

Abstract
Human serum albumin is an endogenous ligand transport protein whose long circulatory half-life is facilitated by engagement with the human cellular recycling neonatal Fc receptor (hFcRn). The single free thiol located at Cys-34 in domain I of albumin has been exploited for monoconjugation of drugs. In this work, we increased the drug-to-albumin ratio potential by engineering recombinant human albumin (rHSA) variants with varying hFcRn affinity to contain three free, conjugation-competent cysteines. Structural analysis was used to identify positions for cysteine introduction to maximize rHSA stability and formation of the conjugated product without affecting hFcRn binding. The thiol rHSA variants exhibited up to 95% monomeric stability over 24 months and retained hFcRn engagement compared with a WT unconjugated control demonstrated by Biolayer Interferometry. The additional cysteines were further introduced into a panel of rHSA variants engineered with different affinities for hFcRn. After conjugation with three Alexa Fluor 680 (AF680) fluorophores, hFcRn binding was similar to that of the original triple-thiol nonconjugated rHSA variants (0.88 and 0.25 μm for WT albumin with or without 3xAF680 respectively, and 0.04 and 0.02 μm for a high hFcRn–binding variant with or without 3xAF680, respectively). We also observed a 1.3-fold increase in the blood circulatory half-life of a high hFcRn–binding triple-thiol variant conjugated with AF680 (t½ = 22.4 h) compared with its WT counterpart (t½ = 17.3 h) in mice. Potential high drug-to-albumin ratios combined with high hFcRn engagement are attractive features of this new class of albumins that offer a paradigm shift for albumin-based drug delivery.
Keywords: albumin, protein engineering, site-directed mutagenesis, Fc receptor, multidrug transporter, multifunctional protein, cysteine-mediated cross-linking, pharmacokinetics, drug delivery, neonatal Fc receptor
Introduction
Human serum albumin (HSA)3 is the most abundant plasma protein in blood. It functions as a transport protein facilitated by multiple noncovalent binding sites for endogenous molecules such as fatty acids, steroids, and bilirubin (1) and a long circulatory half-life (∼19 days) predominately facilitated by engagement with the cellular recycling heterodimeric neonatal Fc receptor (FcRn) (2–5). These properties promote albumin as an attractive drug delivery platform (6–8) that has been utilized for long-acting drugs such as the marketed Levemir® and Victoza® (Novo Nordisk) that reversibly associate with hydrophobic binding pockets in endogenous HSA by inclusion of fatty acid chains in the drug design. An alternative approach to reversible association is covalent drug conjugation that offers more stable drug attachment and a defined drug-to-albumin ratio (DAR). HSA consists of three domains, with the major FcRn binding interface lying in domain III (DIII) (9). Drug attachment distal to this binding interface is thought to be a requirement to retain HSA's cellular recycling capacity and concomitant prolonged circulatory half-life. A single free cysteine at position 34 (Cys-34) located in DI has, therefore, been a focus for site-selective drug conjugation (10–12). For example, Kratz et al. (13) reported maleimide-mediated drug conjugation to Cys-34 of endogenous albumin after intravenous injection of doxorubicin-maleimide. Although an elegant approach, the lack of available Cys-34 on endogenous albumins due to post-translational oxidation (cysteinylation or nitrosylation) (14) may limit reactivity toward the endogenous HSA pool. In vitro conjugation of drug to rHSA variants with Cys-34 in the reduced form (10) offers an alternative strategy to obtain a high conjugation efficiency and an opportunity to utilize albumins engineered for improved pharmacokinetic properties due to different hFcRn engagement (3, 10). Availability of a single free thiol, however, still only permits a DAR of 1:1, limiting the drug-carrying potential.
This present work aims to increase the conjugation capacity by site-selective introduction of additional thiol groups into a range of rHSA variants with different FcRn-binding affinities described previously (3, 15, 16).
Analysis of the co-crystal structure of HSA engagement with hFcRn was used to identify and select desirable sites for the introduction of two additional conjugation-competent cysteine residues, Lys-93 and Glu-294, in DI and DII, respectively (Fig. 1). This engineered triple-thiol variant (DAR3 variant) exhibited high conjugation efficiency and low aggregation and retained hFcRn engagement. When these mutations were introduced into a range of rHSA variants with different hFcRn affinities, tunable blood circulatory time in mice was observed. This work presents an albumin platform that allows enhanced DAR, albumin multifunctionality, and high hFcRn engagement.
Figure 1.
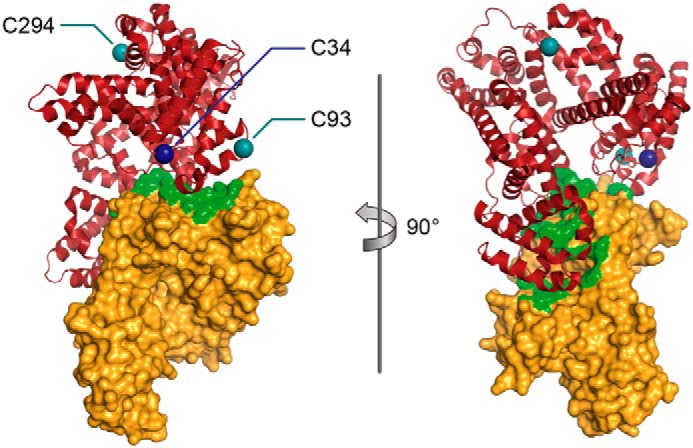
Rotated views of the crystal structure of the engineered triple-thiol human serum albumin in complex with the FcRn receptor. The β-2-microglobulin/FcRn complex is shown in gold, and human serum albumin is shown in red. The location of the naturally occurring cysteine (Cys-34) and the two positions where cysteines with reactive thiol groups have been inserted (K93C and E294C) are shown in dark blue and teal, respectively. Interface residues within a distance of 5 Å from albumin are marked in green on the surface representation of the FcRn receptor. Modified from Protein Data Bank entry 4N0F.
Results
Identification of positions suitable for thiol introduction
Positions for the introduction of cysteines with reactive thiol groups were based on structural analysis of co-crystal complexes of HSA and hFcRn (17, 18). Positions were avoided whereby thiol introduction could potentially negatively impact folding of albumin e.g. due to the number of disulfide bonds required to maintain tertiary structure or its monomeric status. Accessibility of the thiol group for conjugation was also assessed. It was highly desirable to avoid a negative impact on hFcRn binding by direct interference with the intermolecular interface or by the disposition of subsequently conjugated molecules leading to steric hindrance with hFcRn. Therefore, positions in the principal binding interfaces were not selected for screening. Positions identified as meeting all of these criteria are shown in Fig. 1 (Cys-34, K93C, and E294C), Fig. S1, and Table S2 (full panel).
