

Abstract
Background
Major challenges in understanding the functional consequences of genetic risk factors for human disease are which tissues and cell types are affected and the limited availability of suitable tissue. The aim of this study was to evaluate tissue-specific genotype-epigenetic characteristics in DNA samples from both endometrium and blood collected from women at different stages of the menstrual cycle and relate results to genetic risk factors for reproductive traits and diseases.
Results
We analysed DNA methylation (DNAm) data from endometrium and blood samples from 66 European women. Methylation profiles were compared between stages of the menstrual cycle, and changes in methylation overlaid with changes in transcription and genotypes. We observed large changes in methylation (27,262 DNAm probes) across the menstrual cycle in endometrium that were not observed in blood. Individual genotype data was tested for association with methylation at 443,016 and 443,101 DNAm probes in endometrium and blood respectively to identify methylation quantitative trait loci (mQTLs). A total of 4546 sentinel cis-mQTLs (P < 1.13 × 10−10) and 434 sentinel trans-mQTLs (P < 2.29 × 10−12) were detected in endometrium and 6615 sentinel cis-mQTLs (P < 1.13 × 10−10) and 590 sentinel trans-mQTLs (P < 2.29 × 10−12) were detected in blood. Following secondary analyses, conducted to test for overlap between mQTLs in the two tissues, we found that 62% of endometrial cis-mQTLs were also observed in blood and the genetic effects between tissues were highly correlated. A number of mQTL SNPs were associated with reproductive traits and diseases, including one mQTL located in a known risk region for endometriosis (near GREB1).
Conclusions
We report novel findings characterising genetic regulation of methylation in endometrium and the association of endometrial mQTLs with endometriosis risk and other reproductive traits and diseases. The high correlation of genetic effects between tissues highlights the potential to exploit the power of large mQTL datasets in endometrial research and identify target genes for functional studies. However, tissue-specific methylation profiles and genetic effects also highlight the importance of also using disease-relevant tissues when investigating molecular mechanisms of disease risk.
Electronic supplementary material
The online version of this article (10.1186/s13148-019-0648-7) contains supplementary material, which is available to authorized users.
Keywords: DNA methylation, DNA methylation quantitative trait loci (mQTL), Endometrium, Blood, Menstrual cycle, Endometriosis
Background
Genetic risk factors for complex disease mostly reside in non-coding regions of the genome [1, 2] and studies integrating results from genome-wide association studies and the genetic effects on methylation and gene expression provide a powerful approach to understand the functional consequences of these genetic risk factors. DNA methylation (DNAm) is one of the most common forms of epigenetic modification and involves the addition of a methyl group to the carbon-5 position of cytosine, often occurring at CpG sites [3]. Methylation is essential in facilitating embryonic development, chromosomal infrastructure, cell viability, imprinting, X chromosome-inactivation and transcription [3–6]. Methylation patterns in DNA samples from blood are associated with disease pathogenesis and are influenced by underlying genetic variation [7–10]. Difficulty accessing disease-relevant tissues has meant many studies make use of large gene expression and methylation datasets from peripheral blood as a proxy. However, differences in methylation profiles contribute to tissue-specific functions [11–13] and understanding tissue specificity of methylation signals is important to help interpret the role of methylation in disease risk.
The human endometrium is a highly specialised tissue lining the inside of the uterus and is essential to implantation, development of the placenta, and successful pregnancy [14]. Endometrium undergoes a cyclic process of cellular proliferation, differentiation, degradation, and regeneration [14, 15]. This dynamic process is accompanied by marked changes in gene expression that occur in response to changes in circulating concentrations of the steroid hormones oestradiol and progesterone [14, 16–18]. Methylation profiles in human endometrium also change across the menstrual cycle with thousands of genes differentially methylated between cycle stages [19–22].
This study aimed to compare DNA methylation patterns in both endometrium and blood collected from women sampled at different stages across the menstrual cycle. We identified methylation quantitative trait loci (mQTLs) in endometrium and correlated the mQTLs with blood mQTLs in the same women, and with larger mQTL datasets. We then evaluated overlap of mQTLs in endometrium with oestrogen receptor (ESR) binding sites and the overlap of mQTLs in both tissues with genomic regions associated with risk for endometriosis and other reproductive disorders. Results from this study provide novel insight into genetic control of methylation in human endometrium through the identification of endometrial mQTLs. Our work highlights methylation differences between blood and endometrial tissues across the menstrual cycle, and similarities between blood and endometrium in genetic regulation of methylation.
Results
Genome-wide methylation profiles
We analysed genome-wide methylation profiles in endometrium from 66 European women who attended clinics at the Royal Women’s Hospital in Melbourne, Australia. Following quality control (QC) filtering, a total of 443,016 and 443,101 DNAm probes remained for analyses in endometrial tissue samples and blood samples, respectively. Both endometrium and blood had a similar proportion of probes sites consistently hypomethylated (Fig. 1a). However, a larger proportion of probes sites in blood were consistently hypermethylated (Fig. 1a) (Additional file 1: Supplementary Note 1). CpG probe sites were annotated according to their proximity to CpG islands using the Illumina Human Methylation 450 BeadChip manifest file (see Additional file 1: Supplementary Note 1 for definitions and detailed results). Hypomethylated sites were more common in CpG islands and hypermethylated sites were more common in open sea regions in both endometrium and blood (Fig. 1b, c).
Fig. 1.

a Density plot showing the distribution of beta values measured at each DNA methylation (DNAm) probe in endometrium and blood. b Density plot showing the distribution of beta values measured at each DNAm probe in endometrium, values grouped according to location to CPG islands. c Density plot showing the distribution of beta values measured at each DNAm probe in blood, values grouped according to location to CPG islands
We see very similar genome-wide methylation profiles between menstrual (M), proliferative (P) and secretory (S) phases of menstrual cycle in endometrium. In all cycle phases, we observed 35.2–36% of probes consistently hypomethylated in at least 90% of individuals and 22.6–23.9% of probes consistently hypermethylated in individuals (Fig. 2). We also see similar methylation patterns for probes located in CpG islands, shores, shelves and open sea regions across all three phases (Additional file 2: Figure S1).
Fig. 2.
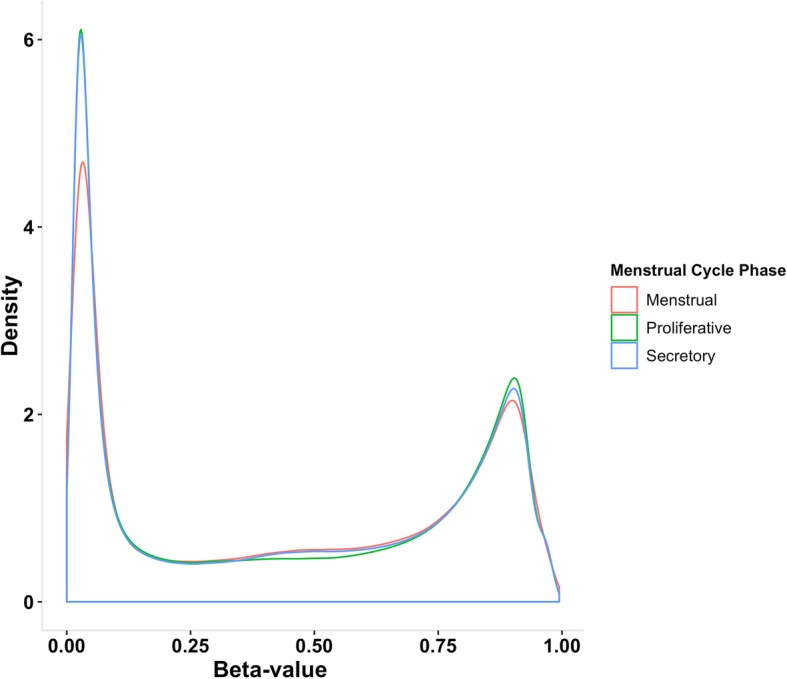
Density plot showing the distribution of beta values measured at each DNA methylation (DNAm) probe in endometrium from three menstrual cycle hases, the menstrual phase, proliferative phase and secretory phase
Differential DNA methylation
To investigate changes in methylation across the menstrual cycle in endometrium and blood, we performed a differential methylation analysis between the proliferative (P) phase and secretory (S) phase of menstrual cycle. Stage of cycle was determined by histological assessment of endometrial tissue. We detected significant differences in methylation in endometrium for 6% of DNAm probe sites (n = 27,262) across the menstrual cycle comparing the P phase and S phases of the cycle (FDR < 0.05, P < 3.07 × 10−3) (Additional file 3: Table S1). Each DNAm probe site was annotated to the gene with the closest transcription start site (11,281 genes). The top 30 differentially methylated DNAm probe sites between the two phases of the menstrual cycle in endometrial tissue are listed in Table 1 and shown in Additional file 2: Figure S2. Marked changes in the methylation of 40 DNAm probe sites with the largest fold change between the proliferative and secretory phase are shown in Additional file 2: Figure S3. The majority of differentially methylated DNAm probe sites (51.9%) were concentrated in open sea locations and CpG island shores (25.3%) throughout the genome (Additional file 3: Table S2, Additional file 2: Figure S4). Differences observed across the cycle in endometrial tissue were not reflected in blood and are likely, in part, to reflect changes in cell composition.
Table 1.
Differentially methylated DNAm probe sites in endometrium. The top 30 significantly differentially methylated DNAm probe sites between the proliferative (P) and secretory (S) phase (PvsS)
| DNAm probe site ID | Log2 fold change | P value | Adjusted P value | Probe start (hg19) | Closest TSS gene name |
|---|---|---|---|---|---|
| cg16201273 | 0.146 | 7.96E-15 | 3.53E-09 | 80,855,803 | ZMIZ1 |
| cg20888995 | 0.146 | 2.74E-11 | 6.06E-06 | 56,822,059 | ARHGEF3 |
| cg01713086 | 0.128 | 1.44E-10 | 1.60E-05 | 28,268,413 | ZNF395 |
| cg07730183 | 0.096 | 1.45E-10 | 1.60E-05 | 2,136,400 | TSC2 |
| cg22934449 | 0.093 | 4.52E-10 | 3.72E-05 | 104,199,723 | ZFYVE21 |
| cg21369890 | 0.137 | 5.28E-10 | 3.72E-05 | 86,099,963 | AK024998 |
| cg06669056 | 0.096 | 6.17E-10 | 3.72E-05 | 5,570,715 | C4orf6 |
| cg25237396 | − 0.092 | 6.71E-10 | 3.72E-05 | 802,148 | MIR4745 |
| cg17900103 | 0.114 | 1.45E-09 | 6.33E-05 | 20,940,931 | PINK1 |
| cg22185879 | − 0.087 | 1.53E-09 | 6.33E-05 | 62,153,192 | PPDPF |
| cg23235622 | 0.096 | 1.57E-09 | 6.33E-05 | 34,039,299 | CEP250 |
| cg25735294 | 0.103 | 2.99E-09 | 1.10E-04 | 186,353,671 | FETUB |
| cg02248729 | − 0.047 | 4.76E-09 | 1.62E-04 | 80,555,018 | FOXK2 |
| cg12082793 | 0.097 | 6.48E-09 | 2.05E-04 | 20,218,923 | OTUD3 |
| cg11224737 | 0.133 | 8.02E-09 | 2.37E-04 | 72,991,072 | LOC728978 |
| cg26479868 | 0.082 | 1.00E-08 | 2.77E-04 | 29,916,194 | TMTC1 |
| cg09714100 | − 0.092 | 1.31E-08 | 3.42E-04 | 44,821,342 | SIK1 |
| cg27133780 | 0.133 | 1.41E-08 | 3.46E-04 | 32,474,743 | CMTM7 |
| cg21163015 | 0.108 | 1.82E-08 | 4.25E-04 | 140,658,386 | FLJ40292 |
| cg02118194 | − 0.040 | 2.37E-08 | 5.09E-04 | 46,404,488 | MYPOP |
| cg03018949 | 0.062 | 2.68E-08 | 5.09E-04 | 127,371,608 | C10orf122 |
| cg26469099 | 0.085 | 2.78E-08 | 5.09E-04 | 4,144,866 | PARP11 |
| cg22182975 | 0.071 | 2.86E-08 | 5.09E-04 | 167,571,122 | GPR31 |
| cg05224671 | 0.090 | 3.15E-08 | 5.09E-04 | 65,435,408 | RAB15 |
| cg09616559 | − 0.066 | 3.22E-08 | 5.09E-04 | 25,921,150 | Y_RNA |
| cg22416376 | 0.092 | 3.33E-08 | 5.09E-04 | 17,395,271 | SLC7A2 |
| cg09100343 | 0.075 | 3.34E-08 | 5.09E-04 | 57,147,152 | CPNE2 |
| cg25420952 | 0.084 | 3.46E-08 | 5.09E-04 | 116,841,084 | AMBP |
| cg18645625 | 0.102 | 3.48E-08 | 5.09E-04 | 79,699,531 | ZFYVE16 |
| cg21642947 | − 0.062 | 3.83E-08 | 5.09E-04 | 62,153,431 | PPDPF |
Gene lists corresponding to the closest transcription start sites (TSS) to differentially methylated DNAm probe sites in endometrial tissue across the cycle were compared to genes found to be differentially expressed between the same stages in endometrial tissue using data from Fung et al. [17]. Over a quarter of genes annotated to differentially methylated sites (3215 genes) were also differentially expressed between the proliferative and secretory phases (Additional file 2: Figure S5). This overlap with differentially expressed genes was significantly different to the proportion expected by chance (chi-square statistic = 5.10, P = 0.02).
