

Abstract
Background
Genetic structural variation underpins a multitude of phenotypes, with significant implications for a range of biological outcomes. Despite their crucial role, structural variants (SVs) are often neglected and overshadowed by single nucleotide polymorphisms (SNPs), which are used in large-scale analysis such as genome-wide association and population genetic studies.
Results
To facilitate the high-throughput analysis of structural variation we have developed an analytical pipeline and visualisation tool, called SV-Pop. The utility of this pipeline was then demonstrated through application with a large, multi-population P. falciparum dataset.
Conclusions
Designed to facilitate downstream analysis and visualisation post-discovery, SV-Pop allows for straightforward integration of multi-population analysis, method and sample-based concordance metrics, and signals of selection.
Keywords: Population genomics, Structural variation, Bioinformatics, Analytics, Python, R, Shiny
Background
Structural variation (SVs) describes changes to a core genome beyond single nucleotide polymorphisms (SNPs) or very short insertions and deletions (indels). Typically, SVs consist of four major types: deletions, insertions, duplications, and inversions. All play an important contribution to human and pathogen diversity and disease susceptibility. For example, duplications of the Plasmodium falciparum malaria parasite gch1 have been associated with antimalarial resistance [1], and deletions of the human Duffy antigen convey resistance to malaria infection [2]. Despite their significant implications, the role of SVs has been overshadowed by SNPs, which can currently be identified easier and faster. Several SV discovery methods, such as DELLY and CNVnator currently exist [3, 4], but there is presently no tool for efficiently identifying concordance between models, up-scaling analysis for multiple populations, or visualising that output.
To assist the identification and investigation of SVs, we have developed a bioinformatics pipeline for high-throughput post-discovery analysis and visualisation that facilitates comparison across multiple populations and between different discovery methods.
Implementation
SV-Pop consists of two core modules: (i) population-based analysis following individual SV discovery, and (ii) visualisation of those variants for dynamic, whole-genome exploration. The analysis module is a Unix command line tool built in Python (v3.3+) with pandas (v0.18+), and numpy (v1.10.4+). The visualisation module is built using the R Shiny web framework [5], and requires R (v3.3+) alongside the shiny, plotly, data.table, and dplyr packages. It can be launched on command line using ‘Rscript easyRun.r’, then explored via your default web browser. Input files should be pre-processed with SV-Pop, using the PREPROCESS mode for full compatibility. An overview of the full pipeline is shown in Fig. 1.
Fig. 1.
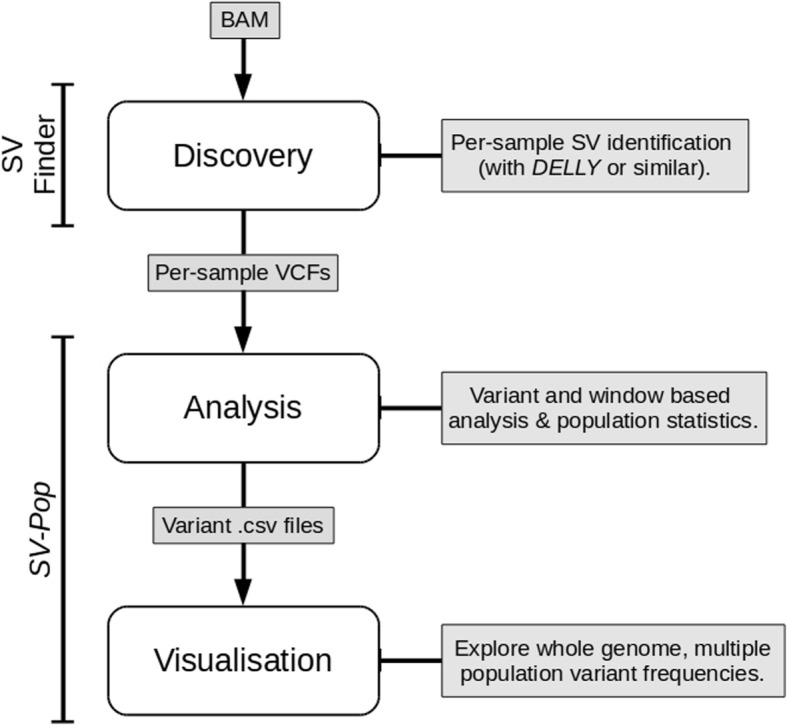
Summary of a typical SV-Pop run
Analysis
Input to SV-Pop consists of an array of post-discovery files (vcf format), one per-individual sample. These are typically the output of a run of DELLY or similar [3]. Variants across all samples are then processed, identifying and combining those specific variants that are shared across multiple samples and performing appropriate summary statistics. If so desired, variants can be filtered according to their concordance with a secondary discovery method by supplying a csv file of those variants with the dirConcordance argument. By default, variants are matched if they overlap at least 80% of the region identified by the primary method.
Once collated, we can consider a rolling window across the sample genome and identify regions with high or low variant overlap. This produces a coverage-like statistic for those underlying SVs. We can then further dissect according to sub-populations, as provided by the user. Specific variant sets can also be annotated, subset, merged, and filtered as required. In addition to core analysis and data processing functionalities, we have structured the pipeline to allow seamless integration of various filters and statistics, including method concordance and fixation indexes (FST).
