

Abstract
A proportion of patients with certain types of interstitial lung disease (ILD), including chronic hypersensitivity pneumonitis and ILDs associated with autoimmune diseases, develop a progressive fibrosing phenotype that shows similarities in clinical course to idiopathic pulmonary fibrosis. Irrespective of the clinical diagnosis, these progressive fibrosing ILDs show commonalities in the underlying pathogenetic mechanisms that drive a self-sustaining process of pulmonary fibrosis. The natural history of progressive fibrosing ILDs is characterized by decline in lung function, worsening of symptoms and health-related quality of life, and early mortality. Greater impairment in forced vital capacity or diffusion capacity of the lungs for carbon monoxide, and a greater extent of fibrotic changes on a computed tomography scan, are predictors of mortality in patients with fibrosing ILDs. However, the course of these diseases is heterogenous and cannot accurately be predicted for an individual patient. Data from ongoing clinical trials and patient registries will provide a better understanding of the clinical course and impact of progressive fibrosing ILDs.
Keywords: Pulmonary fibrosis, Connective tissue diseases, Rheumatic diseases, Systemic sclerosis, Vital capacity, Mortality
Background
Interstitial lung diseases (ILDs) encompass a large and varied group of parenchymal lung disorders, including diseases of unknown cause known as the idiopathic interstitial pneumonias (IIPs), as well as those associated with other diseases or environmental exposures. The most extensively studied type of ILD is idiopathic pulmonary fibrosis (IPF), which occurs mainly in adults aged over 60 years and is characterized by progressive pulmonary fibrosis, decline in lung function and high mortality [1]. A proportion of patients with other types of ILD also develop a progressive fibrosing phenotype that shows similarities in underlying pathogenetic mechanisms and clinical behavior to IPF [2, 3]. ILDs that may be associated with a progressive fibrosing phenotype include connective tissue disease-related ILDs (CTD-ILDs) such as those related to rheumatoid arthritis (RA-ILD) [4], systemic sclerosis (SSc-ILD) [5], and polymyositis/dermatomyositis [6]; ILD related to chronic sarcoidosis [7]; chronic hypersensitivity pneumonitis (HP) [8]; idiopathic non-specific interstitial pneumonia (iNSIP) [9] and unclassifiable ILD [10] (Fig. 1). The proportion of patients with non-IPF ILDs who develop a progressive fibrosing phenotype is not known, but has been estimated by physicians who manage patients with non-IPF ILDs to be up to 40% [11, 12]. In this review, we will describe the natural history of progressive fibrosing ILDs.
Fig. 1.
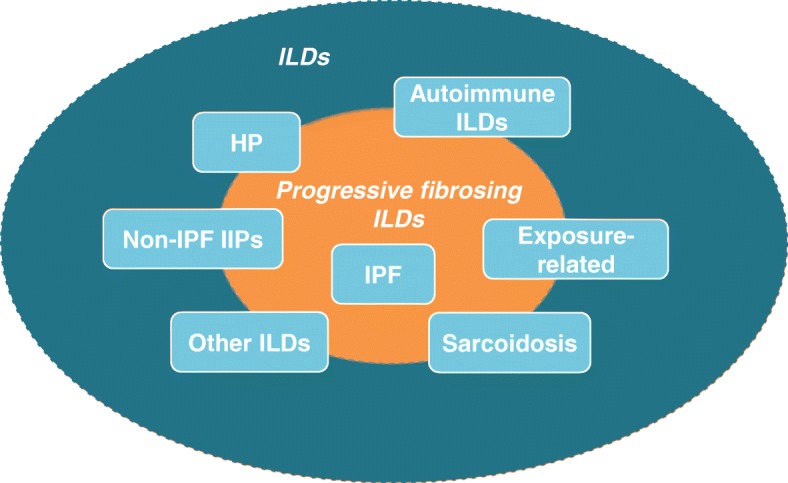
Types of interstitial lung disease associated with a risk of developing a progressive fibrosing phenotype. HP, hypersensitivity pneumonitis; ILD, interstitial lung disease; IIP, idiopathic interstitial pneumonia; IPF, idiopathic pulmonary fibrosis
Pathogenesis of progressive fibrosing ILDs