mQTL analysis
Using genotype information from each individual, we performed an expression quantitative trait loci (eQTL) analysis to identify associations between SNPs and DNAm probes in both endometrial tissue and blood. In endometrial tissue, we observed 4546 sentinel cis-mQTLs (P < 1.13 × 10−10) and 434 sentinel trans-mQTLs (P < 2.29 × 10−12), using a Bonferroni threshold to correct for multiple testing. Sentinel cis-mQTLs were defined as the mQTL with the most significant P value for each DNAm probe and sentinel trans-mQTLs were defined as mQTLs harbouring independent (R2 < 0.5) SNPs on a different chromosome to the associated DNAm site. There were similar numbers in blood DNA with 6615 sentinel cis-mQTLs (P < 1.13 × 10−10) and 590 sentinel trans-mQTLs (P < 2.29 × 10−12) (Fig. 3). The 30 most significant cis-mQTLs identified in endometrial tissue are listed in Table 2 and the 30 most significant cis-mQTLs identified in blood are listed in Table 3. Conditional analysis on cis-mQTLs reaching Bonferroni significance identified secondary cis-mQTL signals for 9 DNAm probe sites in endometrial tissue and 44 DNAm probe sites in blood. Only 23 DNAm probe sites were both differentially methylated across the cycle and had a cis-mQTL in endometrial tissue. We found no interaction between genotype and stage of cycle at these 23 DNAm probe sites.
Fig. 3.

Manhattan plots of endometrial tissue (top; panels a, b) and blood (bottom; panels c, d) cis (left; panels a, c) and trans-mQTLs (right; panels b, d). Each point represents an mSNP, chromosomes are defined by alternating purple and orange points and the red line indicates a Bonferroni threshold of P < 1.13 × 10−10 for cis-eQTLs and P < 2.29 × 10−12 for trans-eQTLs
Table 2.
Top 30 most significant cis-mQTLs in endometrium
| CHR | SNP | BP | A1 | BETA | SE | P value | DNAm probe site ID | Probe start (hg19) | Closest TSS gene name |
|---|---|---|---|---|---|---|---|---|---|
| 3 | rs6783741 | 43,455,926 | T | 0.4525 | 0.00261 | 2.23E-60 | cg11035303 | 43,465,453 | SNRK |
| 4 | rs5856334 | 16,076,153 | AT | 0.3958 | 0.002547 | 1.94E-58 | cg17858192 | 16,077,757 | PROM1 |
| 3 | 3:61237223:T:C | 61,237,223 | T | 0.4387 | 0.002831 | 2.20E-58 | cg17573813 | 61,237,223 | FHIT |
| 14 | rs12436555 | 24,634,825 | A | − 0.4469 | 0.003038 | 1.85E-57 | cg02898977 | 24,662,177 | IPO4 |
| 11 | rs73555593 | 107,462,942 | A | 0.4494 | 0.003092 | 3.00E-57 | cg22355889 | 107,461,585 | ELMOD1 |
| 4 | rs6854452 | 39,446,337 | A | 0.4824 | 0.003458 | 1.62E-56 | cg19311470 | 39,460,490 | RPL9 |
| 17 | rs3833162 | 27,071,442 | G | 0.6435 | 0.004835 | 1.10E-55 | cg04212500 | 27,184,483 | ERAL1 |
| 20 | rs3764715 | 1,287,051 | A | − 0.3891 | 0.002947 | 1.52E-55 | cg17341969 | 1,287,000 | SDCBP2 |
| 1 | rs6697965 | 220,943,801 | C | 0.4621 | 0.003507 | 1.67E-55 | cg12466610 | 220,950,155 | MARCH12 |
| 17 | rs9897355 | 80,078,095 | G | 0.4414 | 0.003403 | 3.19E-55 | cg25388952 | 80,084,596 | CCDC57 |
| 13 | rs1040961 | 40,108,008 | G | 0.4808 | 0.003773 | 6.55E-55 | cg17707870 | 40,107,957 | AK021977 |
| 5 | rs113644940 | 174,915,503 | T | − 0.4505 | 0.003674 | 3.16E-54 | cg20462978 | 174,911,722 | SFXN1 |
| 6 | rs660594 | 31,837,250 | G | 0.3695 | 0.003024 | 3.66E-54 | cg20370184 | 31,838,494 | SLC44A4 |
| 3 | rs76046201 | 15,365,139 | T | − 0.4432 | 0.003671 | 5.98E-54 | cg09627057 | 15,377,670 | SH3BP5 |
| 2 | rs6706223 | 33,944,002 | G | 0.4451 | 0.00374 | 1.07E-53 | cg04131969 | 33,951,597 | MYADML |
| 6 | rs9380143 | 29,802,045 | T | − 0.4279 | 0.003698 | 3.38E-53 | cg03570263 | 30,040,291 | RNF39 |
| 6 | rs138009982 | 170,453,220 | A | 0.4388 | 0.003921 | 1.33E-52 | cg11400162 | 170,455,448 | LOC154449 |
| 6 | rs72860388 | 32,904,703 | T | − 0.3676 | 0.003288 | 1.38E-52 | cg21992044 | 32,918,073 | HLA-DMA |
| 7 | rs7807520 | 2,087,545 | C | − 0.4547 | 0.004083 | 1.63E-52 | cg21598190 | 2,099,404 | MAD1L1 |
| 9 | rs13299342 | 136,141,504 | A | − 0.4277 | 0.003848 | 1.74E-52 | cg13683939 | 136,152,547 | ABO |
| 1 | rs35195267 | 92,398,884 | T | 0.3806 | 0.003438 | 2.08E-52 | cg01081438 | 92,417,998 | BRDT |
| 15 | rs376992916 | 65,245,209 | T | − 0.4207 | 0.003825 | 2.68E-52 | cg25879395 | 65,272,510 | SPG21 |
| 8 | rs11167041 | 142,258,889 | A | − 0.4478 | 0.004167 | 6.95E-52 | cg04123498 | 142,283,514 | SLC45A4 |
| 8 | rs12675160 | 140,918,110 | A | − 0.4302 | 0.004021 | 8.29E-52 | cg16191297 | 140,926,659 | AX748239 |
| 7 | rs1108056 | 101,834,081 | A | − 0.4235 | 0.003977 | 1.01E-51 | cg18088486 | 101,837,098 | SH2B2 |
| 7 | rs798558 | 2,758,935 | G | − 0.4117 | 0.003963 | 2.79E-51 | cg17393140 | 2,764,079 | AMZ1 |
| 1 | rs12074147 | 40,203,722 | C | − 0.4137 | 0.004 | 3.35E-51 | cg07703391 | 40,225,995 | AB075489 |
| 8 | rs7822181 | 10,049,872 | T | − 0.4409 | 0.00427 | 3.57E-51 | cg26077133 | 10,049,821 | MSRA |
| 12 | rs7139321 | 123,719,528 | T | 0.4135 | 0.004077 | 7.40E-51 | cg09084244 | 123,757,810 | CDK2AP1 |
| 11 | rs678679 | 35,608,275 | T | − 0.4807 | 0.004747 | 7.90E-51 | cg26465155 | 35,611,044 | FJX1 |
Table 3.
Top 30 most significant cis-mQTLs in blood
| CHR | SNP | BP | A1 | BETA | SE | P value | DNAm probe Site ID | Probe start (hg19) | Closest TSS gene name |
|---|---|---|---|---|---|---|---|---|---|
| 3 | rs6783741 | 43,455,926 | T | 0.448 | 0.001743 | 2.21E-67 | cg11035303 | 43,465,453 | SNRK |
| 6 | 6:29699301:TGAGAGA :TGAGA | 29,699,301 | TGAGAGA | 0.4433 | 0.002662 | 1.16E-59 | cg27230769 | 29,705,998 | HLA-F-AS1 |
| 8 | rs12675160 | 140,918,110 | A | − 0.4395 | 0.002672 | 1.91E-59 | cg16191297 | 140,926,659 | AX748239 |
| 15 | rs4776894 | 67,416,445 | C | − 0.4491 | 0.002948 | 4.42E-58 | cg07882838 | 67,417,557 | SMAD3 |
| 2 | rs6706223 | 33,944,002 | G | 0.4457 | 0.002985 | 1.00E-57 | cg04131969 | 33,951,597 | MYADML |
| 6 | rs9380143 | 29,802,045 | T | − 0.4285 | 0.002918 | 2.00E-57 | cg03570263 | 30,040,291 | RNF39 |
| 13 | rs1040961 | 40,108,008 | G | 0.4481 | 0.003182 | 1.10E-56 | cg17707870 | 40,107,957 | AK021977 |
| 4 | rs5856334 | 16,076,153 | AT | 0.4034 | 0.002962 | 4.31E-56 | cg17858192 | 16,077,757 | PROM1 |
| 4 | rs6854452 | 39,446,337 | A | 0.4776 | 0.003507 | 4.33E-56 | cg19311470 | 39,460,490 | RPL9 |
| 11 | rs73555593 | 107,462,942 | A | 0.4321 | 0.003249 | 1.15E-55 | cg22355889 | 107,461,585 | ELMOD1 |
| 3 | rs1054190 | 119,536,718 | T | − 0.3963 | 0.002991 | 1.32E-55 | cg12414339 | 119,536,718 | NR1I2 |
| 1 | rs6687657 | 33,599,737 | T | − 0.4317 | 0.003277 | 1.68E-55 | cg12386614 | 33,608,003 | AX747064 |
| 7 | rs62444879 | 2,048,470 | G | − 0.4355 | 0.003316 | 1.90E-55 | cg03723481 | 2,071,723 | MAD1L1 |
| 11 | rs10750097 | 116,664,040 | G | 0.4412 | 0.003384 | 2.59E-55 | cg12556569 | 116,663,989 | APOA5 |
| 19 | rs1433089 | 52,506,985 | C | − 0.435 | 0.003369 | 3.81E-55 | cg01561758 | 52,514,395 | ZNF615 |
| 6 | rs138009982 | 170,453,220 | A | 0.4368 | 0.003437 | 7.35E-55 | cg11400162 | 170,455,448 | LOC154449 |
| 2 | rs61702354 | 25,970,644 | A | 0.3672 | 0.002935 | 1.40E-54 | cg17717333 | 26,101,647 | ASXL2 |
| 14 | rs35595004 | 52,733,244 | A | − 0.4397 | 0.00357 | 2.64E-54 | cg23022053 | 52,733,193 | PTGDR |
| 10 | rs10900074 | 45,071,312 | A | − 0.427 | 0.003469 | 2.70E-54 | cg02113055 | 45,072,470 | CXCL12 |
| 12 | rs928993 | 52,798,364 | A | − 0.4268 | 0.003471 | 2.83E-54 | cg19393008 | 52,798,313 | KRT82 |
| 6 | rs3130978 | 31,082,188 | A | − 0.4033 | 0.003301 | 3.65E-54 | cg24926791 | 31,082,137 | PSORS1C1 |
| 21 | rs1721 | 46,349,496 | T | − 0.4368 | 0.003577 | 3.76E-54 | cg02464073 | 46,349,496 | ITGB2 |
| 9 | rs11789671 | 120,504,614 | A | − 0.414 | 0.003395 | 3.94E-54 | cg21242448 | 120,510,244 | TLR4 |
| 11 | rs11230502 | 60,607,476 | A | − 0.4072 | 0.003374 | 6.09E-54 | cg06394820 | 60,608,241 | CCDC86 |
| 12 | rs7139321 | 123,719,528 | T | 0.4466 | 0.00372 | 7.49E-54 | cg09084244 | 123,757,810 | CDK2AP1 |
| 7 | rs1108056 | 101,834,081 | A | − 0.4386 | 0.003676 | 9.75E-54 | cg18088486 | 101,837,098 | SH2B2 |
| 1 | rs284307 | 10,739,255 | C | − 0.3581 | 0.003024 | 1.33E-53 | cg13387643 | 10,737,562 | Mir_584 |
| 12 | rs10777168 | 76,651,353 | C | 0.407 | 0.003444 | 1.43E-53 | cg26864661 | 76,661,181 | BBS10 |
| 20 | rs6073257 | 42,561,422 | C | 0.3894 | 0.00333 | 2.19E-53 | cg26365090 | 42,574,362 | TOX2 |
| 16 | rs12149056 | 58,690,964 | A | − 0.4447 | 0.003819 | 2.62E-53 | cg05876883 | 58,704,445 | SLC38A7 |
Overlap between endometrial and blood mQTLs
We were able to test how well our blood mQTL dataset reproduced previously identified mQTL signals by overlapping our signals with summary data from a meta-analysis of the Lothian Birth Cohorts (LBC) and Brisbane Systems Genetics Study (BSGS) datasets from 1980 individuals [23]. Approximately 70% of cis-mQTLs identified in blood in this study have been reported in blood previously. This replication shows that our blood data are consistent with larger blood mQTL datasets that themselves can act as a proxy to increase the power of subsequent analyses. Focusing on our matched endometrium and blood data, 60% of endometrial tissue cis-mQTLs were also found in our blood cis-mQTL set. Similarly, when compared to the larger LBC-BSGS blood mQTL dataset, 62% of endometrial tissue cis-mQTLs were also seen in a larger blood dataset. The 30 cis-mQTLs with the largest effect size in endometrial tissue that are also in blood are shown in Additional file 2: Figure S6, the majority displaying effect sizes in the same direction. It is important to note however that the detection of differences in effect size between tissues is dependent on sample size and the power to detect differences [24].
