

Summary
The circadian clock and the hypoxia-signaling pathway are regulated by an integrated interplay of positive and negative feedback limbs that incorporate energy homeostasis and carcinogenesis. We show that the negative circadian regulator CRY1 is also a negative regulator of hypoxia-inducible factor (HIF). Mechanistically, CRY1 interacts with the basic-helix-loop-helix domain of HIF-1α via its tail region. Subsequently, CRY1 reduces HIF-1α half-life and binding of HIFs to target gene promoters. This appeared to be CRY1 specific because genetic disruption of CRY1, but not CRY2, affected the hypoxia response. Furthermore, CRY1 deficiency could induce cellular HIF levels, proliferation, and migration, which could be reversed by CRISPR/Cas9- or short hairpin RNA-mediated HIF knockout. Altogether, our study provides a mechanistic explanation for genetic association studies linking a disruption of the circadian clock with hypoxia-associated processes such as carcinogenesis.
Subject Areas: Biological Sciences, Biochemistry, Molecular Biology, Cell Biology
Graphical Abstract
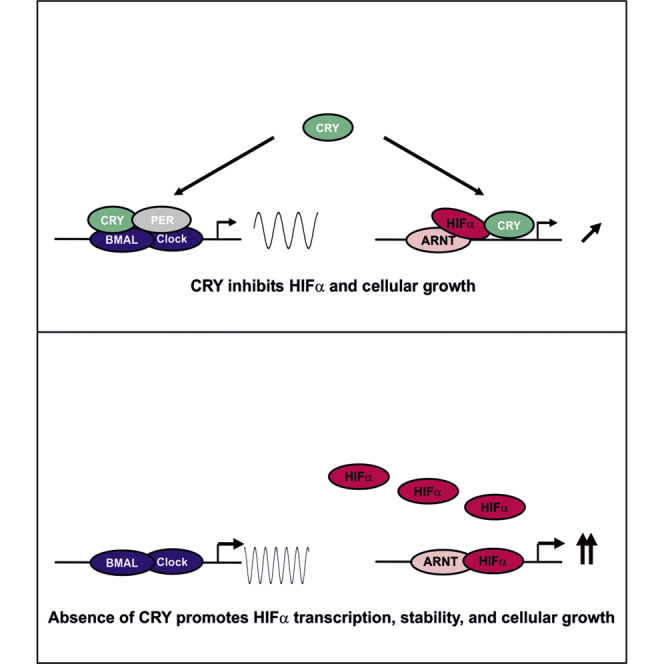
Highlights
-
•
Hypoxia and HIFs affect the circadian rhythm
-
•
CRY1 directly interacts with both HIF-1α and HIF-2α
-
•
CRY1 inhibits binding of HIFs to its target gene promoters
-
•
The CRY1-HIFα interaction has opposite roles on cellular growth and migration
Biological Sciences; Biochemistry; Molecular Biology; Cell Biology
Introduction
A number of epidemiological studies showed that disturbances of the circadian rhythm strongly correlate with carcinogenesis and tumor progression in humans (for review see Fu and Lee, 2003, Filipski and Levi, 2009, Sahar and Sassone-Corsi, 2009, Rana and Mahmood, 2010). Accordingly, the International Agency for Research on Cancer classified shift work as a group 2A carcinogenic factor (Straif et al., 2007).
The circadian rhythm in mammals is maintained by an integrated network of the central (neural or brain) and peripheral (tissue-specific) clocks. The central clock is located in the suprachiasmatic nucleus of the brain, receives light cues to keep in phase with the light-dark cycle, and synchronizes the peripheral clocks in various tissues through a variety of electrical, endocrine, and metabolic signaling pathways (Albrecht, 2012, Richards and Gumz, 2012). At the genetic level, both the central and the peripheral clocks are regulated by an interplay of positive and negative feedback loops involving the same set of clock genes (Yagita et al., 2001). Thereby, the bHLH-PAS transcription factors CLOCK (circadian locomotor output cycles kaput) and BMAL1 (brain-muscle Arnt-like protein 1) represent the major components of the core clock's positive limb. They induce, among others, the expression of the proteins PER (period 1,2) and CRY (cryptochrome 1,2), which constitute the major arm of the negative limb. The induced PER and CRY proteins then form a complex with each other as well as with modulator proteins such as CK1ε, CKII, or FBXL3 and act as repressors of CLOCK/BMAL heterodimers. Subsequently, they inhibit their own expression as well as those of other CLOCK/BMAL-regulated output genes (Griffin et al., 1999, Kume et al., 1999, Yoo et al., 2005, Sato et al., 2006). The core loop is interconnected to several other feedback loops, including those of REV-ERBs or PPARα/RORs, which repress or activate BMAL1 expression, respectively (Albrecht, 2012, Ko and Takahashi, 2006, Asher and Schibler, 2011). Overall, the continuous interplay between the core CLOCK/BMAL1 positive limb and the PER/CRY negative limb in concert with post-translational modifications and interconnected loops results in the oscillation of gene expression in a circadian manner.
Several recent studies have linked clock function with the cell cycle and reported that clock components, such as PER1, PER2, BMAL1, and CRY1/2 decrease cell proliferation or improve the action of anti-cancer drugs in different cancer cell lines. Moreover, certain types of human cancer show an altered expression of circadian clock genes (reviewed in Shostak, 2017).
In addition to disturbances of the circadian clock, most, if not all, solid tumors display hypoxic areas. Hypoxia has been shown to be the major driver of tumor angiogenesis and a critical determinant for proliferation, cell growth, and apoptosis (Semenza, 2017, Masson and Ratcliffe, 2014, Kaelin and Ratcliffe, 2008). Mechanistically, a group of the bHLH-PAS family transcription factors called hypoxia-inducible transcription factors, among which hypoxia-inducible factor (HIF)-1α is the best characterized, mediate the transcriptional adaptation to hypoxia. Thereby, HIF-1α together with its binding partner HIF-1β (also known as aryl hydrocarbon receptor nuclear translocator, ARNT) binds to hypoxia-response elements (HREs) within the promoters of numerous hypoxia-responsive genes (Semenza, 2017, Masson and Ratcliffe, 2014, Kaelin and Ratcliffe, 2008).
The disruption of the circadian rhythm in patients with cancer and the appearance of hypoxia in tumors raises the question whether and how the deregulation of the circadian clock system has an impact on the hypoxia response. Several findings suggested a cross talk between the circadian clock and the hypoxia signaling pathway (Hogenesch et al., 1998, Chilov et al., 2001, Eckle et al., 2012), but the mechanisms behind are not completely understood and have just started to emerge. Recent reports have shown that the modulation of oxygen levels can reset the circadian clock at the positive limb in a HIF-1α-dependent manner (Adamovich et al., 2017) and that HIF-1α and BMAL1 engage in a synergistic cross talk (Wu et al., 2017), which adapts anaerobic metabolism in skeletal muscle (Peek et al., 2017). Although these findings favor the view that this cross talk is bidirectional and would eventually also involve the negative arm of the circadian key players, their participation, in particular that of CRY proteins, remains unknown. In the current study we broaden this view by showing that CRY1, but not CRY2, acts as a repressor of HIFs. This occurs via a specific protein-protein interaction that reduces the binding of HIFs at the HREs of target gene promoters and by altering HIF half-life. Disruption of the CRY1-HIF cross talk at the cellular level shows that CRY1 and HIF-1α have an opposite action on cell growth.
Overall, our study identifies a new level of control that links defects in the circadian clock system with hypoxia signaling and carcinogenesis.
Results
Hypoxia Affects the Circadian Rhythm In Vivo
To study the possible interplay between the circadian clock and hypoxia we measured the circadian behavior of mice under normoxia (21% O2) and hypoxia (17% O2) at 12 h + 12 h light/dark (LD) cycles and at constant darkness (dark/dark; DD). When mice were kept under both normoxia and hypoxia at LD they showed circadian periodicity (Figures 1A and 1B). Intriguingly, we observed that mice at DD lost their circadian rhythm (Figures 1A and 1B) and expressed HIF target genes when exposed to hypoxia (Figure 1C). Together, these data indicate that hypoxia is able to modulate the circadian rhythm.
Figure 1.

Representative Actograms of Individually Monitored Mice under Normoxia and Hypoxia at Light-Dark Cycles (LD) and Constant Darkness (DD)
(A) Mice were kept under normoxia (n = 14) and hypoxia (B) (n = 14) at 12-h light/12-h dark cycles (LD) for a week, which was followed by another week under the same conditions at constant darkness (DD).
(C) Hypoxia-dependent induction of Cited2 and Glut-1 mRNA in liver and kidney of the mice after the DD period. The respective mRNA levels were assessed by qRT-PCR, and the values indicate fold-change hypoxia versus normoxia. Boxes represent the interquartile range. Whiskers represent the minimum and maximum observations. *Significant difference normoxia versus hypoxia.
