 9.6-nm HDL particles, apoA-I samples were added to a POPC/FC solution to final molar ratios of POPC/FC/apoA-I equal to 80:4:1. The reconstituted particles were analyzed by nondenaturing gradient gel electrophoresis (4–20% acrylamide gels) before use.
Statistical analysis
Data are presented as mean ± S.D. Data were analyzed using GraphPad Prism software, by two-tailed t test. Significance is expressed as p < 0.05 (*), p < 0.01 (**), and p < 0.001 (***), unless otherwise indicated. Throughout the paper, the number of determinations is defined as the number of independent replications within the same experiment.
Author contributions
G. C. conceived and coordinated the study. G. C. and A. W. designed, performed, and analyzed all of the experiments except for the human cell experiments, which were designed, executed, and analyzed by S. C. and A. R. R. L. and S. Y. assisted with cytokine level analysis. G. C., A. W., and A. R. wrote the paper.
Supplementary Material
Acknowledgments
We are indebted to Dr. Gordon L. Watson (UCSF BCHO) for contributing the FVB/N mice and to Dorothy Tabron (UCSF BCHO) for assisting with bone marrow extraction. We thank Prof. Andreas Stahl and Prof. Gregory Barton (University of California, Berkeley) for generously providing the CD36−/− (and C57BL/6 controls) and the MyD88−/−TRIF−/− and TLR4−/− mice (and C57BL/6 controls), respectively. We are also grateful to Gary K. L. Chan, Nancy J. Li, Ayuka P. Inoue, and Jaclyn C. Wong for technical assistance. Acquisition of the Agilent 6490 triple quadrupole mass spectrometer was supported by National Institutes of Health Grant 1S10OD018070.
This work was supported in whole or part by National Institutes of Health Grant R01 HL113059 (to G. C.); Japan Society for the Promotion of Science's Invitation Fellowship Program for Research in Japan (L-14559) (to G. C. and S. Y.); Associazione Italiana per la Ricerca sul Cancro (IG 2016–15434) and Fondazione Telethon (GGP14144) (to A. R.); Italian Ministry of Health “Young Investigators” Grant GR-2016-02363630 (to S. C.); MEXT-Supported Program for the Strategic Research Foundation at Private Universities Grant S1201007; and MEXT Japan Grants-in-Aid 24614018, 26461370, and 15H02903 (to S. Y. and R. L.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S6.
- Aβ
- β-amyloid
- LPS
- lipopolysaccharide
- POPC
- 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine
- EU
- endotoxin units
- HBSS
- Hanks' buffered salt solution
- IAPP
- islet amyloid polypeptide
- LDL
- low-density lipoprotein
- TLR
- Toll-like receptor
- DAMP
- danger-associated molecular pattern
- PAMP
- pathogen-associated molecular pattern
- apoA-I
- apolipoprotein A-I
- BMDM
- bone marrow–derived macrophage
- IL
- interleukin
- TNFα
- tumor necrosis factor α
- FC
- free cholesterol
- HDL
- high-density lipoprotein
- iPBS
- isotonic PBS
- HRP
- horseradish peroxidase.