Using the rb method outlined by Qi et al. [24], we estimated the correlation in genetic effects between cis-mQTLs in endometrium and blood, and found a high correlation between tissues from the same individuals (rb = 0.78). This correlation was similar to the correlation in cis-mQTL effects between brain and blood (rb = 0.78) in the Qi et al. [24] study.
Overlap with reproductive traits and pathologies
GWAS overlap
To investigate possible endometriosis-associated disease mechanisms impacted by epigenetic regulation in the endometrium, we identified any cis-mQTL mSNPs (mSNP—SNP with a significant mQTL) in genomic regions previously associated with endometriosis. Five mSNPs associated with DNAm probe sites closest to GREB1, C11orf46, NR2C1, KDR and WNT4 are located within regions associated with endometriosis risk (Table 4).
Table 4.
Endometrial cis-mQTL mSNPs associated with endometriosis
| CHR | SNP | BP | BETA | P value | DNAm probe site ID | Probe start (hg19) | Closest TSS gene name | GWAS P value |
|---|---|---|---|---|---|---|---|---|
| 2 | rs16857668 | 11,723,110 | − 0.3996 | 2.95e-41 | cg16908938 | 11,728,029 | GREB1 | 2.345E-15 |
| 11 | rs11031006 | 30,226,528 | 0.07907 | 2.42e-06 | cg26197155 | 30,344,676 | C11orf46 | 8.558E-08 |
| 12 | rs35223035 | 95,675,326 | 0.05842 | 4.94e-05 | cg06948737 | 95,471,414 | NR2C1 |
R2 = 0.89 with GWAS SNP rs4762326 (P = 2.20E-09) |
| 4 | rs1551641 | 55,993,915 | 0.0592 | 0.0001091 | cg07123701 | 56,024,384 | KDR | 3.736E-11 |
| 1 | rs12405695 | 22,365,689 | − 0.09405 | 0.0001206 | cg03519931 | 22,466,137 | WNT4 | 1.297E-14 |
Using Functional Mapping and Annotation of Genome-Wide Association (FUMA) to test for overlap between mSNPs and SNPs associated with traits and diseases in the GWAS catalogue, we identified 632 mSNPs that matched, or were in linkage disequilibrium with the SNPs in the GWAS catalogue that are significantly associated with 482 different traits and diseases (Additional file 3: Table S3). Some of the overlapping SNPs included those associated with reproductive traits and diseases such as age at first birth, birth weight, endometriosis, ovarian cancer, and age of menarche and menopause.
Summary-data-based Mendelian randomisation
To test for a causal/pleiotropic relationship between methylation status, genotype and endometriosis, we applied summary-data-based Mendelian randomisation (SMR) and heterogeneity in dependent instruments (HEIDI) methods [25] to endometriosis meta-analysis summary data from Sapkota et al. [26] and our endometrial mQTL summary data. A total of five DNAm probe sites passed the SMR test (PSMR < 1.2 × 10−5). These five sites were annotated to growth regulating oestrogen receptor binding 1 (GREB1) and SNP rs59129126, Metazoa_SRP and SNP rs28689909, and kinase insert domain receptor (KDR) (3 DNAm probe sites) and SNPs rs62304733 and rs6554237 (Table 5). Only two of the five DNAm probe sites, those annotated to GREB1 and Metazoa_SRP, were not rejected by the HEIDI test with PHEIDI > 0.01 (Fig. 4).
Table 5.
Results of the SMR analysis conducted using endometrial mQTLs and summary statistics from an endometriosis meta-analysis
| CHR | DNAm probe site ID | Closest TSS gene name | Probe bp | Top SNP | Top SNP bp | A1 | p_SMR | p_HET |
|---|---|---|---|---|---|---|---|---|
| 2 | cg16908938 | GREB1 | 11,728,029 | rs59129126 | 11,728,388 | C | 1.58E-07 | 1.25E-01 |
| 4 | cg10360906 | KDR | 56,023,701 | rs62304733 | 56,024,199 | C | 7.87E-07 | 9.34E-04 |
| 4 | cg09978860 | KDR | 56,023,920 | rs62304733 | 56,024,199 | C | 2.85E-06 | 8.37E-03 |
| 4 | cg01777861 | KDR | 56,023,794 | rs6554237 | 56,025,361 | T | 5.74E-06 | 8.46E-03 |
| 2 | cg07314298 | Metazoa_SRP | 11,723,111 | rs28689909 | 11,735,061 | A | 7.93E-06 | 1.26E-02 |
Fig. 4.

Association of methylation differences near the GREB1 locus and endometriosis risk. a Location of GREB1 transcripts on chromosome 2 with marked locations of the lead endometriosis risk SNP (rs11674184) for the GREB1 locus (pink), the GREB1 gene expression array probe (ILMN_1721170) position (purple), the location of mQTL DNAm probe (cg16908938) (orange) and mQTL SNP (rs59129126) (green) passing the SMR analysis. b Expression of ILMN_1721170 in endometrial samples from women with different genotypes at rs59129126. c Methylation at DNAm probe cg16908938 in endometrial samples from women with different genotypes at rs59129126. d SMR locus plot showing the results at GREB1/Metazoa_SRP locus for endometriosis. Results of the latest GWAS meta-analysis are shown in the top plot, grey dots representing the P values for SNPs and diamonds representing the P values for DNAm probe sites from the SMR test. Sites highlighted in red passed the SMR test. The middle and bottom plots show the endometrial mQTL P values of SNPs from this study for the DNAm probe sites nearest to GREB1 and Metazoa_SRP respectively
The SMR analysis was repeated using blood mQTL summary data from the larger LBC-BSGS blood dataset. Six signals passed the SMR test (PSMR < 5.6 × 10−7) and were not rejected by the HEIDI test (PHEIDI > 3.8 × 10−3) test. These included two DNAm probe sites closest to GREB1, two closest to WNT4, one closest to Metazoa_SRP and one closest to C11orf46 (Table 6).
Table 6.
Results of the SMR analysis conducted using blood mQTLs and summary statistics from an endometriosis meta-analysis
| CHR | DNAm probe Site ID | Closest TSS gene name | Probe bp | Top SNP | Top SNP bp | A1 | b_SMR | p_SMR | p_HET |
|---|---|---|---|---|---|---|---|---|---|
| 2 | cg02584498 | GREB1 | 11,674,057 | rs77294520 | 11,660,955 | C | 0.149038 | 1.01E-11 | 0.009952231 |
| 2 | cg10849854 | GREB1 | 11,674,557 | rs77294520 | 11,660,955 | C | 0.250387 | 4.40E-10 | 0.000121199 |
| 4 | cg10360906 | KDR | 56,023,751 | rs11936364 | 56,019,253 | T | − 0.075528 | 2.43E-09 | 0.000696251 |
| 4 | cg01777861 | KDR | 56,023,843 | rs7696256 | 56,023,747 | G | − 0.096417 | 3.38E-09 | 0.000267448 |
| 4 | cg09978860 | KDR | 56,023,921 | rs11936364 | 56,019,253 | T | − 0.0974429 | 3.70E-09 | 0.000221536 |
| 1 | cg25011003 | WNT4 | 22,470,341 | rs55938609 | 22,470,451 | C | 0.280871 | 4.71E-09 | 0.181277 |
| 4 | cg16572876 | KDR | 56,024,045 | rs11936364 | 56,019,253 | T | − 0.144697 | 1.17E-08 | 0.000408871 |
| 4 | cg20092376 | KDR | 56,023,423 | rs6837695 | 56,015,840 | T | − 0.148622 | 1.17E-08 | 0.000631242 |
| 4 | cg07123701 | KDR | 56,024,434 | rs11936364 | 56,019,253 | T | − 0.151501 | 1.29E-08 | 0.000349096 |
| 2 | cg16908938 | GREB1 | 11,728,079 | rs59129126 | 11,728,388 | C | 0.0779192 | 1.34E-08 | 0.04232295 |
| 2 | cg07314298 |
Metazoa
_SRP |
11,723,111 | rs59129126 | 11,728,388 | C | 0.105449 | 1.88E-08 | 0.1823231 |
| 1 | cg15582954 | WNT4 | 22,470,343 | rs55938609 | 22,470,451 | C | 0.361317 | 7.96E-08 | 0.3437667 |
| 11 | cg26197155 | C11orf46 | 30,344,725 | rs12271187 | 30,319,259 | A | − 0.112429 | 1.71E-07 | 0.8086896 |
Using a multi-omic approach within the SMR software and endometrial eQTL data from Fung et al. [27]; we integrated both our endometrial mQTL dataset and the eQTL dataset to identify any association between genetic regulation of a methylation site and transcription of a gene and vice versa. We used endometrial expression quantitative trait loci (eQTLs) as the outcome and endometrial mQTLs as the exposure (M2T analysis [8]) and identified 472 associations between 414 methylation probes and 186 gene expression probes (Additional file 3: Table S4). Alternatively using endometrial mQTLs as the outcome and endometrial eQTLs as the exposure (T2M analysis), we identified 353 associations between 308 methylation probes and 157 gene expression probes that passed the SMR and HEIDI tests (Additional file 3: Table S5). We observed 275 associations overlapping between M2T and T2M analyses, the majority of loci showing opposite directions of effect (Additional file 1: Supplementary Note 2). This is consistent with both pleiotropy and the hypothesis that genotypes can regulate gene expression by altering the methylation at nearby DNAm probe sites and also can potentially affect methylation at DNAm probe sites via changes in gene expression. An estimated ~ 26% of the DNAm probe sites targeted the closest gene whilst the remaining sites target more distant genes. An example of a DNAm probe site targeting the most immediate gene, threonine synthase like 2 (THNSL2), is shown in Fig. 5, both the DNAm probe site and associated SNP located within the THNSL2 promotor. An example of a DNAm probe site targeting a more distant gene, IGF-like family receptor 1 (IGFLR1/TMEM149), is shown in Fig. 6. We mapped the position of the associated M2T DNAm probe sites, which also have mQTLs in blood, to annotated regulatory regions and found that ~ 90% were within known regulatory elements. M2T DNAm probes were significantly enriched in promoters (fold-change = 1.52, P = 2.18 × 10−8) and were significantly less represented in quiescent regions (fold-change = 0.57, P = 9.78 × 10−9) when compared to randomly sampled probes with matched variance (Additional file 2: Figure S7).
