

Abstract
Background
Microglia play a central role in most neurological disorders, but the impact of microgliosis on brain environment and clinical functions is not fully understood. Mice lacking multifunctional protein-2 (MFP2), a pivotal enzyme in peroxisomal β-oxidation, develop a fatal disorder characterized by motor problems similar to the milder form of MFP2 deficiency in humans. The hallmark of disease in mice is the chronic proliferation of microglia in the brain, but molecular pathomechanisms that drive rapid clinical deterioration in human and mice remain unknown. In the present study, we identified the effects of specific deletion of MFP2 from microglia in the brain on immune responses, neuronal functioning, and behavior.
Methods
We created a novel Cx3cr1-Mfp2−/− mouse model and studied the impact of MFP2 deficiency on microglial behavior at different ages using immunohistochemistry and real-time PCR. Pro- and anti-inflammatory responses of Mfp2−/− microglia were assessed in vitro and in vivo after stimulation with IL-1β/INFγ and IL-4 (in vitro) and LPS and IL-4 (in vivo). Facial nerve axotomy was unilaterally performed in Cx3cr1-Mfp2−/− and control mice, and microglial functioning in response to neuronal injury was subsequently analyzed by histology and real-time PCR. Finally, neuronal function, motor function, behavior, and cognition were assessed using brainstem auditory evoked potentials, grip strength and inverted grid test, open field exploration, and passive avoidance learning, respectively.
Results
We found that Mfp2−/− microglia in a genetically intact brain environment adopt an inflammatory activated and proliferative state. In addition, we found that acute inflammatory and neuronal injury provoked normal responses of Mfp2−/− microglia in Cx3cr1-Mfp2−/− mice during the post-injury period. Despite chronic pro-inflammatory microglial reactivity, Cx3cr1-Mfp2−/− mice exhibited normal neuronal transmission, clinical performance, and cognition.
Conclusion
Our data demonstrate that MFP2 deficiency in microglia causes intrinsic dysregulation of their inflammatory profile, which is not harmful to neuronal function, motor function, and cognition in mice during their first year of life.
Electronic supplementary material
The online version of this article (10.1186/s12974-019-1442-3) contains supplementary material, which is available to authorized users.
Keywords: Microglia, Peroxisomes, Facial nerve axotomy, Conditional mouse model, β-Oxidation, Behavior, Immune response
Background
Inactivation of peroxisomal β-oxidation by the loss of multifunctional protein-2 (MFP2) in human and mice causes a fatal neuropathological phenotype [1–4]. MFP2, encoded by the Hsd17b4 gene, is the key enzyme in peroxisomal β-oxidation, a pathway responsible for chain shortening of carboxylates including very long chain fatty acids and formation of polyunsaturated fatty acids [5]. Dependent on the type of mutation, patients with MFP2 (also called D-bifunctional protein) deficiency display a severe neurodevelopmental disorder leading to death within the first year of life or a milder phenotype with prolonged survival into adolescence or adulthood [3, 6, 7]. Prominent clinical presentations of the milder phenotype are sensorineural hearing loss, leukodystrophy, intellectual decline, ataxia, and sensorimotor neuropathy [3, 8, 9]. Most symptoms are mimicked by the constitutive Mfp2−/− mouse model which develops a progressive fatal phenotype characterized by motor problems, ataxia, weight loss, and lethargy [1, 2]. The pathomechanisms of disease and role of MFP2 in the brain remain however elusive in human and mice.
The most prominent hallmark of Mfp2−/− mice is a strong neuroinflammatory response consisting of proliferating resident microglia in the absence of neuronal loss [2, 10, 11]. Characterization of this excessive microgliosis in the brain of Mfp2−/− mice revealed that resident microglia proliferate, adopt a permanently activated non-phagocytic state, and lose their homeostatic signature [10, 11]. Specific suppression of microgliosis in Mfp2−/− mice by treatment with PLX5622, a selective colony-stimulating factor 1 receptor (CSF1R) inhibitor, failed to prevent neuronal dysfunction and clinical deterioration of Mfp2−/− mice as inflammatory responses and residual reactive microglia remained after treatment [12]. The importance of peroxisomal β-oxidation in innate immune cells is poorly understood, but Mfp2−/− mice do not show systemic inflammation, and there is no infiltration of peripheral immune cells in the brain [10].
Microglia, the primary immune effector cells in the brain, can rapidly respond to disturbances of central nervous system (CNS) homeostasis by adopting an inflammatory activation state which consists of morphological alterations, proliferation, upregulation of cell surface markers, and increased expression of inflammatory molecules [13–15]. The so-called guardians of the brain adopt resting and activated states depending on the brain environment or the insult. Chronic activation of microglia was assumed to be detrimental to proper CNS functioning, but microglial activation is in fact a delicately balanced process that constitutes both harmful and protective effects [16–18].
This early-onset aberrant phenotype of Mfp2−/− microglia gains even more interest as we previously defined that microglia in Nestin-Mfp2−/− mice develop a late-onset and mild inflammatory state [11]. The neural-specific Nestin-Mfp2−/− mouse model lacks MFP2 in neurons, astrocytes, and oligodendrocytes but not in microglia [2]. The chronic and strongly activated microglial phenotype in constitutive Mfp2−/− mice was associated with early-onset deficits in neuronal transmission, explorative behavior, and cognition. In contrast, attenuated microgliosis in Nestin-Mfp2−/− mice was associated with late-onset and minor abnormalities in neuronal function and behavior compared to Mfp2−/− mice. Whereas constitutive Mfp2−/− mice die within 4–6 months, Nestin-Mfp2−/− mice survive up to 8–12 months [2, 19]. Although the progression of microgliosis parallels clinical deterioration, it remains unknown whether the dysregulated microglial phenotype and the behavioral abnormalities are caused by cell-autonomous MFP2 dysfunction in microglia.
Therefore, to investigate the importance of MFP2 function within microglia, we generated a novel mouse model that lacks MFP2 specifically in myeloid cells by Cx3cr1-driven recombination of the Hsd17b4 gene [20]. In the brain parenchyma, the chemokine receptor CX3CR1 is exclusively expressed by microglia [21, 22]. We characterized the Cx3cr1-Mfp2−/− mice with regard to the morphological and immunological properties of microglia and examined responses of Mfp2−/− microglia in vitro and in vivo to immunological challenges and neuronal injury by facial nerve axotomy. In addition, the impact of microglia-specific deletion of MFP2 on neuronal functioning and murine behavior and cognition was assessed. Our study demonstrated that microglia-specific deletion of MFP2 from the Cx3cr1-Mfp2−/− brain leads to intrinsic alterations of microglia that develop a pro-inflammatory and proliferative phenotype but retain proper responses to inflammatory stimuli. This chronic adaptation of Mfp2−/− microglia in a genetically intact brain did however not affect neuronal transmission or murine motor function, cognition, and explorative behavior within the time frame wherein all Mfp2−/− mice have died from the disease [19].
Methods
Mouse breeding
Mfp2loxP/loxP mice in which exon 8 of the Hsd17b4 gene is flanked by LoxP sequences [2] were bred with Cx3cr1-Cre mice, which cause recombination in brain microglia, monocytes, subsets of natural killer, and dendritic cells [20], on a C57Bl6 background. Mfp2−/− mice were obtained by breeding heterozygous mice as described [2]. Genotyping was performed on ear punches. All mice were bred in the animal housing facility of the KU Leuven, had ad libitum access to water and standard rodent food, and were kept on a 12-h light and dark cycle.
Murine behavioral studies
The auditory brainstem response test (BAEP), the open field (OF) exploration, and the grip strength measurement were conducted as previously described [11]. Passive avoidance (PA) learning was examined in a cage consisting of a light and a dark compartment containing a grid floor [23]. After a 30-min adaptation to the dark, the mouse was placed in the light compartment for a training trial. After 5 s, the dark compartment was opened and step-through latency was manually recorded. When all paws were placed on the grid floor, a mild electric footshock (0.2 mA, 2 s) was applied. Retention was tested 24 h later in the dark-adapted mouse, and latency to enter the dark compartment was measured up to a 300-s cutoff value. The inverted grid test or four-limb hang test is a test of combined forepaw and hind paw strength and coordination. Mice are placed on a wired grid which is subsequently inverted. The latency to fall is recorded with a time limit of 300 s. In general, normal mice are able to remain on the inverted grid for at least 300 s. Mice that fall off the grid before the time limit of 300 s were directly given another try, and the best time was recorded. Mice that hang for the 300-s limit were placed back into the cage.
Administration of lipopolysaccharide
Mice aged 5 months and 8 months received an intraperitoneal (i.p.) injection of lipopolysaccharide (LPS) at a dose of 1 mg/kg (Sigma, L4391) or sterile saline vehicle in a total volume of 100 μl. Four hours later, mice were sacrificed, and brainstem was collected and flash frozen in liquid nitrogen for subsequent RNA analysis.
