

Abstract
High mobility group box 1 (HMGB1) is a non-histone chromosomal protein, which can be secreted through a variety of pathways and bind to pattern recognition receptors to release pro-inflammatory cytokines. Previous studies have suggested that HMGB1 is upregulated in numerous inflammatory diseases and that it could be a biomarker for such diseases. However, these studies used immunoassay-based methods to analyze serum HMGB1. Autoantibodies to HMGB1 in serum are found in healthy control subjects as well as in patients with different diseases. HMGB1 also binds to haptoglobin, a highly abundant plasma protein. This means that antibodies used in immunoassays must compete with binding of HMGB1 to endogenous serum HMGB1 autoantibodies and haptoglobin. To overcome these potential problems, we developed and validated a specific and sensitive assay based on stable isotope dilution and immunopurification to quantify HMGB1 in plasma and serum using two-dimensional nano-ultra-high-performance liquid chromatography parallel reaction monitoring/high-resolution mass spectrometry. Using this assay, we found that serum HMGB1 in 24 healthy control subjects (6.0 ± 2.1 ng/mL) was above the mean concentration reported for 18 different diseases (5.4 ± 2.8 ng/mL) where the analyses were conducted with immunoassay methodology. In light of our finding, the role of HMGB1 in these diseases will have to be re-evaluated. The concentration of HMGB1 in citrated and EDTA-treated plasma from the same healthy control subjects was below the limit of detection of our assay (1 ng/mL), confirming that HMGB1 in serum arises when blood is allowed to clot. This means that future studies on the role of HMGB1 in vivo should be conducted on plasma rather than serum.
Amyloid β-peptides1 and high mobility group box 1 (HMGB1),2 a non-histone chromosomal protein, are the two most intensively studied endogenous cellular danger signals known as danger-associated molecular pattern (DAMP) molecules (Figure 1).3,4 DAMP molecules, together with pathogen-associated molecular patterns, alert the innate immune system by activating signal transduction pathways through binding to pattern recognition receptors (PRRs). PRRs include the receptor for advanced glycation end products (RAGE), Toll-like receptors (TLRs), chemokine (C-X-C motif) receptor 3 (CXCR3), and T-cell immunoglobulin mucin 3 (TIM3) (Figure 1).5−7 Binding to PRRs induces pro-inflammatory cascades, which trigger the release of cytokines.8−11 PRRs are expressed by cells of the innate immune system such as macrophages, leukocytes, and dendritic cells (Figure 1).4,12,13 They are also expressed on the surface of vascular cells, fibroblasts, and epithelial cells.14
Figure 1.
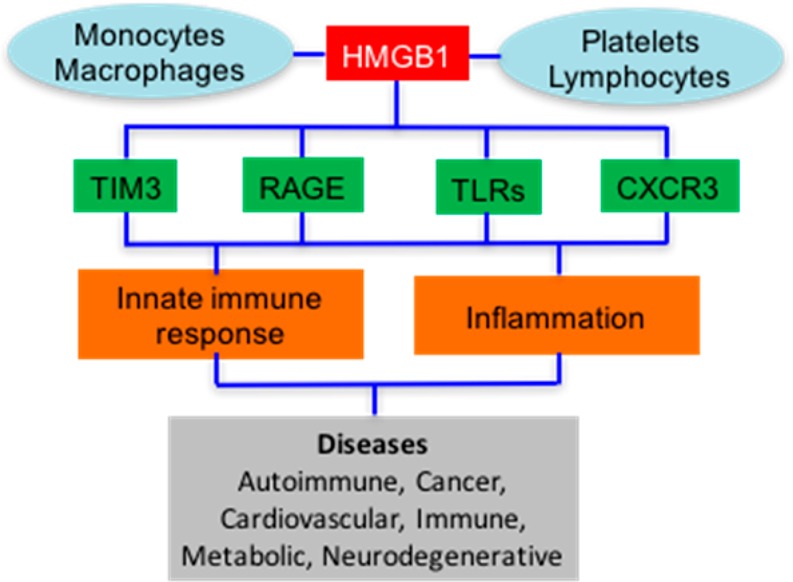
Release of HMGB1 by blood cells, its activation of cell surface PRRs, and its involvement in immunological and inflammatory diseases.
In addition to its role in danger signaling, nuclear HMGB1 binds to the minor groove of nuclear DNA, bending the double helix and altering chromatin structure to recruit transcription-regulating factors;15,16 whereas in the cytosol HMGB1 binds to beclin-2 and induces autophagy.7,17,18 HMGB1 is secreted from cells in four different ways: passive release from necrotic cells;19 active secretion by inflammatory cells, such as monocytes and macrophages;20−22 secretion from natural killer cells to promote dendritic cell maturation during different immune responses;23 and a less well-studied pathway involving secretion from platelets.24
It has been suggested that the involvement of HMGB1 in chronic inflammation serves to promote immunosuppression,14 and so it could play an important role in numerous and diverse diseases (Figure 1). Substantial evidence for this role comes from the comparison of serum HMGB1 levels in diseased individuals with serum HMGB1 levels in healthy control subjects that were determined primarily by enzyme-linked immunosorbent assay (ELISA)-based methodology (Table 1).25−43 Unfortunately, analysis of serum HMGB1 by ELISA poses some fundamental challenges that call into question the results of many of the analyses reported to date. First, autoantibodies to HMGB1 in serum have been reported in healthy control subjects29,44 as well as in serum from patients with different diseases.6,29,44−46 In fact, increased serum HMGB1 autoantibody levels have even been proposed as a potential disease biomarker for systemic lupus erthrythromatosus.44 Second, HMGB1 binds to haptoglobin, a highly abundant plasma protein that is present at levels of 30–200 mg/dL.47 This means that antibodies used in ELISAs must compete with binding to haptoglobin as well as fluctuating levels of endogenous serum HMGB1 autoantibodies, which could result in finding lower HMGB1 concentrations than are actually present. Third, the amino acid sequences of HMGB1 and HMGB2 are 74% similar, so that there is a high likelihood of cross-reactivity between HMGB1 and HMGB2 in ELISA assays.48 Fourth, varying levels of post-translational modification such as acetylation,49,50 methylation,51,52 and glycosylation53 could confound the antibody–antigen interaction that ELISAs rely upon for accurate quantification. As a further complication, anti-HMGB1 autoantibodies from serum do not recognize neutrophil-derived HMGB1, although they do recognize HMGB1 derived from lymphocytes.54
Table 1. Reported Concentrations of Serum HMGB1.
| study | disease type | methoda | control mean (ng/mL) | case mean (ng/mL) | ref |
|---|---|---|---|---|---|
| Autoimmune/Immune | |||||
| 1 | alcoholic liver disease | LC/MS/ELISA | 1.1 | 18 | (25) |
| 2 | asbestos exposure | LC/MS/ELISA | 1.4 | 10 | (26) |
| 3 | drug-induced liver injury | ELISA | 1.1 | 10 | (27) |
| 4 | multiple sclerosis | ELISA | 0.5 | 1.9 | (28) |
| 5 | systemic lupus erythromatosus | ELISA | 2.9 | 6.2 | (29) |
| 6 | sepsis | ELISA | 0.6 | 3.6 | (30) |
| 7 | type 1 diabetes | ELISA | 1.6 | 2.2 | (31) |
| Cancer | |||||
| 8 | breast | ELISA | 2.0 | 4.5 | (32) |
| 9 | pancreatic | ELISA | 1.2 | 2.0 | (33) |
| 10 | laryngeal | ELISA | 3.2 | 4.8 | (34) |
| 11 | mesothelioma (pleural) | ELISA | 5.4 | 27 | (35) |
| 12 | non-small-cell lung cancer | ELISA | 3.0 | 7.1 | (36) |
| Neurodegeneration | |||||
| 13 | Alzheimer’s disease | ELISA | 3.1 | 4.2 | (37) |
| 14 | epilepsy | LC/MS/ELISA | 1.1 | 8.7 | (38) |
| 15 | Parkinson’s disease | ELISA | 1.6 | 2.6 | (39) |
| Cardiovascular/Metabolic Diseases | |||||
| 16 | atrial fibrillation | ELISA | 3.2 | 9.1 | (40) |
| 17 | coronary artery disease | ELISA | 1.7 | 3.3 | (41) |
| 18 | heart failure | ELISA | 2.7 | 7.6 | (42) |
| 19 | stable angina | ELISA | 1.5 | 5.2 | (43) |
| 20 | type 2 diabetes | ELISA | 1.7 | 4.4 | (41) |
| meanb | 1.9 | 5.4 | |||
ELISA = enzyme-linked immunosorbent assay.