The Cell-Autonomous Circadian Oscillation in NIH3T3 Cells Is Affected by HIF-1α or HIF-2α
As the hypoxia-mediated loss of the circadian rhythm in mice at DD stems from systemic and cell-autonomous oscillator effects, we next tested to what extent hypoxia affects the cell-autonomous clock. To do this, we first verified the effect of chronic hypoxia (3% O2 for 32 h) in synchronized Per2:Luc-transfected fibroblasts, which possess a cell-autonomous and self-sustained circadian clock (Yagita et al., 2001, Balsalobre et al., 1998). We found that hypoxia affected the circadian rhythm in two ways, first by reducing the amplitude and second by shortening the period by about 30 min (Figures 2A and 2B). Given that the response to hypoxia is mainly mediated by an increased abundance of the transcription factors HIF-1α and HIF-2α, we next investigated how HIF-1α or HIF-2α affect the circadian clock. To do this, we first cotransfected an expression vector for a hypoxia-mimicking hydroxylation-resistant HIF-1α variant (P/P/N) (Flügel et al., 2012) with the circadian rhythm reporter Bmal1:Luc and analyzed circadian rhythmicity. Similar to hypoxia, overexpression of HIF-1α shortened the circadian period and reduced the amplitude (Figures 2C and 2D). Overexpression of HIF-2α showed a similar response (Figures S1A and S1B). In contrast to hypoxia and HIFs, the non-specific prolyl hydroxylase inhibitor dimethyloxalylglycine (DMOG), which is often considered as a hypoxia mimetic, lengthened the period (Figures S1C and S1D). Together, these data indicate that HIF-1α and HIF-2α mimic the effect of hypoxia on the circadian clock, whereas the DMOG effects appear to be HIF independent.
Figure 2.

The Circadian Oscillation in NIH3T3 Cells Is Affected by HIF-1α
(A and B) (A) Representative examples of the bioluminescence oscillations in NIH3T3mPer2:Luc cells precultured under hypoxia (3% O2) for 32 h. (B) Graphical illustration of the amplitude (y axis) and period length (x axis) upon hypoxia treatment. Data are mean ± SEM. *p < 0.05.
(C) Representative example of the bioluminescence oscillations in NIH3T3 cells cotransfected with an mBmal1:luciferase reporter construct and either empty vector (black line) or HIF-1α P/P/N (green line).
(D) Graphical illustration of the amplitude (y axis) and period length (x axis) upon overexpression of HIF-1α P/P/N. Data are mean ± SEM. *p < 0.05, ***p < 0.001.
The Circadian Oscillation of PAI-1 Is Affected by Hypoxia
The aforementioned observations appear to be important for genes that are responsive to both hypoxia and the circadian clock. The SERPINE1 gene encoding plasminogen activator inhibitor-1 (PAI-1) fulfills these criteria and is well known to be a bona fide hypoxia-responsive and circadian-rhythm-regulated gene (Dimova et al., 2005, Kietzmann et al., 1999, Maemura et al., 2000, Schoenhard et al., 2003, Wang et al., 2006, Oishi et al., 2009, Cheng et al., 2011). Therefore we investigated whether the hypoxia-induced shifts in circadian rhythmicity are transferred to the expression of hypoxia-responsive target genes and to what extent one or the other response prevails. To examine this, we analyzed PAI-1 expression on protein and reporter gene level in synchronized cells. We found that hypoxia enhanced PAI-1 expression and shortened the circadian period of PAI-1 by about 40 min (Figures 3A–3D). Together, these data further substantiate that hypoxia and HIFs can advance the period of clock-dependent genes, whereas the amplitude is likely also modulated depending on the transcriptional context.
Figure 3.

The Circadian Oscillation of PAI-1 Expression Is Affected by Hypoxia
(A) Representative example of the bioluminescence oscillations in cells stably transfected with a PAI-1 promoter-driven luciferase construct under normoxia and hypoxia.
(B and C) (B) Graphical illustration of the amplitude (y axis) and period length (x axis) of PAI-1 promoter-driven luciferase expression under normoxia and hypoxia. Data are mean ± SEM. *p < 0.05. (C) Representative western blot analysis of total cell lysates of synchronized Rat-1 fibroblasts probed with PAI-1 and β-actin antibodies.
(D) Densitometric analysis of the PAI-1 protein levels presented on (C).
CRY1 Interacts with HIF-1α and HIF-2α
Accumulating genetic data indicate that the negative limb of the circadian regulators, in particular CRY1, is responsible for maintaining the period length or amplitude of the circadian clock and that CRY1 is the most potent repressor in the clock's negative limb (Griffin et al., 1999, Kume et al., 1999, Van Der Horst et al., 1999, Vitaterna et al., 1999, Li et al., 2016, Liu et al., 2007, Khan et al., 2012, Matsuo et al., 2003, Koike et al., 2012, Langmesser et al., 2008, Ye et al., 2014, Michael et al., 2017). Based on CRY1's potency and responsibility for period and amplitude maintenance and the recent reports on the HIF-1α-BMAL1 cross talk (Wu et al., 2017, Peek et al., 2017) as well as the aforementioned findings that HIFs shortened the period and affected the amplitude of the circadian clock, we hypothesized that HIFs would interact with CRY1.
To examine this, we performed coimmunoprecipitation analyses with hypoxic mouse embryonic fibroblasts (MEFs). The assays revealed that CRY1 could be detected when HIF-1α was precipitated from the cells (Figure 4A). Furthermore, CRY1 could also be detected upon precipitation with HIF-2α antibodies, although the binding signal was low because of the low abundance of HIF-2α (Figure S2A). Next, we verified these interactions and performed western blot analyses from immunoprecipitates of HEK293 cells expressing Myc-tagged HIF-1α, V5-tagged HIF-2α, and hemagglutinin (HA)-tagged CRY1. The blots from anti-Myc-tagged HIF-1α or anti-V5-tagged HIF-2α immunoprecipitations showed the presence of HA-tagged CRY1 when probed with the HA-tag antibody indicating interaction of HIFα proteins and CRY1 (Figures 4B and S2B). In addition, HIFα proteins were also able to interact with CRY2 (Figures 4C and S2C). We also employed a bimolecular fluorescence complementation (BiFC) assay to verify whether these interactions are direct. The BiFC assay takes advantage from the possibility that a fluorescent protein complex can be formed through association of two per se non-interacting and non-fluorescent N-terminal (YN) and C-terminal (YC) fragments of the yellow fluorescent protein (YFP). The complementation of the fluorescent complex is achieved when YN and YC are brought into proximity owing to a direct interaction of the proteins fused to the YN and YC fragments (Figure 4D) (Kerppola, 2008). We generated vectors allowing expression of fusion proteins consisting of HIF-1α or HIF-2α and the C-terminal part of the YFP (HIF-1α-YC or HIF-2α -YC) as well as of CRY1 and the N-terminal part of the YFP (CRY1-YN). In addition, constructs for the HIFα partner ARNT as well as for the CRY1 partner PER1 served as positive controls to indicate HIF-1α -ARNT or CRY1-PER1 interaction, respectively. The BiFC analyses revealed a strong fluorescent signal when either HIF-1α-YC (Figures 4E and 4F) or HIF-2α-YC (Figure S2D) and CRY1-YN constructs were applied in the assay. In addition, a clear fluorescent signal could be observed when HIF-1α-YC was coexpressed with ARNT-YN or PER1-YC with CRY1-YN as positive controls (Figures 4E, S2D, and S2E). By contrast, when HIF-1α-YC or HIF-2α -YC was coexpressed with a vector encoding only YN (Figures 4E and 4F) no fluorescence was detected. Likewise, the coexpression of YC and YN did not result in the generation of fluorescence, indicating that the separated non-fluorescent fragments did not spontaneously self-assemble (Figures 4E and 4F). Together, these results indicate that HIF-1α and HIF-2α can interact with CRY1 in living cells.
Figure 4.

CRY1 Interacts with HIF-1α
(A) Coimmunoprecipitation (IP) assays from wild-type MEFs cultured under hypoxia in the presence of MG-132. Blots from anti-HIF-1α IPs were probed with the CRY1 and HIF-1α antibody. The blots shown are representative of three independent experiments. IB, immunoblotting.
(B and C) Coimmunoprecipitation studies with HEK293 cells, expressing myc-tagged HIF-1α with either (B) HA-tagged CRY1 or (C) V5-tagged CRY2. Blots from anti-myc-tag IPs were probed with HA-tag or V5-tag antibody. The blots shown are representative of three independent experiments.
(D and E) BiFC analysis. (D) Schematic representation of expressed proteins employed in BiFC. The sequences encoding amino acid residues 1–154 (N-terminus [YN]) or 155–238 (C-terminus [YC]) of the yellow fluorescent protein (YFP) were fused to the 3′ ends of the coding regions for HIF-1α, CRY1, or ARNT to generate HIF-1α -YC, CRY1-YN, and ARNT-YN, respectively. YN and YC are non-fluorescent fragments that do not interact per se with each other. An interaction between the proteins fused to the YN and YC fragments facilitates their association to form a bimolecular fluorescent complex. (E) Visualization of the BiFC signal by confocal microscopy. COS-7 cells cotransfected with the expression vectors for HIF-1α-YC, CRY1-YN, ARNT-YN, and vectors encoding only YN or YC were cultured on glass slides for 24 h. The fluorescence detection was performed using specific filter sets for YFP and DAPI. Scale bar, 10 μm.