References
- 1. Halle A., Hornung V., Petzold G. C., Stewart C. R., Monks B. G., Reinheckel T., Fitzgerald K. A., Latz E., Moore K. J., and Golenbock D. T. (2008) The NALP3 inflammasome is involved in the innate immune response to amyloid-β. Nat. Immunol. 9, 857–865 10.1038/ni.1636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Masters S. L., Dunne A., Subramanian S. L., Hull R. L., Tannahill G. M., Sharp F. A., Becker C., Franchi L., Yoshihara E., Chen Z., Mullooly N., Mielke L. A., Harris J., Coll R. C., Mills K. H., Mok K. H., Newsholme P., Nuñez G., Yodoi J., Kahn S. E., Lavelle E. C., and O'Neill L. A. (2010) Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1β in type 2 diabetes. Nat. Immunol. 11, 897–904 10.1038/ni.1935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Haneklaus M., and O'Neill L. A. (2015) NLRP3 at the interface of metabolism and inflammation. Immunol. Rev. 265, 53–62 10.1111/imr.12285 [DOI] [PubMed] [Google Scholar]
- 4. Wang M., Jiang L., Monticone R. E., and Lakatta E. G. (2014) Proinflammation: the key to arterial aging. Trends Endocrinol. Metab. 25, 72–79 10.1016/j.tem.2013.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Eren E., Ellidag H. Y., Aydin O., and Yilmaz N. (2015) HDL functionality and crystal-based sterile inflammation in atherosclerosis. Clin. Chim. Acta 439, 18–23 10.1016/j.cca.2014.09.024 [DOI] [PubMed] [Google Scholar]
- 6. Ueno H., Koyama H., Shoji T., Monden M., Fukumoto S., Tanaka S., Otsuka Y., Mima Y., Morioka T., Mori K., Shioi A., Yamamoto H., Inaba M., and Nishizawa Y. (2010) Receptor for advanced glycation end-products (RAGE) regulation of adiposity and adiponectin is associated with atherogenesis in apoE-deficient mouse. Atherosclerosis 211, 431–436 10.1016/j.atherosclerosis.2010.04.006 [DOI] [PubMed] [Google Scholar]
- 7. Hirata Y., Kurobe H., Higashida M., Fukuda D., Shimabukuro M., Tanaka K., Higashikuni Y., Kitagawa T., and Sata M. (2013) HMGB1 plays a critical role in vascular inflammation and lesion formation via Toll-like receptor 9. Atherosclerosis 231, 227–233 10.1016/j.atherosclerosis.2013.09.010 [DOI] [PubMed] [Google Scholar]
- 8. Goulopoulou S., McCarthy C. G., and Webb R. C. (2016) Toll-like receptors in the vascular system: sensing the dangers within. Pharmacol. Rev. 68, 142–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. El Khoury J. B., Moore K. J., Means T. K., Leung J., Terada K., Toft M., Freeman M. W., and Luster A. D. (2003) CD36 mediates the innate host response to β-amyloid. J. Exp. Med. 197, 1657–1666 10.1084/jem.20021546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Stewart C. R., Tseng A. A., Mok Y. F., Staples M. K., Schiesser C. H., Lawrence L. J., Varghese J. N., Moore K. J., and Howlett G. J. (2005) Oxidation of low-density lipoproteins induces amyloid-like structures that are recognized by macrophages. Biochemistry 44, 9108–9116 10.1021/bi050497v [DOI] [PubMed] [Google Scholar]