Mean values do not include alcoholic liver disease or peritoneal mesothelioma.
HMGB1 is a very unusual protein, which contains 30 consecutive acidic aspartate and glutamate residues at its C-terminus (amino acids 186–215) together with 43 basic lysine residues spanning amino acids 3–95 (Figure 2). Some 22 of the lysine residues have been reported as being acetylated,49,50,55−57 and two have been reported as being methylated51,52 (Figure 2). In view of the foregoing, there is a compelling need to develop a more specific assay for the quantification of HMGB1 in serum and plasma. This would facilitate more rigorous studies of the role that HMGB1 plays in the inflammation- and immunologically mediated diseases shown in Figure 1. The assay would require an internal standard that fully exchanges with endogenous HMGB1 bound to any plasma or serum autoantibodies to correct for any losses that might occur through noncovalent binding to the autoantibodies. The assay would also need to differentiate HMGB1 from HMGB2, as well as distinguishing HMGB1 from the vast array of potentially acetylated forms (estimated at 100 000).56
Figure 2.

Amino acid sequence of HMGB1, showing the acidic tail in green (residues 186–215) and the 43 basic lysine residues in red. The 22 known sites of acetylation49,50,55−57 are shown in blue, and the two known sites of methylation are shown in brown.51,52 A box = residues 9–79; B box = residues 88–162; nuclear localization sequence 1 (NLS-1) = residues 28–44; NLS-2 = residues 179–185; the oxidized form has a disulfide between C23 and C45 (methionine is amino acid 1).2
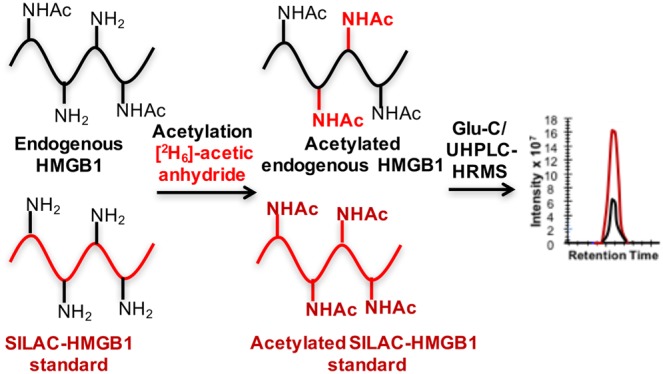
We report a validated, sensitive, and specific assay for plasma and serum HMGB1 using two-dimensional nano-ultra-high-performance liquid chromatography parallel reaction monitoring/high-resolution mass spectrometry (2D-nano-UHPLC-PRM/HRMS). A [13C15N]-HMGB1 internal standard, prepared through stable isotope labeling by amino acids in cell culture (SILAC), was used in combination with initial immunopurification (IP) of the plasma or serum by use of magnetic beads noncovalently bound to an HMGB1 polyclonal antibody (pAb). Further purification was performed by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) coupled with in-gel acetylation with [2H6]acetic anhydride to convert all acetylated and nonacetylated HMGB1 proteins into a single molecular form. In-gel Glu-C digestion was used to generate five peptides suitable for UHPLC/MS analysis. The peptides contained 17 lysine residues, including the eight (residues 28, 29, 30, 180, 182, 183, 184, and 185) that are thought to undergo acetylation to prevent the nuclear localization and facilitate secretion of HMGB1 (Figure 2, Table S1).49,50 By performing simultaneous PRM/full-scan HRMS analysis, it was also possible to distinguish the Glu-C-derived endogenous acetylated or methylated peptides from the corresponding [2H3]-acetylated peptides by their difference in mass.
Experimental Section
Chemicals and Materials
Reagents and solvents were LC/MS-grade quality unless otherwise noted. [13C615N2]lysine, [13C915N1]tyrosine, and [2H6]acetic anhydride were obtained from Cambridge Isotope Laboratories (Andover, MA). Ammonium hydroxide (Optima) was from Fisher Scientific (Pittsburgh, PA). Anti-HMGB1 rabbit pAb (H9539), endoproteinase Glu-C from Staphylococcus aureus V8, ethylenediaminetetraacetoc acid (EDTA)-free protease inhibitor cocktail, and d,l-dithiothreitol (DTT) were purchased from MilliporeSigma (Billerica, MA). LC-grade water and acetonitrile were from Burdick and Jackson (Muskegon, MI). Pierce protein A/G magnetic beads, NuPAGE 12% Bis-Tris protein gels, and colloidal Coomassie blue staining kit were obtained from ThermoFisher Scientific (Grand Island, NY). Tobacco etch virus (TEV) nuclear-inclusion-A endopeptidase (ProTEV Plus) was purchased from Promega (Madison, WI). HMGB1 antibodies used in the IP were obtained from different vendors including Enzo (Farmingdale, NY), Lifespan Biosciences (Seattle, WA), Proteintech (Rosemont, IL), Abcam (Cambridge, MA), and MilliporeSigma (Billerica, MA). Dulbecco’s phosphate-buffered saline (DPBS) was obtained from Corning (Corning, NY). Glutathione (GSH) Sepharose 4B resins were purchased from GE Healthcare Life Sciences (Pittsburgh, PA). HEK293 cells were from American Type Culture Collection (ATCC, Manassas, VA). Dulbecco’s modified Eagle’s medium (DMEM)/F12/SILAC medium was purchased from Athena Enzyme Systems (Baltimore, MD). Western Lightning ECL Pro was from PerkinElmer (Waltham, MA).
Clinical Samples
Blood samples were obtained from healthy human volunteers (12 males, 12 females, average age 50.0 years). They are all enrolled in an ongoing mesothelioma biomarker study at the University of Pennsylvania (IRB 800924).
Serum Preparation
Venous blood was drawn into 8.5 mL red-cap Vacutainer tubes, which were kept at room temperature for 15–30 min to allow the blood to clot. The tubes were spun at 1500g for 5 min and the upper clear layer was carefully transferred to two clean Eppendorf tubes, which were immediately frozen at −80 °C after preparation until analysis.
Plasma Preparation
Venous blood from the same individuals who provided serum samples was transferred to 8.5 mL sodium EDTA-coated or sodium citrate-coated Vacutainer tubes. Blood (4 mL) was transferred to 15 mL polypropylene tubes and spun at 200g for 13 min at room temperature with no brakes. The upper clear plasma was carefully transferred to two clean 1.5 mL Eppendorf tubes, which were spun at 800g for 5 min. The supernatant was carefully transferred to another two clean 1.5 mL Eppendorf tubes that were immediately frozen at −80 °C until analysis.