(F) Quantification of the BiFC signal. Cells were transfected as in (E) and quantified by flow cytometry (cf. Transparent Methods). The cytomegalovirus promoter (CMV) driven-YFP signal was set to 100%. Data are mean ± SEM. *p < 0.05.
The Tail Region of CRY-1 Interacts with HIF-1α
To find out which domain within CRY1 interacts with HIF-1α, we expressed Myc-tagged HIF1α along with different HA-tagged CRY1 protein variants in HEK293 cells (Figure 5A) and analyzed their interaction by coimmunoprecipitation experiments. Full-length CRY1 interacted with HIF-1α, but this interaction was lost when CRY1 proteins lacking either the CC and tail region or only the tail region were employed in the assays (Figure 5B). Thus, these data indicate that the tail region of CRY1 is necessary for the interaction of CRY1 with HIF-1α.
Figure 5.

The Tail Domain of CRY1 Is Responsible for Interaction with HIF-1α
(A) Schematic representation of the HA-tagged CRY1 protein variants.
(B) HEK293 cells were transfected with myc-tagged HIF-1α together with HA-tagged full-length CRY1 or its mutants, cell extracts were prepared and immunoprecipitated with an anti-myc-tag antibody, and precipitates were analyzed by immunoblot with an anti-HA-tag antibody. As a control the expression levels of the CRY1 protein variants as well as myc-tagged HIF-1α were verified in whole-cell extracts (Input). IP, immunoprecipitation; IB, immunoblotting.
CRY1 Interacts with the bHLH Domain of HIF-1α
To map the exact HIF-1α domain that interacts with CRY1, we designed several Myc-tagged HIF-1α deletion mutants comprising one or several domains (Figure 6A) and performed coimmunoprecipitation experiments in HA-CRY1 expressing HEK293 cells. When full-length HIF-1α or its deletion variants were pulled down with anti-Myc-tag antibodies we found that HA-CRY1 coprecipitated full-length HIF-1α and all deletion variants except the HIF-1α mutant lacking the bHLH domain (Figure 6B). Together, these results indicate that CRY1 interacts with the bHLH domain of HIF-1α.
Figure 6.

The bHLH Domain of HIF-1α Is Responsible for Interaction with CRY1
(A) Schematic representation of the myc-tagged HIF-1α protein variants.
(B) Coimmunoprecipitation experiments were performed with total proteins from HEK293 cells expressing HA-tagged CRY1 and myc-tagged full-length HIF-1α or its deletion variants. Anti-myc-tag antibody (IP, immunoprecipitation)-precipitated proteins were analyzed by immunoblot (IB, immunoblotting) using an anti-HA-tag antibody. As a control the expression levels of HIF-1α, its deletion variants, as well as the HA-tagged CRY1 proteins were verified in whole-cell extracts (Input).
CRY1 Inhibits Binding of HIF-1α to Its Target Gene Promoters
As CRY1 interacts with the HIF-1α bHLH domain, which primarily facilitates binding to DNA at target gene promoters, we next analyzed how CRY1 would affect hypoxia and HIF-dependent gene expression. First, we performed a series of functional reporter gene assays in HepG2 cells. Upon cotransfection of these cells with the hypoxia- and HIF-responsive wild-type (WT) human PAI-1 promoter Luc construct (pGL3-hPAI-806/+19) and an expression vector for CRY1 we found that the hypoxia-mediated increase in Luc activity was reduced in the presence of CRY1. This effect was mediated via the hypoxia responsive element (HRE) within the PAI-1 promoter because a construct with a mutated element (pGL3-hPAI-HREm) did not display this effect on Luc activity (Figure 7A). Vice versa, when we employed MEFs deficient for either CRY1 (ΔCRY1) or CRY2 (ΔCRY2) to measure PAI-1 expression, we found that deficiency of CRY1, but not CRY2, enhanced PAI-1 levels already under normoxia to almost the same levels as under hypoxia (Figures S3A and S3C). Reintroduction of CRY1 (Figures S3D and S3E) rescued the hypoxia-dependent PAI-1 expression indicating that these effects are largely CRY1 specific. In line with the interaction assays, these rescue effects were dependent on the tail region of CRY1 since only full-length CRY1, but not CRY1-Δtail, rescued the hypoxia-dependent induction of PAI-1 (Figures S3D and S3E). Thus, CRY1 appears to function as inhibitor of the hypoxia-dependent PAI-1 gene expression.
Figure 7.

CRY1 Inhibits Binding of HIF-1α to Its Target Gene Promoters
(A and B) Cells were cotransfected with an expression vector for CRY1 along with either (A) the wild-type human PAI-1 promoter Luc construct (pGL3-hPAI-806/+19) or a PAI-1 promoter with a mutated hypoxia-responsive element (HRE) (pGL3-hPAI-HREm) as well as with (B) Luc reporter gene constructs containing three HIF-binding HREs (pGL3-HRE) or three mutated HREs (pGL3-HREm) in front of the SV40 promoter and cultured under normoxia (16% O2) and hypoxia (5% O2) for 24 h. The Luc activity of pGL3-hPAI-806/+19-, pGL3-hPAI-HREm-, pGL3-HRE-, or pGL3-HREm-transfected cells at 16% O2 was set to 1. Values are means ± SEM of at least three independent culture experiments, each performed in duplicate. Statistics, Student's t test for paired values: *significant difference, p ≤ 0.05.
(C) Cells were cotransfected with pGL3-HRE or pGL3-HREm Luc reporter gene constructs and expression vectors encoding CRY1, HIF-1α, and ARNT alone or in combination. In each experiment the Luc activity of pGL3-HRE- or pGL3-HREm-transfected cells at 16% O2 was set to 1. Values are means ± SEM of at least three independent culture experiments, each performed in duplicate. Statistics, Student's t test for paired values: ∗significant difference, p ≤ 0.05.
(D) Chromatin immunoprecipitation (ChIP) was performed on cell lysates from formaldehyde cross-linked wild-type or CRY1-deficient MEFs, cultured under normoxia or hypoxia overnight. DNA fragments were co-precipitated with antibodies against mouse HIF-1α and amplified by quantitative PCR using primers specific for the mouse PAI-1, VEGF-A, PGK-1, and GLUT-1 promoters containing the corresponding HRE. Chromatin immunoprecipitation without antibody or using IgG instead of antibody served as additional specificity controls. Differences in the HIF-1α DNA-binding efficiency in wild-type and ΔCRY1 MEFs were calculated by the fold enrichment method relative to the IgG control. Values are means ± SEM of three independent experiments. Statistics, Student's t test for paired values: *significant difference, p ≤ 0.05.
Next, we aimed to substantiate the inhibitory effect of CRY1 for HIF-driven genes and HIFs. To do this, we used bona fide hypoxia reporters containing either three WT HIF-binding HREs or three mutated HREs in front of the SV40 promoter and the luciferase gene. We then cotransfected these reporters together with a CRY1 expression vector or empty control vector into HepG2 cells. Hypoxia induced Luc activity in the control cells, whereas expression of CRY1 predominantly abolished Luc activity under hypoxia (Figure 7B). This effect was specific because mutation of critical nucleotides in the HRE (pGL3-HREm) abolished the induction of Luc activity under hypoxia and the repressive effect of CRY1 (Figure 7B).
As HIFs are dimers consisting of HIF-α and HIF-β (ARNT) subunits, we next investigated whether the CRY1 effect is primarily dependent on each subunit by using the pGL3-HRE Luc construct and the pGL3-HREm construct together with a CRY1 expression vector and HIF-1α, ARNT, or an empty control vector. We found that overexpression of CRY1 together with HIF-1α (Figure 7C) or HIF-2α (Figure S4), but not with ARNT, compromised the ability of HIF-1α to induce HRE-driven Luc activity. Furthermore, cotransfection of the Luc construct with mutant HREs (pGL3-HREm) along with the respective expression vectors alone or in combination resulted in Luc activities comparable with the control (Figure 7C). These results, together with the interaction data, suggest that CRY1 represses HIF-1α function.
To understand whether the inhibitory effect of CRY1 is accompanied by a reduced binding of HIF-1α to target gene promoters in vivo, we performed chromatin immunoprecipitation (ChIP) assays with WT MEFs or CRY1-deficient MEFs cultured under normoxia or hypoxia. The ChIP assays were performed with either unspecific IgG or specific antibodies against mouse HIF-1α or HIF-2α and PCR primers, which allowed amplification of the HIF-binding PAI-1, VEGF-A, PGK-1, and GLUT-1 promoters (Hu et al., 2007, Tanimoto et al., 2010, Wu et al., 2016, Schodel et al., 2011). The assays from WT MEFs reveal that both HIF-1α and HIF-2α preferentially associate with those promoter regions under hypoxia, whereas CRY1-deficient cells bound HIFs already under normoxia (Figures 7D and S5). Thereby, the normoxic binding of HIF-1α to each studied promoter in ΔCRY1 cells was already about 2- to 6-fold higher when compared with WT MEFs (Figure 7D). This binding was further increased in ΔCRY1 cells under hypoxia (Figures 7D and S5). Together, these results indicate that the presence of CRY1 is able to reduce the binding of HIFs to the PAI-1, VEGF-A, PGK-1, and GLUT1 promoters.