- 11. Stewart C. R., Stuart L. M., Wilkinson K., van Gils J. M., Deng J., Halle A., Rayner K. J., Boyer L., Zhong R., Frazier W. A., Lacy-Hulbert A., El Khoury J., Golenbock D. T., and Moore K. J. (2010) CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat. Immunol. 11, 155–161 10.1038/ni.1836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rajamäki K., Lappalainen J., Oörni K., Välimäki E., Matikainen S., Kovanen P. T., and Eklund K. K. (2010) Cholesterol crystals activate the NLRP3 inflammasome in human macrophages: a novel link between cholesterol metabolism and inflammation. PLoS One 5, e11765 10.1371/journal.pone.0011765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Duewell P., Kono H., Rayner K. J., Sirois C. M., Vladimer G., Bauernfeind F. G., Abela G. S., Franchi L., Nuñez G., Schnurr M., Espevik T., Lien E., Fitzgerald K. A., Rock K. L., Moore K. J., Wright S. D., Hornung V., and Latz E. (2010) NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 464, 1357–1361 10.1038/nature08938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Medeiros L. A., Khan T., El Khoury J. B., Pham C. L., Hatters D. M., Howlett G. J., Lopez R., O'Brien K. D., and Moore K. J. (2004) Fibrillar amyloid protein present in atheroma activates CD36 signal transduction. J. Biol. Chem. 279, 10643–10648 10.1074/jbc.M311735200 [DOI] [PubMed] [Google Scholar]
- 15. Tall A. R., and Yvan-Charvet L. (2015) Cholesterol, inflammation and innate immunity. Nat. Rev. Immunol. 15, 104–116 10.1038/nri3793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Andrei C., Dazzi C., Lotti L., Torrisi M. R., Chimini G., and Rubartelli A. (1999) The secretory route of the leaderless protein interleukin 1beta involves exocytosis of endolysosome-related vesicles. Mol. Biol. Cell 10, 1463–1475 10.1091/mbc.10.5.1463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gardella S., Andrei C., Lotti L. V., Poggi A., Torrisi M. R., Zocchi M. R., and Rubartelli A. (2001) CD8+ T lymphocytes induce polarized exocytosis of secretory lysosomes by dendritic cells with release of interleukin-1β and cathepsin D. Blood 98, 2152–2159 10.1182/blood.V98.7.2152 [DOI] [PubMed] [Google Scholar]
- 18. Schroder K., and Tschopp J. (2010) The inflammasomes. Cell 140, 821–832 10.1016/j.cell.2010.01.040 [DOI] [PubMed] [Google Scholar]
- 19. Mitchell J. A., Paul-Clark M. J., Clarke G. W., McMaster S. K., and Cartwright N. (2007) Critical role of Toll-like receptors and nucleotide oligomerisation domain in the regulation of health and disease. J. Endocrinol. 193, 323–330 10.1677/JOE-07-0067 [DOI] [PubMed] [Google Scholar]
- 20. Rubartelli A., and Lotze M. T. (2007) Inside, outside, upside down: damage-associated molecular-pattern molecules (DAMPs) and redox. Trends Immunol. 28, 429–436 10.1016/j.it.2007.08.004 [DOI] [PubMed] [Google Scholar]
- 21. Sheedy F. J., Grebe A., Rayner K. J., Kalantari P., Ramkhelawon B., Carpenter S. B., Becker C. E., Ediriweera H. N., Mullick A. E., Golenbock D. T., Stuart L. M., Latz E., Fitzgerald K. A., and Moore K. J. (2013) CD36 coordinates NLRP3 inflammasome activation by facilitating intracellular nucleation of soluble ligands into particulate ligands in sterile inflammation. Nat. Immunol. 14, 812–820 10.1038/ni.2639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cornwell G. G. 3rd, Murdoch W. L., Kyle R. A., Westermark P., and Pitkänen P. (1983) Frequency and distribution of senile cardiovascular amyloid. A clinicopathologic correlation. Am. J. Med. 75, 618–623 10.1016/0002-9343(83)90443-6 [DOI] [PubMed] [Google Scholar]
- 23. Mucchiano G., Cornwell G. G. 3rd, and Westermark P. (1992) Senile aortic amyloid: evidence for two distinct forms of localized deposits. Am. J. Pathol. 140, 871–877 [PMC free article] [PubMed] [Google Scholar]