Expression and Purification of Unlabeled and SILAC-Labeled HMGB1
The coding sequence of human HMGB1 was amplified by polymerase chain reaction (PCR) from the HMGB1 cDNA plasmid. The amplified HMGB1 fragment was cloned into a pRK5 plasmid and linked to the glutathione S-transferase (GST) tag. After the construct was confirmed by DNA sequencing, the plasmid was expanded in Escherichia coli. The GST–HMGB1 pRK5 plasmid was then transfected into human kidney HEK293 cells (ATCC CRL-1573) by the use of lipofectamine 2000 transfection reagent (Invitrogen) following the manufacturer’s instructions. SILAC-HMGB1 standard was expressed in transfected HEK293 cells that were cultured in DMEM/F12/SILAC medium containing 0.5 mM [13C615N2]lysine and 0.2 mM [13C915N1]tyrosine for at least three passages. Transfected cells were harvested in NP-40 lysis buffer (150 mM NaCl, 50 mM Tris-HCl, pH 7.5, 1 mM EDTA, 0.5% Triton X-100, 0.5% NP-40, and 1 mM DTT) containing protease inhibitor cocktail after 24 h transfection. Cells were lysed by sonication on ice for 20–30 pulses and the lysate was spun at 17200g for 15 min to remove any cell debris. The supernatant was removed from the pellet and incubated with GSH Sepharose beads at 4 °C overnight. After removal of the supernatant, the beads were washed with the following solutions sequentially: IP lysis buffer × 3, 0.5 M KCl in IP lysis buffer × 1, 1 M KCl in IP lysis buffer × 1, and DPBS × 2. TEV enzyme was added to the beads, together with TEV buffer and 1 mM DTT, to conduct the cleavage at 4 °C overnight. Released HMGB1 in the supernatant was collected and its concentration was determined by measuring the absorption at 280 nm and using the extinction coefficient (λmax) = 21 430 M–1·cm–1 (predicted by the amino acid sequence).58 The concentration of the unlabeled standard was verified by comparing HMGB1 bands with bands from known amounts of BSA in a Coomassie blue-stained SDS–polyacrylamide electrophoretic gel and by amino acid analysis. Concentration of the SILAC-labeled HMGB1 internal standard was determined by comparing it with unlabeled HMGB standard bands and bovine serum albumin (BSA) bands in a Coomassie blue-stained SDS–polyacrylamide electrophoretic gel. The acetylated SILAC-HMGB1 standard was prepared by treating SILAC-HMGB1 with excess acetic anhydride in 100 mM aqueous NH4HCO3 at r.t. for 1 h.
Western Blot and Gel Staining
HMGB1 was detected by an HMGB1 pAb (ab18256, Abcam) and anti-rabbit horseradish peroxidase (HRP) (Santa Cruz Biotechnology, Dallas, TX). Western blots were developed by use of electrochemical luminescence (ECL) reagents. Gels were stained with colloidal Coomassie blue staining kit.
Immunopurification
Protein A/G magnetic beads were washed twice with DPBS before use. Anti-HMGB1 rabbit pAb (100 μg) or antiacetyl-lysine pAb (100 μg) was incubated with 10 mg of protein A/G beads at 4 °C overnight. The pAb solution was removed and the beads were washed with 1 mL of DPBS for 3 times before finally being resuspended in 1 mL of DPBS to make the concentration of beads 10 mg/mL. This suspension was aliquoted into 20 clean protein LoBind tubes with each containing 0.5 mg of magnetic beads. The DPBS was removed and 250 μL of IP lysis buffer was added to each tube. The same amount of SILAC-HMGB1 standard (13 ng) was added into each sample (100 μL of serum or plasma) and incubated at room temperature for 15 min. Each sample with SILAC-HMGB1 standard alone or with the acetylated SILAC-HMGB1 standard was then added into the suspension containing 0.5 mg of magnetic beads and 250 μL of IP lysis buffer to carry out IP at 4 °C overnight.
In-Gel Acetylation with [2H6]Acetic Anhydride and Glu-C Digestion
After incubation with 0.5 mg of HMGB1 protein/magnetic beads at 4 °C overnight in protein LoBind tubes, the supernatant was completely removed. Without any wash step, the beads were resuspended in 20 μL of NuPAGE LDS sample buffer (1×) containing 2% β-mercaptoethanol (BME). The beads in the sample buffer were then heated to 95 °C for 10 min before being loaded to 12% NuPAGE Bis-Tris polyacrylamide gel. The gel was run under 150 V for 1.5 h until the blue dye ran to the bottom of the gel. Gels were stained with colloidal Coomassie blue staining kit at room temperature overnight. After destaining with deionized (DI) water for 30–60 min, the gel region between 25 and 37 kDa containing HMGB1 was cut out, sliced into 1 mm2 pieces with a scalpel, and placed into a 1.5 mL protein LoBind tube. In-gel acetylation follows the procedure described previously with minor modification.59 Briefly, the gel pieces were destained by adding 200 μL of 25 mM NH4HCO3 in water/acetonitrile (1:1 v/v) to the gel and shaking at room temperature for 10 min. If blue staining was still visible in the gel, 200 μL of 50 mM aqueous NH4HCO3 was added into the gel, which was shaken at room temperature for 10 min. The destaining step was then repeated. Acetonitrile (200 μL) was added into the gel pieces to dehydrate the gel. The mixture was shaken at room temperature for 5 min, the supernatant was removed, and the dehydration step was repeated. A mixture of [2H6]acetic anhydride (5 μL) and 0.1 M aqueous NH4HCO3 (10 μL) was added to the gel pieces. The mixture was gently mixed with a pipet tip with the lid open. Following the addition of 50 μL of 0.1 M aqueous NH4HCO3, the pH of the mixture was adjusted to ∼8 (determined by pH paper) with concentrated NH4OH. After incubation at 37 °C for 30–60 min, the supernatant was removed, and the gel pieces were washed three times with water (200 μL). The destaining and dehydration steps were repeated after the acetylation. Gel pieces were then placed on ice, to which 2 μg of Glu-C (200 ng/μL in 50 mM ammonium acetate, pH 4) was added. The mixture was incubated on ice until the solution was fully absorbed by the dried gel pieces. Digestion buffer (60 μL of 50 mM ammonium acetate, pH 4) was finally added to cover the gel pieces, and the in-gel digestion was carried out by incubating the mixture at 37 °C for 12–16 h. After the digestion, the supernatant was transferred to a clean 1.5 mL protein LoBind tube, and 200 μL of extraction buffer (3% formic acid in 50% aqueous acetonitrile) was added to the gel pieces. The mixture was sonicated at 37 °C for 30 min. The supernatant was combined and dried under N2. To the residue was added 50 μL of water, and the solution was transferred to deactivated glass inserts (Waters, Milford, MA).
Method Validation
Citrated human plasma was used for preparation of calibration standards and quality controls (QCs). Calibration standards were prepared by spiking appropriate amounts of HMGB1 standard into the plasma (100 μL) to make final concentrations of 1, 2, 4, 8, 20, 40, and 80 ng/mL. The preparation procedures for QC samples at concentrations of 1, 2.4, 32, and 60 ng/mL were the same as the calibration standards. Assay validation was conducted according to U.S. Food and Drug Administration (FDA) guidance.60 The lowest QC sample (1.0 ng/mL) was defined as the lower limit of quantification (LLOQ). The accuracy and precision were determined on five replicates of LLOQ, low quality control (LQC), middle quality control (MQC), and high quality control (HQC). QC samples (n = 5) were analyzed on the same day (intraday) and on three separate days (interday, n = 15) as described below.
Stability Assessment of Reference Standard
The stock solution of reference standard HMGB1 was analyzed on a weekly basis by UV spectroscopy to ensure that there was no degradation. In addition, calibration curves together with the four QC samples (n = 5) were analyzed on two additional days. On each day, calibration solutions were freshly diluted from the same reference standard solution. QC samples were thawed from frozen solutions that were originally prepared and stored in matrix (pooled human plasma).
2D-Nano-UHPLC-PRM/HRMS
Mass spectrometry was conducted on a Q Exactive HF hybrid quadrupole-Orbitrap mass spectrometer coupled to a Dionex Ultimate 3000 RSLCnano with capillary flowmeter chromatographic system (Thermo Fisher Scientific, San Jose, CA). The 2D system was composed of two columns, including a trapping column (Acclaim PepMap C18 cartridge (0.3 mm × 5 mm, 100 Å, Thermo Scientific) for preconcentration purpose and an analytical column (C18 AQ nano-LC column with a 10 μm pulled tip (75 μm × 25 cm, 3 μm particle size; Columntip, New Haven, CT) to separate digested peptides; two pumps, including one nanopump delivering solvents to analytical column and a micropump connecting to the trapping column; and a 10-port valve. The 2D-nano-LC system was controlled by Xcalibur software from the Q-Exactive mass spectrometer.