Absence of CRY1 Increases HIF-1α Half-Life and Induces Its Transcription
Owing to the fact that CRY1 reduces binding of HIF-1α to its target gene promoters and that it is mainly regulated by post-translational stabilization, we hypothesized that CRY1 may promote HIF-1α degradation. To analyze this, we measured the HIF-1α protein half-life in the absence and presence of CRY1. Interestingly, we found that ΔCRY1 cells had higher HIF-1α protein levels than WT MEFs (Figure 8A). In addition, the CRY1-deficient cells displayed a prolonged HIF-1α half-life (t1/2: 57 min versus ∼45 min in WT cells) (Figures 8A and 8B). Furthermore, we examined whether lack of CRY1 would also affect HIF-1α mRNA levels. Concomitant with the increase in HIF-1α protein levels, we also found that the HIF-1α mRNA levels were increased by about 2-fold in the CRY1-deficient cells (Figure 8C). In addition, we found that the HIF-2α mRNA levels were induced by about 10-fold (Figure 8C). Overall, these data indicate that the CRY1-mediated functional repression of HIF-1α DNA binding is also accompanied by reduced transcription and enhanced HIF-1α protein degradation.
Figure 8.

CRY1 Deficiency Increases HIF-1α Protein Half-Life and Transcription
(A and B) Measurement of HIF-1α protein half-life in wild-type (WT) and CRY-deficient MEFs (ΔCRY1). Cells were cultured for 4 h under hypoxia (5% O2) and after inhibition of protein synthesis with cycloheximide (CHX; 10 μg/mL), endogenous HIF-1α protein levels were determined by western blot analysis with an antibody against mouse HIF-1α. Autoradiographic signals were obtained by chemiluminescence and quantified. Values are means ± SEM of three independent experiments. (A) Representative western blots. (B) Quantification of HIF-1α half-life.
(C) Quantitative qRT-PCR data. The values in wild-type MEFs (WT) were set to 1. Values represent means ± SEM of at least three independent experiments. Statistics, Student's t test for paired values: *significant difference WT versus CRY1-deficient cells; p ≤ 0.05.
CRY1 and HIFs Have Opposite Roles in Cell Proliferation and Migration
The aforementioned findings suggest a reciprocal regulation of CRY1 and HIF-1α that would be of importance for cellular growth and survival, in particular during tumor development. To get an overview, we checked CRY1 and HIF-1α mRNA levels in cancer tissues and the corresponding normal tissues from the Oncomine (www.oncomine.org) database by applying a threshold of a >10-fold change and p value of 0.05. The results show that HIF-1α and CRY1 could be oppositely regulated in different tumor entities. In none of the cases HIF1α and CRY1 were regulated in the same manner, i.e., they were never together up- or downregulated in the same dataset. The reason(s) why HIF-1α expression was upregulated and CRY1 expression was not downregulated in the first and sixth microarray datasets and why HIF-1α expression was not upregulated and CRY1 was downregulated in the second and fifth microarray datasets are not known but may reflect the different status of tumor driving mutations, which certainly confound the regulation (Figure 9). Similarly, we observed CRY1 mRNA induction in HIF-1α-deficient cells (Figure S7). Given that HIF-1α overexpression is associated with poor survival in several patients with cancer (Semenza, 2017), these data together with the higher HIF-1α levels in ΔCRY1 cells imply that loss of CRY1 can promote cell growth. Indeed, we found that CRY1-deficient cells showed a higher rate in proliferation, migration, and colony formation than their WT counterparts (Figures 10B–10F and S6). To assess whether these functional differences are, at least in part, mediated by HIFs we re-investigated cellular proliferation, motility, and the ability to form colonies in CRY1-deficient cells with either CRISPR/Cas9-mediated HIF-1α deletion (Figure 10A) or short hairpin RNA (shRNA)-mediated knockdown of HIF-1α or HIF-2α (Figure S6). In contrast to CRY1 deficiency, lack of HIF-1α reduced proliferation (Figures 10B and 10C), motility (Figures 10E and 10F) and colony formation (Figure 10D). Interestingly, the ΔHIF-1α-ΔCRY1 cells harboring the combined knockout of HIF-1α and CRY1 displayed growth, motility, and colony-forming properties resembling those of WT cells (Figures 10B–10E). Similarly, we introduced scrambled non-effective shRNA or HIF-1α shRNA via retroviral infection into WT and ΔCRY1 cells. Again, knockdown of HIF-1α or HIF-2α reduced the positive effects on migration and soft agar colony formation exerted by CRY1 deficiency alone (Figures S6A–S6C).
Figure 9.

CRY-1 and HIF-1α Show an Opposite Expression Pattern in Human Cancer Cohorts
Analysis of CRY1 and HIF1A mRNA expression profiles in the Oncomine database. Six microarray datasets from different cancer entities were analyzed for CRY1 and HIF1A mRNA expression and compared with normal tissue using a threshold of >10-fold change and a p value <0.05. Red, upregulated in cancer versus normal tissue; blue, downregulated in cancer versus normal tissue. Data are presented as log2 median-centered intensity with the median rank of CRY1 and HIF1A mRNA through each dataset analysis. The rank for a gene is the median rank for that gene across each of the analyses. The p value for a gene is its p value for the median-ranked analysis.
Figure 10.

CRY-1 and HIF-1α Show an Opposite Role on Cell Proliferation and Migration
(A) Representative HIF-1α western blots in WT MEFs, ΔCRY1, ΔHIF-1α, and ΔCRY1/ΔHIF-1α MEFs.
(B) Proliferation rate of WT, ΔCRY1, ΔHIF-1α, and ΔCRY1/ΔHIF-1α cells quantified by dynamic imaging using percentage of confluence at 3-h time intervals.
(C) Chart of 24- and 36-h time points. Statistics, Student's t test for paired values: *significant difference, p ≤ 0.01.
(D) Quantification of the colony formation ability of WT, ΔCRY1, ΔHIF-1α, and ΔCRY1/ΔHIF-1α cells as measured by soft agar assay. The cells were plated in soft agar and allowed to grow for 2 weeks under hypoxia. Cell growth was measured using Resazurin staining in a microplate reader with an excitation wavelength of 584 nm and emission at 612 nm. The OD612 in WT MEFs was set to 100%. Statistics, Student's t test for paired values: *significant difference, p < 0.05.
(E) Representative images of the wound recovery of WT, ΔCRY1, ΔHIF-1α, and ΔCRY1/ΔHIF-1α cells at 0, 12, and 24 h after introduction of the wound. Scale bar, 300 μm.
(F) Chart of 12- and 24-h time points. Statistics, Student's t test for paired values: *significant difference, p < 0.01.
Thus concomitant deletion of HIFs in ΔCRY1 cells significantly reduced the stimulating effects exerted by the absence of CRY1 on cellular growth, colony formation, and cell migration. Together, these data indicate that CRY1 and HIFs have an opposite role on cellular growth.
Discussion
The current study extends recently reported findings on the interplay between the circadian clock and the hypoxia pathway (Adamovich et al., 2017, Wu et al., 2017, Peek et al., 2017) by providing detailed mechanistic insights indicating how the circadian clock component CRY1 can regulate the hypoxia response pathway at the level of HIF-1α and HIF-2α. In particular we show that (1) CRY1 directly interacts with both HIF-1α and HIF-2α, (2) that the interaction of CRY1 with HIFs masks the DNA-binding domain of HIFs and consequently changes HIF DNA binding and expression of HIF target genes, and (3) that the CRY1-HIFα interaction is important for cellular growth.
By broadening the knowledge about CRY1's involvement in more than the circadian clock pathway, the current study also extends the existing information about the interplay between the circadian clock and the hypoxia response pathway. Although this cross talk was proposed on the basis of in vitro findings almost 20 years ago (Hogenesch et al., 1998), a functional validation presenting a reciprocal regulation between the positive clock limb protein BMAL1 and HIF-1α (Peek et al., 2017) was just recently reported while this study on the negative limb was ongoing.
Our data show that hypoxia can modulate the circadian rhythm in vivo and in vitro. This is in line with findings of a recent study demonstrating that mimicry of circadian physiological oxygen rhythms as occurring in mouse blood can affect the setting of the clock (Adamovich et al., 2017). In addition, we show that mice under chronic hypoxia become arrhythmic in total darkness (DD) indicating more severe effects of hypoxia on the clock in general (Figure 1). Although the reasons are not known yet, these activity data may be the result of a partial uncoupling of central and peripheral clocks due to disturbed adaptation to chronic hypoxia similar to that seen with restricted feeding (Damiola et al., 2000). This may express as arrhythmicity in periods coinciding with an increased oxygen consumption (Adamovich et al., 2017) such as the onset of activity and food ingestion, which in mice normally occur during the dark phase of the daily rhythm.