Fig. 5.

An mQTL affecting gene expression of THNSL2 immediately adjacent to the DNAm probe. a Location of genes on chromosome 2 surrounding an eQTL for THNSL2 and nearby mQTL. The location of the eQTL gene expression probe is highlighted in purple, the mQTL DNAm probe is highlighted in orange and the mQTL and eQTL SNP is highlighted in green. The arrow indicates the association of the mQTL SNP with expression of the THNSL2 probe. b Expression of the THNSL2 probe (ILMN_2173294) in endometrium from women with different genotypes at rs6547758. c Methylation of the cg24977027 probe in endometrium from women with different genotypes at rs6547758. d SMR locus plot showing the endometrial eQTL P values of SNPs for the THNSL2 probe (ILMN_2173294) (top) and mQTL P values of SNPs from this study for the DNAm probe cg24977027 (bottom)
Fig. 6.

An mQTL affecting gene expression distal to DNAm probe. a Location of genes on chromosome 19 surrounding an mQTL closest to ARHGAP33 and nearby eQTL for IGFLR1(TMEM149). The location of the eQTL gene expression probe is highlighted in purple, the mQTL DNAm probe is highlighted in orange and the mQTL and eQTL SNP is highlighted in green. The arrow indicates the association of the mQTL SNP with expression of the IGFLR1 probe. b Expression of the IGFLR1 probe (ILMN_1786426) in endometrium from women with different genotypes at rs62112162. c Methylation of the cg16569309 probe in endometrium from women with different genotypes at rs62112162. d SMR locus plot showing the endometrial eQTL P values of SNPs for the IGFLR1 probe (ILMN_1786426) (top) and mQTL P values of SNPs from this study for the DNAm probe cg16569309 (bottom)
SMR was also used to test for any associations between endometrial eQTLs and various other traits and diseases. We found pleiotropic associations between 409 probes and 17 traits including those relating to reproductive biology, age at menopause and ovarian cancer (Additional file 3: Table S6). Approximately 63% of mQTLs that passed the SMR test and were not rejected by the HEIDI test for these traits were also present in blood. However, for mQTLs associated with menopause and ovarian cancer, only 6 of the 26 mQTLs were also in blood. This suggests that tissue-specific effects may contribute to these phenotypes.
Functional annotation
Gene pathways potentially impacted by changes in methylation in endometrium were investigated using the pathway enrichment analysis in FUMA. No MsigDB Hallmark pathways were enriched for genes with transcription start sites closest to DNAm probe sites differentially methylated between stages of the menstrual cycle. Significantly enriched pathways for overlapping gene sets between differentially methylated and differentially expressed genes include epithelial mesenchymal transition, oestrogen response, IL2 STAT5 signalling and TNFA signalling via NFKB (Additional file 2: Figure S8).
To identify gene pathways potentially affected by genetic regulation of methylation in endometrium and/or blood, we also conducted a pathway analysis of genes annotated to cis-mQTL probes. Pathway analysis showed that ultraviolet (UV) response, early oestrogen response and epithelial mesenchymal transition were the most significantly enriched hallmark pathways in both endometrial tissue and blood; GO biological processes such as intracellular signal transduction, regulation of cell differentiation and positive regulation of molecular function were also highly enriched in both tissues (Table 7). The majority of enriched hallmark pathways were consistent across both blood and endometrium with the exception of pancreas beta cells, hedgehog signalling and the PI3K/AKT/MTOR signally pathways, which were only enriched in blood mQTLs, and peroxisome and angiogenesis pathways that were only enriched in endometrium (Additional file 3: Table S7 and S8). Similarly, whilst 85% of the enriched GO biological process pathways are shared between blood and endometrium, there are some more biologically relevant pathways that are tissue specific such as artery development, lymphocyte differentiation and cardiac cell development in blood and regulation of meiotic cell cycle, regulation of epithelial structure maintenance and regulation of embryonic development in endometrium (Additional file 3: Table S7 and S8).
Table 7.
Top hallmark and GO biological processes pathways enriched for genes closest to DNAm probe sites in cis-mQTLs in endometrium and blood
| Tissue | GeneSet | N | n | P value | Adjusted P |
|---|---|---|---|---|---|
| Endometrial mQTLs | Go intracellular signal transduction | 1568 | 237 | 1.92E-29 | 5.50E-26 |
| Go regulation of multicellular organismal development | 1667 | 247 | 2.48E-29 | 5.50E-26 | |
| Go regulation of transport | 1799 | 257 | 5.10E-28 | 7.54E-25 | |
| Go regulation of cell differentiation | 1488 | 224 | 1.29E-27 | 1.43E-24 | |
| Go regulation of cell proliferation | 1492 | 221 | 2.73E-26 | 2.42E-23 | |
| Go neurogenesis | 1401 | 211 | 4.37E-26 | 3.23E-23 | |
| Go regulation of cell death | 1471 | 218 | 5.66E-26 | 3.58E-23 | |
| Go locomotion | 1111 | 179 | 1.16E-25 | 6.41E-23 | |
| Go positive regulation of molecular function | 1786 | 248 | 3.00E-25 | 1.48E-22 | |
| Go regulation of transcription from rna polymerase ii promoter | 1780 | 246 | 9.31E-25 | 4.13E-22 | |
| Hallmark uv response dn | 144 | 37 | 9.77E-13 | 4.88E-11 | |
| Hallmark oestrogen response early | 200 | 41 | 1.94E-10 | 4.84E-09 | |
| Hallmark epithelial mesenchymal transition | 199 | 34 | 6.07E-07 | 8.57E-06 | |
| Hallmark complement | 200 | 34 | 6.86E-07 | 8.57E-06 | |
| Hallmark androgen response | 100 | 21 | 1.68E-06 | 9.69E-06 | |
| Hallmark hypoxia | 200 | 33 | 1.94E-06 | 9.69E-06 | |
| Hallmark allograft rejection | 200 | 33 | 1.94E-06 | 9.69E-06 | |
| Hallmark il2 stat5 signalling | 200 | 33 | 1.94E-06 | 9.69E-06 | |
| Hallmark p53 pathway | 200 | 33 | 1.94E-06 | 9.69E-06 | |
| Hallmark myogenesis | 200 | 33 | 1.94E-06 | 9.69E-06 | |
| Blood mQTLs | Go intracellular signal transduction | 1568 | 330 | 9.02e-46 | 4.00e-42 |
| Go positive regulation of molecular function | 1786 | 345 | 2.00e-39 | 4.44e-36 | |
| Go neurogenesis | 1401 | 291 | 3.12e-39 | 4.62e-36 | |
| Go regulation of multicellular organismal development | 1667 | 326 | 2.39e-38 | 2.29e-35 | |
| Go regulation of hydrolase activity | 1325 | 278 | 2.58e-38 | 2.29e-35 | |
| Go regulation of intracellular signal transduction | 1651 | 322 | 1.23e-37 | 9.08e-35 | |
| Go positive regulation of catalytic activity | 1515 | 302 | 3.82e-37 | 2.12e-34 | |
| Go tissue development | 1508 | 301 | 3.91e-37 | 2.12e-34 | |
| Go locomotion | 1111 | 244 | 4.78e-37 | 2.12e-34 | |
| Go regulation of cell differentiation | 1488 | 298 | 4.79e-37 | 2.12e-34 | |
| Hallmark uv response dn | 144 | 48 | 5.31e-16 | 2.66e-14 | |
| Hallmark oestrogen response early | 200 | 56 | 1.74e-14 | 4.35e-13 | |
| Hallmark epithelial mesenchymal transition | 199 | 48 | 3.49e-10 | 5.25e-09 | |
| Hallmark complement | 200 | 48 | 4.20e-10 | 5.25e-09 | |
| Hallmark oestrogen response late | 200 | 46 | 4.04e-09 | 4.04e-08 | |
| Hallmark myogenesis | 200 | 45 | 1.20e-08 | 1.00e-07 | |
| Hallmark mitotic spindle | 200 | 44 | 3.48e-08 | 2.49e-07 | |
| Hallmark p53 pathway | 200 | 43 | 9.80e-08 | 6.12e-07 | |
| Hallmark apical junction | 199 | 42 | 2.32e-07 | 1.22e-06 | |
| Hallmark tnfa signalling via nfkb | 200 | 42 | 2.68e-07 | 1.22e-06 |
Both blood and endometrium cis-mQTLs had very similar methylome patterns in the context of CpG locations; an average of 47% of cis-mQTLs were located in open sea regions of the genome followed by 24% located in shores and 17% in CpG islands (Additional file 3: Table S9, Additional file 2: Figure S9). mQTL DNAm probes were also annotated to predicted regulatory regions, the majority located in promoters and quiescent regions (Additional file 1: Supplementary Note 2).
Using available data on the genomic location of oestrogen receptor (ESR) binding sites, we identified 414 differentially methylated DNAm probes that overlapped ESR binding sites (Additional file 3: Table S10). We also identified 791 cis-mQTL mSNPs that were within ESR binding sites (Additional file 3: Table S11). Pathway analysis identified that the early (P = 5.16 × 10−12) and late (P = 5.43 × 10−5) oestrogen response pathway and the cholesterol homeostasis pathway (P = 9.18 × 10−6) were most significantly enriched for genes closest to these mQTL DNAm probe sites.
Discussion
We analysed genetic control of methylation in human endometrium and compared results with methylation in DNA from blood samples collected from the women at the same time. We observed marked changes in DNAm in the endometrium across the menstrual cycle for some probes in agreement with previous studies [19–21]. The 66 women were sampled at different stages of the menstrual cycle and 6% of DNAm probe sites (27,262 sites) showed evidence of differential methylation across the cycle. The endometrium is a biologically and transcriptionally dynamic tissue with significant changes in gene expression across the menstrual cycle [15, 17, 28]. Genes previously reported as differentially expressed across the cycle that were also assigned to differentially methylated sites across the menstrual cycle were significantly enriched in the oestrogen response pathway and 1.5% of differentially methylated probes are located in ESR binding sites. Oestrogen plays a major role in regulating proliferation of epithelial and stromal cells during the proliferative phase of the menstrual cycle [29–31]. Differences in methylation between cycle stages were not observed in matched blood samples from the same women. Therefore, our findings highlight tissue-specific features of methylation signals in endometrium, although it is not known if the differential methylation is a reflection of differential methylation between cell types and the changes in the cellular composition across the cycle.