Samples (usually 8 μL) were injected via microliter-pickup injection mode. Loading solvent was water/acetonitrile (99.7:0.3 v/v) containing 0.2% formic acid. A 10-port valve was set at the loading position (1–2) with the loading solvent at 10 μL/min for 3 min. The valve was then changed to the analysis position (1–10), at which time the trapping column was connected with the analytical column, and samples loaded on the loading column were back-flushed into the analytical column. The valve was maintained in the analysis position for 10 min before the end of the run, when it was switched to the loading position ready for the next analysis. Samples were eluted with a linear gradient at a flow rate of 0.4 μL/min: 2% B at 2 min, 5% B at 15 min, 35% B at 40 min, 95% B at 45–55 min, 2% B at 58–70 min. Solvent A was water/acetonitrile (99.5:0.5 v/v) containing 0.1% formic acid, and solvent B was acetonitrile/water (98:2 v/v) containing 0.1% formic acid. Nanospray Flex ion source (Thermo Scientific) was used. MS operating conditions were as follows: spray voltage 2500 V, ion transfer capillary temperature 250 °C, ion polarity positive, S-lens RF level 55, in-source collision-induced dissociation (CID) 1.0 eV. Both full-scan and parallel reaction monitoring (PRM) were used. The full-scan parameters were resolution 60 000, automatic gain control (AGC) target 1 × 106, maximum IT 200 ms, scan range m/z 200–1200. The PRM parameters were resolution 60 000, AGC target 2 × 105, maximum IT 80 ms, loop count 5, isolation window 1.0 Da, NCE 25. The PRM was scheduled for 28.5–31.5 min for acetylated E26HKKKHPDASVNFSE40, 28.5–31.5 min for acetylated K57GKFE61, 30.0–33.0 min for acetylated K146KAAKLKE153, and 26.0–29.0 min for acetylated K180SKKKKE186.
Data Analysis
Data analysis was performed by use of Skyline (MacCoss Laboratory, University of Washington, Seattle, WA).60 After selection of the proper transitions, the peak area ratio of each unlabeled/light (L) peptide to SILAC-labeled/heavy (H) peptide pair was calculated by the Skyline software and exported for absolute quantification. The amount defined by each peptide was calculated by averaging L/H ratios of the three PRM transitions. HMGB1 levels were calculated from the average of the four selected quantifying peptides. Statistical analysis was performed with GraphPad Prism (v5.01, GraphPad Software Inc., La Jolla, CA).
Results and Discussion
Preparation of HMGB1 Protein Standard and SILAC-Labeled Internal Standards
Both the endogenous and SILAC-labeled proteins were expressed in HEK293 cells with a short linker containing a recognition site of TEV enzyme and a GST tag at the amino-terminus for purification. After the GST-HMGB1 conjugate was bound to the GSH Sepharose beads, the GST tag was cleaved by TEV to release HMGB1. Glu-C digestion of SILAC-labeled HMGB1 revealed >99.9% {heavy/(light + heavy)} labeling efficiency based on the PRM transitions for the four peptides (Table S1). The acetylated HMGB1 SILAC standard was prepared by adding acetic anhydride to the SILAC standard in 100 mM NH4HCO3 buffer (pH ≈ 8). Glu-C digestion and UPLC-HRMS analysis indicated complete and nonselective acetylation on all the lysines analyzed (data not shown).
Selection of HMGB1 Peptides and Parallel Reaction Monitoring Transitions
To enable the detection of NLS-1 and NLS-2 peptides and the interrogation of acetylation on lysine residues, we chose to utilize Glu-C as the endoproteinase. The expressed and purified HMGB1 and SILAC-HMGB1 standards were acetylated and then digested in solution by Glu-C. The proteolytic peptides were analyzed in both full-scan and PRM modes. The identity of digested peptides was verified by searching the BLAST database in Skyline.61 Five potentially useful acetylated peptides were selected (Table S1), and their precursor and all product ions were assessed for signal intensity in the PRM/MS mode. Acetylated decapeptide A170AKKGVVKAE179 exhibited fluctuating signals among samples, especially at lower levels, and so it was excluded for quantification purposes. NLS-1 was always observed as two peptides, one with one missed cleavage (E26HKKKHPDASVNFSE40) and the fully cleaved form (H27KKKHPDASVNFSE40), even at different protein/protease ratios. We chose the form with one missed cleavage for quantification due to its slightly higher intensity. Although precursor ions always exhibited 5–20-fold higher intensities than their corresponding product ions, they always had interfering signals or were even not detected due to the high interfering background signals. In contrast, product ions were more specific as less interfering signals were present in the channel. For the NLS-1 and NLS-2 peptides, the three most intense product ions were used for PRM of both the endogenous HMGB1 (Table S1) and SILAC-labeled internal standard (Table S1). Due to its short length, K57GKFE61 pentapeptide only generated one product ion with good and consistent intensity (Table S1).
Sample Processing and Immunopurification of HMGB1
The low abundance of endogenous HMGB1 in the serum and plasma necessitated an immunopurification (IP) step before digestion and analysis. Western blot analyses were conducted in order to determine the best pAb to employ for IP of the serum prior to MS analysis. The amount of pAb and magnetic beads (0.5 mg) were identical for each of the Western blot analyses shown in Figure S1. Lanes 7 and 8 exhibited the most intense bands corresponding to HMGB1, which showed that the pAb used (Sigma H9539) had the highest affinity toward HMGB1 (when bound to magnetic beads) among the four different antibodies that were analyzed (Figure S1). Therefore, the Sigma H9539 pAb was used for all IP procedures. HMGB1 is known to noncovalently bind to HMGB1 autoantibodies in serum and plasma. Therefore, the HMGB1-SILAC standard was allowed to equilibrate with HMGB1 bound to autoantibodies and haptoglobin in serum and plasma before IP was conducted. Western blot analysis revealed >80% recovery of HMGB1 after IP even in the presence of serum (Figure S1). However, Coomassie blue staining showed a substantial number of additional proteins were captured by the pAb. In the attempt to remove these nonspecifically bound proteins, we included a wash step with DPBS or used pAbs covalently linked to magnetic beads followed by the stringent washing of the beads, as we reported recently for platelet frataxin protein.62 Unfortunately, >50% of the HMGB1 was eluted by the first DPBS wash and >80% eluted off after three DPBS washes (Figure S2). In addition, covalently linking HMGB1 pAbs to the beads almost completely removed their binding capacity (data not shown). Therefore, SDS–PAGE was used to remove nonspecific bound proteins (Figure 3). Light and heavy chains that arose from the pAb were also removed in this step, reducing any potential suppression of ionization in the mass spectrometer.
Figure 3.
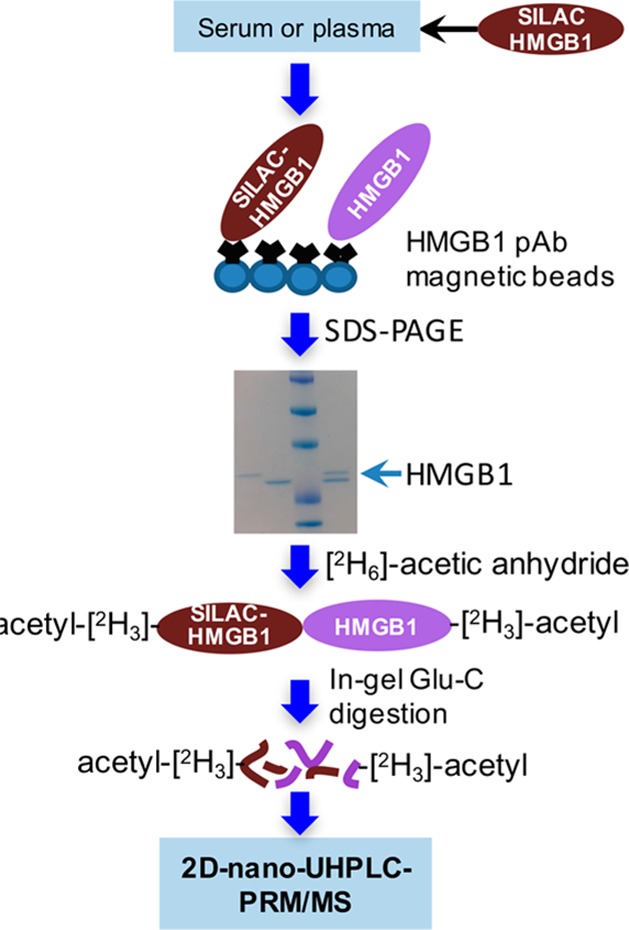
Serum and plasma sample processing procedure.