Intracerebroventricular injection of IL-4
The IL-4 injections were performed on 5-month-old mice as described previously [24]. Briefly, mice were anesthetized with ketamine and xylazine (100 and 10 mg/kg, respectively) and injected with vehicle (0.9% NaCl) or 200 ng IL-4 (R&D) in a total volume of 2.5 μl in the third cerebral ventricle using the following stereotaxic coordinates: bregma, − 0.25 mm; lateral, 1 mm; and depth, 2.25 mm. The animals were allowed to recover for 20 h, after which they were sacrificed with an overdose of Domitor and Nimatek (1 mg and 75 mg/kg, respectively). The frontal cortex contralateral to the injection site was collected and snap frozen in liquid nitrogen.
Facial nerve axotomy
The facial nerve injury experiment was performed as described [25]. Mice were 3-month-old at the time of the surgical procedure. In brief, mice were anesthetized with 3% isoflurane and placed on a 37 °C hot plate during the surgical procedure. Unilateral facial nerve transection at the stylomastoid foramen, posterior from the retroauricular branch point, was performed in Cx3cr1-Mfp2−/− and control mice. The successful outcome of the procedure was verified by ipsilateral whisker paresis immediately upon recovery from mild anesthesia. The facial nerve motor nucleus at the ipsi- and contralateral side of the injury was collected from frozen sections as described [25]. For RNA extraction, the PicoPure RNA isolation kit (Thermo Fisher Scientific) was used.
Immunohistochemical staining and quantification
Mice were anesthetized with a mix of Domitor (1 mg/kg) and Nimatek (75 mg/kg). Tissue processing and IHC staining were performed as described [1, 26, 27]. Briefly, mice were perfused transcardially with PBS (pH 7.4) followed by 4% paraformaldehyde (PFA). The brains were isolated, post-fixed with 4% PFA overnight, and kept in 70% ethanol prior to paraffin embedding. Paraffin sections (7 μm) were used for immunofluorescent stainings with polyclonal rabbit anti-Iba1 (1:500; Wako D19–19741) or rat anti-F4/80 (1:500; Serotec, Oxford, UK). For detection, HRP-labeled secondary antibodies (1:200) and fluorescent labeling with a cyanine 2 (FITC) TSA kit (Perkin Elmer Life Sciences, Boston, USA) were used. Cell nuclei were labeled with Vectashield Antifade Mounting Medium with DAPI (Vector Laboratories, United Kingdom). Images were acquired with a motorized inverted IX-81 microscope connected to a CCD-FV2T digital camera (Olympus, Aartselaar, Belgium) and processed with LSM Image Browser software (Zeiss, Germany).
Microglial cell numbers were counted around the sagittal midline and coronal plane at the height of brainstem and visual cortex. Within one plane (× 20 magnification), only Iba1-positive cells that (1) had fully co-localized with DAPI-positive nuclei, (2) had a clear cell soma, and (3) had at least two clear protrusions were counted in different regions of the brain. Microglial number per frame was corrected for surface area. Three to five different pictures per brain region per mouse were taken. The number of microglia was counted, and the average of all pictures per brain region was used to quantify the number of microglial cells per brain region (n = 4–6/group).
For the facial nerve axotomy model, brains were, after transcardial perfusion (described above), incubated in 30% sucrose in phosphate-buffered saline (PBS) at 4 °C until fully submerged. All tissues were protected from light. Samples were embedded in OCT (Tissue-Tek) for frozen sectioning on a cryostat (Leica). Cryosections from brains with unilateral lesions were prepared on slides and kept at − 20 °C until use. Tissues were rehydrated or permeabilized in blocking solution (0.1% Triton-X 100, 5% bovine albumin, normal goat serum, and PBS) for 1 h at room temperature and then incubated overnight at 4 °C with primary antibody diluted in 5% serum and PBS. The following primary antibodies were used: polyclonal rabbit anti-Iba1 (1:500; Wako 019-19,741) and rat anti-F4/80 (1:500; Serotec, Oxford, UK). Extensive wash steps were performed with PBS, and sections were incubated with goat anti-rabbit secondary antibody conjugated to Alexa Fluor 488 (1:1000; Life Technologies A32731) for 1 h at room temperature. Microglia responses were analyzed 1 day and 5 days post-axotomy. Iba1+ fluorescence intensity and numbers of microglia cells per area were measured in ipsi- and contralateral sides, and the ratio of ipsilateral side relative to their respective contralateral side is shown for both genotypes (Cx3cr1-Mfp2−/− and control mice).
Microglia isolation and cell culture
Microglial cells were isolated from control and Mfp2−/− pups at postnatal day 8 (P8) using magnetic-activated cell sorting (MACS) according to the manufacturer’s instructions (Miltenyi Biotec). Cells were plated in 12-well plates in a Macrophage Serum Free Medium (Thermo Fisher) and stimulated for 24 h with 50 ng/ml IL1β and 20 ng/ml IFNγ or 50 ng/ml IL-4 (all from R and D) to induce a pro- and anti-inflammatory phenotype, respectively.
For confirming recombination of the Mfp2 gene, microglia were isolated from 11-month-old control and Cx3cr1-Mfp2−/− mice. Mice were anesthetized with a mix of Domitor (1 mg/kg) and Nimatek (75 mg/kg) and perfused with approximately 20 ml ice-cold HBSS (without calcium and magnesium). Subsequently, brains were removed and dissociated to a single cell suspension using the Neural Tissue Dissociation Kit (P) according to the manufacturer’s instructions for the automated dissociation using the gentleMACS Dissociator (Miltenyi Biotec). Next, myelin was removed by 22% Percoll gradient and the cell suspension was further processed for microglia separation. Microglia (positive fraction) were separated from other brain cells (negative fraction) by MACS following the manufacturer’s instructions using CD11b MicroBeads (Miltenyi Biotec). Both the positive and negative fractions were further processed for qRT-PCR.
Real-time quantitative PCR
Total RNA was isolated from snap-frozen brain tissue using Trizol reagent (Invitrogen, California, USA) or from isolated or cultured microglia by using the PureLink RNA Mini Kit, both according to the manufacturer’s protocol. Subsequently, cDNA was generated from a 1-μg RNA using the QuantiTect Reverse Transcription Kit (QIAGEN, Venlo, The Netherlands). For real-time PCR, an ABI PRISM 7500 Real-Time PCR instrument (Applied Biosystems, Lennik, Belgium) was used. Primers and probes were ordered from Applied Biosystems as premade Taqman Gene Expression Assays (Il1b, Mm011336189_m1; Cx3cr1, Mm0262011_s1; Tgfbr1, Mm00436964_m1; arginase 1, Mm00475991_m1; Mrc1, Mm00485148_m1) and used as previously described [27]. Alternatively, the following genes were tested in triplicate using the PowerUp SYBR Green Master Mix (Thermo Fisher) with primers ordered from Integrated DNA Technologies (Leuven, Belgium): cd200, Mm.PT.58.33215550; Cx3cl1, Mm.PT.58.8767901; Csf1r, Mm.PT.58.12811749; Csf1, Mm.PT.58.11661276; Cxcl1, Mm.PT.58.8767901; Il34, Mm.PT.58.32379406; Fizz-1, Mm.PT.58.43062398; Il4, Mm.PT.58.7882098; Ym-1, Mm.PT.58.33370435; Tlr2, Mm.PT.58.45820113; Tspo, Mm.PT.58.43313736; F4/80 or Emr1, Mm.PT.58.11087779; Tmem119, Mm.PT.58.6766267; P2ry12, Mm.PT.58.43542033; and Mfp2, Mm.PT.58.16985875. The data were analyzed using the 2-ΔΔCT method [28]. The relative expression levels of the target genes were calculated as a ratio to the housekeeping gene β-actin except for the facial nerve injury experiment for which adaptor-related protein complex 3, delta 1 subunit (Ap3d1), F-box protein 38 (Fbxo38), and MON2 homolog (Mon2) were used that remain unaltered after neuronal injury [25]. The following primers were used: Ap3d1 (forward, 5′-CAAGGGCAGTATCGACCGC-3′; reverse, 5′-GATCTCGTCAATGCACTGGGA- 3′), Mon2 (forward, 5′-CTACAGTCCGACAG GTCGTGA-3′; reverse, 5′-CGGCACTGGAGGTTCTATATCTC-3′), and Fbxo38 (forward, 5′-ATGGGACCACGAAAG AAAAGTG-3′; reverse, 5′-TAGCTTCCGAGAGAGGCATTC-3′).
MFP2 activity measurements
The forebrain of 11-month-old mice was homogenized in 5 mM MOPS pH 7.2, 1 mM EDTA, 250 mM sucrose, and 0.1% (v/v) ethanol. After the appropriate dilution of the samples, the dehydratase activity of MFP2 was measured as previously described [29] with 3S-hydroxy-3-phenylproprionyl-CoA as a substrate, except that Thesit was increased to 0.05% (w/v).