Free thiol profiles
Conjugation to Cys-34 was prevented by replacing the cysteine with an alanine (C34A), and conjugation profiles were assessed for 36 new introduced cysteine mutations in this C34A rHSA. To detect the number of free thiols, 5,5-dithio-bis-(2-nitrobenzoic acid) (DTNB) was incubated with the variants, and mass increase (+197 Da for 5-thio-2-nitrobenzoic acid (TNB)) was detected using MS. An increase in mass of 197 ± 15 Da measured by MS upon DTNB incubation was indicative of the presence of one free thiol group on the protein in the sample. All variants exhibited successful binding of a TNB molecule, as shown in Table 1 (a sample selection) and Table S2 (all samples).
Table 1.
Selection of samples for free thiol profile detection by DTNB addition (+197 ± 15 Da increase in mass) of the new mutations in the thiol variants
Shown are predicted molecular mass post-DTNB treatment for conjugation efficiency measurements using MS spectra. Difference shows actual measured mass minus theoretical mass. (For the complete sample set, see Table S2).
| Variant description | Theoretical mass | Mass post-DTNB treatment |
||
|---|---|---|---|---|
| Theoretical | Actual measured | Difference | ||
| Da | Da | |||
| C34A/K93C | 66,382 | 66,579 | 66,588 | 9 |
| C34A/E294C | 66,381 | 66,578 | 66,581 | 3 |
Aggregation characteristic profiles
The tendency of WT rHSA or the thiol rHSA variants to aggregate was determined by measurement of monomer, dimer, and higher-order oligomer quantities in solutions of the single-thiol rHSA variants. Gel-permeation high-performance liquid chromatography (GP-HPLC) and ultrahigh-performance liquid chromatography (UHPLC) were used to determine the aggregation of the single-thiol rHSA variants. Results are typically expressed as “percentage monomer,” which is calculated as follows: amount of monomeric albumin by mass × 100/(amount of monomeric albumin by mass + amount of dimeric albumin by mass + amount of higher-order oligomer by mass). The majority of shake flask–derived rHSA variants after purification by chromatography using AlbuPure® matrix (9) had a monomer percentage equivalent to or higher than that of the WT rHSA control at t = 0, which had a monomer percentage of 86–87%. The variants maintained their monomeric protein stability during 6 months of storage at 2–8 °C, with no significant evidence of aggregation observed (Table 2 and Tables S3 and S4). However, it was evident that variants L66C, E277C, and E311C had a relatively low percentage monomer at t = 0 and consequently had a propensity to form aggregates.
Table 2.
Selection of samples for GP-HPLC aggregation screening indicates monomeric stability over 6 months at 2–8 °C
Table shows percentage of monomers at timepoints 0 weeks, 7 weeks and 6 months. Parentheses shows percentage monomer compared to WT sample at the given timepoint. For the complete sample set, see Tables S3 and S4.
| Sample | GP-HPLC concentration | Monomer percentage |
Change in monomer percentage |
|||
|---|---|---|---|---|---|---|
| t = 0 | t = 7 weeks | t = 6 months | 0–7 weeks | 0–6 months | ||
| mg/ml | % | Δ% | ||||
| WT rHSA | 1.1 | 87 (100) | 88 (100) | 89 (100) | 1 | 2 |
| C34A/K93C | 0.7 | 91 (105) | 92 (105) | 92 (103) | 1 | 1 |
| C34A/E294C | 0.9 | 96 (110) | 96 (109) | 96 (108) | 0 | 0 |
Conjugation efficiency profiles
The MS spectra for the single-thiol rHSA variants L66C, A92C, Q94C, A226C, D259C, L275C, and L284C indicated no conjugation following overnight incubation with maleimide-PEG2-biotin (Table S5). The MS spectra for the single-thiol rHSA variants E230C and I271C indicated that conjugation had occurred after overnight incubation, with both giving 72% monoconjugate when comparing the relative peak heights of conjugated and unconjugated species. The MS spectrum for single-thiol rHSA variant K93C exhibited a single species at 66908 Da (Table 3), indicating 100% conjugation and confirming that K93C has only one single free thiol group (data not shown). Also, the MS spectra for the WT rHSA control and the single-thiol rHSA variants L24C, V54C, H128C, E227C, K240C, K262C, Q268C, E277C, E294C, K317C, A322C, E358C, K359C, and A362C all indicated that >90% conjugation was achieved with maleimide-PEG2-biotin (Table 3 and Table S5).
Table 3.
Single conjugation of maleimide-PEG2-biotin (+525 Da) during overnight incubation
Shown is a selection of samples for conjugation efficiency measured by the weight increase using mass spectrometry. (For the complete sample set, see Table S5.)
| Sample description | Reference molecular mass unconjugated | Theoretical conjugate mass | Conjugate intact mass | Conjugation |
|---|---|---|---|---|
| Da | Da | Da | % | |
| WT rHSA | 66,439 | 66,964 | 66,966 | 93 |
| C34A/K93C | 66,382 | 66,907 | 66,908 | 100 |
| C34A/E294C | 66,381 | 66,906 | 66,909 | >95 |
Variant stability over long-term storage
The double-thiol (DAR2) and triple-thiol (DAR3) rHSA variants and the DAR3 combined with a high-binding hFcRn (HBI) mutation (K573P) (3) were assessed with a single-thiol WT rHSA for stability during long-term storage at 5 °C for 24 months in 25 mm sodium phosphate, 215 mm sodium chloride, pH 6.5. The aggregation profiles were determined at various time points by GP-HPLC (Table 4 and Fig. S3). All rHSA variants had a monomer percentage greater than 98% at t = 0. The variants maintained a monomeric protein percentage ≥ 95% after 24 months of storage at 5 °C.
Table 4.
Monomeric stability of combinations of cysteine mutations in the thiol variants during long-term storage at 5 °C by GP-HPLC, maintaining >95% monomer over 24 months
| Sample description | Percentage monomer |
||||||
|---|---|---|---|---|---|---|---|
| 0 months | 1 month | 2 months | 3 months | 6 months | 12 months | 24 months | |
| % | % | % | % | % | % | % | |
| WT rHSA | 100 | 99 | 100 | 100 | 100 | 99 | 100 |
| Double-thiol (C34 + K93C) | 100 | 99 | 99 | 99 | 98 | 97 | 97 |
| Double-thiol (C34 + E294C) | 100 | 99 | 99 | 99 | 99 | 97 | 98 |
| Triple-thiol (C34 + K93C + E294C) | 99 | 99 | 98 | 98 | 97 | 95 | 95 |
| Triple-thiol HBI variant (C34 + K93C + E294C + K573P) | 100 | 99 | 99 | 99 | 98 | 98 | 96 |
Conjugation with Alexa Fluor 680
The K93C and E294C mutations were combined with null FcRn-binding (NB) (3, 9) and a high FcRn-binding hFcRn HBII variant described previously (16). The thiol variants were conjugated with Alexa Fluor 680 (AF680) dye, and the labeling efficiency was determined by the ratio of absorbance of the fluorophore at 680 nm and albumin at 280 nm. The 680 nm/280 nm ratio for the single-thiol WT rHSA variant was 1.3. The ratios for the triple-thiol rHSA variants NB, WT, and HBII were shown to be 4.3, 3.7, and 3.7, respectively, which indicated that the triple-thiol rHSA variants were ∼3 times more fluorescent than the single-thiol WT conjugate (Table 5).