The possibility that plasma and serum HMGB1 could be acetylated posed a challenge for the quantification because the endogenous acetylated HMGB1 peptides would have different retention times and different masses from SILAC-HMGB1-derived peptides. To prevent this potential problem, an in-gel acetylation step was introduced to convert HMGB1 and SILAC-HMGB1 to the same molecular forms. Acetylation was conducted with [2H6]acetic anhydride in order to differentiate the in-gel chemical acetylation from the endogenous post-translational acetylation (Figure 3). Endoproteinase Glu-C was then used to conduct in-gel protease digestion (Figure 3). UPLC-HRMS analysis revealed that the acetylation was quantitative and that the acetylation reaction was nonselective (data not shown).
Method Validation by Use of IP-UHPLC-PRM/HRMS and SILAC-Labeled HMGB1
Human citrated plasma was selected as the matrix for method validation because the test experiment results indicated that the endogenous HMGB1 level in human citrated plasma was under the limit of detection of our method. Calibration curves were constructed at seven different concentrations ranging from 1 to 80 ng/mL with human citrated plasma (100 μL) as the matrix. Citrated plasma pooled from 10 healthy control subjects was used to minimize preanalytical variability. Linear standard curves were obtained for each of the four peptides, with r2 values between 0.9854 and 0.9959 (Figure S3A–D). Furthermore, similar values were found when mean area ratios of analyte to internal standard for the four peptides were plotted against HMGB1 concentrations (Figure S3E). The LLOQ was set at 1 ng/mL, which is below the mean concentration reported in 20 different studies of serum from healthy control subjects and serum from diseased individuals (Table 1). Accuracy and precision for the LLOQ were well within the limits of acceptance: intraday (n = 5), precision 12.8%, accuracy 91.9% (Table S2). Therefore, this LLOQ readily met the criteria required by the FDA of precision better than 20% and accuracy of between 80% and 120%.
Additional validation was performed with quality control (QC) samples at three different concentrations according to FDA guidance63 including low (LQC, 2.4 ng/mL), middle (MQC, 32 ng/mL), and high (HQC, 60 ng/mL) QC samples prepared in human citrated plasma. Precision for intraday (n = 5) QC analysis was 2.1–5.3% and accuracy was 87.6–96.6% (Table S2A). Precision for interday (n = 15) QC analysis was 4.8–7.8% and accuracy was 100.0%-108.6.% (Table S2B). Freeze–thawing through three cycles of the LQC (n = 5), MQC (n = 5), and HQC (n = 5) plasma samples did not affect the precision and accuracy (data not shown).
HMGB1 Levels in Plasma and Serum from Healthy Control Subjects
Endogenous HMGB1 levels were quantified in several human matrices from 24 healthy control subjects to evaluate the utility of this IP 2D-nano-UHPLC-PRM/MS method. Typical chromatograms for endogenous HMGB1 and SILAC standard in citrated plasma and in serum are shown in Figure 4 panels A and B, respectively.
Figure 4.

2D-Nano-UHPLC-PRM/MS chromatograms of Glu-C peptides containing eight of the important acetylation sites.49,50 (A) Citrated plasma sample. (B) Serum sample with a determined HMGB1 concentration of 4.7 ng/mL. K* = [13C615N2]lysine, Ac* = C[2H3]CO.
No acetylated peptides were detected when relevant ions were reconstructed for individual endogenous acetylated peptides under full-scan mode (Figures S4 and S5). This is consistent with previous studies because acetylated HMGB1 has never been detected in serum from healthy control subjects. However, it is conceivable that the HMGB1 pAb did not fully recognize the acetylated HMGB1. Therefore, additional proof of the lack of endogenous lysine acetylation in serum HMGB1 from healthy control individuals was obtained by repeating the assay with anti-acetyllysine pAb bound to the magnetic beads together with the HMGB1 mAb magnetic beads. No additional HMGB1 was detected even though an acetylated HMGB1 SILAC standard was recovered in excellent yield (Figures S4 and S5).
Five healthy control serum samples were randomly selected for repeat analysis. The percent deviation from the mean (Mean Dev %) of the first analysis and repeat analysis ranged from +2.7% to −8.0% (Table S3). The mean value for the five repeat samples of 6.4 ng/mL was −2.4% of the mean for the five samples analyzed originally (6.6 ng/mL) and not significantly different (p = 0.313). This confirms that the repeat analyses were well within the guidance of 30% suggested by the FDA for repeat samples.
Conclusion
Serum HMGB1 has been reported to be a biomarker for a variety of diseases (Table 1).25−43 Most of these previous studies quantified HMGB1 by ELISA, which is problematic due to the presence of HMGB1 autoantibodies in serum and the ability of haptoglobin to bind HMGB1. This led us to develop a more specific stable isotope dilution IP 2D-nano-UHPLC-PRM/MS method. A SILAC-labeled standard was used to account for any losses during IP, gel purification, and in-gel digestion. The requirement for stable isotopically labeled protein internal standards to improve precision of protein analysis stimulated the development of methodology based on that used for NMR spectroscopy.64 At the same time, the concept of SILAC was developed, in which labeling was conducted in experimental cells and endogenous proteins were derived from control cells in culture.65 However, cells can grow more slowly when stable isotopically labeled amino acids are substituted for endogenous amino acids, leading to differential protein expression. In addition, conventional SILAC methodology cannot be used for analysis of proteins in tissues and biofluids. This stimulated new strategies termed stable isotope labeled proteome (SILAP) standard and absolute SILAC, which were introduced in 200566 and 2008,67 respectively, to circumvent these problems. SILAC-labeled recombinant proteins produced in vitro or in vivo are used as internal standards, which are directly mixed into lysates of cells or tissues,67−69 or appropriate biofluids such as cervicovaginal fluid,70 serum,71,72 and platelet lysates.62 We have shown previously for the analysis of drugs,73 DNA adducts,74 coenzymes,75 lipids,76 peptides,77 and proteins62,72 that it is not necessary to determine exactly how much internal standard is being added to the biofluid. However, it is very important to add exactly the same amount of internal standard to the biofluid standards, QCs, and study samples. It is also extremely important to have an accurate determination of the amount of endogenous analyte used in the standard curves. Therefore, the HMGB1 standard was carefully purified and quantified by PAGE with Coomassie staining and UV and amino acid analysis. We also rigorously determined that there were no post-translational modifications on the endogenous HMGB1 protein that we expressed.