- 24. Westermark P., Mucchiano G., Marthin T., Johnson K. H., and Sletten K. (1995) Apolipoprotein A1-derived amyloid in human aortic atherosclerotic plaques. Am. J. Pathol. 147, 1186–1192 [PMC free article] [PubMed] [Google Scholar]
- 25. Mucchiano G. I., Häggqvist B., Sletten K., and Westermark P. (2001) Apolipoprotein A-1-derived amyloid in atherosclerotic plaques of the human aorta. J. Pathol. 193, 270–275 10.1002/1096-9896(2000)9999:9999<::AID-PATH753>3.0.CO;2-S [DOI] [PubMed] [Google Scholar]
- 26. Mucchiano G. I., Jonasson L., Häggqvist B., Einarsson E., and Westermark P. (2001) Apolipoprotein A-I-derived amyloid in atherosclerosis: its association with plasma levels of apolipoprotein A-I and cholesterol. Am. J. Clin. Pathol. 115, 298–303 10.1309/PJE6-X9E5-LX6K-NELY [DOI] [PubMed] [Google Scholar]
- 27. Röcken C., Tautenhahn J., Bühling F., Sachwitz D., Vöckler S., Goette A., and Bürger T. (2006) Prevalence and pathology of amyloid in atherosclerotic arteries. Arterioscler. Thromb. Vasc. Biol. 26, 676–677 10.1161/01.ATV.0000201930.10103.be [DOI] [PubMed] [Google Scholar]
- 28. Kristen A. V., Schnabel P. A., Winter B., Helmke B. M., Longerich T., Hardt S., Koch A., Sack F. U., Katus H. A., Linke R. P., and Dengler T. J. (2010) High prevalence of amyloid in 150 surgically removed heart valves–a comparison of histological and clinical data reveals a correlation to atheroinflammatory conditions. Cardiovasc. Pathol. 19, 228–235 10.1016/j.carpath.2009.04.005 [DOI] [PubMed] [Google Scholar]
- 29. Audet A., Côté N., Couture C., Bossé Y., Després J. P., Pibarot P., and Mathieu P. (2012) Amyloid substance within stenotic aortic valves promotes mineralization. Histopathology 61, 610–619 10.1111/j.1365-2559.2012.04265.x [DOI] [PubMed] [Google Scholar]
- 30. Howlett G. J., and Moore K. J. (2006) Untangling the role of amyloid in atherosclerosis. Curr. Opin. Lipidol. 17, 541–547 10.1097/01.mol.0000245260.63505.4f [DOI] [PubMed] [Google Scholar]
- 31. Shao B., Pennathur S., and Heinecke J. W. (2012) Myeloperoxidase targets apolipoprotein A-I, the major high density lipoprotein protein, for site-specific oxidation in human atherosclerotic lesions. J. Biol. Chem. 287, 6375–6386 10.1074/jbc.M111.337345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zheng L., Settle M., Brubaker G., Schmitt D., Hazen S. L., Smith J. D., and Kinter M. (2005) Localization of nitration and chlorination sites on apolipoprotein A-I catalyzed by myeloperoxidase in human atheroma and associated oxidative impairment in ABCA1-dependent cholesterol efflux from macrophages. J. Biol. Chem. 280, 38–47 10.1074/jbc.M407019200 [DOI] [PubMed] [Google Scholar]
- 33. Nicholls S. J., Zheng L., and Hazen S. L. (2005) Formation of dysfunctional high-density lipoprotein by myeloperoxidase. Trends Cardiovasc. Med. 15, 212–219 10.1016/j.tcm.2005.06.004 [DOI] [PubMed] [Google Scholar]
- 34. Chan G. K., Witkowski A., Gantz D. L., Zhang T. O., Zanni M. T., Jayaraman S., and Cavigiolio G. (2015) Myeloperoxidase-mediated methionine oxidation promotes an amyloidogenic outcome for apolipoprotein A-I. J. Biol. Chem. 290, 10958–10971 10.1074/jbc.M114.630442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Witkowski A., Chan G. K. L., Boatz J. C., Li N. J., Inoue A. P., Wong J. C., van der Wel P. C. A., and Cavigiolio G. (2018) Methionine oxidized apolipoprotein A-I at the crossroads of HDL biogenesis and amyloid formation. FASEB J. 32, 3149–3165 10.1096/fj.201701127R [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Liang H. Q., Rye K. A., and Barter P. J. (1995) Cycling of apolipoprotein A-I between lipid-associated and lipid-free pools. Biochim. Biophys. Acta 1257, 31–37 10.1016/0005-2760(95)00055-H [DOI] [PubMed] [Google Scholar]