We identified 4546 sentinel cis-mQTLs and 434 sentinel trans-mQTLs in endometrial tissue samples. There was a high correlation of genetic effects (rb = 0.78) and overlap (~ 60%) in mQTLs between endometrium and blood samples from the same women and results were similar when comparing with a much larger sample of mQTLs in blood samples from unrelated individuals [23]. Of interest were the subset of mQTLs not present in blood and that overlapped oestrogen receptor binding sites, suggesting possible tissue-specific effects. Two examples were mQTLs at loci near the G protein-coupled oestrogen receptor 1 (GPER) and Plectin (PLEC). GPER is a membrane protein from the seven-transmembrane (7TM) GPCR family, localised to the endoplasmic reticulum [32]. This receptor mediates both rapid non-genomic signalling cascades and transcriptional changes that regulate cell proliferation and apoptosis in response to oestrogen [32, 33]. GPER has the potential to play an important regulatory role in the proliferation and regeneration of endometrium in response to an increase in circulating oestrogen during the proliferative phase of the menstrual cycle. PLEC belongs to a family of proteins that function as cytolinkers/plakins and play an important role in maintaining cytoskeleton structure and subsequently cell and tissue integrity, and cell adhesion [34]. PLEC is upregulated from the early to mid-secretory stage of the cycle in normal women and decreases again from mid to late-secretory stage during end of receptive period [35]. PLEC has been reported as downregulated in endometrium of women with repeated embryo implantation failure [36], and in women with endometriosis during the window of implantation [37], suggesting an important role in female fertility. The differences in DNA methylation across the menstrual cycle and mQTLs specific to the endometrium support the need for both tissue-specific studies and comparisons between tissues to understand regulation of epigenetic signals and their role in disease. However, much larger studies in target tissues such as endometrium will be necessary to have sufficient power to detect the tissue-specific mQTLs that may be associated with genetic effects on disease risk.
SMR analysis identified significant overlap of mQTLs with five endometriosis GWAS signals. Results include new evidence that the risk SNPs on chromosome 2 alter methylation at DNAm probe sites located within 350 bp of the GREB1 transcription start site in blood and another within the 3′UTR of a GREB1 transcript in both endometrium and blood. GREB1 is an oestrogen-responsive gene involved in the oestrogen receptor-regulated pathway, essential for oestrogen receptor transcription [38]. GREB1 has also been reported to regulate proliferation in breast, prostate, and ovarian cancers [39–42].
Evidence for mQTLs near GREB1 is an interesting result as we did not detect any genetic effects on GREB1 gene or protein expression in endometrium previously [17, 43]. Changes in methylation can result in alternative splicing [44] and the risk SNPs may alter methylation and expression of particular GREB1 transcripts that could not be individually identified from the microarray. The absence of an eQTL for GREB1 could also suggest epigenetic regulation of post-transcriptional modifications through mechanisms including microRNA regulation of GREB1 [45, 46], association with protein QTLs (pQTLs) independent of mRNA expression [47, 48], RNA folding, accessibility of functional sites [49, 50] or post-transcriptional modifications such as N6-methyladenosine (m6A) methylation which are enriched in stop codons and 3′UTRs [45, 51, 52]. Investigation into transcript-specific and post-transcriptional effects at this locus are needed to confirm any effects on GREB1. The absence of one of the SMR significant mQTLs (cg02584498- rs77294520) in endometrium may indicate tissue-specific effects or limited power in our endometrial dataset to detect the mQTL.
Three probes nearest to KDR also passed the SMR analysis in both endometrium and blood, but were rejected by the HEIDI test, suggesting significant heterogeneity and the possibility of multiple causal variants. KDR is a vascular endothelial growth factor receptor involved in the proliferation and differentiation of endothelial cells with a potential role in implantation and successful pregnancy [53–55]. The association of GREB1 and KDR mQTLs with endometriosis risk in both endometrium and blood may suggest the biological mechanisms that increase endometriosis risk may not be specific to endometrium.
The SMR analyses also detected target genes for DNAm probe sites with pleiotropic associations for mQTLs associated with age at menopause and ovarian cancer, many of which were not replicated in blood. These associations may implicate tissue-specific mQTLs, such as those in oestrogen-responsive tissues, in reproductive disease. Some examples of instances whereby genomic regulation of methylation may influence reproductive traits and pathologies include the ZNF346/UIMC1, SYCP2L and HOX gene loci. The ZNF346/UIMC1 locus is strongly associated with age at menopause and forms part the BRCA1-A complex, which regulates oestrogen receptor transcription and DNA repair, both of which are important in regulating endometrial oestrogen response and meiosis [56, 57]. The SYCP2L locus associated with age at menopause also had a pleiotropic relationship with an endometrial mQTL in this region. SYCP2L is a paralog of the synaptonemal complex protein 2 and is known to localise to centromeres in oocytes and promote primordial oocyte survival [57, 58]. Finally, a locus surrounded by HOX genes on chromosome 2 and containing an endometrial mQTL has been associated with epithelial ovarian cancer; many of the HOX genes in the region are known to regulate embryogenesis and neoplastic development [59]. The significant associations between endometrial mQTLs and various traits and diseases highlight the importance of our findings in the broader scientific community, identifying genetic regulatory mechanisms that are contributing to disease phenotypes.
We also identified pleiotropic associations between methylation at 414 methylation probes and altered expression for 186 gene expression probes, where both are associated with a shared causal variant. DNAm probes associated with gene expression were enriched in promoters, supporting the hypothesis that DNAm probe sites located in regulatory regions can affect gene expression of the associated genes in endometrium, as shown previously in blood [8]. The high proportion of DNAm probe sites (> 70%) mapping to distant genes is important when interpreting the mechanisms behind transcription regulation. DNAm probe sites located further away from their target gene may reside in regulatory regions such as enhancers that can interact with distant target genes through mechanisms such as chromatin looping [60, 61]. One example of a distant target, IGFLR1(TMEM149), has expression associated with a SNP and DNAm site ~ 35 kb downstream. The locus containing the SNP and DNAm is located within 400 bp of a regulatory region, a CTCF binding site, annotated in the Ensembl regulatory build [62]. CTCF protein is known to mediate chromatin looping and may represent a possible mechanism of regulation of IGFLR1 expression [63, 64]. Most disease-associated loci are within non-coding regions [65–67], including loci associated with the endometrial-related diseases endometriosis [26] and endometrial cancer [68]. Therefore, understanding how regulatory regions in these non-coding regions can affect distant target genes is important in understanding both endometrial biology and disease mechanisms.
An important limitation of this study is the small sample size which limits the statistical power to detect robust differences in methylation in endometrium. Previous epigenetic studies using the 450 k methylation beadchip and a significance level of P = 2.4 × 10−7 reported that 59 and 186 samples would have 80% power to detect mean differences in methylation of 15% and 8% respectively [69]. Power calculations by Rahmioglu et al. [21] show that 500 samples are needed to detect mean changes in methylation of 2% with 80% power in endometrium. Using the pwr.t2n.test function in R, we calculated that we had 80% power to detect a mean difference in methylation of 10% at a significance level of P = 0.05 and 25% at a genome-wide significance level of P = 1.13 × 10−7. Using variance estimates from our data, we estimated that 155 samples could detect a mean difference of 8% and ~ 3000 samples would be required to detect a mean methylation differences of 2%, similar to estimates by Saffari et. el [69] (Additional file 3: Table S12). Larger sample sizes would be needed to detect smaller effects of disease and menstrual cycle phase on methylation in endometrium. However, even with a limited sample size, we were able to detect over 4000 significant endometrial cis-mQTLs, the majority of which were previously reported in larger blood studies [23].
Another limitation of this study is results have not been adjusted for change in cell composition. There is currently no published method of estimating cell type composition in endometrial samples. Differences in methylation between menstrual cycle phases reported in this study are also likely to reflect changes in cell composition. Due to the complexity of endometrial tissue and the presence of several different cell types including stromal cells, epithelial cells and infiltrating immune cells, development of an accurate cell composition correction requires extensive research to characterise omic signatures of the individual cell types and validate a method to correct for differences in cell composition in endometrium.
Conclusions
This is the first study to identify mQTLs in human endometrium, and shows significant overlap and correlation between mQTLs seen in endometrium with those observed in blood from the same and independent individuals. The high degree of overlap supports the use of large blood mQTL datasets as a proxy for endometrium to increase power to detect target genes for endometrial traits and diseases. There was evidence for variation in genome-wide methylation profiles across the menstrual cycle for a proportion of sites in human endometrium, changes not observed in blood, and our results highlight possible tissue-specific effects for mQTLs and enriched pathways not shared between blood and endometrium. We show that genomic regulation of methylation in endometrium has the potential to influence endometrial biology and overlap of mQTLs with risk loci for endometriosis and ovarian cancer indicate a role of methylation in reproductive diseases. Larger sample sizes are needed to identify effects of disease on methylation in endometrium and identify tissue-specific mQTLs that may be involved in endometrial biology and disease.
Methods
Sample collection
A total of 66 women of reproductive age (31.08 ± 6.64 years) and from European ancestry were selected for inclusion in the study. Women were recruited when attending clinics at the Royal Women’s Hospital in Melbourne, Australia, following informed written consent. The study was approved by the Human Research Ethics Committees of the Royal Women’s Hospital, Melbourne, the QIMR Berghofer Medical Research Institute and The University of Queensland (Projects 11-24 and 16-43). All sample and data collection was performed in accordance with institutional approved guidelines and regulations.
The clinical history for each participant was obtained alongside surgical notes and pathology results. Whole blood samples were collected prior to surgery. Endometrial tissue was collected by curettage during laparoscopic surgery for investigation of recurring pelvic pain and/or infertility. Forty-five of the 66 women were diagnosed with endometriosis. All women were free from exogenous hormone treatment in the 3 months prior to surgery. Menstrual cycle stage for each participant was categorised by an experienced pathologist into menstrual (M) = 3, early proliferative (EP) = 1, mid proliferative (MP) = 27, late proliferative (LP) = 5, early secretory (ES) = 6, mid secretory (MS) = 14 and late secretory (LS) = 10.
DNA extraction, methylation array and genotyping
Buffy coat was isolated from whole blood for DNA extraction using a salting out method [70]. Endometrial tissue samples were stored in RNAlater (Life Technologies, Grand Island, NY, USA) at − 80 °C until RNA/DNA extraction. Genomic DNA was extracted from homogenised endometrial tissues using the AllPrep DNA/RNA mini kit according to the manufacturer’s instructions (QIAGEN, Valencia, CA).
Bisulfite conversions were performed in 96-well plates using the EZ-96 DNA Methylation Direct Kit (Zymo Research, Irvine, CA, USA). Prior to conversion, DNA concentrations were determined by Quant-iT™ PicoGreen™ dsDNA Reagent (Life Technologies, Carlsbad, CA, USA) and standardised to 500 ng DNA per sample. Bisulfite converted DNA samples were hybridised to Illumina Human Methylation 450 BeadChips using the Infinium HD Methylation protocol and Tecan robotics (Illumina, San Diego, CA, USA). Samples were scanned using an Illumina iScan Reader. Methylation at each site was measured as a ratio of the intensities of methylated and un-methylated alleles at the DNAm probe site represented as β values [71].
Genomic DNA extracted from whole blood was genotyped on the HumanCoreExome chips and Infinium PsychArray (Illumina Inc., San Diego) [27]. Genotype data was filtered using the program PLINK ver 1.9 [72, 73]. SNPs not genotyped in at least 95% of individuals were removed (-geno 0.05 command) along with SNPs with a minor allele frequency (MAF) < 0.05 (-maf 0.05 command) and with Hardy-Weinberg Equilibrium (HWE) P < 1 × 10−6 (-hwe 0.000001 command). A total of 282,625 SNPs were used for imputation using the 1000 Genomes Phase 3 V5 reference panel. Genotypes were phased with ShapeIt V2 prior to imputation on the Michigan Imputation Server [74]. Additional quality control was performed on imputed genotypes to remove SNPs of poor quality (R2 < 0.8) or low MAF < 0.05, leaving 5,162,603 autosomal SNPs for subsequent analysis.