Chemically acetylating all unmodified lysines simplified the 100 000 possible modified forms of HMGB150 to one fully acetylated molecular form (Figure 3). Concomitant full-scan MS1 data generated during PRM analysis were used to show that endogenous acetylation had not occurred on 17 of the potential acetylation sites on HMGB1 (Figures S4 and S5), including the eight present on NLS-1 and NLS-2 that are thought to be critical for HMGB1 secretion (Figure 2).49,50 If necessary, the specific endogenous acetylation sites could have been identified by LC/MS/MS analysis. The mean serum HMGB1 level reported in 18 studies of healthy control subjects was 1.9 ± 0.9 ng/mL, and serum HMGB1 from 18 different diseases was 5.4 ± 2.8 ng/mL (Figure 5). In contrast, the mean serum HMGB1 concentration found in 24 healthy control subjects determined by stable isotope dilution IP 2D-nano-UHPLC-PRM/MS was 6.0 ± 2.1 ng/mL. Furthermore, HMGB1 levels remained unchanged when anti-acetyllysine pAb was used in addition to the HMGB1 pAb, demonstrating that endogenous HMGB1 was not acetylated in serum from healthy control subjects. In contrast to serum, citrated and EDTA-treated plasma had HMGB1 concentrations of <1 ng/mL in the same individuals (Figure 5), which confirmed that HMGB1 is released when blood is allowed to clot. It is noteworthy that an ELISA assay also showed that plasma HMGB1 concentrations were lower than those in serum.78 In light of these findings, 18 of the previous studies of serum HMGB1 in immune, inflammatory, and cardiovascular diseases and cancer will have to be re-evaluated (Table 1). Until the HMGB1 concentrations in these diseases have been rigorously established, the role of HMGB1 should be considered questionable. Two studies (alcoholic liver disease and peritoneal mesothelioma) have reported serum HMGB1 concentrations significantly higher (18 and 27 ng/mL, Table 1) than the healthy control serum concentrations determined by UHPLC-HRMS (Table 1). This indicates that HMGB1 could be upregulated in these diseases. However, the concentrations of serum HMGB1 concentrations reported in the alcoholic liver disease (ALD) study for healthy control subjects of 1.1 ng/mL were significantly lower than the concentrations determined by UPLC-HRMS (6.0 ng/mL, Figure 5). This suggests that there could be a problem with the assay that was used in the alcoholic liver disease study and that circulating HMGB1 concentrations in this disease should also be re-evaluated. Our study has provided clear evidence that HMGB1 is released during the clotting process (Figure 5). Furthermore, it is known that platelets release HMGB1 when they aggregate.79 This situation is reminiscent of serum thromboxane B2 analysis, where ex vivo platelet activation is used to assess the capacity for cyclooxygase 1-mediated thromboxane production.80 There has been substantial interest in the analysis of serum HMGB1 for monitoring systemic HMGB1 production in numerous diseases (Table 1). A review of citations in PubMed revealed that over the last 15 years, there have been 897 published studies on the analysis of serum HMGB1, with a 28-fold increase from four in 2002 to 113 in 2017. Our study has clearly shown that serum HMGB1 is not a biomarker of systemic disease, and so all of the previous studies reporting serum HMGB1 levels will have to be re-evaluated by use of citrated or EDTA-treated plasma instead of serum.
Figure 5.
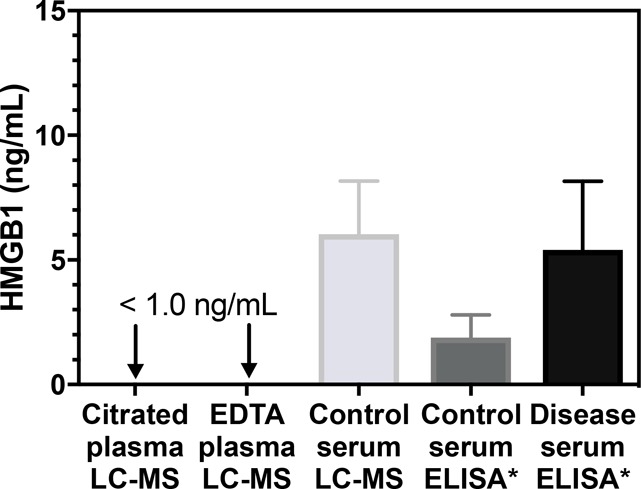
HMGB1 levels in citrated plasma (<1.0 ng/mL, n = 24), EDTA-treated plasma (<1.0 ng/mL, n = 24), and serum (mean = 6.0 ± 2.1 ng/mL, n = 24) from healthy control subjects, determined by stable isotope dilution IP 2D-nano-UHPLC-PRM/MS, and reported levels of serum HMGB1 determined by ELISA in healthy control subjects (mean = 2.1 ± 0.9 ng/mL) and cases (mean = 5.4 ± 2.8 ng/mL) for 18 of the studies shown in Table 1 (indicated with asterisks). Control plasma and serum samples from healthy subjects for the LC/MS analyses were obtained from the same individuals. Data are shown as means ± stndard deviation (SD).
Acknowledgments
We gratefully acknowledge the support of NIH Grants P42ES023720, P30ES013508, and T32ES019851.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.analchem.8b01175.
Three tables listing PRM transitions for acetylated GluC peptides from HMGB1, accuracy and precision for determination of serum HMGB1, and repeat analysis of serum samples; five figures showing Western blot images for screening and washing of antibodies, calibration curves for HMGB1 constructed in plasma, and LC/MS chromatograms for acetylated peptides (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Oe T.; Ackermann B. L.; Inoue K.; Berna M. J.; Garner C. O.; Gelfanova V.; Dean R. A.; Siemers E. R.; Holtzman D. M.; Farlow M. R.; Blair I. A. Rapid Commun. Mass Spectrom. 2006, 20 (24), 3723–3735. 10.1002/rcm.2787. [DOI] [PubMed] [Google Scholar]
- Lotze M. T.; Tracey K. J. Nat. Rev. Immunol. 2005, 5 (4), 331–342. 10.1038/nri1594. [DOI] [PubMed] [Google Scholar]
- Yang H.; Antoine D. J.; Andersson U.; Tracey K. J. J. Leukocyte Biol. 2013, 93 (6), 865–873. 10.1189/jlb.1212662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venegas C.; Heneka M. T. J. Leukocyte Biol. 2017, 101 (1), 87–98. 10.1189/jlb.3MR0416-204R. [DOI] [PubMed] [Google Scholar]
- Lotze M. T.; Deisseroth A.; Rubartelli A. Clin. Immunol. 2007, 124 (1), 1–4. 10.1016/j.clim.2007.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims G. P.; Rowe D. C.; Rietdijk S. T.; Herbst R.; Coyle A. J. Annu. Rev. Immunol. 2010, 28, 367–388. 10.1146/annurev.immunol.021908.132603. [DOI] [PubMed] [Google Scholar]
- Tang D.; Kang R.; Coyne C. B.; Zeh H. J.; Lotze M. T. Immunol Rev. 2012, 249 (1), 158–175. 10.1111/j.1600-065X.2012.01146.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L.-C.; Gao J.; Li J. J. Cell Mol. Med. 2014, 18 (12), 2331–2339. 10.1111/jcmm.12419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinotti S.; Patrone M.; Ranzato E. ImmunoTargets Ther. 2015, 4, 101–109. 10.2147/ITT.S58064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi M. E.; Crippa M. P.; Manfredi A. A.; Mezzapelle R.; Rovere Querini P.; Venereau E. Immunol Rev. 2017, 280 (1), 74–82. 10.1111/imr.12601. [DOI] [PubMed] [Google Scholar]