A complimentary approach with Per2:Luc cells exposed to hypoxia as well as with mBmal1:Luc circadian reporter cells in which we overexpressed variants of HIF-1α and HIF-2α revealed that hypoxia and HIFs can not only shorten the period but also decrease the amplitude of the clock (Figures 2 and S1). The amplitude effects of the HIFs seen here are in line with previous studies in which C2C12 and U2OS cells were treated with DMOG (Wu et al., 2017, Peek et al., 2017). Interestingly, our data appeared to be in contrast to one study (Wu et al., 2017) wherein overexpression of a degradation-resistant HIF1α (HIF1α-P402A/P564A) resulted in a lengthening of the period of up to 4 h in U2OS cells. The reasons for the discrepancies might be multiple and could be attributable to the different model systems used (U2OS osteosarcoma cells there versus non-cancer mammalian fibroblasts in our study) or the different way of cell synchronization (dexamethasone versus forskolin in our study). In addition, the other study used the doxycycline-dependent tetR system to overexpress HIF-1α in U2OS cells. Hence, secondary effects of the doxycycline treatment, which are known to inhibit protein translation in mitochondria and to impair metabolism may lead to effects that can confound experimental results (Moullan et al., 2015, Chatzispyrou et al., 2015). Nonetheless, and despite the differences, both studies underline the important roles of HIF-1α as circadian modulator.
Although no study, including the present, can yet explain the detailed mechanism by which HIFs and DMOG affect the period, it is tempting to speculate that this may be mediated by the proposed effects of HIF on BMAL1 (Peek et al., 2017). Indeed, deletion of HIF-1α resulted in a dramatic reduction in BMAL1 mRNA levels (Figure S7) in line with another study wherein overexpression of HIF-1α induced it (Yu et al., 2015). However, HIFs and DMOG appear to act by a different mechanism when seeing the effects on the period. In the present and previous studies (Wu et al., 2017, Peek et al., 2017) DMOG lengthened the period (Figure S1), whereas overexpression of HIFs shortened it (Figures 2 and S1). The difference in period length between DMOG and HIFs is explainable by the rather pleiotropic effects of DMOG, which is a synthetic analog of α-ketoglutarate/2-oxoglutarate. As such, DMOG can act as a pan-inhibitor of α-ketoglutarate/2-oxoglutarate-dependent dioxygenases, a family consisting of about ∼70 enzymes in humans, among them the HIF-prolyl 4-hydroxylases (Hirsila et al., 2003, Kietzmann et al., 2017). Consequently, a number of HIF-independent effects may influence the data achieved upon usage of DMOG. In line are the results obtained upon knockdown of the α-ketoglutarate/2-oxoglutarate-dependent dioxygenases FIH-1 and the EglN family, which also resulted in a lengthened period (Wu et al., 2017). To circumvent the pleiotropic DMOG effects, we used a hydroxylation-resistant HIF-1α variant (PPN) to specify whether it can mimic the shorter period as seen during chronic hypoxia. Importantly, both HIF-1α and HIF-2α could resemble the effects of hypoxia on the period (Figures 2 and S1). Thus the difference in period length upon DMOG usage or EglN and FIH-1 knockdown versus hypoxia or HIF overexpression as seen in this study can be attributed to non-HIF targets of which several, so far unknown, may be involved in clock regulation (Ivan and Kaelin, 2017). Together, all these data suggest that the hypoxia-signaling pathway may have different layers that can affect the circadian period and amplitude.
Intrigued by the HIF effects on the circadian period, and given that CRY proteins represent dominant critical regulators of the circadian period length in humans and mice (Van Der Horst et al., 1999, Vitaterna et al., 1999, Li et al., 2016, Hirota et al., 2012, Ode et al., 2017, Oshima et al., 2015, Siepka et al., 2007, Patke et al., 2017) we hypothesized a connection between HIFs and CRYs. Therefore, we probed whether CRY1 and CRY2 can interact with HIF-1α and HIF-2α, and indeed our experiments revealed that both CRY proteins were able to undergo interaction with the two HIF proteins (Figures 4 and S2).
The CRY1 protein contains a conserved photolyase homology region crucially involved in repression of CLOCK/BMAL1; a C-terminal helix also known as the predicted coiled coil (CC), which interacts with PER2 and FBXL3 in a mutually exclusive manner; and a C-terminal extension also referred to as the “tail” (Figure 5) (Chaves et al., 2011, Merbitz-Zahradnik and Wolf, 2015). Our mapping experiments showed that HIF-1α interacts with the CRY1 “tail” region, a part of the protein (Figure 5), which has the ability to modulate the period length and amplitude of the resulting oscillation (Khan et al., 2012, Xu et al., 2015). In line, a recent report showed that a dominant CRY1 allele coding for a protein with a deletion of 24 residues in the tail region (Patke et al., 2017) caused lengthening of the circadian period and could be linked to familial delayed sleep phase disorder. Thus the effects seen under hypoxia on the period length are in accord with the interaction of HIF-1α with CRY1's tail region. Furthermore, the tail has been shown to affect CRY1 translocation, to interact with the BMAL1 transactivation domain possibly in an acetylation-dependent fashion, and to be phosphorylated in a manner that involves regulation by DNA-PK (Xu et al., 2015, Chaves et al., 2006, Czarna et al., 2011, Hirayama et al., 2007). In addition, the existence of a nuclear localization signal in the CRY1 tail coincides well with the nuclear localization of its interaction with HIFs as seen in the BiFC assays (Figures 4 and S2). In line, the bHLH domain of HIF-1α, which appears to work as the CRY1 interaction module (Figure 6), also contains a nuclear localization sequence (Kallio et al., 1998). Although not directly tested in this study, a similar mode of interaction is likely occurring with HIF-2α because both share an amino acid sequence identity of ∼85% in the bHLH domain.
As the HIF bHLH domain is primarily involved in DNA binding, it is conceivable that the CRY1 tail could affect binding of HIFs to target gene promoters causing a repression of their expression. Thereby the repressive effect of CRY1 may not only depend solely on HIF-1α but also on the promoter context. This is exemplified with the PAI-1 promoter where CRY1 did not repress its activity under normoxia, whereas it clearly suppressed its hypoxia-dependent induction. To minimize the influence of the promoter context and to generalize the findings with respect to HIFs, we then used a bona fide hypoxia reporter containing three HREs as enhancers in front of the SV40 promoter and the luciferase gene. Indeed, these HRE reporter gene assays revealed that CRY1 suppressed their activity under normoxia and their hypoxia and HIF-1-dependent induction. Vice versa, the ChIP experiments from CRY1-deficient cells showed enhanced binding of HIFs to target gene promoters under both normoxia and hypoxia (Figures 7D and S5). Together, these effects need to be seen in context with our data showing that absence of CRY1 regulates HIF-1α at two levels, namely, by increasing its transcription and its stability (Figure 8). Consequently, the net effect is an increase in HIF-1α binding to its enhancers under both conditions. Overall, these data point to a suppressive role of CRY1, and in particular its tail, in the transcriptional response toward hypoxia at various stages of the circadian cycle.
The current data are also in agreement with another recent study also showing enhanced HIF-1α levels in CRY-deficient cells, whereas BMAL1 absence decreased HIF-1α levels (Peek et al., 2017). Similarly, Per2 supported recruitment of HIF-1α to the VEGF promoter (Kobayashi et al., 2017). Together with reports from mouse liver and cardiac tissue, showing that Hif1α mRNA levels cycled in a circadian manner (Eckle et al., 2012, Wu et al., 2017) and data showing a rhythmic appearance of nuclear HIF1α in mouse brain and kidney (Adamovich et al., 2017), the current findings substantiate the existence of a feedback mechanism in which the circadian clock pathway can regulate HIF-1α expression.
Earlier findings from different cancer entities such as chronic myeloid leukemia (Yang et al., 2011), B cell chronic lymphocytic leukemia (Jantus Lewintre et al., 2009), prostate cancer (Zhu et al., 2009), epithelial ovarian cancer (Tokunaga et al., 2008), and colon cancer (Mazzoccoli et al., 2016, Huisman et al., 2016) indicate a reduced CRY1 expression and associated this, as well as a deregulation in Bmal1 or Per2 expression (Huisman et al., 2016), to be a negative prognostic marker.
Vice versa, HIFs are frequently found to be enriched in the hypoxic areas of solid tumors (Talks et al., 2000, Zhong et al., 1998). By showing that CRY1 deficiency induces HIF levels (Figures 10 and S6) and accelerates proliferation and migration, two key aspects relevant to carcinogenesis, and that knocking out HIFs either by CRISPR/Cas9 or by the use of shRNAs reversed these effects, our study now provides a mechanistic link between the above-mentioned genetic association studies.
Although these findings underline the connection between CRY1 and HIFs, it appears that other regulators involved in carcinogenesis can also be regulated by CRY1. For example, several findings from both Per and Cry knockout mice strongly suggest that their dysfunction cooperates with loss of p53 during carcinogenesis (Fu and Kettner, 2013). This is also in agreement with studies showing the complex interconnections between the individual components of the circadian clock with nucleotide excision repair and DNA damage in mice (Kang and Sancar, 2009, Kang et al., 2009) which affect not only carcinogenesis but also aging.