Methylation quality control and normalisation
Quality control and normalisation of raw methylation data was performed separately for blood and tissue samples using the R package “Meffil” [75]. Genotype data present in the methylation array data was compared to genotypes of the same samples run on the HumanCoreExome chips and Infinium PsychArray (Illumina Inc., San Diego). Genotypes for all samples matched 65 corresponding SNP probes on the microarray confirming no sample error between the methylation profiles for endometrium and blood. QC parameters outlined in the Meffil manual were used for the blood and endometrial dataset (https://github.com/perishky/meffil). DNAm probes that did not exceed the background signal and met a detection P value of < 0.01 in > 10% of samples were removed (220 DNAm probes in endometrial tissue and 184 DNAm probes in blood) alongside probes with low bead numbers in > 10% of samples (418 DNAm probes in endometrial tissue and 346 DNAm probes in blood). There were no sample outliers with poor probe detection with > 90% of DNAm probes detected in all samples. Using control probes, ten principal components were used to adjust the methylation levels for technical effects. DNAm probe sites found to target multiple genomic regions as previously annotated by Price et al. [76] were also removed. Functional normalisation was applied to remove global differences in methylation data and to extend quantile normalisation of control probes across the data.
DNAm principle component analysis and covariate effects
The presence of potentially cofounding sources of variation in the data was investigated through principal component (PC) analysis of DNAm profiles and association of top PCs with known covariates for both blood and tissue datasets. Principal components were computed from normalised methylation profiles for endometrium and blood. A significant association between methylation beadchip (P = 2.14 × 10−6) and PC two and between stage of cycle (P = 3.19 × 10−4) and PC one was observed in endometrium. We also detected a significant association between methylation beadchip (P = 1.92 × 10−15) and age (P = 0.008), with PC two and three respectively, in blood. No significant effect of endometriosis status was observed. All covariates were corrected for in later analyses where appropriate.
Differential DNA methylation
To identify changes in the methylation state of DNAm probe sites between stages of the menstrual cycle, we performed a differential methylation analysis on both the blood and endometrial tissue methylation datasets. To increase the number of samples within each group, and power for subsequent analyses, we combined menstrual cycle stages into three main phases; menstrual (M) stage (n = 3), EP, MP and LP stages were merged into the proliferative (P) phase (n = 33) and the ES, MS and LS stages were merged into the secretory (S) phase (n = 30). We subsequently removed M stage samples from the differential analysis due to the small sample size and limited power. Following QC of the methylation data, 443,101 DNAm probe sites for blood and 443,016 DNAm probe sites for endometrial tissue were retained for inclusion in the cycle stage analysis. We used the eBayes method implemented in the limma package to compute a moderated t statistic and fold change between P and S phases.
To test for any confounding effects of endometriosis status, we also tested for differently methylated DNAm probes between women with and without endometriosis. Tests were conducted using the eBayes method for individuals in the same menstrual cycle phase and using all samples by including stage of cycle as an additional covariate. No differentially methylated DNAm probes were detected.
mQTL analysis
We tested the association between genotype and DNAm probe site methylation in both blood and endometrial tissue datasets to identify mQTLs. All 443,101 DNAm probe sites in blood and 443,016 DNAm probe sites in endometrium passing QC were included in the mQTL analysis. Associations between 5,162,603 SNP genotypes and normalised methylation intensities were tested using a linear regression model in the software PLINK ver 1.9 (-linear command). Covariates, including age, presence of endometriosis, stage of cycle and methylation chip, were adjusted for in the analysis. The distance distribution of significant (FDR < 0.05) mQTL SNPs from their associated DNAm probes within 1 Mb showed that the vast majority of SNPs (92%) were within 250 kb of the probe site (Additional file 2: Figure S10). To capture the majority of cis-mQTLs and limit multiple testing, cis-mQTLs were defined as ± 250 kb between the SNP and the DNAm probe site start position. Trans-mQTLs were defined as associations between a SNP and DNAm probe site located on different chromosomes. To identify secondary independent cis-mQTL signals, we performed conditional analysis on sentinel cis-mQTLs that met a Bonferroni significance threshold of P < 1.13 × 10−10. The conditional analysis was conducted by repeating the association analysis between genotype and DNAm probe site methylation conditioning on the primary SNP.
Context-specific mQTL analysis and overlap with differentially methylated DNAm probe sites
We tested for overlap between the differentially methylated DNAm probe sites and mQTLs. To test for interaction between genotype and stage of cycle on DNAm probe site methylation, we used the context-specific analysis method outlined by Fung et al. [17]. Briefly, we used linear regression to test for interaction between stage and genotype using the observed normalised methylation level of a probe as the dependent variable and fitting the regression coefficient of the genotype, regression coefficient of the stage of cycle and the regression coefficient of the interaction between genotype and stage of cycle. We tested 23 cis-mQTL probes passing Bonferroni correction in endometrial tissue that corresponded to genes differentially methylated between P and S phases of the menstrual cycle.
Overlap between endometrial and blood mQTLs
Using blood mQTLs detected in a large meta-analysis of the Lothian Birth Cohorts (LBC) and Brisbane Systems Genetics Study (BSGS) datasets consisting of 1980 individuals [23], we were able to assess the overlap between our endometrial and blood mQTLs and those from a more highly powered study in blood. The LBC-BSGS dataset consists of 94,338 sentinel cis-mQTLs with a significance of at least P < 5 × 10−8 and SNPs within 2 Mb distance from each probe. mQTLs were considered to overlap if they had the same probe and associated SNP. Additionally, overlap was defined in terms of linkage disequilibrium (LD) r2 > 0.7 between the mSNP in the LBC-BSGS dataset and the endometrial mSNP based on the 1000 Genome phase 3 reference panel.
Tissue specificity of endometrial mQTLs was investigated by identifying the presence of mQTLs found in endometrial tissue in blood. Tissue mQTLs were tested for overlap with both the blood mQTL set from this study and the larger LBC-BSGS dataset. Overlap was defined in the same manner as described previously.
The correlation of cis-mQTL effects between endometrium and blood from the same individuals was estimated using the rb method developed by Qi et al. [24]. mQTL effect sizes and standard errors were standardised between tissues based on z-statistics using the method described in Zhu et al. [25]. Top cis-mQTLs (P < 5 × 10−8) were taken from the LBC-BSGS blood dataset as an independent reference set. The top cis-mQTLs from the reference set were then extracted from our endometrium and blood set; cis-mQTLs not present in our sets were excluded and the remaining cis-mQTLs were used to estimate the correlation.
Overlap with endometriosis risk loci and reproductive traits
GWAS overlap
Summary data available from Sapkota et al. [26] generated from ~ 15,000 European endometriosis cases were used to test overlap with our endometrial mQTLs. Overlap was determined if sentinel mQTL mSNPs matched those identified at the 19 endometriosis risk loci or if sentinel mQTL mSNPs had a minimum LD of r2 > 0.7 with the GWAS SNP. The Functional Mapping and Annotation of Genome-Wide Association (FUMA) SNP2GENE function was also used to test mSNPs for association with other traits and diseases from the GWAS catalogue.
Summary-data-based Mendelian randomisation
Using SMR software developed by Zhu et al. [25], we tested for pleiotropic association between DNAm probe site methylation and endometriosis. Summary data from the Sapkota et al. [26] meta-analysis was used alongside summary data from mQTLs identified in this study as input for the analysis. A total of 4546 DNAm probe sites reaching Bonferroni significance were included in the analysis and a study-wide significance threshold of PSMR = 1.10 × 10−5 was applied. Heterogeneity of SMR estimates at surrounding SNPs (in LD with the top cis-mQTL) was tested using HEIDI which is incorporated in the SMR software. A PHEIDI of < 0.05/m, where m is the number of probes passing the SMR test, was used to suggest heterogeneity of SMR estimates in the cis-region. The SMR analysis was repeated in blood using summary data from the large blood LBC-BSGS mQTL dataset [23].
The SMR software also allows the integration of multiple-omic datasets to infer a likely regulatory mechanism. We used this multi-omic analysis option to test the association between endometrial mQTLs from this study and eQTLs from our previous study [27], using only probes that passed Benjamini-Hochberg false discovery rate (FDR) cut off of 0.05. This analysis was performed using gene expression as the outcome and methylation as the exposure in a M2T analysis and was performed again using expression as the exposure and methylation as the outcome in a T2M analysis, as previously described in Wu et al. [8].
To investigate the possible impact of endometrial mQTLs in other phenotypes, we conducted further SMR analyses using GWAS summary datasets for a range of traits including body mass index (BMI), body fat percentage, leptin, lipid levels including high-density lipoprotein (HDL), low-density lipoprotein (LDL), total cholesterol (TC) and triglycerides (TG), coronary artery disease, heart rate, rheumatoid arthritis, celiac disease, inflammatory bowel disease, ulcerative colitis, type 1 diabetes, type 2 diabetes, glucose levels, insulin levels, attention deficit hyperactivity disorder (ADHD), Alzheimer’s, schizophrenia, bipolar disorder, major depressive disorder, autism, motor neurone disease, age-related macular degeneration and osteoporosis. We also included reproductive traits such as maternal birth weight, age of menopause, maternal gestational weight gain and epithelial ovarian cancer [57].
Functional annotation
Applying locational data for ESR binding sites previously identified by Carrol et al. [77], we sought to identify any overlaps between differentially methylated DNAm probes and sentinel mSNPs for cis-mQTLs and ESR binding sites. Regulatory elements within which mQTL loci may act were annotated using data available from the Roadmap Epigenomics Mapping Consortium (REMC) and ENCODE [78, 79]. Due to the absence of chromatin state information for endometrium, we used chromatin state model based imputation data for 23 blood cell lines from 127 epigenomes in which 12 histone-modification marks were used to predict 25 chromatin states [80]. Functionally similar annotations were combined into 14 categories as suggested by Wu et al. [8]. Endometrial mQTLs also identified in blood were annotated to the 14 categories of functionally similar chromatin states. Using the method outlined in Wu et al. [8], we performed an enrichment analysis to test for enrichment of DNAm probes significantly associated with gene expression in the M2T analysis, in the 14 functional categories.
Pathway analysis
To identify pathways potentially affected by or regulating changes in methylation across the cycle, we performed a pathway analysis using the “GENE2FUNC” function on the FUMA GWAS web-based platform [81]. DNAm probe sites differentially methylated between phases of the menstrual cycle were annotated to the nearest TSS, and the resulting gene lists were used as input for the pathway analysis. The same was done for cis-mQTLs meeting Bonferroni genome-wide significance.
Additional files
Supplementary Notes. (PDF 65 kb)
Supplementary Figures. (PDF 4540 kb)
Supplementary Tables. (XLSX 4830 kb)
Acknowledgements
We thank the women who participated in the study, research nurses Ranita Charitra, Tracy Middleton and Irene Bell who recruited and consented all the endometrial biopsy patients at the Royal Women’s Hospital and the surgeons and anaesthetists who collected tissue and blood samples. We thank Associate Professor Joseph Powell for his guidance during the analysis of our omic datasets and sharing his knowledge in QTL analysis.
Funding
This work was supported by the National Health and Medical Research Council (NHMRC) under project grants GNT1026033, GNT1049472, GNT1046880, GNT1050208, GNT1105321, GNT1083405 and GNT1107258 and was supported in part by the NIH Eunice Kennedy Shriver National Institute of Child Health & Human Development R01HD089511. A.M and G.W.M are supported by NHMRC Fellowships (GNT1083656, GNT1078399). J.Y is supported by the Sylvia and Charles Viertel Charitable Foundation.
Availability of data and materials
The mQTL datasets generated during the current study are available at http://reproductivegenomics.com.au/shiny/mqtl_endo/ and http://reproductivegenomics.com.au/shiny/mqtl_blood/. Other data generated and/or analysed during this study are included in this article and its supplementary information files or are available on reasonable request.