- Messer J. S. Cell. Mol. Life Sci. 2017, 74 (7), 1281–1296. 10.1007/s00018-016-2403-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey S.; Singh S.; Anang V.; Bhatt A. N.; Natarajan K.; Dwarakanath B. S. Cancer Growth Metastasis 2015, 8, 25–34. 10.4137/CGM.S24314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magna M.; Pisetsky D. S. Clin. Ther. 2016, 38 (5), 1029–1041. 10.1016/j.clinthera.2016.02.029. [DOI] [PubMed] [Google Scholar]
- Li G.; Liang X.; Lotze M. T. Front. Immunol. 2013, 4, 68. 10.3389/fimmu.2013.00068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulloa L.; Messmer D. Cytokine Growth Factor Rev. 2006, 17 (3), 189–201. 10.1016/j.cytogfr.2006.01.003. [DOI] [PubMed] [Google Scholar]
- Pallier C.; Scaffidi P.; Chopineau-Proust S.; Agresti A.; Nordmann P.; Bianchi M. E.; Marechal V. Mol. Biol. Cell 2003, 14 (8), 3414–3426. 10.1091/mbc.e02-09-0581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang R.; Livesey K. M.; Zeh H. J. III; Loze M. T.; Tang D. Autophagy 2011, 7 (10), 1256–1258. 10.4161/auto.7.10.16753. [DOI] [PubMed] [Google Scholar]
- Martins J. D.; Liberal J.; Silva A.; Ferreira I.; Neves B. M.; Cruz M. T. DNA Cell Biol. 2015, 34 (4), 274–281. 10.1089/dna.2014.2752. [DOI] [PubMed] [Google Scholar]
- Scaffidi P.; Misteli T.; Bianchi M. E. Nature 2002, 418 (6894), 191–195. 10.1038/nature00858. [DOI] [PubMed] [Google Scholar]
- Kalinina N.; Agrotis A.; Antropova Y.; DiVitto G.; Kanellakis P.; Kostolias G.; Ilyinskaya O.; Tararak E.; Bobik A. Arterioscler., Thromb., Vasc. Biol. 2004, 24 (12), 2320–2325. 10.1161/01.ATV.0000145573.36113.8a. [DOI] [PubMed] [Google Scholar]
- Tang D.; Shi Y.; Kang R.; Li T.; Xiao W.; Wang H.; Xiao X. J. Leukocyte Biol. 2007, 81 (3), 741–747. 10.1189/jlb.0806540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmblad K.; Schierbeck H.; Sundberg E.; Horne A.-C.; Harris H. E.; Henter J.-I.; Antoine D. J.; Andersson U. Mol. Med. 2015, 20, 538–547. 10.2119/molmed.2014.00183. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Cerwenka A.; Kopitz J.; Schirmacher P.; Roth W.; Gdynia G. Mol. Cell Oncol 2016, 3 (4), e1175538. 10.1080/23723556.2016.1175538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel S.; Rath D.; Borst O.; Mack A.; Loughran P.; Lotze M. T.; Neal M. D.; Billiar T. R.; Gawaz M. Biochem. Biophys. Res. Commun. 2016, 478 (1), 143–148. 10.1016/j.bbrc.2016.07.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge X.; Antoine D. J.; Lu Y.; Arriazu E.; Leung T.-M.; Klepper A. L.; Branch A. D.; Fiel M. I.; Nieto N. J. Biol. Chem. 2014, 289 (33), 22672–22691. 10.1074/jbc.M114.552141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napolitano A.; Antoine D. J.; Pellegrini L.; Baumann F.; Pagano I.; Pastorino S.; Goparaju C. M.; Prokrym K.; Canino C.; Pass H. I.; Carbone M.; Yang H. Clin. Cancer Res. 2016, 22 (12), 3087–3096. 10.1158/1078-0432.CCR-15-1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antoine D. J.; Jenkins R. E.; Dear J. W.; Williams D. P.; McGill M. R.; Sharpe M. R.; Craig D. G.; Simpson K. J.; Jaeschke H.; Park B. K. J. Hepatol. 2012, 56 (5), 1070–1079. 10.1016/j.jhep.2011.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Malhotra S.; Fissolo N.; Tintore M.; Wing A. C.; Castillo J.; Vidal-Jordana A.; Montalban X.; Comabella M. J. Neuroinflammation 2015, 12, 48. 10.1186/s12974-015-0269-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdulahad D. A.; Westra J.; Bijzet J.; Limburg P. C.; Kallenberg C. G. M.; Bijl M. Arthritis Res. Ther 2011, 13 (3), R71. 10.1186/ar3332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson S.; Pettila V.; Tenhunen J.; Laru-Sompa R.; Hynninen M.; Ruokonen E. Intensive Care Med. 2008, 34 (6), 1046–1053. 10.1007/s00134-008-1032-9. [DOI] [PubMed] [Google Scholar]
- Skrha J. Jr.; Kalousova M.; Svarcova J.; Muravska A.; Kvasnicka J.; Landova L.; Zima T.; Skrha J. Exp. Clin. Endocrinol. Diabetes 2012, 120 (5), 277–281. 10.1055/s-0031-1283161. [DOI] [PubMed] [Google Scholar]
- Sun S.; Zhang W.; Cui Z.; Chen Q.; Xie P.; Zhou C.; Liu B.; Peng X.; Zhang Y. OncoTargets Ther. 2015, 8, 413–419. 10.2147/OTT.S73366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittwer C.; Boeck S.; Heinemann V.; Haas M.; Stieber P.; Nagel D.; Holdenrieder S. Int. J. Cancer 2013, 133 (11), 2619–2630. 10.1002/ijc.28294. [DOI] [PubMed] [Google Scholar]
- Qiu G.; Li Y.; Liu Z.; Wang M.; Ge J.; Bai X. Med. Oncol. 2014, 31 (12), 316. 10.1007/s12032-014-0316-x. [DOI] [PubMed] [Google Scholar]
- Tabata C.; Shibata E.; Tabata R.; Kanemura S.; Mikami K.; Nogi Y.; Masachika E.; Nishizaki T.; Nakano T. BMC Cancer 2013, 13, 205. 10.1186/1471-2407-13-205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niki M.; Yokoi T.; Kurata T.; Nomura S. Lung Cancer: Targets Ther. 2017, 8, 91–99. 10.2147/LCTT.S138887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Festoff B. W.; Sajja R. K.; van Dreden P.; Cucullo L. J. Neuroinflammation 2016, 13 (1), 194. 10.1186/s12974-016-0670-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker L. E.; Frigerio F.; Ravizza T.; Ricci E.; Tse K.; Jenkins R. E.; Sills G. J.; Jorgensen A.; Porcu L.; Thippeswamy T.; Alapirtti T.; Peltola J.; Brodie M. J.; Park B. K.; Marson A. G.; Antoine D. J.; Vezzani A.; Pirmohamed M. J. Clin. Invest. 2017, 127 (6), 2118–2132. 10.1172/JCI92001. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Santoro M.; Maetzler W.; Stathakos P.; Martin H. L.; Hobert M. A.; Rattay T. W.; Gasser T.; Forrester J. V.; Berg D.; Tracey K. J.; Riedel G.; Teismann P. Neurobiol. Dis. 2016, 91, 59–68. 10.1016/j.nbd.2016.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y.; Zhang K.; Zhao L.; Guo J.; Hu X.; Chen Z. J. Int. Med. Res. 2013, 41 (6), 1796–1802. 10.1177/0300060513503917. [DOI] [PubMed] [Google Scholar]
- Yan X. X.; Lu L.; Peng W. H.; Wang L. J.; Zhang Q.; Zhang R. Y.; Chen Q. J.; Shen W. F. Atherosclerosis 2009, 205 (2), 544–548. 10.1016/j.atherosclerosis.2008.12.016. [DOI] [PubMed] [Google Scholar]
- Wang L. J.; Lu L.; Zhang F. R.; Chen Q. J.; De Caterina R.; Shen W. F. Eur. J. Heart Failure 2011, 13 (4), 440–449. 10.1093/eurjhf/hfq231. [DOI] [PubMed] [Google Scholar]
- Hu X.; Jiang H.; Bai Q.; Zhou X.; Xu C.; Lu Z.; Cui B.; Wen H. Clin. Chim. Acta 2009, 406 (1–2), 139–142. 10.1016/j.cca.2009.06.016. [DOI] [PubMed] [Google Scholar]
- Hwang C.-S.; Liu G.-T.; Chang M. D.-T.; Liao I.-L.; Chang H.-T. Neurobiol. Dis. 2013, 58, 13–18. 10.1016/j.nbd.2013.04.013. [DOI] [PubMed] [Google Scholar]
