

Abstract
Desmoplakin is encoded by DSP gene, whose altered function leads to skin and hair abnormalities, and heart diseases. The whole triad of these traits characterizes the Carvajal syndrome (CS).
CS is an autosomal recessive genetic disorder, mapping on chromosome 6q24 and caused by mutations in DSP gene.
We report a patient with CS caused by two novel mutations in DSP gene, inherited from his parents, both asymptomatic. The same phenotype was present in his younger sister who showed skin abnormality and woolly hairs. The segregation analysis of the known loci in DSP gene performed by genetic testing, was able to established the trans position of the two mutations (c.6986T > C and c.7123G > C) in the patient and his sister. The first mutation has been inherited from the mother, the other one from the father. The resulting compound heterozygous mutation in the siblings, is likely the cause of the disease.
Key words: Carvajal syndrome, cardiocutaneous phenotype, desmoplakin mutations
Desmosomes are cell junctional complexes involved in the regulation of homeostasis. They participate in signalling cascades and provide resistance against mechanical stress, maintaining the strength and rigidity of the cells (1). This property is especially important for cardiac and epidermal tissues, where adjacent cell contacts largely contribute to correct functioning (2). A crucial component of desmosomes is desmoplakin encoded by the DSP gene (3), whose mutations have been associated to the Carvajal syndrome (CS). This syndrome is an autosomal recessive genetic disorder characterized by the following manifestations: woolly hair, striate palmoplantar keratoderma and left ventricular dilated cardiomyopathy (DCM). Additional phenotypic signs include dental abnormalities and leukonychia (4, 5). The symptoms of the disease usually manifest over time. Woolly hair is present from birth, while palmoplantar keratoderma develops after infancy. Left ventricular dilatation is usually asymptomatic at an early age and cannot be diagnosed without instrumental cardiologic examination. Complaints of chest pain appear in 8-12 years. DCM in CS progresses rapidly, leading to heart failure or sudden death in adolescence. CS is caused by homozygous or compound heterozygous mutations in the DSP gene often occurring in hot spots of the gene, exons 23 and 24 (6, 7).
In this article, we report a familial CS with two affected sibs sharing a compound heterozygous mutation in DSP gene, inherited from asymptomatic parents.
Case Presentation
The proband is a 11-year-old boy presenting focal keratoderma, specifically affecting the palmoplantar epidermis, woolly hair, and DCM (Fig. 1), consistent with a diagnosis of Carvajal Syndrome. Woolly hair has been observed from birth, while the keratoderma appeared during the second year of life. The manifestation of the DCM occurred at the age of 10 years with complaints of dyspnea and weakness. The cardiomyopathy was confirmed by cardiological examination including electrocardiography (ECG) echocardiography (Echo) and computed tomography (CT). A year later, despite the pharmacological treatment, the physicians observed a significant and rapid evolution of biatrial and biventricular dilatation with severe systolic dysfunction of both ventricles. The patient was hospitalized to receive optimal therapy for severe heart failure and then, at the age of 11 years, he underwent successful orthotopic heart transplantation.
Figure 1.
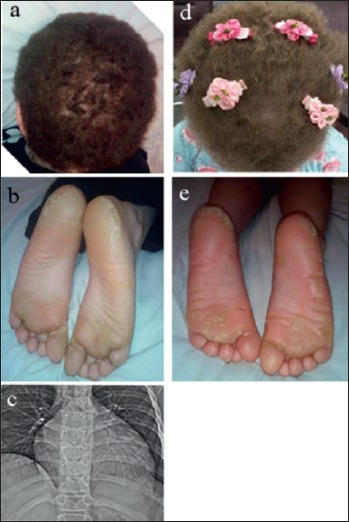
Clinical findings. Manifestations of the Carvajal syndrome in the proband (a, b) and his sister (d, e.); CT scan of the patient’s chest with signs of cardiomegaly (c).