Statistical analysis
All data except some behavioral tests (see below) were analyzed with GraphPad Prism software (version 5.0 and 6.0, San Diego, CA). Statistical analyses were carried out using unpaired and paired, two-sided Student’s t test, one-way ANOVA, two-way ANOVA, or two-way repeated measure (RM) ANOVA followed by the Bonferroni post hoc test. Data are shown as mean ± standard error of the mean (SEM), and statistical significance was set at p < 0.05. SPSS Statistics software was used for three-way ANOVA and three-way RM ANOVA.
BAEP test
Two-way ANOVA with genotype and (inter)peak as sources of variation was used to evaluate neuronal transmission in Cx3cr1-Mfp2−/− mice. The Holm-Sidak and Bonferroni methods were used for multiple comparisons. Unpaired t test was used to evaluate the peak amplitudes.
OF and PA test
Independent sample t test and Mann-Whitney U test were used to compare performance between different genotypes.
Results
Generation of microglia/monocyte-specific Cx3cr1-Mfp2−/− mice
In order to investigate whether inactivation of peroxisomal β-oxidation in microglia impacts on microglial behavior and may contribute to the neuropathology that we observed in Mfp2−/− mice, we generated a microglia/monocyte-specific knockout, by crossbreeding Cx3cr1-Cre mice [20] with floxed Mfp2 mice [2]. First, we analyzed whether insertion of the Cre-recombinase gene in the genome and haploinsufficiency of CX3CR1 did not negatively influence microglial behavior in the brain. Microglia were investigated in the brain of both Cre-positive and Cre-negative control mice at 5, 8, and 12 months of age. No differences in Iba1+ cells regarding morphology and cell numbers were observed in the brain of Cre-positive (Cre Mfp2Wt/LoxP) versus Cre-negative (Mfp2Wt/LoxP) control mice (Additional file 1: Figure S1). There were no signs of microglial reactivity, and the F4/80 marker was absent in both Cre-positive and Cre-negative control mice at all ages (data not shown). In addition, no differences in GFAP expression were observed in mice of both genotypes indicating that insertion of Cre did not influence astroglia (data not shown). Therefore, we used both Cre-positive and Cre-negative mice as control animals. Because reliable antibodies for immunohistochemical detection of MFP2 are not available [30], we confirmed the recombination of Mfp2 in microglia of Cx3cr1-Mfp2−/− mice using transcript analysis on MACS-isolated microglia. The selectivity of Mfp2 recombination was further confirmed by the normal expression of MFP2 in the non-microglia fraction and by the fact that the activity of MFP2 was not significantly reduced in whole brain homogenates (Additional file 2: Figure S2). Cx3cr1-Mfp2−/− mice were indistinguishable from their control littermates during the first year of life, were fertile and survived past the age of 14 months.
Development of microglial proliferation and morphological transformation in the brain of Cx3cr1-Mfp2−/− mice
Constitutive Mfp2−/− mice develop severe and extensive microgliosis from 6 weeks of age that progressively increases. In contrast, Nestin-Mfp2−/− mice only develop microgliosis from 12 to 17 weeks of age, which is never as extensive as in constitutive Mfp2−/− mice [11], suggesting that intrinsic loss of MFP2 from microglia might be involved in the development of microgliosis. In order to address this, we analyzed cell numbers and morphology of microglia in Cx3cr1-Mfp2−/− mice at different ages. IHC analysis and quantification of the microglial marker Iba1 revealed that numbers of microglial cells progressively increased from the age of 3 months in all brain regions, both in gray and white matter, of Cx3cr1-Mfp2−/− mice compared to age-matched control mice (Fig. 1a–l). In the control mice, microglial cell numbers were similar across all ages that were investigated, so the data were combined (Fig. 1k, l). The proliferation of microglial cells was associated with progressive morphological transformation (Fig. 1m–q). At 3 months of age, several microglia in the Cx3cr1-Mfp2−/− brain developed thicker and shorter protrusions and a mildly enlarged cell soma (Fig. 1n). These hypertrophic features became gradually more pronounced with age (Fig. 1o–q).
Fig. 1.

Development of microgliosis in Cx3cr1-Mfp2−/− brain. a–e Overview pictures of Iba1+ cells (green) in visual cortex of control (a) and Cx3cr1-Mfp2−/− mice (b–e) at the indicated ages and at higher magnification (f–j). k, l Quantification of microglial cells in the cortex (visual and motor cortex) and the brainstem of Cx3cr1-Mfp2−/− mice (n = 4 mice/age) compared to control mice. Data of control mice across different ages (3, 5, 8, 12 months) is combined (n = 16 mice). m–q Gradual morphological transformation of microglia in Cx3cr1-Mfp2−/− brain at the same ages. r–v Gene expression analysis by qRT-PCR of markers related to the proliferation of microglia in 8-month-old mice. Cx3cr1-Mfp2−/− mice compared to control: **p < 0.05, ***p < 0.001, ****p < 0.0001. ns, not significant. Error bars indicate SEM. m, months. n = 4 mice/group. Representative pictures are shown
Microglial proliferation is typically induced by the cytokines colony-stimulating factor 1 (CSF1) or interleukin-34 (IL-34) that are ligands of the CSF1R. We analyzed whether the gene expression of these cytokines was changed in the brain of Cx3cr1-Mfp2−/− mice by qPCR analysis. Transcript levels of Csf1 were not altered (Fig. 1r), but transcript levels of Il34 and Csf1r were induced (Fig. 1s, t) in 8-month-old Cx3cr1-Mfp2−/− mice in comparison to age-matched control mice.
In the healthy brain, neurons chronically restrain microglia in order to maintain their surveilling state and prevent microglial proliferation. To investigate whether the microglial proliferation was related to the loss of neuronal restraint signals, we analyzed pivotal markers in the brains of Cx3cr1-Mfp2−/− mice. We found that transcript levels of Cx3cl (fractalkine) (Fig. 1u) and Cd200 (Fig. 1v) were equal to those in age-matched control brains. Taken together, the deletion of the peroxisomal β-oxidation enzyme MFP2 from microglia induces progressive morphological changes and IL-34-driven proliferation, typical features of microglial activation. In comparison to constitutive Mfp2−/− mice, the microgliosis is less pronounced and delayed in Cx3cr1-Mfp2−/− brain [10, 11].
Microglia in Cx3cr1-Mfp2−/− mice are inflammatory activated and adopt a pro-inflammatory state at later stages
The microglia/macrophage marker F4/80 becomes upregulated on the microglial membrane when microglia get activated in response to the disruption of CNS homeostasis, such as neuronal injury, aging, or infectious pathogens in the brain parenchyma. Numbers of activated F4/80+ microglia increased with age in Cx3cr1-Mfp2−/− mice during the first year of life (Fig. 2a–f), whereas F4/80+ cells were absent in the control mice at all ages (Fig. 2a). Microglial activation is accompanied with the induction of inflammatory markers in pathological conditions. The transcript levels of the pro-inflammatory markers Tnfa, Il1b, and Tlr2 were not significantly changed at 5 months of age but were induced at 8 months of age in Cx3cr1-Mfp2−/− mice (Fig. 2g–i). We assessed whether induction of pro-inflammatory markers was associated with the downregulation of anti-inflammatory markers. Transcripts of Il4 (Fig. 2j) and Fizz (Fig. 2k) were downregulated at 8 months, but not at 5 months of age. Transcript levels of Ym1 (Fig. 2l) and Arginase-1 (Arg1) (Fig. 2m) were not significantly decreased. Together, this indicates that Mfp2−/− microglia in Cx3cr1-Mfp2−/− mice adopt an activated pro-inflammatory phenotype.
Fig. 2.

Inflammatory characteristics of Cx3cr1-Mfp2−/− brain. a–e F4/80+ cells (green) are absent in the brains of control mice (a), but F4/80+ cells increase with age in Cx3cr1-Mfp2−/− brains (b–e). Cell nuclei are stained blue with DAPI. Representative pictures of the brainstem are shown. f Quantification of F4/80+ cells in the brainstem of Cx3cr1-Mfp2−/− mice (n = 4 mice/age) compared to control mice. Data of control mice across different ages (3, 5, 8, 12 months) is combined (n = 16). g–i Transcript levels of pro-inflammatory markers in Cx3cr1-Mfp2−/− brain at 5 and 8 months of age. j–m Transcript levels of anti-inflammatory markers in Cx3cr1-Mfp2−/− brain at 5 and 8 months of age. Cx3cr1-Mfp2−/− mice compared to control: **p < 0.01, ***p < 0.001, ****p < .0001. ns, not significant. Error bars indicate SEM. n = 4–8 mice/group; m, months
Normal responses of Mfp2−/− microglia to pro- and anti-inflammatory challenges in vivo and in vitro
We subsequently assessed how microglia in Cx3cr1-Mfp2−/− mice respond to pro- and anti-inflammatory stimuli. LPS was systemically administered to 5-month-old (Fig. 3a, b) and 8-month-old (Fig. 3c, d) mice, and transcript levels of pro-inflammatory markers were monitored. Expression of Tnfa (Fig. 3a, c) and Il1b (Fig. 3b, d) were similarly induced in the Cx3cr1-Mfp2−/− and control mice after LPS injection at both ages, indicating that microglia in Cx3cr1-Mfp2−/− mice are not primed. Subsequently, we investigated responses to an anti-inflammatory stimulus by performing i.c.v. injections of IL-4 in the Cx3cr1-Mfp2−/− and control mice at 5 months of age. Both the Cx3cr1-Mfp2−/− and control mice exhibited an anti-inflammatory brain environment post-injection, evident by increased expression of anti-inflammatory markers (Arg1 and Fizz1) (Fig. 3e, f) and unaltered expression of pro-inflammatory markers (Tnf, Il1b, and Tlr2, data not shown). The data demonstrate normal responsiveness of Mfp2−/− microglia to an anti-inflammatory challenge as anti-inflammatory markers in Cx3cr1-Mfp2−/− mice were elevated to the same extent as in control mice.