Some ILDs are primarily considered fibro-proliferative disorders, in which alveolar epithelial injury and fibroblastic proliferation lead to fibrosis, while other ILDs are considered primarily inflammatory disorders in which the pathogenic process shifts to a fibro-proliferative pathway under certain conditions [13, 14]. Regardless of the trigger, fibrosing ILDs show commonalities in the mechanisms involved in their pathogenesis and progression. Repeated chronic epithelial or vascular injuries lead to cell destruction and to unregulated repair [15–17]. Fibroblasts proliferate, migrate from different sources to the site of injury and are activated to become myofibroblasts, which secrete increased amounts of extracellular matrix. This, together with reduced matrix degradation, results in increased tissue stiffness and loss of function of the alveolar tissue [18–22]. Macrophages and lymphocytes are recruited to the site of injury and release pro-fibrotic mediators that further promote fibroblast activation [18]. In a feed-forward loop, the increased lung tissue stiffness further activates and stimulates fibroblasts to drive a self-sustaining process of fibrosis [23, 24]. As an increasing extent of the lung is lost to fibrosis, the volume of the lung is reduced and gas exchange impaired, resulting in worsening breathlessness and capacity for exertion, and ultimately in respiratory failure.
Lung function decline in progressive fibrosing ILDs
Progression of fibrosing ILD is reflected in a decline in lung function, worsening of symptoms and deterioration in health-related quality of life (Fig. 2) [25–28]. In clinical studies, disease progression is most commonly assessed through measurement of forced vital capacity (FVC) and diffusion capacity of the lungs for carbon monoxide (DLco). The course of lung function decline in patients with IPF is variable, but IPF is, by definition, a progressive disease [1]. Other fibrosing ILDs are also heterogeneous but progressive in their clinical course. In patients with SSc, ILD is more common in patients who have diffuse cutaneous disease or are positive for anti-topoisomerase I antibodies [29]. The risk of development and progression of ILD seems to be greatest in the few years after diagnosis. Among 695 patients with SSc in the European League Against Rheumatism (EULAR) Scleroderma Trials and Research group (EUSTAR) cohort, approximately one third of patients had a DLco < 50% predicted within three years of the onset of Raynaud’s phenomenon [30]. A recent study of 81 patients at a specialized SSc-ILD clinic who were followed for ≥8 years after diagnosis or died during follow-up identified three distinct subgroups with different rates of FVC decline and mortality (Fig. 3). In all the subgroups, FVC decline was largely linear over the long term, but was too variable over the short term to enable recent change to be used to predict future change [31]. Patients with other CTD-ILDs may also suffer rapid loss of lung function after ILD develops. In an analysis of 167 patients with RA-ILD referred to a single tertiary care center, the proportion of patients with FVC < 50% predicted increased from 14% at diagnosis to 22% after 5 years (Fig. 4) [4]. In an analysis of 107 patients with ILD associated with polymyositis/dermatomyositis, 16% had a decline in FVC of ≥10% predicted and/or a decline in DLco of ≥15% predicted over a median follow-up of 34 months, despite treatment [6].
Fig. 2.