Typically, an analysis module run follows calling SVs across multiple models for a population of samples, inputting those individual output vcf files into SV-Pop, and producing per-variant or per-window based statistics (as csv files) for input into the visualisation module.
Visualisation
Post-analysis, per-window files can be brought forward to the visualisation module, facilitating dynamic investigation of whole genome structural variation across multiple populations. By default, the visualisation module will identify variant frequencies and difference metrics (e.g. FST values) for all populations if present within your provided files, allowing the user to easily specify those they are interested in viewing. Similarly, the chromosomes and their sizes are detected allowing the user to specify regions of interest. Users are also able to subset and download specified genomic regions of interest for further analysis.
Results
To demonstrate the utility of SV-Pop, P. falciparum malaria parasite alignment files from 3110 samples across 21 countries with published sequence data [6] were processed with SV-Pop and loaded into the visualiser. As shown in Fig. 2, both elevated frequencies and a spike in the FST metric highlight the previously identified gch1 promoter duplication.
Fig. 2.

Screenshot of the visualization module displaying region-based FST values and window-based duplication frequencies for samples from Malawi, South America, and Asia. a Variant viewer, displaying per-window frequencies and statistical metrics. b Region summary, statistics regarding the region highlighted in the viewer. c Variant and Chromosome selector. d Population selection. e Location selection and download. The highlighted region demonstrated the presence of shorter three-window duplications in Malawi in contrast to an absence of duplications in South America and longer but less frequent duplications in Asia
The spike in the Malawi track (red) is the previously identified gch1 promoter region duplication, whilst the ridge in the Asia track (cyan) indicates whole gene duplications. The FST track (purple) highlights frequency differences between region groups.
Conclusions
SV-Pop dramatically increases the accessibility of large, population-based SV studies, allowing for a greater volume of downstream analysis and visualisation. It also establishes a core pipeline upon which to incorporate existing and future metrics such as method concordance and selection statistics. This implementation, which has been demonstrated on a P. falciparum dataset, is species-agnostic ensuring that it can be applied in a wide range of biological and geographical contexts.
Availability and requirements
Project name: SV-Pop.
Project home page: https://github.com/mattravenhall/SV-Pop
Operating system(s): Unix (MacOS, Linux) or Windows 10.
Programming language: Python, R.
Other requirements: Python (3.3+): numpy (v1.10.4), pandas (v0.18); R (3.3+): shiny, plotly, dplyr, data.table. Included setup scripts will attempt to install all packages. Running on Windows 10 required use of the Bash shell.
License: MIT.
Acknowledgements
The Medical Research Council UK funded eMedLab computing resource was used to support development.
Funding
MR is funded by the Biotechnology and Biological Sciences Research Council (Grant Number BB/J014567/1). TGC and SC are supported by the Medical Research Council UK (MR/M01360X/1, MR/N010469/1) and BBSRC (BB/R013063/1).
Availability of data and materials
Further documentation and the SV-Pop source code are available at https://github.com/mattravenhall/SV-Pop.
Abbreviations
- FST
Fixation Index
- SNP
Single Nucleotide Polymorphism
- SV
Structural Variant
Authors’ contributions
MR developed SV-Pop and co-wrote the manuscript. SC advised on package functionality. TC advised on package functionality and co-wrote the manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Matt Ravenhall, Email: matt.ravenhall@lshtm.ac.uk.
Susana Campino, Email: susana.campino@lshtm.ac.uk.
Taane G. Clark, Email: taane.clark@lshtm.ac.uk
References
- 1.Heinberg A, Kirkman L. The molecular basis of antifolate resistance in plasmodium falciparum: looking beyond point mutations. Ann N Y Acad Sci. 2015;1342. 10.1111/nyas.12662. [DOI] [PMC free article] [PubMed]
- 2.Miller LH, Mason SJ, Clyde DF, McGinniss MH. The resistance factor to plasmodium vivax in blacks. N Engl J Med. 1976;295:302–304. doi: 10.1056/NEJM197608052950602. [DOI] [PubMed] [Google Scholar]
- 3.Rausch T, Zichner T, Schlattl A, Stutz AM, Benes V, Korbel JO, et al. DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics. 2012;28:i333–i339. doi: 10.1093/bioinformatics/bts378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Abyzov A, Urban AE, Snyder M, Gerstein M. CNVnator: an approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res. 2011;21:974–984. doi: 10.1101/gr.114876.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chang W, Cheng J, Allaire J, Xie Y, McPherson J. shiny: Web Application Framework for R. R package shiny version 1.2.0. 2017. https://cran.r-project.org/package=shiny. Accessed 2 Nov 2018.
- 6.Ravenhall M, Benavente ED, Mipando M, Jensen ATR, Sutherland CJ, Roper C, et al. Characterizing the impact of sustained sulfadoxine/pyrimethamine use upon the plasmodium falciparum population in Malawi. Malar J. 2016;15. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Further documentation and the SV-Pop source code are available at https://github.com/mattravenhall/SV-Pop.