- 37. Liang H. Q., Rye K. A., and Barter P. J. (1994) Dissociation of lipid-free apolipoprotein A-I from high density lipoproteins. J. Lipid Res. 35, 1187–1199 [PubMed] [Google Scholar]
- 38. Kee P., Rye K. A., Taylor J. L., Barrett P. H., and Barter P. J. (2002) Metabolism of apoA-I as lipid-free protein or as component of discoidal and spherical reconstituted HDLs: studies in wild-type and hepatic lipase transgenic rabbits. Arterioscler. Thromb. Vasc. Biol. 22, 1912–1917 10.1161/01.ATV.0000038485.94020.7F [DOI] [PubMed] [Google Scholar]
- 39. Miyazaki O., Ogihara J., Fukamachi I., and Kasumi T. (2014) Evidence for the presence of lipid-free monomolecular apolipoprotein A-1 in plasma. J. Lipid Res. 55, 214–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. DiDonato J. A., Huang Y., Aulak K. S., Even-Or O., Gerstenecker G., Gogonea V., Wu Y., Fox P. L., Tang W. H., Plow E. F., Smith J. D., Fisher E. A., and Hazen S. L. (2013) Function and distribution of apolipoprotein A1 in the artery wall are markedly distinct from those in plasma. Circulation 128, 1644–1655 10.1161/CIRCULATIONAHA.113.002624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Huang Y., DiDonato J. A., Levison B. S., Schmitt D., Li L., Wu Y., Buffa J., Kim T., Gerstenecker G. S., Gu X., Kadiyala C. S., Wang Z., Culley M. K., Hazen J. E., Didonato A. J., et al. (2014) An abundant dysfunctional apolipoprotein A1 in human atheroma. Nat. Med. 20, 193–203 10.1038/nm.3459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. DiDonato J. A., Aulak K., Huang Y., Wagner M., Gerstenecker G., Topbas C., Gogonea V., DiDonato A. J., Tang W. H., Mehl R. A., Fox P. L., Plow E. F., Smith J. D., Fisher E. A., and Hazen S. L. (2014) Site-specific nitration of apolipoprotein A-I at tyrosine 166 is both abundant within human atherosclerotic plaque and dysfunctional. J. Biol. Chem. 289, 10276–10292 10.1074/jbc.M114.556506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Shao B., Tang C., Sinha A., Mayer P. S., Davenport G. D., Brot N., Oda M. N., Zhao X. Q., and Heinecke J. W. (2014) Humans with atherosclerosis have impaired ABCA1 cholesterol efflux and enhanced high-density lipoprotein oxidation by myeloperoxidase. Circ. Res. 114, 1733–1742 10.1161/CIRCRESAHA.114.303454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wong Y. Q., Binger K. J., Howlett G. J., and Griffin M. D. (2010) Methionine oxidation induces amyloid fibril formation by full-length apolipoprotein A-I. Proc. Natl. Acad. Sci. U.S.A. 107, 1977–1982 10.1073/pnas.0910136107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lavieri R., Piccioli P., Carta S., Delfino L., Castellani P., and Rubartelli A. (2014) TLR costimulation causes oxidative stress with unbalance of proinflammatory and anti-inflammatory cytokine production. J. Immunol. 192, 5373–5381 10.4049/jimmunol.1303480 [DOI] [PubMed] [Google Scholar]