In summary, the current study presents a mechanistic link by which the lack of circadian control at the CRY1 level can favor the action of HIFs during cellular growth, which underpins that chronic misalignment between our lifestyle and the rhythm dictated by our endogenous circadian clock is associated with an increased risk for various diseases including cancer.
Limitations of the Study
In this study we made use of mice and MEFs to investigate the cross talk between HIFs and CRY1. Although this study provided novel insights into how CRY1 affects the HIF system in MEFs, subsequent studies are necessary to investigate the implication for cancer cells and cancer treatment. It is likely, however, that other circadian proteins (such as tCK2) could contribute to the regulation as well. As different regulators may affect CRY1 expression and activity in different ways, their exact roles and contributions need to be further studied.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
This work was supported by grants from the Finnish Academy of Science (SA 296027), Jane and Aatos Erkko Foundation, the Finnish Cancer Foundation, the Finnish Center of International Mobility (CIMO), Biocenter Oulu, and University of Oulu to T.K.
Author Contributions
Conceptualization, T.K. and E.Y.D.; Methodology, E.Y.D., M.J., K.K., D.M., T.F.C., F.T., M.O., J.H., N.B., K.A.M., K.-H.H., P.K., I.C., G.v.d.H., and T.K.; Investigation, E.Y.D., M.J., K.K., D.M., T.F.C., F.T., M.O., J.H., N.B., K.A.M., I.C.; Data Curation, E.Y.D., M.J., K.K., D.M., T.F.C., F.T., M.O., J.H., N.B., and K.A.M.; Visualization, E.Y.D., M.J., K.K., D.M., T.F.C., M.O., I.C., G.v.d.H., and T.K.; Writing – Original Draft, E.Y.D., M.J., G.v.d.H., and T.K.; Writing – Review & Editing, E.Y.D., I.C., G.v.d.H., and T.K.; Funding Acquisition, G.v.d.H., P.K., and T.K.; Resources, G.v.d.H., P.K., K.-H.H., and T.K.; Supervision, E.Y.D., and T.K.
Declaration of Interests
The authors declare no competing interests.
Published: March 29, 2019
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2019.02.027.
Contributor Information
Elitsa Y. Dimova, Email: elitsa.dimova@oulu.fi.
Thomas Kietzmann, Email: thomas.kietzmann@oulu.fi.
Supplemental Information
References
- Adamovich Y., Ladeuix B., Golik M., Koeners M.P., Asher G. Rhythmic oxygen levels reset circadian clocks through HIF1a. Cell Metab. 2017;25:93–101. doi: 10.1016/j.cmet.2016.09.014. [DOI] [PubMed] [Google Scholar]
- Albrecht U. Timing to perfection: the biology of central and peripheral circadian clocks. Neuron. 2012;74:246–260. doi: 10.1016/j.neuron.2012.04.006. [DOI] [PubMed] [Google Scholar]
- Asher G., Schibler U. Crosstalk between components of circadian and metabolic cycles in mammals. Cell Metab. 2011;13:125–137. doi: 10.1016/j.cmet.2011.01.006. [DOI] [PubMed] [Google Scholar]
- Balsalobre A., Damiola F., Schibler U. A serum shock induces circadian gene expression in mammalian tissue culture cells. Cell. 1998;93:929–937. doi: 10.1016/s0092-8674(00)81199-x. [DOI] [PubMed] [Google Scholar]
- Chatzispyrou I.A., Held N.M., Mouchiroud L., Auwerx J., Houtkooper R.H. Tetracycline antibiotics impair mitochondrial function and its experimental use confounds research. Cancer Res. 2015;75:4446–4449. doi: 10.1158/0008-5472.CAN-15-1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaves I., Pokorny R., Byrdin M., Hoang N., Ritz T., Brettel K., Essen L., Van Der Horst G.T.J., Batschauer A., Ahmad M. The cryptochromes: blue light photoreceptors in plants and animals. Annu. Rev. Plant Biol. 2011;62:335–364. doi: 10.1146/annurev-arplant-042110-103759. [DOI] [PubMed] [Google Scholar]
- Chaves I., Yagita K., Barnhoorn S., Okamura H., Van Der Horst G.T.J., Tamanini F. Functional evolution of the photolyase/cryptochrome protein family: importance of the C terminus of mammalian CRY1 for circadian core oscillator performance. Mol. Cell. Biol. 2006;26:1743–1753. doi: 10.1128/MCB.26.5.1743-1753.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng S., Jiang Z., Zou Y., Chen C., Wang Y., Liu Y., Xiao J., Guo H., Wang Z. Downregulation of Clock in circulatory system leads to an enhancement of fibrinolysis in mice. Exp. Biol. Med. (Maywood) 2011;236:1078–1084. doi: 10.1258/ebm.2011.010322. [DOI] [PubMed] [Google Scholar]
- Chilov D., Hofer T., Bauer C., Wenger R.H., Gassmann M. Hypoxia affects expression of circadian genes PER1 and CLOCK in mouse brain. FASEB J. 2001;15:2613–2622. doi: 10.1096/fj.01-0092com. [DOI] [PubMed] [Google Scholar]
- Czarna A., Breitkreuz H., Mahrenholz C.C., Arens J., Strauss H.M., Wolf E. Quantitative analyses of cryptochrome-mBMAL1 interactions: mechanistic insights into the transcriptional regulation of the mammalian circadian clock. J. Biol. Chem. 2011;286:22414–22425. doi: 10.1074/jbc.M111.244749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damiola F., Le Minli N., Preitner N., Kornmann B., Fleury-Olela F., Schibler U. Restricted feeding uncouples circadian oscillators in peripheral tissues from the central pacemaker in the suprachiasmatic nucleus. Genes Dev. 2000;14:2950–2961. doi: 10.1101/gad.183500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimova E.Y., Moller U., Herzig S., Fink T., Zachar V., Ebbesen P., Kietzmann T. Transcriptional regulation of plasminogen activator inhibitor-1 expression by insulin-like growth factor-1 via MAP kinases and hypoxia-inducible factor-1 in HepG2 cells. Thromb. Haemost. 2005;93:1176–1184. [Google Scholar]
- Eckle T., Hartmann K., Bonney S., Reithel S., Mittelbronn M., Walker L.A., Lowes B.D., Han J., Borchers C.H., Buttrick P.M. Adora2b-elicited Per2 stabilization promotes a HIF-dependent metabolic switch crucial for myocardial adaptation to ischemia. Nat. Med. 2012;18:774–782. doi: 10.1038/nm.2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filipski E., Levi F. Circadian disruption in experimental cancer processes. Integr. Cancer Ther. 2009;8:298–302. doi: 10.1177/1534735409352085. [DOI] [PubMed] [Google Scholar]
- Flügel D., Görlach A., Kietzmann T. GSK-3beta regulates cell growth, migration, and angiogenesis via Fbw7 and USP28-dependent degradation of HIF-1alpha. Blood. 2012;119:1292–1301. doi: 10.1182/blood-2011-08-375014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu L., Kettner N.M. The circadian clock in cancer development and therapy. Prog. Mol. Biol. Transl. Sci. 2013;119:221–282. doi: 10.1016/B978-0-12-396971-2.00009-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu L., Lee C.C. The circadian clock: pacemaker and tumour suppressor. Nat. Rev. Cancer. 2003;3:350–361. doi: 10.1038/nrc1072. [DOI] [PubMed] [Google Scholar]
- Griffin E.A., Jr., Staknis D., Weitz C.J. Light-independent role of CRY1 and CRY2 in the mammalian circadian clock. Science. 1999;286:768–771. doi: 10.1126/science.286.5440.768. [DOI] [PubMed] [Google Scholar]
- Hirayama J., Sahar S., Grimaldi B., Tamaru T., Takamatsu K., Nakahata Y., Sassone-Corsi P. CLOCK-mediated acetylation of BMAL1 controls circadian function. Nature. 2007;450:1086–1090. doi: 10.1038/nature06394. [DOI] [PubMed] [Google Scholar]
- Hirota T., Lee J.W., St. John P.C., Sawa M., Iwaisako K., Noguchi T., Pongsawakul P.Y., Sonntag T., Welsh D.K., Brenner D.A. Identification of small molecule activators of cryptochrome. Science. 2012;337:1094–1097. doi: 10.1126/science.1223710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsila M., Koivunen P., Gunzler V., Kivirikko K.I., Myllyharju J. Characterization of the human prolyl 4-hydroxylases that modify the hypoxia-inducible factor. J. Biol. Chem. 2003;278:30772–30780. doi: 10.1074/jbc.M304982200. [DOI] [PubMed] [Google Scholar]