Fig. 3.
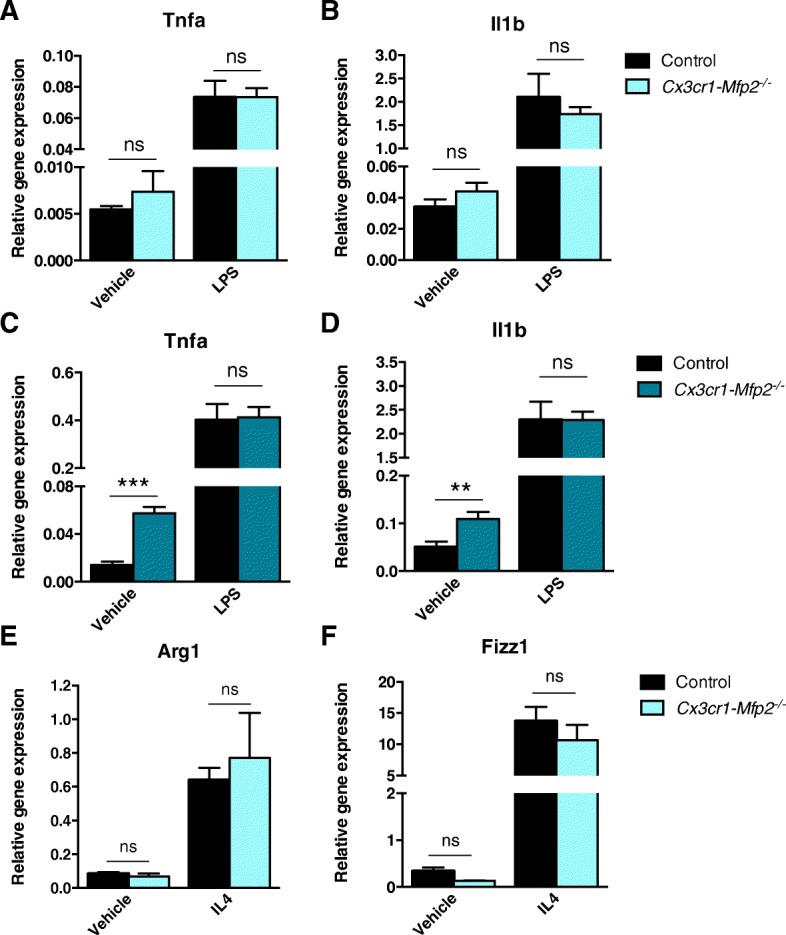
Cx3cr1-Mfp2−/−mice respond adequately to pro- and anti-inflammatory challenges. a–d Five-month-old (a, b) and 8-month-old (c, d) Cx3cr1-Mfp2−/− and control mice were challenged with i.p. LPS or vehicle, and the brainstem was analyzed after 6 h for transcript levels of pro-inflammatory markers. n = 4–6 mice/group. e, f Five-month-old Cx3cr1-Mfp2−/− and control mice were challenged with i.c.v. IL-4 or vehicle, and the frontal cortex contralateral to the injection site was analyzed after 20 h for transcript levels of anti-inflammatory markers. n = 3 mice/group. Cx3cr1-Mfp2−/− mice compared to control: **p < 0.05, ***p < 0.001. ns, not significant. Error bars indicate SEM
Finally, we tested whether cultured Mfp2−/− microglia derived from P8 Mfp2−/− mice react normally to cytokine exposure. Under basal conditions, no differences in expression levels of pro- or anti-inflammatory markers were observed (Additional file 3: Figure S3). Likewise, when challenged with a pro- or anti-inflammatory stimulus, Mfp2−/− microglia responded similarly as compared to the control microglia (Additional file 3: Figure S3). Taken together, our results show that Mfp2−/− microglia are not primed and respond normally to pro- and anti-inflammatory stimuli.
Mfp2−/− microglia in Cx3cr1-Mfp2−/− mice exhibit a normal response to neuronal injury
In order to elucidate how Mfp2−/− microglia respond to neuronal injury, we induced unilateral transection of the facial nerve (FN) in 3-month-old Cx3cr1-Mfp2−/− and control mice. The contralateral side remained intact and was considered as the control side. Facial nerve axotomy provokes a local microglial response in the facial motor nucleus (FMN) in the brainstem [31–33] which reaches a maximum at 5 days post-injury [25]. The microglial response in FMN was analyzed at day 1 (Fig. 4a–j) and at day 5 post-axotomy (Fig. 4k–t). As microglia are activated and cell numbers increased in intact 3-month-old Cx3cr1-Mfp2−/− relative to control brains (Fig. 1), Iba1+ fluorescence intensity and cell numbers in ipsilateral FMNs were measured relative to their contralateral FMNs (ratio) in both genotypes. Iba1+ intensity and cell numbers were similarly increased in Cx3cr1-Mfp2−/− and control mice after facial nerve axotomy at day 1 (Fig. 4e, j) and at day 5 post-axotomy (Fig. 4o, t). At day 1 post-injury, fold change measurements of Iba1+ fluorescence in ipsilateral FMN relative to contralateral FMN indicated that microglial response to acute neurodegeneration is similar in Cx3cr1-Mfp2−/− (1.6-fold) and control (1.4-fold) mice, in parallel with similarly increased microglial numbers in ipsilateral FMN in control (1.5-fold) and Cx3cr1-Mfp2−/− (1.7-fold) mice. This indicates that Mfp2−/− microglia respond normally to neuronal injury immediately after lesion was generated.
Fig. 4.

Microglia in Cx3cr1-Mfp2−/− brain react normally to nerve injury. a–t Facial nerve was axotomized at the left side (ipsilateral) of the brain, whereas the right facial nerve remained intact (contralateral) in 3-month-old Cx3cr1-Mfp2−/− and control mice (n = 3–5 mice/group). Microgliosis was analyzed in facial motor nucleus (FMN) in the brainstem at 1 day (a–j) and 5 days post-axotomy. a–d, k–n Overview pictures of the brainstem and FMN regions (marked by a white line) in control and Cx3cr1-Mfp2−/− mice are shown at 1 day (a–d) and 5 days (k–n) post-axotomy. f–i, p–s Magnifications of FMN in control and Cx3cr1-Mfp2−/− mice at 1 day (f–i) and 5 days (k–n) post-axotomy. Both ipsilateral and contralateral sides are shown. e, o Quantification of Iba1+ fluorescence intensity in FMN in ipsilateral sides relative to their respective contralateral sides at 1 day (e) and 5 days (o) post-axotomy. j, t Quantification of Iba1+ cell numbers in FMN in ipsilateral relative to their respective contralateral sides at 1 day (j) and 5 days (t) post-axotomy. Representative pictures are shown. Cx3cr1-Mfp2−/− mice compared to control: * p < 0.05. ns, not significant. Error bars indicate SEM
At 5 days post-axotomy, an extensive microglial response was generated in the ipsilateral FMN of both control (Fig. 4l, q) and Cx3cr1-Mfp2−/− (Fig. 4n, s) mice. We found that there was no significant difference in the induction of microglial response in the ipsilateral FMN of control (16-fold) and Cx3cr1-Mfp2−/− (13-fold) mice (Fig. 4o). Accordingly, microglial numbers are equally increased in the ipsilateral FMN of control (5.1-fold) and Cx3cr1-Mfp2−/− (4.3-fold) mice (Fig. 4t). Taken together, the neuronal injury did not provoke an exaggerated or diminished inflammatory response in Mfp2−/− microglia during the acute post-injury period. Accordingly, no differences were observed in F4/80+ microglial cells and GFAP+ astroglial cells in the ipsilateral FMNs of Cx3cr1-Mfp2−/− and control brain at day 1 and 5 post-axotomy (data not shown).