The 5-year-old sister of the proband had similar clinical manifestations as skin abnormality and woolly hairs, appearing from birth (Fig. 1d-e). She was asymptomatic and showed no signs of cardiac involvement. Since the age of 4 she underwent a comprehensive cardiac examination every 6 months, without any pathological result. ECG, Holter, and Echo investigation.
Genetics study
The NGS analysis was performed in the Laboratory of molecular pathology “Genomed Ltd.” (Moscow). using a panel for inherited heart diseases. Two heterozygous mutations in exon 24 of the DSP gene were found – c.7123G > C and c.6986T > C (NM_004415.2), not represented in the gnomAD database, so they can be defined as truly rare variants.
The first mutation – c.7123G > C – leads to the amino acid substitution p.Gly2375Arg (rs376923069). Only GO-ESP database reports 0.0001 (1/13006) frequency for this mutation; however the G > A change in the same position, that leads to Gly > Arg substitution too, has a 0.00006 (2/31394) frequency, according to gnomAD. c.7123G > C was found in one individual with DCM, woolly hair, keratoderma and classified as variant of uncertain significance (VUS) in the “Actionable exomic incidental findings in 6503 participants”, and it was previously described by Alcalai et al. (2003) in a case study of a patient with familial autosomal recessive arrhythmogenic right ventricular dysplasia, woolly hair and a pemphigous-like skin disorder (8).
The second mutation – c.6986T > C – that results in p.Leu2329Pro substitution, is not reported neither in the population databases nor mentioned in published reports.
To follow the segregation of the variants, we performed the segregation analysis in the whole family. The research was carried out according to the Declaration of Helsinki Principles. The parents gave written consent for genetic research after being informed. Their DNA was extracted from buccal epithelium by the phenol-chloroform method. The 24th exon of DSP gene was amplified and sequenced using the Sanger method. The mother was found to be carrier of the c.6986T > C mutation, the father carrier of the 7123G > C mutation (Fig. 2). Both parents of the proband are asymptomatic.
Figure 2.
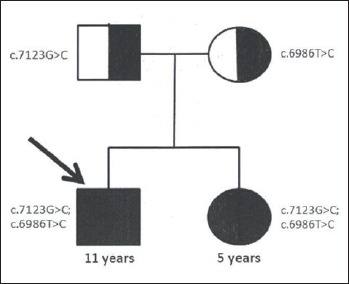
Results of the segregation analysis. The arrow indicates the proband; ■= affected; ◑ = carrier.
The analisys of segregation in the family established the trans position for c.7123G > C and c.6986T > C variants. As a consequence, the structure of the DSP gene is disrupted on both chromosomes 6 in the proband and his sister.
The combination of these variants forms a compound heterozygous conditon having a high probability to be the cause of the disease. Currently, the ClinVar database includes more than 108 pathogenic and 78 likely pathogenic variations in the DSP gene that are associated with defects in epidermal tissue (skin, hair) in a wide range of phenotypes, frequently with heart abnormalities (ClinVar 2018).
The evaluation of the c.7123G > C and c.6986T > C variants according to the algorithm ACMG/AMP (9) is likely pathogenic. The following criteria have been assigned: PM1 (variant is located in a mutational hot spot), PM2 (absent fromcontrols/extremely low frequency), PM3 (detected in trans with a pathogenic variant), PP1 (co-segregation with disease), PP4 (patient’s phenotype is highly specific for a disease), PP3 (results of in silico analysis). Computational methods predict a deleterious impact of the mutations on the protein functions (Table 1). The authors of the ACMG/AMP guideline recommend to use a PP1 as stronger evidence under specific conditions. Based on the facts outlined above, we consider the segregation of the two mutations in this family as a strong evidence of pathogenicity.
Table 1.