Table 5.
Labeling efficiency of AF680 dye post-conjugation, as measured by GP-HPLC dual-wavelength ratio determination (680 nm/280 nm)
| Sample description | Monomer concentration | 280-nm monomer peak area [mAU] | 680-nm monomer peak area [mAU] | Ratio |
|---|---|---|---|---|
| mg/ml | ||||
| WT-1xAF680 | 1.989 | 3,162,386 | 4,222,436 | 1.34 |
| WT-3xAF680 | 2.009 | 3,185,808 | 11,835,297 | 3.72 |
| NB-3xAF680 | 1.991 | 3,117,158 | 13,434,647 | 4.31 |
| HBII-3xAF680 | 2.029 | 3,238,326 | 11,848,510 | 3.66 |
hFcRn affinity of albumin variants
The hFcRn-binding affinities of the selected triple-thiol NB, WT, and HBII variants were determined by biolayer interferometry (Table 6 and Fig. 2). Triple-thiol rHSA variants conjugated to three AF680 fluorophores showed slightly lower affinity compared with the unconjugated counterpart. For the triple-thiol WT rHSA, the conjugation of 3xAF680 increased the dissociation constant (KD) from 0.25 to 0.88 μm. For the triple-thiol HBII rHSA, the KD increased from 0.02 to 0.04 μm after conjugation of 3xAF680. The triple-thiol NB rHSA did not show any hFcRn binding with or without 3xAF680 conjugated. The triple-thiol rHSA variants, conjugated or unconjugated, all showed the same tendency of KD value following the ranking WT > HBII, with NB not detectable. These findings show that the hFcRn affinity is not diminished due to the K93C and E294C mutations and is only marginally affected when fluorophores are conjugated to the three free thiols.
Table 6.
Binding affinities for triple-thiol rHSA variants toward hFcRn, pH 5.5
KD (koff/kon) is shown as mean ± S.D. Average Biolayer Interferometry data from three replicates are given, from two-fold dilution series from 3 to 0.1875 μm albumin. (For binding curves, see Fig. 2.) NA, not available.
| Albumin | KD average | Kon average | Koff average |
|---|---|---|---|
| μm | m−1 s−1 | s−1 | |
| Single-thiol WT | 0.55 ± 0.20 | 1.38 × 104 ± 1.69 × 103 | 7.76 × 10−3 ± 2.50 × 10−3 |
| Triple-thiol WT | 0.25 ± 0.05 | 1.79 × 104 ± 2.70 × 103 | 4.46 × 10−3 ± 1.48 × 10−3 |
| Triple-thiol WT-3xAF680 | 0.88 ± 0.01 | 7.57 × 103 ± 1.82 × 103 | 6.45 × 10−3 ± 5.52 × 10−4 |
| Triple-thiol NB | NA | NA | NA |
| Triple-thiol NB -3xAF680 | NA | NA | NA |
| Triple-thiol HBII | 0.02 ± 0.006 | 2.01 × 104 ± 4.13 × 103 | 3.08 × 10−4 ± 9.96 × 10−5 |
| Triple-thiol HBII -3xAF680 | 0.04 ± 0.017 | 1.25 × 104 ± 1.70 × 103 | 4.53 × 10−4 ± 2.33 × 10−4 |
Figure 2.

Representative fitted binding affinity curves (black) from three replicates for triple-thiol rHSA variants binding to hFcRn from Biolayer Interferometry measurements. The two-fold dilution series from 3 to 0.1875 μm albumin is shown. Raw data curves are depicted in gray. The vertical dashed line indicates transition between association phase and dissociation phase. Top (left to right), standard WT HSA control (single-thiol), triple-thiol WT rHSA, and triple-thiol WT rHSA 3xAF680. Middle (left to right), triple-thiol NB rHSA and triple-thiol NB rHSA 3xAF680. Bottom (left to right), triple-thiol HBII rHSA and triple-thiol HBII rHSA 3xAF680.
Pharmacokinetics of albumin variants in mice
The triple-thiol rHSA 3xAF680 fluorescence–labeled variants (NB, WT, and HBII) were administered by intravenous injection into BALB/cAnNRj mice (n = 7); blood samples were taken at 1 min, 30 min, 4 h, 24 h, 48 h, 72 h, and 96 h; and the level of fluorescence in blood detected by In Vivo Image System (IVIS) (PerkinElmer Life Sciences) bioimaging (Table 7, Fig. 3, and Fig. S4). The blood circulatory half-life (t½) for the variants showed an increase from 17.3 h for the triple-thiol WT rHSA 3xAF680 variant to 22.4 h for the triple-thiol HBII rHSA 3xAF680 variant. The triple-thiol NB rHSA 3xAF680 variant showed a slightly decreased half-life (15.4 h) compared with the triple-thiol WT rHSA 3xAF680 variant.
Table 7.
Elimination half-life (t½) calculated from the exponential fit curves (Fig. 3) and the fit values (R2) for each triple-thiol rHSA variant
| Variant | t½ | R2 |
|---|---|---|
| h | ||
| WT -3xAF680 | 17.3 | 0.9999 |
| NB-3xAF680 | 15.4 | 0.9986 |
| HBII-3xAF680 | 22.4 | 0.9759 |
Figure 3.