To assess the inflammatory profile of proliferated microglia, FMN at contra- and ipsilateral sides was isolated at 5 days post-axotomy from Cx3cr1-Mfp2−/− and control brain, and transcript levels of several inflammatory molecules were measured by qPCR analysis. In basal conditions, represented by the contralateral side, only the transcript levels of Iba1 were significantly induced in Cx3cr1-Mfp2−/− compared to control mice. At the axotomized ipsilateral side, the expression of inflammatory markers being the microglial marker Iba1 (Fig. 5a); activation marker F4/80 (Fig. 5b); neuroinflammatory marker Tspo (Fig. 5c); pro-inflammatory markers Tlr2, Tnfa, and Il1b (Fig. 5d, e, f); proliferative microglial receptor Csf1r (Fig. 5g); and homeostatic microglial marker Tgfbr1 (Fig. 5h) were similarly induced versus the intact contralateral FMNs in both genotypes. These results show that Mfp2−/− microglia in the Cx3cr1-Mfp2−/− brain exhibit a normal inflammatory reaction to acute neuronal injury.
Fig. 5.

Microglia in Cx3cr1-Mfp2−/− brain adopt a normal inflammatory phenotype after nerve injury. Facial nerve was axotomized at the left side (ipsilateral) of the brain, whereas the right facial nerve remained intact in 3-month-old Cx3cr1-Mfp2−/− and control. Transcript levels of inflammatory and microglial markers were analyzed in facial nuclei at 5 days post-axotomy. The expression of all markers significantly increased at the axotomized ipsilateral side (ipsi) of the brain versus intact contralateral side (contra) in both Cx3cr1-Mfp2−/− and control mice (significance levels not shown). There were no differences between genotypes at the affected ipsilateral side. Cx3cr1-Mfp2−/− mice compared to control: *p < 0.05, **p < 0.01, ****p < 0.0001. ns, not significant. n = 5 mice/group. Error bars indicate SEM
Cx3cr1-Mfp2−/− mice exhibit intact neuronal functioning and a normal clinical phenotype and cognition
We found previously that neuronal transmission of auditory signals in the brain was severely delayed and peak amplitudes were reduced in constitutive Mfp2−/− mice after evoking auditory potentials in the brainstem (BAEPs). Decreased neuronal transmission progressed in parallel with aggravating neuroinflammation [11]. In contrast, there was only a minor delay in transmission and normal peak responses of evoked auditory signals in Nestin-Mfp2−/− mice [11]. In order to reveal whether a pro-inflammatory state of microglia in Cx3cr1-Mfp2−/− mice affects auditory brainstem responses, BAEPs were analyzed in 8-month-old Cx3cr1-Mfp2−/− and control mice. Peak latencies show the time when the auditory signal evokes a response in a specific auditory nucleus in the brain [34]. Mean peak and interpeak latencies were similar in Cx3cr1-Mfp2−/− and control mice (Fig. 6a,b), and peak amplitudes are normal in Cx3cr1-Mfp2−/− versus control mice (Fig. 6c). These results demonstrate that neuronal signal transmission and amplitudes of brainstem responses are intact in Cx3cr1-Mfp2−/− mice.
Fig. 6.

Intact neuronal functioning in Cx3cr1-Mfp2−/− brain. a–c BAEP test shows normal brainstem responses in Cx3cr1-Mfp2−/− versus control mice at 8 months of age. a Mean peak latencies show that all peaks assigned to specific brainstem regions (peak 2–4) and thalamus/cortex regions (peak 5) show normal latencies of the auditory signal in Cx3cr1-Mfp2−/− mice. b Normal brain responses to the auditory stimulus were shown by similar interpeak latencies in Cx3cr1-Mfp2−/− and control mice. c Peak amplitudes are normal in Cx3cr1-Mfp2−/− versus control mice. n = 5–9 mice/group. ns, not significant. Error bars indicate SEM
Whereas constitutive Mfp2−/− mice succumb before the age of 6 months and show severely impaired locomotor activity, exploration, and fear conditioning as an index of cognition at early age, the neural-specific Nestin-Mfp2−/− mice survive up to 1 year and clinical impairments were delayed and less pronounced at the end stage of disease [11]. In order to investigate whether microglia-restricted loss of MFP2 affects grip strength, motor abilities, and behavior, the same tests were performed on Cx3cr1-Mfp2−/− mice at the age of 8 months when inflammatory activation is manifested. We found that grip strength of front paws (Fig. 7a) and all paws together (Fig. 7b) remains intact in Cx3cr1-Mfp2−/− mice. Grip strength and coordination on an inverted grid did not differ in Cx3cr1-Mfp2−/− mice and age-matched control mice (Fig. 7c). Cx3cr1-Mfp2−/− mice displayed normal locomotor activity in an open field environment as they display a similar amount of corner entries (Fig. 7d) and similar path length (Fig. 7e) as compared to control mice. Cx3cr1-Mfp2−/− mice exhibited a normal explorative behavior as the mean distance to the center (Fig. 7f), number of center entries (Fig. 7g), and time in the center (not shown) were similar to the control mice. Fear-conditioned memory was analyzed by passive avoidance test. There was no significant difference in time when Cx3cr1-Mfp2−/− and control mice enter into the dark room during training (data not shown). During the test phase, Cx3cr1-Mfp2−/− and control mice showed a similar delay in time to enter the dark room (Fig. 7h), demonstrating that the 8-month-old Cx3cr1-Mfp2−/− mice have a normal cognition. Taken together, we show that Mfp2−/− microglia in a genetically intact brain environment do not induce abnormalities in motor function, explorative behavior, and cognition before 8 months of age.
Fig. 7.

Cx3cr1-Mfp2−/−mice show a normal clinical phenotype. a, b Normal grip strength in front paws (a) and all paws together (b) in 3-, 5-, and 8-month-old Cx3cr1-Mfp2−/− mice compared to control mice. c Inverted grid test shows similar performances regarding grip strength and coordination in 8-month-old Cx3cr1-Mfp2−/− mice and control mice. d, e Eight-month-old Cx3cr1-Mfp2−/− mice show normal locomotor activity as the number of entries in the corners (d) and total path length (e) in an open field environment is similar to control mice. f, g Eight-months-old Cx3cr1-Mfp2−/− mice exhibit normal exploratory behavior as the mean distance to the center (f) and number of entries in the center (g) are similar to control mice in an open field environment. h Fear-conditioned memory was assessed by means of the passive avoidance test. Cx3cr1-Mfp2−/− show a similar delay to traverse to the dark compartment as control mice indicative of a normal cognition. ns, not significant. Error bars indicate SEM. m, months. n = 5–12 mice/group
Discussion
In this study, we investigated the impact of deletion of the pivotal peroxisomal β-oxidation enzyme MFP2 from microglia on immune response and neural functioning. Previous data demonstrated that global loss of MFP2 elicited an early-onset and extensive microgliosis in the CNS that was accompanied by quick deterioration of the mice [2, 10]. Here, we established that Mfp2−/− microglia in a genetically intact CNS environment adopt an inflammatory activated and proliferative state. Despite the pro-inflammatory microglial state, Cx3cr1-Mfp2−/− mice exhibited normal clinical performance and cognition. In addition, we found that acute inflammatory and neuronal injury provoked normal responses of Mfp2−/− microglia in Cx3cr1-Mfp2−/− mice during the post-injury period. Our data strongly suggest that MFP2 deficiency in microglia causes intrinsic pro-inflammatory deregulation, which is not harmful for neuronal function, motor function, and cognition in mice during their first year of life.
Histological examination of the Cx3cr1-Mfp2−/− brain demonstrated progressive development of microgliosis in the absence of neuronal degeneration. Our results indicate that microglial proliferation is stimulated by IL-34 rather than by CSF1 signaling. The CSF1R is ligated by both CSF1 and IL-34 which play redundant roles in developing and adult brain [35]. It was demonstrated that IL-34 binding on CSF1R promotes maintenance and proliferation of microglia in the brain [36–39]. Although both IL-34 and CSF1 may induce microglial proliferation upon neurodegeneration, IL-34 has a stronger proliferation-inducing capacity and higher expression in the brain compared to CSF1 [35, 40]. Despite identical signaling pathways induced by CSF1 and IL-34 downstream CSF1R, a recent study found that IL-34-stimulated monocytes produce different cytokines/chemokines in an inflammatory context and exhibit a distinct polarization potential compared to CSF1-derived macrophages [41]. There is indeed increasing evidence that IL-34 expression is upregulated in pathological conditions and plays important roles in autoimmune disorders, infections, and inflammatory conditions [42]. Previous results showed that Il34 levels were also increased in constitutive Mfp2−/− mice (eightfold) and neural-specific Nestin-Mfp2−/− mice (fivefold) [11], whereas Csf1 levels were unchanged in both mouse models at the end stage of disease. Further research is however necessary to elucidate the distinct biological profile and effects of both cytokines.