Abbreviations
- ADHD
Attention deficit hyperactivity disorder
- BMI
Body mass index
- BSGS
Brisbane Systems Genetics Study
- DNAm
DNA methylation
- EP
Early proliferative
- eQTL
Expression quantitative trait loci
- ES
Early secretory
- ESR
Oestrogen receptor
- FDR
False discovery rate
- HDL
High-density lipoprotein
- HEIDI
Heterogeneity in dependent instruments
- HWE
Hardy-Weinberg equilibrium
- LBC
Lothian Birth Cohorts
- LD
Linkage disequilibrium
- LDL
Low-density lipoprotein
- LP
Late proliferative
- LS
Late secretory
- M
Menstrual
- MAF
Minor allele frequency
- MP
Mid proliferative
- mQTL
Methylation quantitative trait loci
- MS
Mid secretory
- P
Proliferative
- PC
Principal component
- SMR
Summary-data-based Mendelian randomisation
- TC
Total cholesterol
- TG
Triglycerides
- TSS
Transcription start site
Authors’ contributions
S.M, J.N.F, P.A.W.R and G.W.M designed the study with input from the other authors. S.M, R.R, J.E.G, S.J.H-C and J.N.F coordinated data collection and quality control of data with support, input and oversight from M.H, Z.Z, Y.W,T.Q, P.A.W.R, J.Y, A.M and G.W.M. Data analysis was performed by S.M, R.R, R.L and J.N.F which was interpreted by all authors. S.W.L built the web browser for summary results. S.M, R.L, J.N.F and G.W.M drafted the report with input from all other authors. The final manuscript has been critically revised and approved by all authors.
Ethics approval and consent to participate
The study was approved by the Royal Women’s Hospital Human Research Ethics Committee (Projects 11-24 and 16-43) and the University of Queensland. Informed consent was obtained from all participants.
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Sally Mortlock, Phone: +617 3346 2077, Email: s.mortlock@imb.uq.edu.au.
Restuadi Restuadi, Email: r.restuadi@uq.edu.au.
Rupert Levien, Email: rupert.levien@uqconnect.edu.au.
Jane E. Girling, Email: jane.girling@otago.ac.nz
Sarah J. Holdsworth-Carson, Email: scarson@unimelb.edu.au
Martin Healey, Email: kathandmutt@bigpond.com.
Zhihong Zhu, Email: z.zhu@imb.uq.edu.au.
Ting Qi, Email: t.qi@imb.uq.edu.au.
Yang Wu, Email: y.wu2@uq.edu.au.
Samuel W. Lukowski, Email: s.lukowski@imb.uq.edu.au
Peter A. W. Rogers, Email: parogers@unimelb.edu.au
Jian Yang, Email: jian.yang@imb.uq.edu.au.
Allan F. McRae, Email: a.mcrae@imb.uq.edu.au
Jenny N. Fung, Email: j.fung@imb.uq.edu.au
Grant W. Montgomery, Email: g.montgomery1@uq.edu.au
References
- 1.Roussos P, Mitchell Amanda C, Voloudakis G, Fullard John F, Pothula Venu M, Tsang J, et al. A role for noncoding variation in schizophrenia. Cell Rep. 2014;9(4):1417–1429. doi: 10.1016/j.celrep.2014.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Farh KK-H, Marson A, Zhu J, Kleinewietfeld M, Housley WJ, Beik S, et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature. 2014;518:337. doi: 10.1038/nature13835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002;16(1):6–21. doi: 10.1101/gad.947102. [DOI] [PubMed] [Google Scholar]
- 4.Robertson KD, Wolffe AP. DNA methylation in health and disease. Nat Rev Genet. 2000;1:11. doi: 10.1038/35049533. [DOI] [PubMed] [Google Scholar]
- 5.Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet. 2003;33:245. doi: 10.1038/ng1089. [DOI] [PubMed] [Google Scholar]
- 6.Reik W. Stability and flexibility of epigenetic gene regulation in mammalian development. Nature. 2007;447:425. doi: 10.1038/nature05918. [DOI] [PubMed] [Google Scholar]
- 7.Ciuculete DM, Boström AE, Voisin S, Philipps H, Titova OE, Bandstein M, et al. A methylome-wide mQTL analysis reveals associations of methylation sites with GAD1 and HDAC3 SNPs and a general psychiatric risk score. Transl Psychiatry. 2017;7:e1002. doi: 10.1038/tp.2016.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu Y, Zeng J, Zhang F, Zhu Z, Qi T, Zheng Z, et al. Integrative analysis of omics summary data reveals putative mechanisms underlying complex traits. Nat Commun. 2018;9(1):918. doi: 10.1038/s41467-018-03371-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Joo JE, Dowty JG, Milne RL, Wong EM, Dugué P-A, English D, et al. Heritable DNA methylation marks associated with susceptibility to breast cancer. Nat Commun. 2018;9(1):867. doi: 10.1038/s41467-018-03058-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Freytag V, Vukojevic V, Wagner-Thelen H, Milnik A, Vogler C, Leber M, et al. Genetic estimators of DNA methylation provide insights into the molecular basis of polygenic traits. Transl Psychiatry. 2018;8(1):31. doi: 10.1038/s41398-017-0070-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lokk K, Modhukur V, Rajashekar B, Märtens K, Mägi R, Kolde R, et al. DNA methylome profiling of human tissues identifies global and tissue-specific methylation patterns. Genome Biology. 2014;15(4):r54–r5r. doi: 10.1186/gb-2014-15-4-r54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Slieker RC, Bos SD, Goeman JJ, Bovée JVMG, Talens RP, van der Breggen R, et al. Identification and systematic annotation of tissue-specific differentially methylated regions using the Illumina 450k array. Epigenetics Chromatin. 2013;6(1):26. doi: 10.1186/1756-8935-6-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Muangsub T, Samsuwan J, Tongyoo P, Kitkumthorn N, Mutirangura A. Analysis of methylation microarray for tissue specific detection. Gene. 2014;553(1):31–41. doi: 10.1016/j.gene.2014.09.060. [DOI] [PubMed] [Google Scholar]
- 14.Evans J, Salamonsen LA, Winship A, Menkhorst E, Nie G, Gargett CE, et al. Fertile ground: human endometrial programming and lessons in health and disease. Nat Rev Endocrinol. 2016;12:654. doi: 10.1038/nrendo.2016.116. [DOI] [PubMed] [Google Scholar]
- 15.Ruiz-Alonso M, Blesa D, Simón C. The genomics of the human endometrium. Biochim Biophys Acta (BBA) - Mol Basis Dis. 2012;1822(12):1931–1942. doi: 10.1016/j.bbadis.2012.05.004. [DOI] [PubMed] [Google Scholar]
- 16.Gargett CE, Masuda H. Adult stem cells in the endometrium. MHR: Basic Sci Reprod Med. 2010;16(11):818–834. doi: 10.1093/molehr/gaq061. [DOI] [PubMed] [Google Scholar]
- 17.Fung JN, Girling JE, Lukowski SW, Sapkota Y, Wallace L, Holdsworth-Carson SJ, et al. The genetic regulation of transcription in human endometrial tissue. Hum Reprod. 2017;32(4):893–904. doi: 10.1093/humrep/dex006. [DOI] [PubMed] [Google Scholar]
- 18.Ponnampalam AP, Weston GC, Trajstman AC, Susil B, Rogers PAW. Molecular classification of human endometrial cycle stages by transcriptional profiling. MHR: Basic Sci Reprod Med. 2004;10(12):879–893. doi: 10.1093/molehr/gah121. [DOI] [PubMed] [Google Scholar]
- 19.Houshdaran S, Zelenko Z, Irwin JC, Giudice LC. Human endometrial DNA methylome is cycle-dependent and is associated with gene expression regulation. Mol Endocrinol. 2014;28(7):1118–1135. doi: 10.1210/me.2013-1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kukushkina V, Modhukur V, Suhorutšenko M, Peters M, Mägi R, Rahmioglu N, et al. DNA methylation changes in endometrium and correlation with gene expression during the transition from pre-receptive to receptive phase. Sci Rep. 2017;7:3916. doi: 10.1038/s41598-017-03682-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rahmioglu N, Drong AW, Lockstone H, Tapmeier T, Hellner K, Saare M, et al. Variability of genome-wide DNA methylation and mRNA expression profiles in reproductive and endocrine disease related tissues. Epigenetics. 2017;12(10):897–908. doi: 10.1080/15592294.2017.1367475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Saare M, Modhukur V, Suhorutshenko M, Rajashekar B, Rekker K, Sõritsa D, et al. The influence of menstrual cycle and endometriosis on endometrial methylome. Clin Epigenetics. 2016;8:2. doi: 10.1186/s13148-015-0168-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McRae AF, Marioni RE, Shah S, Yang J, Powell JE, Harris SE, et al. Identification of 55,000 Replicated DNA Methylation QTL. Sci Rep. 2018;8(1):17605. [DOI] [PMC free article] [PubMed]
- 24.Qi T, Wu Y, Zeng J, Zhang F, Xue A, Jiang L, et al. Identifying gene targets for brain-related traits using transcriptomic and methylomic data from blood. Nat Commun. 2018;9(1):2282. [DOI] [PMC free article] [PubMed]
- 25.Zhu Z, Zhang F, Hu H, Bakshi A, Robinson MR, Powell JE, et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat Genet. 2016;48(5):481–487. doi: 10.1038/ng.3538. [DOI] [PubMed] [Google Scholar]
- 26.Sapkota Y, Steinthorsdottir V, Morris AP, Fassbender A, Rahmioglu N, De Vivo I, et al. Meta-analysis identifies five novel loci associated with endometriosis highlighting key genes involved in hormone metabolism. Nat Commun. 2017;8:15539. [DOI] [PMC free article] [PubMed]
- 27.Fung JN, Mortlock S, Girling JE, Holdsworth-Carson SJ, Teh WT, Zhu Z, et al. Genetic regulation of disease risk and endometrial gene expression highlights potential target genes for endometriosis and polycystic ovarian syndrome. Sci Rep. 2018;8(1):11424. doi: 10.1038/s41598-018-29462-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hawkins SM, Matzuk MM. Menstrual cycle: basic biology. Ann N Y Acad Sci. 2008;1135:10–18. doi: 10.1196/annals.1429.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ferenczy A, Bertrand G, Gelfand MM. Proliferation kinetics of human endometrium during the normal menstrual cycle. Am J Obstet Gynecol. 1979;133(8):859–867. doi: 10.1016/0002-9378(79)90302-8. [DOI] [PubMed] [Google Scholar]
- 30.Slayden OD, Brenner RM. Hormonal regulation and localization of estrogen, progestin and androgen receptors in the endometrium of nonhuman primates: effects of progesterone receptor antagonists. Arch Histol Cytol. 2004;67(5):393–409. doi: 10.1679/aohc.67.393. [DOI] [PubMed] [Google Scholar]
- 31.Matsuzaki S, Fukaya T, Suzuki T, Murakami T, Sasano H, Yajima A. Oestrogen receptor α and β mRNA expression in human endometrium throughout the menstrual cycle. MHR: Basic Sci Reprod Med. 1999;5(6):559–564. doi: 10.1093/molehr/5.6.559. [DOI] [PubMed] [Google Scholar]
- 32.Prossnitz ER, Arterburn JB, Sklar LA. GPR30: a G protein-coupled receptor for estrogen. Mol Cell Endocrinol. 2007;265–266:138–142. doi: 10.1016/j.mce.2006.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Prossnitz ER, Arterburn JB, Smith HO, Oprea TI, Sklar LA, Hathaway HJ. Estrogen signaling through the transmembrane G protein–coupled receptor GPR30. Annu Rev Physiol. 2008;70(1):165–190. doi: 10.1146/annurev.physiol.70.113006.100518. [DOI] [PubMed] [Google Scholar]
- 34.Ortega E, Manso JA, Buey RM, Carballido AM, Carabias A, Sonnenberg A, et al. The structure of the plakin domain of plectin reveals an extended rod-like shape. J Biol Chem. 2016;291(36):18643–18662. doi: 10.1074/jbc.M116.732909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Talbi S, Hamilton AE, Vo KC, Tulac S, Overgaard MT, Dosiou C, et al. Molecular phenotyping of human endometrium distinguishes menstrual cycle phases and underlying biological processes in normo-ovulatory women. Endocrinology. 2006;147(3):1097–1121. doi: 10.1210/en.2005-1076. [DOI] [PubMed] [Google Scholar]