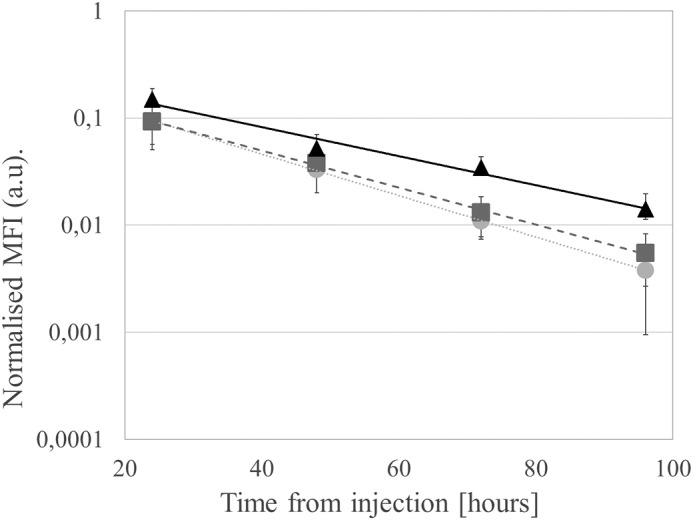
Plasma levels of fluorescently labeled 3xAF680-triple-thiol rHSA variants at different time points (elimination phase: 24–96 h). Fluorescence was measured using the IVIS scanner and normalized to fluorescence level at 1 min (not depicted). Graphs show mean fluorescence intensity (●, NB; ■, WT; ▴, HBII, n = 7) with fitted exponential curves. Error bars, S.D. (For 1 min to 96 h, see Fig. S4.)
Discussion
A high drug load, site-selectively conjugated to rHSA without compromising hFcRn binding, is important for harnessing albumin's physiological transport properties for effective drug delivery. We present, for the first time, rHSA variants site-selectively engineered by a structural screen to contain three free cysteines for improved DAR without affecting hFcRn engagement.
Candidate positions were selected for screening to avoid negatively impacting binding of albumin to hFcRn, based on co-crystal structures. Additional factors included accessibility for conjugation and predicted minimal impact on the monomeric or folded status of albumin. Surface exposure and flexibility of the backbone in the regions of introduced thiols were noted as promoting these latter features.
Following identification of positions with desirable conjugation features, the spatial disposition of the sites on rHSA was considered before production of variants containing multiple free thiol groups, to limit possible steric interactions from conjugated moieties. The spatial distribution of the natural and the two engineered thiols have been illustrated based on a previously published crystal structure of FcRn in complex with HSA (Fig. 1). Positions selected for mutations were Lys-93 (to give K93C) and Glu-294 (to give E294C), which, when combined with the Cys-34, gives the triple-thiol rHSA a monomer stability > 95%. Further combining with K573P gives high hFcRn binding (HBI) (3) (Table 4). The single-thiol K93C and E294C (C34A versions) rHSA variants each showed promising conjugation potential (100 and >95%, respectively) when conjugated to maleimide-PEG2-biotin.
These two additional thiol mutations, K93C and E294C, were introduced in DI and DII distal from the predominant hFcRn binding interface in DIII. However, DI has a contributory role in binding to FcRn, and Cys-34 is proximal to this interface (19). Conjugation to the free cysteine at Cys-34 in DI can, therefore, influence the hFcRn binding affinity as observed in earlier studies (11, 20). In this work, the hFcRn-binding affinity of the triple-thiol WT-FcRn–binding rHSA variant was retained and in the same range as the WT HSA single-thiol control (KD was 0.25 μm compared with 0.55 μm, respectively). This was also observed for the fluorophore-conjugated triple-thiol WT rHSA version carrying three AF680 molecules (KD = 0.88 μm). The K93C and E294C mutations were further introduced into rHSA variants either engineered for null-FcRn binding (NB; H464Q/K500A) or for high FcRn binding (HBII; E492G/K573P/K574H/Q580K). The NB rHSA variant contains the K500A mutation combined with a key histidine residue known to reduce FcRn binding (9), where the HBII rHSA variant is a combination variant comprising K573P and additional mutated positions from domain III in regions that have previously been shown to impact FcRn binding (3, 9) and exhibit a long circulatory half-life (16). Neither of the triple-thiol NB rHSA variants (with or without 3xAF680) exhibited hFcRn binding, whereas the triple-thiol HBII rHSA variant (with or without 3xAF680) bound with >10-fold higher affinity to hFcRn compared with triple-thiol WT rHSA variants (with or without 3xAF680). There was a slight decrease in binding affinity after conjugation of three AF680 molecules observed as a KD shift from 0.02 to 0.04 μm for the triple-thiol HBII rHSA variants and from 0.25 to 0.88 μm for the triple-thiol WT rHSA variant. Conjugation of nucleic acids or PEG to rHSA has previously been shown to decrease hFcRn binding affinity, but hFcRn engagement could be rescued by using a high hFcRn–binding rHSA (HBI) variant (11, 20). The high-binding (HBII) triple-thiol rHSA described here exhibited higher hFcRn affinity when bearing three AF680 molecules compared with the unconjugated single-thiol WT rHSA control; therefore, both the DAR and hFcRn engagement are improved overall.
Three-AF680 molecule conjugation creates a fluorescent detection system for analysis of the pharmacokinetic properties. Furthermore, conjugation of 3xAF680 (∼1000 Da/molecule) mimics transportation and distribution of small molecular drugs conjugated to HSA. The Cys-34–conjugated nucleic acids and PEG molecules (presented in Refs. 11 and 20, respectively) are all considered macromolecular drugs due to their size (10 and 5–30 kDa, respectively) and are more likely to create steric hindrance in the binding of HSA to hFcRn, as observed in the decreased binding affinity toward hFcRn. Small molecular drugs, such as the anticancer therapeutics paclitaxel and doxorubicin (800 and 500 Da, respectively) may exhibit minimal alteration of HSA-hFcRn binding, as observed with the 3xAF680 conjugation. The application of fluorophore conjugation creates a platform for easy measurement of the circulatory half-life of rHSA, by scanning fluorescent signal in blood samples as described previously by our group (21). The relative fluorescent signal in heparinized plasma was detected, and half-lives were calculated. The circulatory half-life of the NB and that of the WT triple-thiol rHSA were similar (t½ = 15.4 and 17.3 h, respectively). This similarity for NB and WT rHSA was shown by Andersen et al. (3) in NMRI mice, where the half-lives of unconjugated NB and WT rHSA were 19.1 and 21.0 h, respectively. The lack of a larger difference in circulatory half-lives of the NB and the WT is most likely due to the effect of competition from the endogenous MSA for mouse FcRn (mFcRn) binding. This is supported by work in MSA-deficient mice, where HSA showed a longer half-life compared with the half-life observed in WT mice (22). The cross-species difference in HSA/MSA mFcRn-binding affinity has been observed by Andersen et al. (3, 23), showing a ∼25-fold increase of the KD of MSA binding to mFcRn (KD = 0.8 μm) compared with the KD of HSA binding to mFcRn (KD = 25 μm). This was further demonstrated in the work of Viuff et al. (15). The use of a humanised mouse model, such as the humanised albumin/FcRn double transgenic mouse introduced in our previous work (15), has shown longer circulatory half-life of rHSA variants, however, the same tendency of a longer half-life for the rHSA variants engineered for higher FcRn affinity is also shown in the wild type model presented in the present work. Furthermore, the same binding trend HBII > HBI > WT of the single-thiol albumin variants toward hFcRn was observed for mFcRn at pH 5.5 (Table S6), which supports the use of the WT model to identify improved pharmacokinetics of HBII.