- 46. Piccini A., Carta S., Tassi S., Lasiglié D., Fossati G., and Rubartelli A. (2008) ATP is released by monocytes stimulated with pathogen-sensing receptor ligands and induces IL-1β and IL-18 secretion in an autocrine way. Proc. Natl. Acad. Sci. U.S.A. 105, 8067–8072 10.1073/pnas.0709684105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Semino C., Carta S., Gattorno M., Sitia R., and Rubartelli A. (2018) Progressive waves of IL-1β release by primary human monocytes via sequential activation of vesicular and gasdermin D-mediated secretory pathways. Cell Death Dis. 9, 1088 10.1038/s41419-018-1121-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Molteni M., Gemma S., and Rossetti C. (2016) The role of Toll-like receptor 4 in infectious and noninfectious inflammation. Mediators Inflamm. 2016, 6978936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Libby P. (2012) Inflammation in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 32, 2045–2051 10.1161/ATVBAHA.108.179705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kirii H., Niwa T., Yamada Y., Wada H., Saito K., Iwakura Y., Asano M., Moriwaki H., and Seishima M. (2003) Lack of interleukin-1β decreases the severity of atherosclerosis in ApoE-deficient mice. Arterioscler. Thromb. Vasc. Biol. 23, 656–660 10.1161/01.ATV.0000064374.15232.C3 [DOI] [PubMed] [Google Scholar]
- 51. Bursill C. A., Castro M. L., Beattie D. T., Nakhla S., van der Vorst E., Heather A. K., Barter P. J., and Rye K. A. (2010) High-density lipoproteins suppress chemokines and chemokine receptors in vitro and in vivo. Arterioscler. Thromb. Vasc. Biol. 30, 1773–1778 10.1161/ATVBAHA.110.211342 [DOI] [PubMed] [Google Scholar]
- 52. Navab M., Yu R., Gharavi N., Huang W., Ezra N., Lotfizadeh A., Anantharamaiah G. M., Alipour N., Van Lenten B. J., Reddy S. T., and Marelli D. (2007) High-density lipoprotein: antioxidant and anti-inflammatory properties. Curr. Atheroscler. Rep. 9, 244–248 10.1007/s11883-007-0026-3 [DOI] [PubMed] [Google Scholar]
- 53. Hyka N., Dayer J. M., Modoux C., Kohno T., Edwards C. K. 3rd, Roux-Lombard P., and Burger D. (2001) Apolipoprotein A-I inhibits the production of interleukin-1β and tumor necrosis factor-α by blocking contact-mediated activation of monocytes by T lymphocytes. Blood 97, 2381–2389 10.1182/blood.V97.8.2381 [DOI] [PubMed] [Google Scholar]
- 54. Ansell B. J., Navab M., Hama S., Kamranpour N., Fonarow G., Hough G., Rahmani S., Mottahedeh R., Dave R., Reddy S. T., and Fogelman A. M. (2003) Inflammatory/antiinflammatory properties of high-density lipoprotein distinguish patients from control subjects better than high-density lipoprotein cholesterol levels and are favorably affected by simvastatin treatment. Circulation 108, 2751–2756 10.1161/01.CIR.0000103624.14436.4B [DOI] [PubMed] [Google Scholar]
- 55. Barter P. J., Nicholls S., Rye K. A., Anantharamaiah G. M., Navab M., and Fogelman A. M. (2004) Antiinflammatory properties of HDL. Circ. Res. 95, 764–772 10.1161/01.RES.0000146094.59640.13 [DOI] [PubMed] [Google Scholar]
- 56. Ansell B. J., Fonarow G. C., and Fogelman A. M. (2007) The paradox of dysfunctional high-density lipoprotein. Curr. Opin. Lipidol. 18, 427–434 10.1097/MOL.0b013e3282364a17 [DOI] [PubMed] [Google Scholar]