Different predictors’ scores provided by Condel.
| Predictor | The substitution | |
|---|---|---|
| p.Gly2375Arg | p.Leu2329Pro | |
| SIFT 1¦0 | 0.08 | 0.0 |
| PPH2 0¦1 | 1.0 | 1.0 |
| MA 0¦4 | 3.63 | 3.31 |
| FATHMM +8¦-5 | -2.23 | -2.64 |
| Condel 0¦1 | 0.73 | 0.68 |
| Condel label | Deleterious | Deleterious |
The direction of the arrows shows the way of increasing pathogenicity strength of the variants.
Discussion
An unfavorable prognosis of the Carvajal syndrome is associated with early onset of the disease, high risk of sudden cardiac death and a rapidly developing heart failure consequent to the progressive dilatation of the heart chambers (10). Heart failure develops in patients with CS in early childhood, and an extreme dilatation of the left ventricle is detected in more than 90% of patients in their early teens.
Although the young sister of the proband does not show cardiac involvement, considering her young age an onset of the heart disease in future cannot be excluded.
In this regard, international experts recommend to perform in children presenting palmoplantar keratoderma and woolly hair a comprehensive cardiological evaluation by ECG, Holter monitoring and Echo as soon as possible in order to detect possible cardiac anomalies and children at risk of premature death. An early diagnosis may be lifesaving in these patients (10).
Conclusions
We describe two cases of CS syndrome due to novel mutations in the DSP gene. The pathogenicity of the two variants – c.6986T > C and c.7123G > C – was assumed by the clear segregation of the disease with mutations in exon 24 of the DSP gene, and by the presence in the family of heterozygous unaffected parents who generated two children with Carvajal’s syndrome, sharing mutations in a compound heterozygous condition.
References
- 1.Green K, Parry D, Steinert D, et al. Structures of the human desmoplakins: implications for function in the desmosomal plaque. J Biol Chem 1990;265:2603-12. [PubMed] [Google Scholar]
- 2.Cowin P, Bruke B. Cytoskeleton-membrane interactions. Curr Opin Cell Bio 1996;8:56-65. [DOI] [PubMed] [Google Scholar]
- 3.Nitoiu D, Etheridge SL, Kelsell DP. Insights into desmosome biology from inherited human skin disease and cardiocutaneous syndromes. Cell Commun Adhes 2014;21:129-40. [DOI] [PubMed] [Google Scholar]
- 4.Boule S, Fressart V, Laux D, et al. Expanding the phenotype associated with a desmoplakin dominant mutation: Carvajal/Naxos syndrome associated with leukonychia and oligodontia. Int J Cardiol 2012;161:50-2. [DOI] [PubMed] [Google Scholar]
- 5.Carvajal-Huerta L. Epidermolytic palmo-plantar keratoderma with woolly hair and dilated cardiomyopathy. J Am Acad Dermatol 1998;39:418-21. [DOI] [PubMed] [Google Scholar]
- 6.Protonotarios N, Tsatsopoulou A. Naxos disease and Carvajal syndrome: cardiocutaneous disorders that highlight the pathogenesis and broaden the spectrum of arrhythmogenic right ventricular cardiomyopathy. Cardiovasc Pathol 2004;13:185-94. [DOI] [PubMed] [Google Scholar]
- 7.Yesudian PD, Cabral RM, Ladusans E, et al. Novel compound heterozygous mutations in the desmoplakin gene cause hair shaft abnormalities and culminate in lethal cardiomyopathy. Clin Exp Dermatol 2014;39:506-8. [DOI] [PubMed] [Google Scholar]
- 8.Alcalai R, Metzger S, Rosenheck S, et al. A recessive mutation in desmoplakin causes arrhythmogenic right ventricular dysplasia, skin disorder, and woolly hair. J Am Coll Cardiol 2003;42:319-27. [DOI] [PubMed] [Google Scholar]
- 9.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17:405-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Malčić I, Buljević B. Arrhythmogenic right ventricular cardiomyopathy, Naxos island disease and Carvajal syndrome. Central Eur J Paed 2017;13:93-106. [Google Scholar]