The circulatory half-life of the 3xAF680-conjugated triple-thiol HBII rHSA variant was increased to 22.4 h in this present work, which is a 1.3-fold change compared with 3xAF680-conjugated triple-thiol WT rHSA. This is comparable with the 1.65-fold change observed between WT and HBII rHSA fusions in the work of Larsen et al. (16).
In our laboratory, we have used nucleic acid assemblies to multifunctionalize HSA at Cys-34 (12). These could be further utilized for the triple-thiol rHSA variants introduced in this work, to further maximize multifunctionalization. Additionally, use of the HBII rHSA triple-thiol engineered variant should allow a longer blood circulatory time. Conjugation of drugs to the three available cysteines is a future focus of our laboratory.
The potential for enhanced DAR, multifunctionality, and high hFcRn engagement promotes this new class of engineered albumins as a highly attractive drug delivery platform.
Experimental procedures
HSA variant engineering: Protein design
A range of candidate positions for replacement with cysteine were identified in surface-exposed regions of HSA. Crystal structures of HSA complexed with hFcRn are available (17, 18) and were inspected using Yasara software (24). Positions that could directly impact the hFcRn binding interface when replaced with cysteine were avoided. Consideration was also given to avoid positions where introduction of a free thiol group could adversely affect the HSA structure or function or where conjugation of an additional moiety may sterically hinder hFcRn binding.
Plasmid construction
Escherichia coli 5 α (New England Biolabs) was used for propagation and preparation of the thiol rHSA DNA. Expression cassettes contained Saccharomyces cerevisiae PRB1 promoter, leader sequence, HSA gene, and S. cerevisiae ADH1 terminator sequence (9). An HSA overexpression plasmid pDB5102 (Fig. S2), which contains the HSA gene codon optimized for expression in S. cerevisiae and a unique NheI site for cloning, was mutated to C34A to remove the single free thiol at position 34. Briefly, the template, pDB5102, was dam-methylated following the manufacturer's instructions (New England Biolabs); 5 ng was used in a round plasmid PCR using primers C34AForward (5′-TTCGCTCAATACTTGCAACAAGCTCCATTCGAAGATCACGTCAAG-3′) and C34AReverse (5′-TTGTTGCAAGTATTGAGCGAAAGCGATCAAGACCAA-3′); and the PCR product was treated with DpnI and transformed into E. coli. The resulting plasmid, pDB5155, was used as the backbone for the construction of the single-thiol variants; the primers used to generate the variants are listed in Table S1. Expression cassettes were generated by digesting the single-thiol plasmids with NsiI and PvuI, and the required DNA fragment was purified from an agarose gel using the Qiagen gel extraction kit. Gap repair into S. cerevisiae was performed as described previously (9).
Microtiter plate (MTP) analysis
The S. cerevisiae transformants were grown in 500 μl of buffered minimal media sucrose (BMMS) + CSM-Leu in a 48-well MTP. The cultures were grown in a humidity chamber at 30 °C and 200 rpm for 96 h. The plate was then centrifuged at 1500 rpm for 5 min, and the supernatant was removed for DTNB analysis.
Free thiol profiles
The number of free thiols on a protein can be determined using mass spectrometric analysis of the protein sample treated with Ellman's (DTNB) reagent. TNB has a molecular mass of 199 Da; thus, an increase in mass of 197 Da, when TNB loses H2 during disulfide bond formation with the free thiol group on the test protein, indicates the presence of one free thiol group on the test protein. Thus, 4 μl of buffer (4 mg/ml DTNB, 500 mm sodium phosphate, pH 7.0) was added to 200 μl of the test protein culture sample in a 96-well MTP format. The preparation was incubated for 25 min at ambient temperature (20 ± 5 °C) to allow TNB labeling. Protein intact mass was determined by UHPLC-MS. UHPLC separation was carried out on 10 μl of sample using a Waters Acquity system on a BEH 50 × 2.1-mm ACQUITY BEH 1.7-μm 300A C4 column and a 5-min analytical gradient of buffer A (0.1% formic acid) and buffer B (100% acetonitrile, 0.1% formic acid). Eluted proteins were directly introduced to a Bruker MicrOTOF II mass spectrometer via an electrospray ionization source. Instrument control and sample acquisition was performed using BioPharma CompassTM.All data were manually processed over the leading edge of the protein peak between 2.9–3.0 minutes within BioPharma CompassTM DataAnalysis software. This included spectral smoothing using a Gauss smoothing algorithm set at 0.0765 Da and a baseline correction setting of 0.8 flatness. Deconvoluted intact mass spectra were obtained using the Max. Entropy algorithm.
Shake flask expression and purification
BMMS medium (10 ml) was inoculated with S. cerevisiae and grown for 2 days at 30 °C with orbital shaking at 200 rpm. An aliquot of each starter culture (5 ml) was used to inoculate two flasks of 200 ml of BMMS medium and grown for a further 5 days at 30 °C with orbital shaking at 200 rpm. Cells were harvested by filtration through a 0.2-μm vacuum filter membrane (Nalgene Sterile Top Filter), and the supernatant was retained for purification. A single-step chromatography procedure was used to prepare purified material from the thiol-rHSA variants. The purification step used a column (bed volume ∼2 ml) packed with AlbuPure® matrix (ProMetic Biosciences Ltd., Cambridge, UK). This was equilibrated with 50 mm sodium acetate, pH 5.3, and loaded with neat shake flask culture supernatants, at pH ∼5.5–6.5. The column was washed with a ∼10 column volumes each of 50 mm sodium acetate, pH 5.3, and 50 mm ammonium acetate, pH 8.0, respectively. Bound protein was eluted using ∼10 column volumes of 50 mm ammonium acetate, 10 mm octanoate, pH 7.0. The flow rate throughout was 240 cm/h using an AKTA Explorer system (GE Healthcare).
Aggregation characteristic profiles
rHSA variants were analyzed for the tendency to remain as a monomer in solution. Individual rHSA variants had a single free thiol group (single-thiol) and were compared with WT HSA. The concentration and percentage monomer of the eluate samples were determined by GP-HPLC. Injections of 25 μl were made onto a 7.8-mm internal diameter × 300-mm length TSK G3000SWXL column (Tosoh Bioscience), with a 6.0-mm internal diameter × 40-mm length TSK SW guard column (Tosoh Bioscience) at 1 ml/min, with a run time of 15 min. Alternatively, using an Agilent 1260 isocratic UHPLC instrument, injections of 4 μl were made onto a 4.6-mm inner diameter × 150-mm length BEH 200A, 1.7-μm column (Waters), at 0.5 ml/min, with a run time of 5 min. Samples were analyzed by chromatography in 25 mm sodium phosphate, 100 mm sodium sulfate, 0.05% (w/v) sodium azide, pH 7.0. Samples were quantified by UV detection at 280 nm, by peak area, relative to a 10 mg/ml rHSA standard. The samples were reanalyzed to determine the change in percentage monomer after storage at 2–8 °C.