- 57. Van Lenten B. J., Hama S. Y., de Beer F. C., Stafforini D. M., McIntyre T. M., Prescott S. M., La Du B. N., Fogelman A. M., and Navab M. (1995) Anti-inflammatory HDL becomes pro-inflammatory during the acute phase response. Loss of protective effect of HDL against LDL oxidation in aortic wall cell cocultures. J. Clin. Invest. 96, 2758–2767 10.1172/JCI118345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Undurti A., Huang Y., Lupica J. A., Smith J. D., DiDonato J. A., and Hazen S. L. (2009) Modification of high density lipoprotein by myeloperoxidase generates a pro-inflammatory particle. J. Biol. Chem. 284, 30825–30835 10.1074/jbc.M109.047605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Navab M., Reddy S. T., Van Lenten B. J., and Fogelman A. M. (2011) HDL and cardiovascular disease: atherogenic and atheroprotective mechanisms. Nat. Rev. Cardiol. 8, 222–232 10.1038/nrcardio.2010.222 [DOI] [PubMed] [Google Scholar]
- 60. Rosenson R. S., Brewer H. B. Jr, Ansell B. J., Barter P., Chapman M. J., Heinecke J. W., Kontush A., Tall A. R., and Webb N. R. (2016) Dysfunctional HDL and atherosclerotic cardiovascular disease. Nat. Rev. Cardiol. 13, 48–60 10.1038/nrcardio.2015.124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Venereau E., Casalgrandi M., Schiraldi M., Antoine D. J., Cattaneo A., De Marchis F., Liu J., Antonelli A., Preti A., Raeli L., Shams S. S., Yang H., Varani L., Andersson U., Tracey K. J., et al. (2012) Mutually exclusive redox forms of HMGB1 promote cell recruitment or proinflammatory cytokine release. J. Exp. Med. 209, 1519–1528 10.1084/jem.20120189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ait-Oufella H., Taleb S., Mallat Z., and Tedgui A. (2011) Recent advances on the role of cytokines in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 31, 969–979 10.1161/ATVBAHA.110.207415 [DOI] [PubMed] [Google Scholar]
- 63. Kusters P. J., and Lutgens E. (2015) Cytokines and immune responses in murine atherosclerosis. Methods Mol. Biol. 1339, 17–40 10.1007/978-1-4939-2929-0_2 [DOI] [PubMed] [Google Scholar]
- 64. Jiang Y., Wang M., Huang K., Zhang Z., Shao N., Zhang Y., Wang W., and Wang S. (2012) Oxidized low-density lipoprotein induces secretion of interleukin-1β by macrophages via reactive oxygen species-dependent NLRP3 inflammasome activation. Biochem. Biophys. Res. Commun. 425, 121–126 10.1016/j.bbrc.2012.07.011 [DOI] [PubMed] [Google Scholar]
- 65. Estruch M., Rajamäki K., Sanchez-Quesada J. L., Kovanen P. T., Öörni K., Benitez S., and Ordoñez-Llanos J. (2015) Electronegative LDL induces priming and inflammasome activation leading to IL-1β release in human monocytes and macrophages. Biochim. Biophys. Acta 1851, 1442–1449 10.1016/j.bbalip.2015.08.009 [DOI] [PubMed] [Google Scholar]
- 66. Ferrari D., Pizzirani C., Adinolfi E., Lemoli R. M., Curti A., Idzko M., Panther E., and Di Virgilio F. (2006) The P2X7 receptor: a key player in IL-1 processing and release. J. Immunol. 176, 3877–3883 10.4049/jimmunol.176.7.3877 [DOI] [PubMed] [Google Scholar]
- 67. Garner B., Witting P. K., Waldeck A. R., Christison J. K., Raftery M., and Stocker R. (1998) Oxidation of high density lipoproteins. I. Formation of methionine sulfoxide in apolipoproteins AI and AII is an early event that accompanies lipid peroxidation and can be enhanced by α-tocopherol. J. Biol. Chem. 273, 6080–6087 10.1074/jbc.273.11.6080 [DOI] [PubMed] [Google Scholar]
- 68. Cavigiolio G., Geier E. G., Shao B., Heinecke J. W., and Oda M. N. (2010) Exchange of apolipoprotein A-I between lipid-associated and lipid-free states: a potential target for oxidative generation of dysfunctional high density lipoproteins. J. Biol. Chem. 285, 18847–18857 10.1074/jbc.M109.098434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Kirkitadze M. D., Bitan G., and Teplow D. B. (2002) Paradigm shifts in Alzheimer's disease and other neurodegenerative disorders: the emerging role of oligomeric assemblies. J. Neurosci. Res. 69, 567–577 10.1002/jnr.10328 [DOI] [PubMed] [Google Scholar]