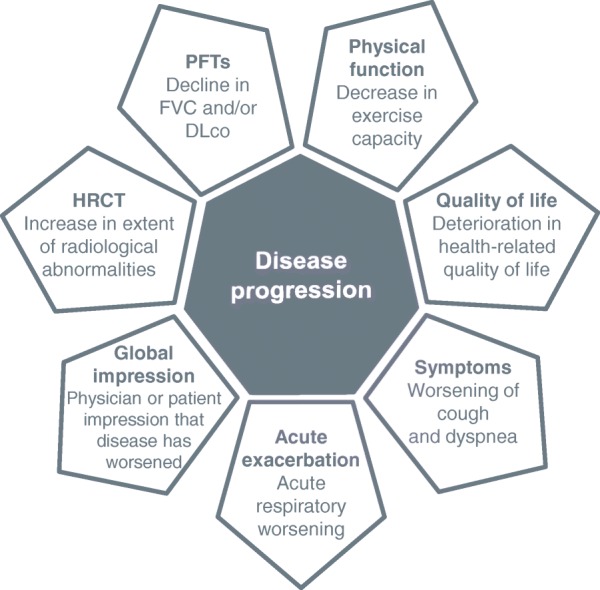
Factors that reflect progression of interstitial lung diseases. DLco, diffusion capacity of the lungs for carbon monoxide; FVC, forced vital capacity; HRCT, high-resolution computed tomography; PFTs, pulmonary function tests
Fig. 3.

Decline in FVC % predicted in patients with SSc-ILD categorized by survival time from diagnosis of ILD. Adapted from [31]. Reprinted with permission of the American Thoracic Society. Copyright© 2019 American Thoracic Society. Guler SA et al. 2018. Does systemic sclerosis-associated interstitial lung disease burn out? Specific phenotypes of disease progression. Ann Am Thorac Soc 2018;15:1427–1433. Annals of the American Thoracic Society is an official journal of the American Thoracic Society
Fig. 4.

The proportions of patients with FVC < 50% predicted and DLco < 40% predicted in the 10 years from diagnosis of ILD associated with RA. Adapted from [4]
Although some patients with chronic HP experience partial recovery, patients with chronic fibrosing HP may experience rapid disease progression, particularly if the inciting antigen cannot be identified and removed [32, 33]. In a recent longitudinal cohort analysis, the monthly decline in FVC % predicted over 1 year in 119 patients with chronic HP was similar to that of 286 patients with IPF [34]. In patients with stage IV pulmonary sarcoidosis, both increases and decreases in FVC and DLco may be observed over time as the disease relapses and remits [35]. Little is known about the clinical course of fibrotic iNSIP, but it seems to progress more slowly than other forms of fibrosing ILD [36].
Acute exacerbations of fibrosing ILDs
Acute exacerbations - episodes of rapid respiratory worsening accompanied by evidence of new ground glass opacities on HRCT - are a common feature of the natural history of IPF, believed to occur in 5–10% of patients per year [37]. Acute exacerbations of IPF may be idiopathic or triggered by an insult such as infection or aspiration. Irrespective of their cause, acute exacerbations of IPF are associated with very high morbidity and mortality, with a median post-event survival of only 3 to 4 months [37]. Far fewer data are available on acute exacerbations of fibrosing ILDs other than IPF, and most come from retrospective reviews of medical records. However, it appears that acute exacerbations do occur in patients with fibrosing ILDs other than IPF and are often fatal. An analysis of medical records from 84 patients with RA-ILD from two tertiary referral centers found that 14 patients (17%) had an idiopathic acute exacerbation over a median follow-up of 33 months, and 13 of those 14 patients died within 1.5 months of the event [38]. A study of 51 patients with RA-ILD at another center found that 22% had an acute exacerbation over a follow-up period of about 8 years [39]. In patients with SSc-ILD, acute exacerbations appear to be more common in patients with a usual interstitial pneumonia (UIP) pattern on computed tomography or histology [40, 41]. A study of 100 patients with chronic HP found that 14 patients were hospitalized for an acute exacerbation over a 2-year period, of whom 11 died within one month; patients with a UIP pattern on histology were more likely to experience acute exacerbations than those with other patterns [42].