The proliferation of microglia in Cx3cr1-Mfp2−/− mice was accompanied with morphological transformation characterized by a swollen cell soma and thicker and shorter protrusions, both typical features of microglial activation [15, 43]. A progressive expansion of F4/80+ cells in Cx3cr1-Mfp2−/− brains verified that increasing numbers of Mfp2−/− microglia are activated from at least 3 months of age. In contrast to Mfp2−/− microglia in constitutive Mfp2−/− mice, which adopt a mixed pro- and anti-inflammatory phenotype, microglia in Cx3cr1-Mfp2−/− mice downregulate anti-inflammatory and induce pro-inflammatory cytokines from 8 months of age. This demonstrates that Mfp2−/− microglia in an intact CNS environment adopt a pro-inflammatory state several months after microglial proliferation and activation were initiated. These observations are in line with the pro-inflammatory profile of BV2 microglia in which another peroxisomal β-oxidation enzyme, ACOX1, was inactivated [44].
It should be noted that transient Cx3cr1 promoter activity has been detected in neurons during the development in some mouse models [45]. However, in Cx3cr1-Mfp2−/− mice, we did not find evidence for the neuronal inactivation of MFP2 based on normal Mfp2 transcripts in non-microglial cells and the preservation of MFP2 activity in whole brain homogenates of Cx3cr1-Mfp2−/− mice. The latter is compatible with the fact that microglia only constitute approximately 10% of brain cells [46, 47] and that MFP2 expression in microglia is lower or similar to the more abundant cell types [48]. An additional argument supporting that the reactive microglial phenotype does not depend on the neuronal inactivation of MFP2 is the fact that neuronal deficits such as Purkinje cell degeneration were not observed in Cx3cr1-Mfp2−/− mice, in contrast to constitutive Mfp2 knockouts [2]. Finally, microglial proliferation and transformation were not only seen in gray but also in white matter regions.
We also examined how Mfp2−/− microglia respond to acute inflammatory stimuli or to neuronal injury at an early disease stage, before inflammatory polarization was established. Mfp2−/− microglia show normal responses to acute inflammatory stimuli in vitro and in vivo. Mfp2−/− microglia acquired anti-inflammatory properties in response to an IL-4 stimulus which were indistinguishable from control microglia. An acute systemic pro-inflammatory stimulus elicited a similar response in microglia in Cx3cr1-Mfp2−/− compared to control mice, proving that microglia in Cx3cr1-Mfp2−/− mice are not primed despite their inflammatory activated state in unstimulated conditions. In contrast, Mfp2−/− microglia in Mfp2−/− mice are primed in the absence of systemic inflammation, and neuronal transmission is severely disturbed, suggesting that dysfunctional microglia-neuron bidirectional communication might trigger microglial priming and rapid progression of disease in constitutive Mfp2−/− mice [11]. Neuronal injury was induced by unilateral axotomy of the facial nerve of Cx3cr1-Mfp2−/− mice. We found that microglial responses in the axotomized relative to the intact FMN in Cx3cr1-Mfp2−/− and control brain were comparable. Similar expression levels of several pro- and anti-inflammatory molecules in the ipsilateral FMN of Cx3cr1-Mfp2−/− and control mice confirmed that Mfp2−/− microglia do not elicit an exaggerated response to nerve injury during the acute post-injury period. In conclusion, microglia in Cx3cr1-Mfp2−/− mice respond properly to acute pro- and anti-inflammatory challenges and to neuronal injury.
Our previous study demonstrated clear differences in the neuropathology of constitutive Mfp2−/− mice versus neural-specific Nestin-Mfp2−/− mice that were paralleled by distinct microglial phenotypes, indicating that Mfp2−/− microglia play a role in the severe neuropathology of Mfp2−/− mice [11]. However, we found that selective deletion of MFP2 from microglia in the Cx3cr1-Mfp2−/− brain did not affect grip strength, locomotor activity, explorative behavior, and fear conditioning as an index of cognitive function within the time frame wherein all Mfp2−/− mice and Nestin-Mfp2−/− mice have died from the disease [19]. Hence, the early-onset clinical and behavioral abnormalities in Mfp2−/− mice can neither be assigned purely to neuronal deficits nor to intrinsic microglial pathology. Likewise, the BAEP test demonstrated that brainstem responses and peak amplitudes are normal in Cx3cr1-Mfp2−/− mice in contrast to affected responses and amplitudes in Mfp2−/− mice and to a lesser extent in Nestin-Mfp2−/− mice [11]. In the healthy brain, neurons persistently restrain microglia in order to maintain their surveilling state. Neurons in danger downregulate these restraint signals and send out “help me” signals that trigger microglial activation in pathological situations [15, 49, 50]. In Mfp2−/− brain, the impaired neuronal signaling in the BAEP test was associated with lowered expression of the neuronal restraint signals Cx3cl and Cd200. In accordance to intact neuronal functioning in Cx3cr1-Mfp2−/− mice, the expression of these neuron-microglia signaling molecules was normal in Cx3cr1-Mfp2−/− brain. This indicates that intrinsic microglial pathology by itself is not sufficient to cause early-onset dysfunctional neuronal transmission in Mfp2−/− mice. Taking into account all data on microglial reactivity, neuronal functioning, and neuropathological features, we hypothesize that impaired functioning of the CNS and clinical deterioration in Mfp2−/− deficiency occur through synergistic instability of distinct brain cell types in Mfp2−/− mice.
Conclusion
We demonstrated in this study that Mfp2−/− microglia in a genetically intact brain environment intrinsically adopt a proliferative and modified inflammatory state. The mild pro-inflammatory phenotype acquired by Mfp2−/− microglia does not give rise to neuronal dysfunction nor to abnormal clinical behavior. Nevertheless, we cannot exclude that microglia become neurotoxic at a later stage. Our data indicate that microglia can develop a chronically proliferative and pro-inflammatory phenotype through cell-autonomous dysfunction without affecting the CNS environment and murine clinical behavior.
Additional files
Figure S1. No microgliosis in both Cre-positive and Cre-negative control mice. (A-D) No differences in microglia number and shape are observed between Cre-positive (Cre Mfp2Wt/LoxP) and Cre-negative (Mfp2Wt/LoxP) control mice at 5 months of age (A-D) and 12 months of age (not shown). Representative pictures are shown. n = 3–5 mice/group. (PPTX 988 kb)
Figure S2. Efficient and selective inactivation of MFP2 in microglia in Cx3cr1-Mfp2−/− mice. (A) Microglia were isolated from 11-month-old control and Cx3cr1-Mfp2−/− mice, and microglia purity was confirmed by the high expression of microglial markers (Tmem119 and P2ry12) in the positive (microglia) versus the negative fraction (neurons, astrocytes, and oligodendrocytes). Transcript expression of Mfp2 was determined in control and Cx3cr1-Mfp2−/− in both positive and negative fraction. Representative experiment out of two with similar results. (B) MFP2 activity in brain homogenates of control and Cx3cr1-Mfp2−/− mice. n = 2–3 mice/group. Mean ± SD is shown. (PPTX 46 kb)
Figure S3. Inflammatory properties of cultured Mfp2−/− and control microglia. MACS-isolated microglia from P8 mice were kept either in basal conditions or polarized to a pro-inflammatory state (Il1β/IFNγ) or an anti-inflammatory state (IL4). Transcript expression of pro-inflammatory (Tnfa, iNOS, Cxcl1) and anti-inflammatory cytokines (Arg1, Fizz1, Ym1) were determined. Significance levels: Φ p < 0.05, ΦΦ p < 0.01, ΦΦΦΦ p < 0.0001; ns, not significant. n = 8–11 mice/group. (PPTX 59 kb)
Acknowledgements
The authors are grateful to Steffen Jung (Weizmann Institute of Science, Rehovot, Israel) for generously providing Cx3cr1-Cre mice. They thank Yannick Das and Prof Paul Van Veldhoven for assistance with measuring MFP2 enzyme activity, and Benny Das and Ann Bouché for excellent technical assistance.
Funding
This work was funded by grants from Fonds Wetenschappelijk Onderzoek Vlaanderen (G.0675.12 and G.0A15.13), KU Leuven (OT12/78), and ERA-Net Neuron (MICRO-MET).
Availability of data and materials
All data generated or analysed during this study are included in this published article and its supplementary information files.
Abbreviations
- BAEP
Brainstem auditory evoked potentials
- CNS
Central nervous system
- CSF1R
Colony-stimulating factor 1 receptor
- FMN
Facial motor nucleus
- i.c.v.
Intracerebroventricular
- IHC
Immunohistochemistry
- Il
Interleukin
- LPS
Lipopolysaccharide
- MFP2
Multifunctional protein 2
- OF
Open field
- PA
Passive avoidance
Authors’ contributions
LB performed most of the experiments, analyzed and interpreted the data, and wrote the manuscript. SB assisted with the analysis of the facial nerve axotomy experiment. IG performed, analyzed, and reported the i.c.v. IL-4 experiment, microglia isolations, and culturing. SS and RH supervised the behavioral experiments. MB guided the project and was a major contributor in writing the manuscript. All authors read and approved the final manuscript.