- 36.Tapia-Pizarro A, Figueroa P, Brito J, Marín JC, Munroe DJ, Croxatto HB. Endometrial gene expression reveals compromised progesterone signaling in women refractory to embryo implantation. Reprod Biol Endocrinol. 2014;12(1):92. doi: 10.1186/1477-7827-12-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kao LC, Germeyer A, Tulac S, Lobo S, Yang JP, Taylor RN, et al. Expression profiling of endometrium from women with endometriosis reveals candidate genes for disease-based implantation failure and infertility. Endocrinology. 2003;144(7):2870–2881. doi: 10.1210/en.2003-0043. [DOI] [PubMed] [Google Scholar]
- 38.Mohammed H, D’Santos C, Serandour Aurelien A, Ali HR, Brown Gordon D, Atkins A, et al. Endogenous purification reveals GREB1 as a key estrogen receptor regulatory factor. Cell Rep. 2013;3(2):342–349. doi: 10.1016/j.celrep.2013.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ghosh MG, Thompson DA, Weigel RJ. PDZK1 and GREB1 are estrogen-regulated genes expressed in hormone-responsive breast cancer. Cancer Res. 2000;60(22):6367. [PubMed] [Google Scholar]
- 40.Hodgkinson K, Forrest LA, Vuong N, Garson K, Djordjevic B, Vanderhyden BC. GREB1 is an estrogen receptor-regulated tumour promoter that is frequently expressed in ovarian cancer. Oncogene. 2018;37(44):5873–86. [DOI] [PMC free article] [PubMed]
- 41.Rae JM, Johnson MD, Scheys JO, Cordero KE, Larios JM, Lippman ME. GREB1 is a critical regulator of hormone dependent breast cancer growth. Breast Cancer Res Treat. 2005;92(2):141–149. doi: 10.1007/s10549-005-1483-4. [DOI] [PubMed] [Google Scholar]
- 42.Rae JM, Johnson MD, Cordero KE, Scheys JO, Larios JM, Gottardis MM, et al. GREB1 is a novel androgen-regulated gene required for prostate cancer growth. Prostate. 2006;66(8):886–894. doi: 10.1002/pros.20403. [DOI] [PubMed] [Google Scholar]
- 43.Fung JN, Holdsworth-Carson SJ, Sapkota Y, Zhao ZZ, Jones L, Girling JE, et al. Functional evaluation of genetic variants associated with endometriosis near GREB1. Hum Reprod. 2015;30(5):1263–1275. doi: 10.1093/humrep/dev051. [DOI] [PubMed] [Google Scholar]
- 44.Lev Maor G, Yearim A, Ast G. The alternative role of DNA methylation in splicing regulation. Trends Genet. 2015;31(5):274–280. doi: 10.1016/j.tig.2015.03.002. [DOI] [PubMed] [Google Scholar]
- 45.Nachtergaele S, He C. The emerging biology of RNA post-transcriptional modifications. RNA Biol. 2017;14(2):156–163. doi: 10.1080/15476286.2016.1267096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tan S, Ding K, Li R, Zhang W, Li G, Kong X, et al. Identification of miR-26 as a key mediator of estrogen stimulated cell proliferation by targeting CHD1, GREB1 and KPNA2. Breast Cancer Res. 2014;16(2):R40. doi: 10.1186/bcr3644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wu L, Candille SI, Choi Y, Xie D, Li-Pook-Than J, Tang H, et al. Variation and genetic control of protein abundance in humans. Nature. 2013;499(7456):79–82. doi: 10.1038/nature12223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hause Ronald J, Stark Amy L, Antao Nirav N, Gorsic Lidija K, Chung Sophie H, Brown Christopher D, et al. Identification and validation of genetic variants that influence transcription factor and cell signaling protein levels. Am J Hum Genet. 2014;95(2):194–208. doi: 10.1016/j.ajhg.2014.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wan Y, Qu K, Zhang QC, Flynn RA, Manor O, Ouyang Z, et al. Landscape and variation of RNA secondary structure across the human transcriptome. Nature. 2014;505(7485):706–709. doi: 10.1038/nature12946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mulin Jun L, Yan B, Chung Sham P, Wang J. Exploring the function of genetic variants in the non-coding genomic regions: approaches for identifying human regulatory variants affecting gene expression 2014. [DOI] [PubMed]
- 51.Meyer Kate D, Saletore Y, Zumbo P, Elemento O, Mason Christopher E, Jaffrey SR. Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell. 2012;149(7):1635–1646. doi: 10.1016/j.cell.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.e GP Enhancing GTEx by bridging the gaps between genotype, gene expression, and disease. Nat Genet. 2017;49:1664. doi: 10.1038/ng.3969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sugino N, Kashida S, Karube-Harada A, Takiguchi S, Kato H. Expression of vascular endothelial growth factor (VEGF) and its receptors in human endometrium throughout the menstrual cycle and in early pregnancy. Reproduction. 2002;123(3):379–387. doi: 10.1530/rep.0.1230379. [DOI] [PubMed] [Google Scholar]
- 54.Meduri G, Bausero P, Perrot-Applanat M. Expression of vascular endothelial growth factor receptors in the human endometrium: modulation during the menstrual cycle1. Biol Reprod. 2000;62(2):439–447. doi: 10.1095/biolreprod62.2.439. [DOI] [PubMed] [Google Scholar]
- 55.Girling JE, Rogers PAW. Regulation of endometrial vascular remodelling: role of the vascular endothelial growth factor family and the angiopoietin–TIE signalling system. Reproduction. 2009;138(6):883–893. doi: 10.1530/REP-09-0147. [DOI] [PubMed] [Google Scholar]
- 56.Hu Y, Scully R, Sobhian B, Xie A, Shestakova E, Livingston DM. RAP80-directed tuning of BRCA1 homologous recombination function at ionizing radiation-induced nuclear foci. Genes Dev. 2011;25(7):685–700. doi: 10.1101/gad.2011011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Day FR, Ruth KS, Thompson DJ, Lunetta KL, Pervjakova N, Chasman DI, et al. Large-scale genomic analyses link reproductive aging to hypothalamic signaling, breast cancer susceptibility and BRCA1-mediated DNA repair. Nat Genet. 2015;47:1294. doi: 10.1038/ng.3412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhou J, Stein P, Leu NA, Chmátal L, Xue J, Ma J, et al. Accelerated reproductive aging in females lacking a novel centromere protein SYCP2L. Hum Mol Genet. 2015;24(22):6505–6514. doi: 10.1093/hmg/ddv359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Goode EL, Chenevix-Trench G, Song H, Ramus SJ, Notaridou M, Lawrenson K, et al. A genome-wide association study identifies susceptibility loci for ovarian cancer at 2q31 and 8q24. Nat Genet. 2010;42:874. doi: 10.1038/ng.668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Whalen S, Truty RM, Pollard KS. Enhancer–promoter interactions are encoded by complex genomic signatures on looping chromatin. Nat Genet. 2016;48:488. doi: 10.1038/ng.3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Krijger PHL, de Laat W. Regulation of disease-associated gene expression in the 3D genome. Nat Rev Mol Cell Biol. 2016;17:771. doi: 10.1038/nrm.2016.138. [DOI] [PubMed] [Google Scholar]
- 62.Zerbino DR, Wilder SP, Johnson N, Juettemann T, Flicek PR. The Ensembl regulatory build. Genome Biol. 2015;16(1):56. doi: 10.1186/s13059-015-0621-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Holwerda SJB, de Laat W. CTCF: the protein, the binding partners, the binding sites and their chromatin loops. Philos Trans R Soc B. 2013;368(1620):20120369. doi: 10.1098/rstb.2012.0369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Handoko L, Xu H, Li G, Ngan CY, Chew E, Schnapp M, et al. CTCF-mediated functional chromatin interactome in pluripotent cells. Nat Genet. 2011;43:630. doi: 10.1038/ng.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Maurano MT, Humbert R, Rynes E, Thurman RE, Haugen E, Wang H, et al. Systematic localization of common disease-associated variation in regulatory DNA. Science. 2012;337(6099):1190. doi: 10.1126/science.1222794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schaub MA, Boyle AP, Kundaje A, Batzoglou S, Snyder M. Linking disease associations with regulatory information in the human genome. Genome Res. 2012;22(9):1748–1759. doi: 10.1101/gr.136127.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Welter D, MacArthur J, Morales J, Burdett T, Hall P, Junkins H, et al. The NHGRI GWAS catalog, a curated resource of SNP-trait associations. Nucleic Acids Res. 2014;42(D1):D1001–D10D6. doi: 10.1093/nar/gkt1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chen MM, O'Mara TA, Thompson DJ, Painter JN, Attia J, Black A, et al. GWAS meta-analysis of 16 852 women identifies new susceptibility locus for endometrial cancer. Hum Mol Genet. 2016;25(12):2612–2620. doi: 10.1093/hmg/ddw092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Saffari A, Silver MJ, Zavattari P, Moi L, Columbano A, Meaburn EL, et al. Estimation of a significance threshold for epigenome-wide association studies. Genet Epidemiol. 2018;42(1):20–33. doi: 10.1002/gepi.22086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988;16(3):1215. doi: 10.1093/nar/16.3.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bibikova M, Lin Z, Zhou L, Chudin E, Garcia EW, Wu B, et al. High-throughput DNA methylation profiling using universal bead arrays. Genome Res. 2006;16(3):383–393. doi: 10.1101/gr.4410706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ. Second-generation PLINK: rising to the challenge of larger and richer datasets. GigaScience. 2015;4(1):7. doi: 10.1186/s13742-015-0047-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Purcell S, Chang C. PLINK V1.9 Available from: www.cog-genomics.org/plink/1.9.
- 74.Das S, Forer L, Schonherr S, Sidore C, Locke AE, Kwong A, et al. Next-generation genotype imputation service and methods. Nat Genet. 2016;48(10):1284–1287. doi: 10.1038/ng.3656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Relton C, Davey Smith G, Hemani G, Min JL, Suderman M. Meffil: efficient normalization and analysis of very large DNA methylation datasets. Bioinformatics. 2018;34(23):3983–9. [DOI] [PMC free article] [PubMed]
- 76.Price EM, Cotton AM, Lam LL, Farré P, Emberly E, Brown CJ, et al. Additional annotation enhances potential for biologically-relevant analysis of the Illumina Infinium HumanMethylation450 BeadChip array. Epigenetics Chromatin. 2013;6(1):4. doi: 10.1186/1756-8935-6-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Carroll JS, Meyer CA, Song J, Li W, Geistlinger TR, Eeckhoute J, et al. Genome-wide analysis of estrogen receptor binding sites. Nat Genet. 2006;38(11):1289–1297. doi: 10.1038/ng1901. [DOI] [PubMed] [Google Scholar]
- 78.The EPC An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bernstein BE, Stamatoyannopoulos JA, Costello JF, Ren B, Milosavljevic A, Meissner A, et al. The NIH Roadmap Epigenomics mapping consortium. Nat Biotechnol. 2010;28(10):1045–1048. doi: 10.1038/nbt1010-1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Roadmap Epigenomics C, Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, et al. Integrative analysis of 111 reference human epigenomes. Nature. 2015;518:317. doi: 10.1038/nature14248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Watanabe K, Taskesen E, van Bochoven A, Posthuma D. Functional mapping and annotation of genetic associations with FUMA. Nat Commun. 2017;8(1):1826. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Notes. (PDF 65 kb)
Supplementary Figures. (PDF 4540 kb)
Supplementary Tables. (XLSX 4830 kb)
Data Availability Statement
The mQTL datasets generated during the current study are available at http://reproductivegenomics.com.au/shiny/mqtl_endo/ and http://reproductivegenomics.com.au/shiny/mqtl_blood/. Other data generated and/or analysed during this study are included in this article and its supplementary information files or are available on reasonable request.