Conjugation efficiency profiles
rHSA variants were conjugated with biotin (Thermo Scientific, EZ-Link maleimide-PEG2-biotin) using a 3.2-fold molar excess of maleimide-PEG2-biotin to protein. Eluates from the AlbuPure® chromatography were diluted with PBS buffer, pH 7.4, to give 10-ml solutions at 0.3 mg/ml (45.15 nmol). Stock solutions of 2 mg/ml biotin were prepared by the addition of 5 × 200-μl aliquots of PBS buffer, pH 7.4, to each of two 2-mg preweighed EZ-Link microtubes, and the vials were rinsed to maximize recovery of the lyophilized product. The two 1-ml volumes were pooled into a 7-ml container with a lid. From the biotin stock solution, 38 μl (144.5 nmol) was added to the 10-ml rHSA samples to give a ∼3.2-fold molar excess of biotin over each rHSA variant. Samples were gently mixed and incubated at ambient temperature overnight. Following incubation, the samples were subjected to MS to determine the intact protein mass after conjugation according to the method described previously, but using a 15-min analytical gradient, and processing data for the protein peak between ∼7 and 10 min.
Combining thiol variants with rHSA variants with different hFcRn affinity
The triple-thiol rHSA variant was constructed by a two-step cloning strategy. First, the single-thiol variants K93C (pDB5623) and E294C (pDB5624) were combined; this was performed by DNA fragment swaps. Both plasmids were digested with NheI and SphI, and the insert containing the E294C position was cloned into the backbone containing the K93C position, resulting in plasmid pDB5625 (C34A/K93C/E294C). For the second step of the cloning, both plasmids pDB5102 and pDB5625 were digested with PstI and SacII, and the Cys-34 insert from pDB5102 was cloned into the pDB5625 backbone, resulting in the plasmid pDB5628 (Cys-34, K93C, and E294C). Expression cassettes used for in vivo homologous recombination were generated by digesting the plasmid with NsiI and PvuI, and the required DNA fragment was purified from an agarose gel using the Qiagen gel extraction kit. S. cerevisiae transformations and albumin purifications were performed as described above. The triple-thiol rHSA variant was further modified to create expression cassettes for proteins that also exhibit modified hFcRn-binding affinity: NB, HBI, and HBII, described previously (16). S. cerevisiae transformations and albumin purification using AlbuPure® matrix and diethylaminoethyl-Sepharose Fast Flow matrix (GE Healthcare) were performed as described previously (9, 25).
Variant stability over long-term storage
rHSA variants at 5 mg/ml in 25 mm sodium phosphate, 215 mm sodium chloride, pH 6.5, were analyzed for monomer content over 24 months, using GP-HPLC as described earlier.
Conjugation with Alexa Fluor 680
Triple-thiol rHSA variants were further modified with AF680 for developing a detection system for in vivo studies. Variants were conjugated with AF680 dye with a maleimide-reactive group (Molecular Probes), using a 3.1-fold excess of AF680-maleimide to thiol. Combination variants in 25 mm sodium phosphate, 215 mm sodium chloride, pH 6.5, were diluted with PBS, pH 7.4, to give 20-ml solutions at 1 mg/ml (300 nmol). AF680 C2-maleimide (1 mg) was solubilized in 200 μl of DMSO, and 186 μl (930 nmol) was added to the 20-ml rHSA samples to give an ∼3.1 molar excess of fluorescence dye over rHSA variant. Samples were gently mixed and incubated at 2–8 °C overnight, wrapped in foil to protect against light. The unreacted fluorescence–maleimide compound was removed by two cycles of concentration and dilution with PBS in VivaSpin-20 centrifugal concentrators. The conjugation was stabilized by hydrolysis (ring-opening) of the maleimide ring (26) by the addition of 20 mm glycine buffer, pH 9.0. The hydrolysis was performed for 6 h at 37 °C and terminated by neutralizing to pH 7.4. Because the overall yield after hydrolysis is ∼50%, the material was subjected to another cycle of conjugation and hydrolysis. Following the second round of hydrolysis, unreacted and hydrolyzed fluorescence–maleimide compound was removed by ultra/diafiltration in a VivaSpin-20 centrifugal concentrator with repeated cycles of concentration and dilution with PBS, pH 7.4. The degree of labeling was determined based on the ratio of absorbance from the fluorophore at 680 nm and albumin at 280 nm using the GP-HPLC method described previously, with a dual-wavelength collection at 280 and 680 nm.
hFcRn affinity by Biolayer Interferometry
A BLItz® system (Fortebio, Pall Life Sciences, Menlo Park, CA) was used to detect the affinity between selected triple-thiol rHSA variants (K93C and E294C mutations combined with WT, NB mutations, or HBII mutations) or WT control rHSA (Sigma-Aldrich, A6608) and hFcRn. Streptravidin-coated biosensors were hydrated in Dulbecco's PBS (Sigma-Aldrich, D8537) supplemented with 0.01% Tween 20 (PBST) (pH 7.4) for 10 min and incubated in 5 μl of 70 nm hFcRn-biotin (Immunitrack, Copenhagen, Denmark) for 15 min followed by a wash in PBST. For kinetic measurements, 2-fold dilution series of the rHSA and triple-thiol rHSA variants were made (0.1875–3 μm) and performed in PBST association buffer supplemented with 25 mm sodium acetate, 25 mm NaH2PO4, and 150 mm NaCl, pH 5.5. A 120-s association phase followed by a 300-s dissociation phase were subtracted from a baseline phase of 60 s in association buffer. Biosensors were regenerated by a 30-s wash in PBST to allow multiple use. Binding curves were fitted to a 1:1 binding model after intercept correction using global fit in the BLItz PRO version 1.2 software.