Mortality in patients with progressing fibrosing ILDs
Progressive fibrosing ILDs are associated with high mortality. In patients with IPF who are not receiving an antifibrotic therapy, median post-diagnosis survival is 3–4 years [43–45]. ILD is one of the leading causes of death in patients with SSc [46]. Estimates of survival time vary depending on the population studied, but a recent study of patients at a specialized SSc-ILD clinic found that median survival was 11.2 years from the date of the first HRCT showing evidence of ILD [31]. RA-ILD is also associated with high mortality, particularly in patients with a UIP pattern on HRCT [47–49]. A retrospective analysis of 82 patients with RA-ILD at two tertiary referral centers found a median survival time of 5 years from the initial clinic visit (3.2 years in patients with a UIP pattern on HRCT) [48]. Patients with features consistent with the research construct known as interstitial pneumonia with autoimmune features (IPAF), i.e., who have interstitial pneumonia with features suggestive of an autoimmune disease but do not meet criteria for a defined disease [50] appear to have a mortality rate that is intermediate between patients with IPF and CTD-ILDs [51]. Fibrotic HP has a poor prognosis. A recent analysis of US claims data found that only 58% of patients with fibrotic HP were still alive approximately 7 years after diagnosis [52]. Fibrotic iNSIP appears to have a better prognosis than other forms of progressive fibrosing ILD: considering only disease-related death, 5-year survival is approximately 75% [53]. Assessment of the mortality associated with unclassifiable IIP is hampered by the wide variability in case definition across studies, but a systematic literature review found estimates for 5-year survival in patients with unclassifiable ILD, based on four studies of 46 to 70% [54].
Predictors of disease progression in patients with fibrosing ILDs
Studies in patients with progressive fibrosing ILDs have identified several factors that predict mortality, but these need to be interpreted carefully given the variation in the methodology used and the retrospective nature of most of the studies. Lower FVC is an established predictor of mortality in patients with progressive fibrosing ILDs, as evidenced by numerous studies spanning IPF [55–57], RA-ILD [4, 49], SSc-ILD [58–60], chronic HP [61, 62] and fibrotic iNSIP [53]. The same is true of DLco [55, 56, 60, 63–65], although this is harder to assess in multi-center studies due to a lack of standardization in its measurement. A decline in FVC > 10% predicted is another well-established predictor of mortality [49, 56, 62, 65], but smaller declines in FVC have also been shown to be associated with a worse prognosis, at least in patients with IPF [66–68]. Further, it is important to bear in mind that “small” annual declines in FVC may become substantial when summed over a period of years. This is particularly relevant when considering ILDs with a relatively early age of onset.
Composite scoring systems have been developed to predict mortality in patients with progressive fibrosing ILDs. One of the most widely used is the GAP (gender, age, physiology) model, which was developed to predict mortality in patients with IPF based on gender, age, FVC % predicted and DLco % predicted [69], and has since been shown to predict mortality in patients with RA-ILD [70], SSc-ILD [71], unclassifiable ILD [72] and a mixed cohort [73]. Composite scoring systems have also been developed specifically for prediction of progression of SSc-ILD [74, 75]. Unfortunately, none of the scoring systems developed to date provides an accurate prediction of the way that ILD will progress in an individual patient, creating challenges for therapeutic decision-making and patient counselling. Recently a cluster analysis in a cohort of 770 patients with diverse ILDs (IPF, CTD-ILDs, chronic HP, IPAF) identified four phenotypic groups based on factors such as age, race, smoking, and radiological features that differed in FVC decline and survival [34]. Whilst interesting from an academic perspective, the use of such groupings in clinical practice remains to be established.
There is increasing interest in the use of radiological markers as predictors of disease progression in patients with fibrosing ILDs. A greater extent of fibrotic changes on HRCT is known to be predictive of mortality. This has been demonstrated in several studies in IPF [76], RA-ILD [48, 77], SSc-ILD [5, 58], chronic HP [61, 78], pulmonary sarcoidosis [79] and unclassifiable ILD [63]. In addition, specific radiological features such as honeycombing and traction bronchiectasis have been associated with worse prognosis [33, 80]. However, the use of radiological scoring systems as predictors of prognosis in clinical practice is hampered by subjectivity and large inter-observer variability in reading HRCT scans, and will probably not be widely implemented until validated, low-cost, automated scoring systems are available [81].