- Hogenesch J.B., Gu Y.Z., Jain S., Bradfield C.A. The basic-helix-loop-helix-PAS orphan MOP3 forms transcriptionally active complexes with circadian and hypoxia factors. Proc. Natl. Acad. Sci. U S A. 1998;95:5474–5479. doi: 10.1073/pnas.95.10.5474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu C.J., Sataur A., Wang L., Chen H., Simon M.C. The N-terminal transactivation domain confers target gene specificity of hypoxia-inducible factors HIF-1alpha and HIF-2alpha. Mol. Biol. Cell. 2007;18:4528–4542. doi: 10.1091/mbc.E06-05-0419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huisman S.A., Ahmadi A.R., IJzermans J.N., Verhoef C., van der Horst G.T., de Bruin R.W. Disruption of clock gene expression in human colorectal liver metastases. Tumour Biol. 2016;37:13973–13981. doi: 10.1007/s13277-016-5231-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivan M., Kaelin W.G., Jr. The EGLN-HIF O2-sensing system: multiple inputs and feedbacks. Mol. Cell. 2017;66:772–779. doi: 10.1016/j.molcel.2017.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jantus Lewintre E., Reinoso Martin C., Montaner D., Marin M., Jose Terol M., Farras R., Benet I., Calvete J.J., Dopazo J., Garcia-Conde J. Analysis of chronic lymphotic leukemia transcriptomic profile: differences between molecular subgroups. Leuk. Lymphoma. 2009;50:68–79. doi: 10.1080/10428190802541807. [DOI] [PubMed] [Google Scholar]
- Kaelin W.G., Jr., Ratcliffe P.J. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol. Cell. 2008;30:393–402. doi: 10.1016/j.molcel.2008.04.009. [DOI] [PubMed] [Google Scholar]
- Kallio P.J., Okamoto K., O'Brien S., Carrero P., Makino Y., Tanaka H., Poellinger L. Signal transduction in hypoxic cells: inducible nuclear translocation and recruitment of the CBP/p300 coactivator by the hypoxia-inducible factor-1alpha. EMBO J. 1998;17:6573–6586. doi: 10.1093/emboj/17.22.6573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang T.H., Reardon J.T., Kemp M., Sancar A. Orcadian oscillation of nucleotide excision repair in mammalian brain. Proc. Natl. Acad. Sci. U S A. 2009;106:2864–2867. doi: 10.1073/pnas.0812638106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang T., Sancar A. Circadian regulation of DNA excision repair: implications for chrono-chemotherapy. Cell Cycle. 2009;8:1665–1667. doi: 10.4161/cc.8.11.8707. [DOI] [PubMed] [Google Scholar]
- Kerppola T.K. Bimolecular fluorescence complementation (BiFC) analysis as a probe of protein interactions in living cells. Annu. Rev. Biophys. 2008;37:465–487. doi: 10.1146/annurev.biophys.37.032807.125842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan S.K., Xu H., Ukai-Tadenuma M., Burton B., Wang Y., Ueda H.R., Liu A.C. Identification of a novel cryptochrome differentiating domain required for feedback repression in circadian clock function. J. Biol. Chem. 2012;287:25917–25926. doi: 10.1074/jbc.M112.368001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kietzmann T., Petry A., Shvetsova A., Gerhold J.M., Gorlach A. The epigenetic landscape related to reactive oxygen species formation in the cardiovascular system. Br. J. Pharmacol. 2017;174:1533–1554. doi: 10.1111/bph.13792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kietzmann T., Roth U., Jungermann K. Induction of the plasminogen activator inhibitor-1 gene expression by mild hypoxia via a hypoxia response element binding the hypoxia-inducible factor-1 in rat hepatocytes. Blood. 1999;94:4177–4185. [PubMed] [Google Scholar]
- Ko C.H., Takahashi J.S. Molecular components of the mammalian circadian clock. Hum. Mol. Genet. 2006;15(Suppl. 2):R271–R277. doi: 10.1093/hmg/ddl207. [DOI] [PubMed] [Google Scholar]
- Kobayashi M., Morinibu A., Koyasu S., Goto Y., Hiraoka M., Harada H. A circadian clock gene, PER2, activates HIF-1 as an effector molecule for recruitment of HIF-1alpha to promoter regions of its downstream genes. FEBS J. 2017;284:3804–3816. doi: 10.1111/febs.14280. [DOI] [PubMed] [Google Scholar]
- Koike N., Yoo S.H., Huang H.C., Kumar V., Lee C., Kim T.K., Takahashi J.S. Transcriptional architecture and chromatin landscape of the core circadian clock in mammals. Science. 2012;338:349–354. doi: 10.1126/science.1226339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kume K., Zylka M.J., Sriram S., Shearman L.P., Weaver D.R., Jin X., Maywood E.S., Hastings M.H., Reppert S.M. mCRY1 and mCRY2 are essential components of the negative limb of the circadian clock feedback loop. Cell. 1999;98:193–205. doi: 10.1016/s0092-8674(00)81014-4. [DOI] [PubMed] [Google Scholar]
- Langmesser S., Tallone T., Bordon A., Rusconi S., Albrecht U. Interaction of circadian clock proteins PER2 and CRY with BMAL1 and CLOCK. BMC Mol. Biol. 2008;9:41. doi: 10.1186/1471-2199-9-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., Xiong W., Zhang E.E. The ratio of intracellular CRY proteins determines the clock period length. Biochem. Biophys. Res. Commun. 2016;472:531–538. doi: 10.1016/j.bbrc.2016.03.010. [DOI] [PubMed] [Google Scholar]
- Liu A.C., Welsh D.K., Ko C.H., Tran H.G., Zhang E.E., Priest A.A., Buhr E.D., Singer O., Meeker K., Verma I.M. Intercellular coupling confers robustness against mutations in the SCN circadian clock network. Cell. 2007;129:605–616. doi: 10.1016/j.cell.2007.02.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maemura K., de la Monte S.M., Chin M.T., Layne M.D., Hsieh C.M., Yet S.F., Perrella M.A., Lee M.E. CLIF, a novel cycle-like factor, regulates the circadian oscillation of plasminogen activator inhibitor-1 gene expression. J. Biol. Chem. 2000;275:36847–36851. doi: 10.1074/jbc.C000629200. [DOI] [PubMed] [Google Scholar]
- Masson N., Ratcliffe P.J. Hypoxia signaling pathways in cancer metabolism: the importance of co-selecting interconnected physiological pathways. Cancer Metab. 2014;2:3. doi: 10.1186/2049-3002-2-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuo T., Yamaguchi S., Mitsui S., Emi A., Shimoda F., Okamura H. Control mechanism of the circadian clock for timing of cell division in vivo. Science. 2003;302:255–259. doi: 10.1126/science.1086271. [DOI] [PubMed] [Google Scholar]
- Mazzoccoli G., Colangelo T., Panza A., Rubino R., De Cata A., Tiberio C., Valvano M.R., Pazienza V., Merla G., Augello B. Deregulated expression of cryptochrome genes in human colorectal cancer. Mol. Cancer. 2016;15:6. doi: 10.1186/s12943-016-0492-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merbitz-Zahradnik T., Wolf E. How is the inner circadian clock controlled by interactive clock proteins?: structural analysis of clock proteins elucidates their physiological role. FEBS Lett. 2015;589:1516–1529. doi: 10.1016/j.febslet.2015.05.024. [DOI] [PubMed] [Google Scholar]
- Michael A.K., Fribourgh J.L., Chelliah Y., Sandate C.R., Hura G.L., Schneidman-Duhovny D., Tripathi S.M., Takahashi J.S., Partch C.L. Formation of a repressive complex in the mammalian circadian clock is mediated by the secondary pocket of CRY1. Proc. Natl. Acad. Sci. U S A. 2017;114:1560–1565. doi: 10.1073/pnas.1615310114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moullan N., Mouchiroud L., Wang X., Ryu D., Williams E.G., Mottis A., Jovaisaite V., Frochaux M.V., Quiros P.M., Deplancke B. Tetracyclines disturb mitochondrial function across eukaryotic models: a call for caution in biomedical research. Cell Rep. 2015;10:1681–1691. doi: 10.1016/j.celrep.2015.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ode K.L., Ukai H., Susaki E.A., Narumi R., Matsumoto K., Hara J., Koide N., Abe T., Kanemaki M.T., Kiyonari H., Ueda H.R. Knockout-rescue embryonic stem cell-derived mouse reveals circadian-period control by quality and quantity of CRY1. Mol. Cell. 2017;65:176–190. doi: 10.1016/j.molcel.2016.11.022. [DOI] [PubMed] [Google Scholar]
- Oishi K., Miyazaki K., Uchida D., Ohkura N., Wakabayashi M., Doi R., Matsuda J., Ishida N. PERIOD2 is a circadian negative regulator of PAI-1 gene expression in mice. J. Mol. Cell. Cardiol. 2009;46:545–552. doi: 10.1016/j.yjmcc.2009.01.001. [DOI] [PubMed] [Google Scholar]
- Oshima T., Yamanaka I., Kumar A., Yamaguchi J., Nishiwaki-Ohkawa T., Muto K., Kawamura R., Hirota T., Yagita K., Irle S. C-H activation generates period-shortening molecules that target cryptochrome in the mammalian circadian clock. Angew. Chem. Int. Ed. 2015;54:7193–7197. doi: 10.1002/anie.201502942. [DOI] [PubMed] [Google Scholar]