The binding of single-thiol rHSA variants (WT, HBI, and HBII) to hFcRn and mFcRn was compared using an Octet RED96 system. Biotinylated soluble hFcRn and mFcRn (Immunitrack) were immobilized on streptavidin-coated biosensors (PALL/Fortebio) in PBST. Nonspecific binding to the sensors was minimized by preblocking in albumin (diluted to 0.5 mg/ml in PBS, pH 7.4) for 5 min followed by a 5-min PBS rinse, pH 7.4, and then MilliQ water rinse. The sensors were kept in storage by prior soaking in 15% sucrose and air-dried. For kinetic characterization, a seven-step 2-fold dilution series was prepared in association buffer, pH 5.5. Binding kinetics were performed at 30 °C with a 120-s association phase and 300-s dissociation phase. The sensors were regenerated with PBST, pH 7.4, and equilibrated between samples in association buffer, pH 5.5. All data were referenced with FcRn-streptavidin sensors in association buffer without HSA. The Octet data analysis software version 8.0 (PALL/Fortebio) was used for data analysis using curve fitting to a 1-1 model for estimation of kinetic parameters.
Animals
Twenty-four 8–12-week-old female BALB/cAnNRj mice were purchased from Janvier Labs (Le Genest-Saint-Isle, France). All mouse experiments were performed at the biomedical animal facilities of Aarhus University. All animal studies were approved and performed according to the guidelines from the Danish Animal Experiments Inspectorate under the auspices of the Danish Ministry of Environment and Food.
Blood circulation time of Alexa Fluor 680 rHSA variants
Mice were intravenously injected in the tail vein with the 2 mg/ml triple-thiol rHSA (K93C and E294C mutations combined with WT, NB, or HBII mutations) variants (10 mg/kg) bearing three AF680 molecules each (n = 7). A PBS-injected control group was also included. Blood samples (15 μl) were taken after 1 min from vena sublingualis and from the tail vein at 30 min, 4 h, 24 h, 48 h, 72 h, and 96 h and placed into heparin-coated 20-μl capillaries. Blood samples were centrifuged and then scanned using Xenogen IVIS (PerkinElmer Life Sciences). Spectral unmixing was performed to subtract autofluorescence (from the control plasma) from the AF680 signal on the rHSA variants using Living Image version 4.4 (PerkinElmer Life Sciences). Signal intensity from plasma was obtained from regions of interest (ROIs) using equivalent area selection and surface radiance within the ROIs measured as the number of photons per second that leave a square centimeter of tissue and radiate into a solid angle of one steradian (photons/s/cm2/sr). Surface radiance from plasma from each time point was normalised to the surface radiance from the 1 minute plasma sample for each mouse.
Author contributions
C. J. A. F., J. C., K. B., K. N., and K. A. H. conceived the research and planned the studies; K. N., H. R., M. W., B. A., K. K. S., and F. D.-H. performed experiments and analyzed and interpreted data. K. A. H., K. K. S., K. N., and H. R. drafted the manuscript. All authors read and approved the final manuscript.
Supplementary Material
This work was supported by Innovation Fund Denmark Grant 102-2014-3. K. N., H. R., and J. C. are employees of Albumedix Ltd., who manufacture and supply albumin for pharmaceutical use.
This article contains Tables S1–S6 and Figs. S1–S4.
- HSA
- human serum albumin
- rHSA
- recombinant human albumin
- MSA
- murine serum albumin
- FcRn
- neonatal Fc receptor
- hFcRn
- human FcRn
- mFcRn
- murine FcRn
- DAR
- drug-to-albumin ratio
- AF680
- Alexa Fluor 680
- NB
- null-FcRn binding affinity variant
- HB
- high FcRn–binding affinity variant
- HBI
- HB variant I
- HBII
- HB variant II
- DI
- DII, and DIII, domains I, II, and III, respectively
- GP-HPLC
- gel-permeation HPLC
- UHPLC
- ultrahigh-performance liquid chromatography
- DTNB
- 5,5-dithio-bis-(2-nitrobenzoic acid)
- TNB
- 5-thio-2-nitrobenzoic acid
- ROI
- region of interest
- t1/2
- half-life
- KD
- dissociation constant
- BMMS
- buffered minimal media sucrose.
References
- 1. Curry S., Brick P., and Franks N. P. (1999) Fatty acid binding to human serum albumin: new insights from crystallographic studies. Biochim. Biophys. Acta 1441, 131–140 10.1016/S1388-1981(99)00148-1 [DOI] [PubMed] [Google Scholar]
- 2. Chaudhury C., Mehnaz S., Robinson J. M., Hayton W. L., Pearl D. K., Roopenian D. C., and Anderson C. L. (2003) The major histocompatibility complex–related Fc receptor for IgG (FcRn) binds albumin and prolongs its lifespan. J. Exp. Med. 197, 315–322 10.1084/jem.20021829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Andersen J. T., Dalhus B., Viuff D., Ravn B. T., Gunnarsen K. S., Plumridge A., Bunting K., Antunes F., Williamson R., Athwal S., Allan E., Evans L., Bjørås M., Kjærulff S., and Sleep D. (2014) Extending serum half-life of albumin by engineering neonatal Fc receptor (FcRn) binding. J. Biol. Chem. 289, 13492–13502 10.1074/jbc.M114.549832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Schmidt E. G. W., Hvam M. L., Antunes F., Cameron J., Viuff D., Andersen B., Kristensen N. N., and Howard K. A. (2017) Direct demonstration of a neonatal Fc receptor (FcRn)-driven endosomal sorting pathway for cellular recycling of albumin. J. Biol. Chem. 292, 13312–13322 10.1074/jbc.M117.794248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kim J., Bronson C. L., Hayton W. L., Radmacher M. D., Roopenian D. C., Robinson J. M., and Anderson C. L. (2006) Albumin turnover: FcRn-mediated recycling saves as much albumin from degradation as the liver produces. Am. J. Physiol. Gastrointest. Liver Physiol. 290, G352–G360 10.1152/ajpgi.00286.2005 [DOI] [PubMed] [Google Scholar]
- 6. Sleep D. (2015) Albumin and its application in drug delivery. Expert Opin. Drug Deliv. 12, 793–812 10.1517/17425247.2015.993313 [DOI] [PubMed] [Google Scholar]
- 7. Howard K. A. (2015) Albumin: the next-generation delivery technology. Ther. Deliv. 6, 265–268 10.4155/tde.14.124 [DOI] [PubMed] [Google Scholar]