Ethics approval
All experiments were conducted in accordance with “Guidelines for Care and Use of Experimental Animals” and fully approved by the Research Ethical committee of the KU Leuven (#190/2012, #181/2015).
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Lien Beckers, Email: Lien.beckers@hotmail.com.
Ivana Geric, Email: Ivana.geric@kuleuven.be.
Stijn Stroobants, Email: Stijn.stroobants@kuleuven.be.
Sander Beel, Email: sander_beel@hotmail.com.
Philip Van Damme, Email: Philip.vandamme@kuleuven.be.
Rudi D’Hooge, Email: rudi.dhooge@kuleuven.be.
Myriam Baes, Phone: + 32 16 330853, Email: Myriam.Baes@kuleuven.be.
References
- 1.Huyghe S, Schmalbruch H, Hulshagen L, Veldhoven PV, Baes M, Hartmann D. Peroxisomal multifunctional protein-2 deficiency causes motor deficits and glial lesions in the adult central nervous system. Am J Pathol. 2006;168(4):1321–1334. doi: 10.2353/ajpath.2006.041220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Verheijden S, Bottelbergs A, Krysko O, Krysko DV, Beckers L, De Munter S, Van Veldhoven PP, Wyns S, Kulik W, Nave KA, et al. Peroxisomal multifunctional protein-2 deficiency causes neuroinflammation and degeneration of Purkinje cells independent of very long chain fatty acid accumulation. Neurobiol Dis. 2013;58:258–269. doi: 10.1016/j.nbd.2013.06.006. [DOI] [PubMed] [Google Scholar]
- 3.Ferdinandusse S, Denis S, Mooyer PA, Dekker C, Duran M, Soorani-Lunsing RJ, Boltshauser E, Macaya A, Gartner J, Majoie CB, et al. Clinical and biochemical spectrum of D-bifunctional protein deficiency. Ann Neurol. 2006;59(1):92–104. doi: 10.1002/ana.20702. [DOI] [PubMed] [Google Scholar]
- 4.Huyghe S, Mannaerts GP, Baes M, Van Veldhoven PP. Peroxisomal multifunctional protein-2: the enzyme, the patients and the knockout mouse model. Biochim Biophys Acta. 2006;1761(9):973–994. doi: 10.1016/j.bbalip.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 5.Van Veldhoven PP. Biochemistry and genetics of inherited disorders of peroxisomal fatty acid metabolism. J Lipid Res. 2010;51(10):2863–2895. doi: 10.1194/jlr.R005959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lines MA, Jobling R, Brady L, Marshall CR, Scherer SW, Rodriguez AR, Lee L, Lang AE, Mestre TA, Wanders RJ, et al. Peroxisomal D-bifunctional protein deficiency: three adults diagnosed by whole-exome sequencing. Neurology. 2014;82(11):963–968. doi: 10.1212/WNL.0000000000000219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lieber DS, Hershman SG, Slate NG, Calvo SE, Sims KB, Schmahmann JD, Mootha VK. Next generation sequencing with copy number variant detection expands the phenotypic spectrum of HSD17B4-deficiency. BMC Med Genet. 2014;15:30. doi: 10.1186/1471-2350-15-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Khan A, Wei XC, Snyder FF, Mah JK, Waterham H, Wanders RJ. Neurodegeneration in D-bifunctional protein deficiency: diagnostic clues and natural history using serial magnetic resonance imaging. Neuroradiology. 2010;52(12):1163–1166. doi: 10.1007/s00234-010-0768-4. [DOI] [PubMed] [Google Scholar]
- 9.Pierce SB, Walsh T, Chisholm KM, Lee MK, Thornton AM, Fiumara A, Opitz JM, Levy-Lahad E, Klevit RE, King MC. Mutations in the DBP-deficiency protein HSD17B4 cause ovarian dysgenesis, hearing loss, and ataxia of Perrault syndrome. Am J Hum Genet. 2010;87(2):282–288. doi: 10.1016/j.ajhg.2010.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Verheijden S, Beckers L, Casazza A, Butovsky O, Mazzone M, Baes M. Identification of a chronic non-neurodegenerative microglia activation state in a mouse model of peroxisomal beta-oxidation deficiency. Glia. 2015;63(9):1606–1620. doi: 10.1002/glia.22831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Beckers L, Stroobants S, D'Hooge R, Baes M. Neuronal dysfunction and behavioral abnormalities are evoked by neural cells and aggravated by inflammatory microglia in peroxisomal beta-oxidation deficiency. Front Cell Neurosci. 2018;12:136. doi: 10.3389/fncel.2018.00136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Beckers L, Stroobants S, Verheijden S, West B, D'Hooge R, Baes M. Specific suppression of microgliosis cannot circumvent the severe neuropathology in peroxisomal beta-oxidation-deficient mice. Mol Cell Neurosci. 2017;80:123–133. doi: 10.1016/j.mcn.2017.03.004. [DOI] [PubMed] [Google Scholar]
- 13.Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, Littman DR, Dustin ML, Gan WB. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci. 2005;8(6):752–758. doi: 10.1038/nn1472. [DOI] [PubMed] [Google Scholar]
- 14.Wyss-Coray T, Mucke L. Inflammation in neurodegenerative disease--a double-edged sword. Neuron. 2002;35(3):419–432. doi: 10.1016/S0896-6273(02)00794-8. [DOI] [PubMed] [Google Scholar]
- 15.Saijo K, Glass CK. Microglial cell origin and phenotypes in health and disease. Nat Rev Immunol. 2011;11(11):775–787. doi: 10.1038/nri3086. [DOI] [PubMed] [Google Scholar]
- 16.Crotti A, Ransohoff RM. Microglial physiology and pathophysiology: insights from genome-wide transcriptional profiling. Immunity. 2016;44(3):505–515. doi: 10.1016/j.immuni.2016.02.013. [DOI] [PubMed] [Google Scholar]
- 17.Neumann H, Kotter MR, Franklin RJ. Debris clearance by microglia: an essential link between degeneration and regeneration. Brain. 2009;132(Pt 2):288–295. doi: 10.1093/brain/awn109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Michell-Robinson MA, Touil H, Healy LM, Owen DR, Durafourt BA, Bar-Or A, Antel JP, Moore CS. Roles of microglia in brain development, tissue maintenance and repair. Brain. 2015;138(Pt 5):1138–1159. doi: 10.1093/brain/awv066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Verheijden S, Beckers L, De Munter S, Van Veldhoven PP, Baes M. Central nervous system pathology in MFP2 deficiency: insights from general and conditional knockout mouse models. Biochimie. 2014;98:119–126. doi: 10.1016/j.biochi.2013.08.009. [DOI] [PubMed] [Google Scholar]
- 20.Yona S, Kim KW, Wolf Y, Mildner A, Varol D, Breker M, Strauss-Ayali D, Viukov S, Guilliams M, Misharin A, et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity. 2013;38(1):79–91. doi: 10.1016/j.immuni.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cardona AE, Pioro EP, Sasse ME, Kostenko V, Cardona SM, Dijkstra IM, Huang D, Kidd G, Dombrowski S, Dutta R, et al. Control of microglial neurotoxicity by the fractalkine receptor. Nat Neurosci. 2006;9(7):917–924. doi: 10.1038/nn1715. [DOI] [PubMed] [Google Scholar]
- 22.Wolf Y, Yona S, Kim KW, Jung S. Microglia, seen from the CX3CR1 angle. Front Cell Neurosci. 2013;7:26. doi: 10.3389/fncel.2013.00026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.D'Hooge R, Lullmann-Rauch R, Beckers T, Balschun D, Schwake M, Reiss K, von Figura K, Saftig P. Neurocognitive and psychotiform behavioral alterations and enhanced hippocampal long-term potentiation in transgenic mice displaying neuropathological features of human alpha-mannosidosis. J Neurosci. 2005;25(28):6539–6549. doi: 10.1523/JNEUROSCI.0283-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pepe G, Calderazzi G, De Maglie M, Villa AM, Vegeto E. Heterogeneous induction of microglia M2a phenotype by central administration of interleukin-4. J Neuroinflammation. 2014;11:211. doi: 10.1186/s12974-014-0211-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Beel S, Moisse M, Damme M, De Muynck L, Robberecht W, Van Den Bosch L, Saftig P, Van Damme P. Progranulin functions as a cathepsin D chaperone to stimulate axonal outgrowth in vivo. Hum Mol Genet. 2017;26(15):2850–2863. doi: 10.1093/hmg/ddx162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hulshagen L, Krysko O, Bottelbergs A, Huyghe S, Klein R, Van Veldhoven PP, De Deyn PP, D'Hooge R, Hartmann D, Baes M. Absence of functional peroxisomes from mouse CNS causes dysmyelination and axon degeneration. J Neurosci. 2008;28(15):4015–4027. doi: 10.1523/JNEUROSCI.4968-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bottelbergs A, Verheijden S, Van Veldhoven PP, Just W, Devos R, Baes M. Peroxisome deficiency but not the defect in ether lipid synthesis causes activation of the innate immune system and axonal loss in the central nervous system. J Neuroinflammation. 2012;9:61. doi: 10.1186/1742-2094-9-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods. 2001;25(4):402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 29.Baes M, Huyghe S, Carmeliet P, Declercq PE, Collen D, Mannaerts GP, Van Veldhoven PP. Inactivation of the peroxisomal multifunctional protein-2 in mice impedes the degradation of not only 2-methyl-branched fatty acids and bile acid intermediates but also of very long chain fatty acids. J Biol Chem. 2000;275(21):16329–16336. doi: 10.1074/jbc.M001994200. [DOI] [PubMed] [Google Scholar]