- Patke A., Murphy P.J., Onat O.E., Krieger A.C., Özçelik T., Campbell S.S., Young M.W. Mutation of the human circadian clock gene CRY1 in familial delayed sleep phase disorder. Cell. 2017;169:203–215.e13. doi: 10.1016/j.cell.2017.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peek C.B., Levine D.C., Cedernaes J., Taguchi A., Kobayashi Y., Tsai S.J., Bonar N.A., McNulty M.R., Ramsey K.M., Bass J. Circadian clock interaction with HIF1a mediates oxygenic metabolism and anaerobic glycolysis in skeletal muscle. Cell Metab. 2017;25:86–92. doi: 10.1016/j.cmet.2016.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rana S., Mahmood S. Circadian rhythm and its role in malignancy. J. Circadian Rhythms. 2010;8:3. doi: 10.1186/1740-3391-8-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards J., Gumz M.L. Advances in understanding the peripheral circadian clocks. FASEB J. 2012;26:3602–3613. doi: 10.1096/fj.12-203554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahar S., Sassone-Corsi P. Metabolism and cancer: the circadian clock connection. Nat. Rev. Cancer. 2009;9:886–896. doi: 10.1038/nrc2747. [DOI] [PubMed] [Google Scholar]
- Sato T.K., Yamada R.G., Ukai H., Baggs J.E., Miraglia L.J., Kobayashi T.J., Welsh D.K., Kay S.A., Ueda H.R., Hogenesch J.B. Feedback repression is required for mammalian circadian clock function. Nat. Genet. 2006;38:312–319. doi: 10.1038/ng1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schodel J., Oikonomopoulos S., Ragoussis J., Pugh C.W., Ratcliffe P.J., Mole D.R. High-resolution genome-wide mapping of HIF-binding sites by ChIP-seq. Blood. 2011;117:e207–e217. doi: 10.1182/blood-2010-10-314427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoenhard J.A., Smith L.H., Painter C.A., Eren M., Johnson C.H., Vaughan D.E. Regulation of the PAI-1 promoter by circadian clock components: differential activation by BMAL1 and BMAL2. J. Mol. Cell. Cardiol. 2003;35:473–481. doi: 10.1016/s0022-2828(03)00051-8. [DOI] [PubMed] [Google Scholar]
- Semenza G.L. Hypoxia-inducible factors: coupling glucose metabolism and redox regulation with induction of the breast cancer stem cell phenotype. EMBO J. 2017;36:252–259. doi: 10.15252/embj.201695204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shostak A. Circadian clock, cell division, and cancer: from molecules to organism. Int. J. Mol. Sci. 2017;18 doi: 10.3390/ijms18040873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siepka S.M., Yoo S.H., Park J., Song W., Kumar V., Hu Y., Lee C., Takahashi J.S. Circadian mutant overtime reveals F-box protein FBXL3 regulation of cryptochrome and period gene expression. Cell. 2007;129:1011–1023. doi: 10.1016/j.cell.2007.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straif K., Baan R., Grosse Y., Secretan B., Ghissassi F.E., Bouvard V., Altieri A., Benbrahim-Tallaa L., Cogliano V., WHO International Agency for Research on Cancer Monograph Working Group Carcinogenicity of shift-work, painting, and fire-fighting. Lancet Oncol. 2007;8:1065–1066. doi: 10.1016/S1470-2045(07)70373-X. [DOI] [PubMed] [Google Scholar]
- Talks K.L., Turley H., Gatter K.C., Maxwell P.H., Pugh C.W., Ratcliffe P.J., Harris A.L. The expression and distribution of the hypoxia-inducible factors HIF-1alpha and HIF-2alpha in normal human tissues, cancers, and tumor-associated macrophages. Am. J. Pathol. 2000;157:411–421. doi: 10.1016/s0002-9440(10)64554-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanimoto K., Tsuchihara K., Kanai A., Arauchi T., Esumi H., Suzuki Y., Sugano S. Genome-wide identification and annotation of HIF-1alpha binding sites in two cell lines using massively parallel sequencing. Hugo J. 2010;4:35–48. doi: 10.1007/s11568-011-9150-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokunaga H., Takebayashi Y., Utsunomiya H., Akahira J., Higashimoto M., Mashiko M., Ito K., Niikura H., Takenoshita S., Yaegashi N. Clinicopathological significance of circadian rhythm-related gene expression levels in patients with epithelial ovarian cancer. Acta Obstet. Gynecol. Scand. 2008;87:1060–1070. doi: 10.1080/00016340802348286. [DOI] [PubMed] [Google Scholar]
- Van Der Horst G.T.J., Muijtjens M., Kobayashi K., Takano R., Kanno S., Takao M., De Wit J., Verkerk A., Eker A.P.M., Van Leenen D. Mammalian Cry1 and Cry2 are essential for maintenance of circadian rhythms. Nature. 1999;398:627–630. doi: 10.1038/19323. [DOI] [PubMed] [Google Scholar]
- Vitaterna M.H., Selby C.P., Todo T., Niwa H., Thompson C., Fruechte E.M., Hitomi K., Thresher R.J., Ishikawa T., Miyazaki J. Differential regulation of mammalian period genes and circadian rhythmicity by cryptochromes 1 and 2. Proc. Natl. Acad. Sci. U S A. 1999;96:12114–12119. doi: 10.1073/pnas.96.21.12114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Yin L., Lazar M.A. The orphan nuclear receptor Rev-erb alpha regulates circadian expression of plasminogen activator inhibitor type 1. J. Biol. Chem. 2006;281:33842–33848. doi: 10.1074/jbc.M607873200. [DOI] [PubMed] [Google Scholar]
- Wu H.T., Kuo Y.C., Hung J.J., Huang C.H., Chen W.Y., Chou T.Y., Chen Y., Chen Y.J., Chen Y.J., Cheng W.C. K63-polyubiquitinated HAUSP deubiquitinates HIF-1alpha and dictates H3K56 acetylation promoting hypoxia-induced tumour progression. Nat. Commun. 2016;7:13644. doi: 10.1038/ncomms13644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y., Tang D., Liu N., Xiong W., Huang H., Li Y., Ma Z., Zhao H., Chen P., Qi X., Zhang E.E. Reciprocal regulation between the circadian clock and hypoxia signaling at the genome level in mammals. Cell Metab. 2017;25:73–85. doi: 10.1016/j.cmet.2016.09.009. [DOI] [PubMed] [Google Scholar]
- Xu H., Gustafson C.L., Sammons P.J., Khan S.K., Parsley N.C., Ramanathan C., Lee H.-., Liu A.C., Partch C.L. Cryptochrome 1 regulates the circadian clock through dynamic interactions with the BMAL1 C terminus. Nat. Struct. Mol. Biol. 2015;22:476–484. doi: 10.1038/nsmb.3018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagita K., Tamanini F., Van der Horst G.T.J., Okamura H. Molecular mechanisms of the biological clock in cultured fibroblasts. Science. 2001;292:278–281. doi: 10.1126/science.1059542. [DOI] [PubMed] [Google Scholar]
- Yang M.Y., Yang W.C., Lin P.M., Hsu J.F., Hsiao H.H., Liu Y.C., Tsai H.J., Chang C.S., Lin S.F. Altered expression of circadian clock genes in human chronic myeloid leukemia. J. Biol. Rhythms. 2011;26:136–148. doi: 10.1177/0748730410395527. [DOI] [PubMed] [Google Scholar]
- Ye R., Selby C.P., Chiou Y.Y., Ozkan-Dagliyan I., Gaddameedhi S., Sancar A. Dual modes of CLOCK: BMAL1 inhibition mediated by Cryptochrome and period proteins in the mammalian circadian clock. Genes Dev. 2014;28:1989–1998. doi: 10.1101/gad.249417.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo S.H., Ko C.H., Lowrey P.L., Buhr E.D., Song E.J., Chang S., Yoo O.J., Yamazaki S., Lee C., Takahashi J.S. A noncanonical E-box enhancer drives mouse Period2 circadian oscillations in vivo. Proc. Natl. Acad. Sci. U S A. 2005;102:2608–2613. doi: 10.1073/pnas.0409763102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu C., Yang S.L., Fang X., Jiang J.X., Sun C.Y., Huang T. Hypoxia disrupts the expression levels of circadian rhythm genes in hepatocellular carcinoma. Mol. Med. Rep. 2015;11:4002–4008. doi: 10.3892/mmr.2015.3199. [DOI] [PubMed] [Google Scholar]
- Zhong H., Agani F., Baccala A.A., Laughner E., Rioseco-Camacho N., Isaacs W.B., Simons J.W., Semenza G.L. Increased expression of hypoxia inducible factor-1alpha in rat and human prostate cancer. Cancer Res. 1998;58:5280–5284. [PubMed] [Google Scholar]
- Zhu Y., Stevens R.G., Hoffman A.E., Fitzgerald L.M., Kwon E.M., Ostrander E.A., Davis S., Zheng T., Stanford J.L. Testing the circadian gene hypothesis in prostate cancer: a population-based case-control study. Cancer Res. 2009;69:9315–9322. doi: 10.1158/0008-5472.CAN-09-0648. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.