- Andersson U.; Harris H. E. Biochim. Biophys. Acta, Gene Regul. Mech. 2010, 1799 (1–2), 141–148. 10.1016/j.bbagrm.2009.11.003. [DOI] [PubMed] [Google Scholar]
- Gougeon M.-L.; Poirier-Beaudouin B.; Durant J.; Lebrun-Frenay C.; Saidi H.; Seffer V.; Ticchioni M.; Chanalet S.; Carsenti H.; Harvey-Langton A.; Laffon M.; Cottalorda J.; Pradier C.; Dellamonica P.; Vassallo M. Heliyon 2017, 3 (2), e00245. 10.1016/j.heliyon.2017.e00245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H.; Wang H.; Wang Y.; Addorisio M.; Li J.; Postiglione M. J.; Chavan S. S.; Al-Abed Y.; Antoine D. J.; Andersson U.; Tracey K. J. J. Intern. Med. 2017, 282 (1), 76–93. 10.1111/joim.12619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada S.; Yakabe K.; Ishii J.; Imaizumi H.; Maruyama I. Clin. Chim. Acta 2006, 372 (1–2), 173–178. 10.1016/j.cca.2006.04.016. [DOI] [PubMed] [Google Scholar]
- Zhang Q.; Wang Y. Biochim. Biophys. Acta, Proteins Proteomics 2008, 1784 (9), 1159–1166. 10.1016/j.bbapap.2008.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabadi M. M.; Xavier S.; Vasko R.; Kaur K.; Goligorksy M. S.; Ratliff B. B. Kidney Int. 2015, 87 (1), 95–108. 10.1038/ki.2014.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito I.; Fukazawa J.; Yoshida M. J. Biol. Chem. 2007, 282 (22), 16336–16344. 10.1074/jbc.M608467200. [DOI] [PubMed] [Google Scholar]
- Tang Y.; Zhao X.; Antoine D.; Xiao X.; Wang H.; Andersson U.; Billiar T. R.; Tracey K. J.; Lu B. Antioxid. Redox Signaling 2016, 24 (12), 620–634. 10.1089/ars.2015.6409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y. H.; Kwak M. S.; Park J. B.; Lee S.-A.; Choi J. E.; Cho H.-S.; Shin J.-S. J. Cell Sci. 2016, 129 (1), 29–38. 10.1242/jcs.176412. [DOI] [PubMed] [Google Scholar]
- Ito I.; Mitsuoka N.; Sobajima J.; Uesugi H.; Ozaki S.; Ohya K.; Yoshida M. J. Biochem. 2004, 136 (2), 155–162. 10.1093/jb/mvh107. [DOI] [PubMed] [Google Scholar]
- Sterner R.; Vidali G.; Allfrey V. G. J. Biol. Chem. 1979, 254 (22), 11577–11583. (http://www.jbc.org/content/254/22/11577.full.pdf). [PubMed] [Google Scholar]
- Bonaldi T.; Talamo F.; Scaffidi P.; Ferrera D.; Porto A.; Bachi A.; Rubartelli A.; Agresti A.; Bianchi M. E. EMBO J. 2003, 22 (20), 5551–5560. 10.1093/emboj/cdg516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasheva E.; Sarov M.; Bidjekov K.; Ugrinova I.; Sarg B.; Lindner H.; Pashev I. G. Biochemistry 2004, 43 (10), 2935–2940. 10.1021/bi035615y. [DOI] [PubMed] [Google Scholar]
- Arnold M. E.; Booth B.; King L.; Ray C. AAPS J. 2016, 18 (6), 1366–1372. 10.1208/s12248-016-9946-6. [DOI] [PubMed] [Google Scholar]
- Weng L.; Greenberg M. M. J. Am. Chem. Soc. 2015, 137 (34), 11022–11031. 10.1021/jacs.5b05478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLean B.; Tomazela D. M.; Shulman N.; Chambers M.; Finney G. L.; Frewen B.; Kern R.; Tabb D. L.; Liebler D. C.; MacCoss M. J. Bioinformatics 2010, 26 (7), 966–968. 10.1093/bioinformatics/btq054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill S. C.; von Hippel P. H. Anal. Biochem. 1989, 182 (2), 319–326. 10.1016/0003-2697(89)90602-7. [DOI] [PubMed] [Google Scholar]
- Guo L.; Wang Q.; Weng L.; Hauser L. A.; Strawser C. J.; Rocha A. G.; Dancis A.; Mesaros C.; Lynch D. R.; Blair I. A. Anal. Chem. 2018, 90 (3), 2216–2223. 10.1021/acs.analchem.7b04590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bansal S.; DeStefano A. AAPS J. 2007, 9 (1), E109–E114. 10.1208/aapsj0901011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H.; Pan S.; Gu S.; Bradbury E. M.; Chen X. Rapid Commun. Mass Spectrom. 2002, 16 (22), 2115–2123. 10.1002/rcm.831. [DOI] [PubMed] [Google Scholar]
- Ong S.-E.; Blagoev B.; Kratchmarova I.; Kristensen D. B.; Steen H.; Pandey A.; Mann M. Mol. Cell. Proteomics 2002, 1 (5), 376–386. 10.1074/mcp.M200025-MCP200. [DOI] [PubMed] [Google Scholar]
- Yan Y.; Weaver V. M.; Blair I. A. J. Proteome Res. 2005, 4 (6), 2007–2014. 10.1021/pr050175d. [DOI] [PubMed] [Google Scholar]
- Hanke S.; Besir H.; Oesterhelt D.; Mann M. J. Proteome Res. 2008, 7 (3), 1118–1130. 10.1021/pr7007175. [DOI] [PubMed] [Google Scholar]
- Rangiah K.; Tippornwong M.; Sangar V.; Austin D.; Tetreault M.-P.; Rustgi A. K.; Blair I. A.; Yu K. H. J. Proteome Res. 2009, 8 (11), 5153–5164. 10.1021/pr900518v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccimaro E.; Hanks S. K.; Yu K. H.; Blair I. A. Anal. Chem. 2009, 81 (9), 3304–3313. 10.1021/ac900204f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah S. J.; Yu K. H.; Sangar V.; Parry S. I.; Blair I. A. J. Proteome Res. 2009, 8 (5), 2407–2417. 10.1021/pr8010342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehr A. Y.; Hwang W.-T.; Blair I. A.; Yu K. H. J. Proteome Res. 2012, 11 (3), 1749–1758. 10.1021/pr201011f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q.; Zhang S.; Guo L.; Busch C. M.; Jian W.; Weng N.; Snyder N. W.; Rangiah K.; Mesaros C.; Blair I. A. Bioanalysis 2015, 7 (22), 2895–2911. 10.4155/bio.15.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oe T.; Tian Y.; O’Dwyer P. J.; Roberts D. W.; Malone M. D.; Bailey C. J.; Blair I. A. Anal. Chem. 2002, 74 (3), 591–599. 10.1021/ac010792v. [DOI] [PubMed] [Google Scholar]
- Williams M. V.; Lee S.-H.; Pollack M.; Blair I. A. J. Biol. Chem. 2006, 281 (15), 10127–10133. 10.1074/jbc.M600178200. [DOI] [PubMed] [Google Scholar]
- Basu S. S.; Mesaros C.; Gelhaus S. L.; Blair I. A. Anal. Chem. 2011, 83 (4), 1363–1369. 10.1021/ac1027353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazaleuskaya L. L.; Salamatipour A.; Sarantopoulou D.; Weng L.; FitzGerald G. A.; Blair I. A.; Mesaros C. J. Lipid Res. 2018, 59 (3), 564–575. 10.1194/jlr.D081414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oe T.; Ackermann B. L.; Inoue K.; Berna M. J.; Garner C. O.; Gelfanova V.; Dean R. A.; Siemers E. R.; Holtzman D. M.; Farlow M. R.; Blair I. A. Rapid Commun. Mass Spectrom. 2006, 20 (24), 3723–3735. 10.1002/rcm.2787. [DOI] [PubMed] [Google Scholar]
- Lehner J.; Wittwer C.; Fersching D.; Siegele B.; Holdenrieder S.; Stoetzer O. J. Anticancer Res. 2012, 32 (5), 2059–2062. [PubMed] [Google Scholar]
- Yang X.; Wang H.; Zhang M.; Liu J.; Lv B.; Chen F. Diagn. Pathol. 2015, 10, 134. 10.1186/s13000-015-0348-3. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Mazaleuskaya L. L.; Lawson J. A.; Li X.; Grant G.; Mesaros C.; Grosser T.; Blair I. A.; Ricciotti E.; FitzGerald G. A. JCI Insight 2016, 1 (12), e87031 10.1172/jci.insight.87031. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.