- 8. Larsen M. T., Kuhlmann M., Hvam M. L., and Howard K. A. (2016) Albumin-based drug delivery: harnessing nature to cure disease. Mol. Cell Ther. 4, 3 10.1186/s40591-016-0048-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Andersen J. T., Dalhus B., Cameron J., Daba M. B., Plumridge A., Evans L., Brennan S. O., Gunnarsen K. S., Bjørås M., Sleep D., and Sandlie I. (2012) Structure-based mutagenesis reveals the albumin-binding site of the neonatal Fc receptor. Nat. Commun. 3, 610 10.1038/ncomms1607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Caspersen M. B., Kuhlmann M., Nicholls K., Saxton M. J., Andersen B., Bunting K., Cameron J., and Howard K. A. (2017) Albumin-based drug delivery using cysteine 34 chemical conjugates: important considerations and requirements. Ther. Deliv. 8, 511–519 10.4155/tde-2017-0038 [DOI] [PubMed] [Google Scholar]
- 11. Schmøkel J., Voldum A., Tsakiridou G., Kuhlmann M., Cameron J., Sørensen E. S., Wengel J., and Howard K. A. (2017) Site-selective conjugation of an anticoagulant aptamer to recombinant albumins and maintenance of neonatal Fc receptor binding. Nanotechnology 28, 204004 10.1088/1361-6528/aa6a9b [DOI] [PubMed] [Google Scholar]
- 12. Kuhlmann M., Hamming J. B. R., Voldum A., Tsakiridou G., Larsen M. T., Schmøkel J. S., Sohn E., Bienk K., Schaffert D., Sørensen E. S., Wengel J., Dupont D. M., and Howard K. A. (2017) An albumin-oligonucleotide assembly for potential combinatorial drug delivery and half-life extension applications. Mol. Ther. Nucleic Acids 9, 284–293 10.1016/j.omtn.2017.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kratz F., Müller-Driver R., Hofmann I., Drevs J., and Unger C. (2000) A novel macromolecular prodrug concept exploiting endogenous serum albumin as a drug carrier for cancer chemotherapy. J. Med. Chem. 43, 1253–1256 10.1021/jm9905864 [DOI] [PubMed] [Google Scholar]
- 14. Miyamura S., Imafuku T., Anraku M., Taguchi K., Yamasaki K., Tominaga Y., Maeda H., Ishima Y., Watanabe H., Otagiri M., and Maruyama T. (2016) Comparison of posttranslational modification and the functional impairment of human serum albumin in commercial preparations. J. Pharm. Sci. 105, 1043–1049 10.1016/j.xphs.2015.12.015 [DOI] [PubMed] [Google Scholar]
- 15. Viuff D., Antunes F., Evans L., Cameron J., Dyrnesli H., Thue Ravn B., Stougaard M., Thiam K., Andersen B., Kjærulff S., and Howard K. A. (2016) Generation of a double transgenic humanized neonatal Fc receptor (FcRn)/albumin mouse to study the pharmacokinetics of albumin-linked drugs. J. Control Release 223, 22–30 10.1016/j.jconrel.2015.12.019 [DOI] [PubMed] [Google Scholar]
- 16. Larsen M. T., Rawsthorne H., Schelde K. K., Dagnæs-Hansen F., Cameron J., and Howard K. A. (2018) Cellular recycling-driven in vivo half-life extension using recombinant albumin fusions tuned for neonatal Fc receptor (FcRn) engagement. J. Control Release 287, 132–141 10.1016/j.jconrel.2018.07.023 [DOI] [PubMed] [Google Scholar]
- 17. Schmidt M. M., Townson S. A., Andreucci A. J., King B. M., Schirmer E. B., Murillo A. J., Dombrowski C., Tisdale A. W., Lowden P. A., Masci A. L., Kovalchin J. T., Erbe D. V., Wittrup K. D., Furfine E. S., and Barnes T. M. (2013) Crystal structure of an HSA/FcRn complex reveals recycling by competitive mimicry of HSA ligands at a pH-dependent hydrophobic interface. Structure 21, 1966–1978 10.1016/j.str.2013.08.022 [DOI] [PubMed] [Google Scholar]
- 18. Oganesyan V., Damschroder M. M., Cook K. E., Li Q., Gao C., Wu H., and Dall'Acqua W. F. (2014) Structural insights into neonatal Fc receptor-based recycling mechanisms. J. Biol. Chem. 289, 7812–7824 10.1074/jbc.M113.537563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sand K. M. K., Bern M., Nilsen J., Dalhus B., Gunnarsen K. S., Cameron J., Grevys A., Bunting K., Sandlie I., and Andersen J. T. (2014) Interaction with both domain I and III of albumin is required for optimal pH-dependent binding to the neonatal Fc receptor (FcRn). J. Biol. Chem. 289, 34583–34594 10.1074/jbc.M114.587675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Petersen S. S., Kläning E., Ebbesen M. F., Andersen B., Cameron J., Sørensen E. S., and Howard K. A. (2016) Neonatal Fc receptor binding tolerance toward the covalent conjugation of payloads to cysteine 34 of human albumin variants. Mol. Pharm. 13, 677–682 10.1021/acs.molpharmaceut.5b00605 [DOI] [PubMed] [Google Scholar]
- 21. Bienk K., Hvam M. L., Pakula M. M., Dagnæs-Hansen F., Wengel J., Malle B. M., Kragh-Hansen U., Cameron J., Bukrinski J. T., and Howard K. A. (2016) An albumin-mediated cholesterol design-based strategy for tuning siRNA pharmacokinetics and gene silencing. J. Control Release 232, 143–151 10.1016/j.jconrel.2016.04.013 [DOI] [PubMed] [Google Scholar]
- 22. Roopenian D. C., Low B. E., Christianson G. J., Proetzel G., Sproule T. J., and Wiles M. V. (2015) Albumin-deficient mouse models for studying metabolism of human albumin and pharmacokinetics of albumin-based drugs. MAbs 7, 344–351 10.1080/19420862.2015.1008345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Andersen J. T., Daba M. B., Berntzen G., Michaelsen T. E., and Sandlie I. (2010) Cross-species binding analyses of mouse and human neonatal Fc receptor show dramatic differences in immunoglobulin G and albumin binding. J. Biol. Chem. 285, 4826–4836 10.1074/jbc.M109.081828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Krieger E., and Vriend G. (2014) YASARA View—molecular graphics for all devices—from smartphones to workstations. Bioinformatics 30, 2981–2982 10.1093/bioinformatics/btu426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Evans L., Hughes M., Waters J., Cameron J., Dodsworth N., Tooth D., Greenfield A., and Sleep D. (2010) The production, characterisation and enhanced pharmacokinetics of scFv-albumin fusions expressed in Saccharomyces cerevisiae. Protein Expr. Purif. 73, 113–124 10.1016/j.pep.2010.05.009 [DOI] [PubMed] [Google Scholar]
- 26. Smith M. E. B., Caspersen M. B., Robinson E., Morais M., Maruani A., Nunes J. P. M., Nicholls K., Saxton M. J., Caddick S., Baker J. R., and Chudasama V. (2015) A platform for efficient, thiol-stable conjugation to albumin's native single accessible cysteine. Org. Biomol. Chem. 13, 7946–7949 10.1039/C5OB01205H [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.