- 30.De Munter S, Bamps D, Malheiro AR, Kumar Baboota R, Brites P, Baes M. Autonomous Purkinje cell axonal dystrophy causes ataxia in peroxisomal multifunctional protein-2 deficiency. Brain Pathol. 2018;28(5):631–643. doi: 10.1111/bpa.12586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moran LB, Graeber MB. The facial nerve axotomy model. Brain Res Brain Res Rev. 2004;44(2–3):154–178. doi: 10.1016/j.brainresrev.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 32.Tay TL, Mai D, Dautzenberg J, Fernandez-Klett F, Lin G, Sagar DM, Drougard A, Stempfl T, Ardura-Fabregat A, et al. A new fate mapping system reveals context-dependent random or clonal expansion of microglia. Nat Neurosci. 2017;20(6):793–803. doi: 10.1038/nn.4547. [DOI] [PubMed] [Google Scholar]
- 33.Krasemann S, Madore C, Cialic R, Baufeld C, Calcagno N, El Fatimy R, Beckers L, O'Loughlin E, Xu Y, Fanek Z, et al. The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity. 2017;47(3):566–581. doi: 10.1016/j.immuni.2017.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Singer W, Panford-Walsh R, Knipper M. The function of BDNF in the adult auditory system. Neuropharmacology. 2014;76(Pt C):719–728. doi: 10.1016/j.neuropharm.2013.05.008. [DOI] [PubMed] [Google Scholar]
- 35.Wei S, Nandi S, Chitu V, Yeung YG, Yu W, Huang M, Williams LT, Lin H, Stanley ER. Functional overlap but differential expression of CSF-1 and IL-34 in their CSF-1 receptor-mediated regulation of myeloid cells. J Leukoc Biol. 2010;88(3):495–505. doi: 10.1189/jlb.1209822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zelante T, Ricciardi-Castagnoli P. The yin-yang nature of CSF1R-binding cytokines. Nat Immunol. 2012;13(8):717–719. doi: 10.1038/ni.2375. [DOI] [PubMed] [Google Scholar]
- 37.Greter M, Lelios I, Pelczar P, Hoeffel G, Price J, Leboeuf M, Kundig TM, Frei K, Ginhoux F, Merad M, et al. Stroma-derived interleukin-34 controls the development and maintenance of langerhans cells and the maintenance of microglia. Immunity. 2012;37(6):1050–1060. doi: 10.1016/j.immuni.2012.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mizuno T, Doi Y, Mizoguchi H, Jin S, Noda M, Sonobe Y, Takeuchi H, Suzumura A. Interleukin-34 selectively enhances the neuroprotective effects of microglia to attenuate oligomeric amyloid-beta neurotoxicity. Am J Pathol. 2011;179(4):2016–2027. doi: 10.1016/j.ajpath.2011.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang Y, Szretter KJ, Vermi W, Gilfillan S, Rossini C, Cella M, Barrow AD, Diamond MS, Colonna M. IL-34 is a tissue-restricted ligand of CSF1R required for the development of Langerhans cells and microglia. Nat Immunol. 2012;13(8):753–760. doi: 10.1038/ni.2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gomez-Nicola D, Fransen NL, Suzzi S, Perry VH. Regulation of microglial proliferation during chronic neurodegeneration. J Neurosci. 2013;33(6):2481–2493. doi: 10.1523/JNEUROSCI.4440-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Boulakirba S, Pfeifer A, Mhaidly R, Obba S, Goulard M, Schmitt T, Chaintreuil P, Calleja A, Furstoss N, Orange F, et al. IL-34 and CSF-1 display an equivalent macrophage differentiation ability but a different polarization potential. Sci Rep. 2018;8(1):256. doi: 10.1038/s41598-017-18433-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Baghdadi M, Umeyama Y, Hama N, Kobayashi T, Han N, Wada H, Seino KI. Interleukin-34, a comprehensive review. J Leukoc Biol. 2018;104(5):931–951. doi: 10.1002/JLB.MR1117-457R. [DOI] [PubMed] [Google Scholar]
- 43.Salter MW, Beggs S. Sublime microglia: expanding roles for the guardians of the CNS. Cell. 2014;158(1):15–24. doi: 10.1016/j.cell.2014.06.008. [DOI] [PubMed] [Google Scholar]
- 44.Raas Q, Saih FE, Gondcaille C, Trompier D, Hamon Y, Leoni V, Caccia C, Nasser B, Jadot M, Menetrier F, et al. A microglial cell model for acyl-CoA oxidase 1 deficiency. Biochim Biophys Acta Mol Cell Biol Lipids. 2018;1864(4):567–76. doi: 10.1016/j.bbalip.2018.10.005. [DOI] [PubMed] [Google Scholar]
- 45.Haimon Z, Volaski A, Orthgiess J, Boura-Halfon S, Varol D, Shemer A, Yona S, Zuckerman B, David E, Chappell-Maor L, et al. Re-evaluating microglia expression profiles using RiboTag and cell isolation strategies. Nat Immunol. 2018;19(6):636–644. doi: 10.1038/s41590-018-0110-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lawson LJ, Perry VH, Dri P, Gordon S. Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience. 1990;39(1):151–170. doi: 10.1016/0306-4522(90)90229-W. [DOI] [PubMed] [Google Scholar]
- 47.Pelvig DP, Pakkenberg H, Stark AK, Pakkenberg B. Neocortical glial cell numbers in human brains. Neurobiol Aging. 2008;29(11):1754–1762. doi: 10.1016/j.neurobiolaging.2007.04.013. [DOI] [PubMed] [Google Scholar]
- 48.Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O'Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci. 2014;34(36):11929–11947. doi: 10.1523/JNEUROSCI.1860-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Polazzi E, Monti B. Microglia and neuroprotection: from in vitro studies to therapeutic applications. Prog Neurobiol. 2010;92(3):293–315. doi: 10.1016/j.pneurobio.2010.06.009. [DOI] [PubMed] [Google Scholar]
- 50.Li Y, Du XF, Liu CS, Wen ZL, Du JL. Reciprocal regulation between resting microglial dynamics and neuronal activity in vivo. Dev Cell. 2012;23(6):1189–1202. doi: 10.1016/j.devcel.2012.10.027. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. No microgliosis in both Cre-positive and Cre-negative control mice. (A-D) No differences in microglia number and shape are observed between Cre-positive (Cre Mfp2Wt/LoxP) and Cre-negative (Mfp2Wt/LoxP) control mice at 5 months of age (A-D) and 12 months of age (not shown). Representative pictures are shown. n = 3–5 mice/group. (PPTX 988 kb)
Figure S2. Efficient and selective inactivation of MFP2 in microglia in Cx3cr1-Mfp2−/− mice. (A) Microglia were isolated from 11-month-old control and Cx3cr1-Mfp2−/− mice, and microglia purity was confirmed by the high expression of microglial markers (Tmem119 and P2ry12) in the positive (microglia) versus the negative fraction (neurons, astrocytes, and oligodendrocytes). Transcript expression of Mfp2 was determined in control and Cx3cr1-Mfp2−/− in both positive and negative fraction. Representative experiment out of two with similar results. (B) MFP2 activity in brain homogenates of control and Cx3cr1-Mfp2−/− mice. n = 2–3 mice/group. Mean ± SD is shown. (PPTX 46 kb)
Figure S3. Inflammatory properties of cultured Mfp2−/− and control microglia. MACS-isolated microglia from P8 mice were kept either in basal conditions or polarized to a pro-inflammatory state (Il1β/IFNγ) or an anti-inflammatory state (IL4). Transcript expression of pro-inflammatory (Tnfa, iNOS, Cxcl1) and anti-inflammatory cytokines (Arg1, Fizz1, Ym1) were determined. Significance levels: Φ p < 0.05, ΦΦ p < 0.01, ΦΦΦΦ p < 0.0001; ns, not significant. n = 8–11 mice/group. (PPTX 59 kb)
Data Availability Statement
All data generated or analysed during this study are included in this published article and its supplementary information files.