- 70. Benilova I., Karran E., and De Strooper B. (2012) The toxic Aβ oligomer and Alzheimer's disease: an emperor in need of clothes. Nat. Neurosci. 15, 349–357 10.1038/nn.3028 [DOI] [PubMed] [Google Scholar]
- 71. Clark I. A., and Vissel B. (2015) Amyloid β: one of three danger-associated molecules that are secondary inducers of the proinflammatory cytokines that mediate Alzheimer's disease. Br. J. Pharmacol. 172, 3714–3727 10.1111/bph.13181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Poltorak A., He X., Smirnova I., Liu M. Y., Van Huffel C., Du X., Birdwell D., Alejos E., Silva M., Galanos C., Freudenberg M., Ricciardi-Castagnoli P., Layton B., and Beutler B. (1998) Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science 282, 2085–2088 10.1126/science.282.5396.2085 [DOI] [PubMed] [Google Scholar]
- 73. Hoshino K., Takeuchi O., Kawai T., Sanjo H., Ogawa T., Takeda Y., Takeda K., and Akira S. (1999) Cutting edge: Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR4 as the Lps gene product. J. Immunol. 162, 3749–3752 [PubMed] [Google Scholar]
- 74. Balducci C., Frasca A., Zotti M., La Vitola P., Mhillaj E., Grigoli E., Iacobellis M., Grandi F., Messa M., Colombo L., Molteni M., Trabace L., Rossetti C., Salmona M., and Forloni G. (2017) Toll-like receptor 4-dependent glial cell activation mediates the impairment in memory establishment induced by β-amyloid oligomers in an acute mouse model of Alzheimer's disease. Brain Behav. Immun. 60, 188–197 10.1016/j.bbi.2016.10.012 [DOI] [PubMed] [Google Scholar]
- 75. Jayaraman S., Abe-Dohmae S., Yokoyama S., and Cavigiolio G. (2011) Impact of self-association on function of apolipoprotein A-I. J. Biol. Chem. 286, 35610–35623 10.1074/jbc.M111.262485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Ryan J. (2008) Endotoxins and cell culture. Corning Technical Bulletin TC-305, Lowell, MA [Google Scholar]
- 77. Arpaia N., Godec J., Lau L., Sivick K. E., McLaughlin L. M., Jones M. B., Dracheva T., Peterson S. N., Monack D. M., and Barton G. M. (2011) TLR signaling is required for Salmonella typhimurium virulence. Cell 144, 675–688 10.1016/j.cell.2011.01.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Febbraio M., Abumrad N. A., Hajjar D. P., Sharma K., Cheng W., Pearce S. F., and Silverstein R. L. (1999) A null mutation in murine CD36 reveals an important role in fatty acid and lipoprotein metabolism. J. Biol. Chem. 274, 19055–19062 10.1074/jbc.274.27.19055 [DOI] [PubMed] [Google Scholar]
- 79. Cavigiolio G., Shao B., Geier E. G., Ren G., Heinecke J. W., and Oda M. N. (2008) The interplay between size, morphology, stability, and functionality of high-density lipoprotein subclasses. Biochemistry 47, 4770–4779 10.1021/bi7023354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Yazdi A. S., Guarda G., Riteau N., Drexler S. K., Tardivel A., Couillin I., and Tschopp J. (2010) Nanoparticles activate the NLR pyrin domain containing 3 (Nlrp3) inflammasome and cause pulmonary inflammation through release of IL-1α and IL-1β. Proc. Natl. Acad. Sci. U.S.A. 107, 19449–19454 10.1073/pnas.1008155107 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.