Several blood biomarkers have been investigated as predictors of disease progression in patients with fibrosing ILDs. These include Krebs von den Lungen-6 protein (KL-6), which has been associated with a higher rate of disease progression in patients with IPF and CTD-ILDs [82–85], and surfactant protein-D (SP-D) [86, 87]. Several studies have shown that levels of matrix metalloproteinase-7 (MMP-7), an enzyme involved in remodeling of the extracellular matrix, are negatively correlated with lung function and survival in patients with IPF [88–91]. Protein fragments generated by breakdown of the extracellular matrix have also been investigated as predictors of disease progression [92, 93]. To date, however, no serum biomarker has been shown to be a sufficiently robust prognostic marker to justify its use in clinical practice, although KL-6 is routinely used at some ILD centers in Japan.
Genetic mutations and telomere length have also been investigated as predictors of disease progression in patients with fibrosing ILDs. In patients with IPF, rs35705950, the minor allele of a single nucleotide polymorphism (SNP) in MUC5B, a gene encoding a component of mucus secretions, has been associated with improved survival [94] while rs5743890, the minor allele of an SNP of TOLLIP (toll interacting protein), has been associated with worse survival [95]. However, in patients with chronic HP, neither of these alleles was associated with survival [96]. In patients with features consistent with IPAF, MUC5B rs35705950 was associated with worse survival, while there was no association between TOLLIP genotype and survival [97]. Recently, MUC5B rs35705950 was associated with the development of RA-ILD [98]. In addition, mutations in several genes related to telomere maintenance, such as telomerase reverse transcriptase (TERT) and telomerase RNA component (TERC), have been identified in patients with IPF [99], and patients with short telomeres have reduced survival [100, 101]. Short telomere length has also been associated with worse survival in patients with features consistent with IPAF [97] and in patients with chronic HP [96]. Variants in telomere-related genes have been identified in patients with RA-ILD [102] and SSc-ILD [103], but further investigation is needed into the association between these variants and the development and progression of ILD.
Further research into the natural history of progressive fibrosing ILDs
Data from clinical trials of investigational therapies made a large contribution to our understanding of the clinical course of IPF [104]. Ongoing large clinical trials conducted in patients with non-IPF fibrosing ILDs [105–108] will provide valuable insights into the progression of these diseases in well-characterized populations. In addition, the natural history of fibrosing ILDs is being investigated in many patient registries including the Pulmonary Fibrosis Foundation Patient Registry [109] and IPF-PRO/ILD-PRO Registry [110] in the USA, the Canadian Registry for Pulmonary Fibrosis (CARE-PF) [111], the INSIGHTS-IPF [112] and EXCITING-ILD [113] registries in Germany, and the EMPIRE registry in central and Eastern Europe [60]. These and other registries will provide valuable information on the course of fibrosing ILDs diagnosed and managed in clinical practice, including their impact on lung function, patient-reported outcomes, hospitalizations and mortality. In addition, further research is needed into the impact that comorbidities such as pulmonary hypertension and ischemic heart disease have on outcomes in patients with ILDs [114].
Conclusions
A proportion of patients with fibrosing ILDs develop a progressive fibrosing phenotype that is similar to IPF in clinical behavior and in many of the underlying pathogenetic mechanisms that drive a self-sustaining process of pulmonary fibrosis. The natural history of progressive fibrosing ILDs is characterized by deterioration in lung function, worsening dyspnea and high mortality. A greater extent of fibrosis on HRCT and decline in lung function are predictors of mortality, but the course of disease for an individual patient cannot accurately be predicted using the currently available tools. Ongoing research will provide a better understanding of the clinical course of progressive fibrosing ILDs and their impact on patients.
Acknowledgements
Medical writing assistance, supported financially by Boehringer Ingelheim, was provided by Elizabeth Ng and Wendy Morris of FleishmanHillard Fishburn, London, UK during preparation of this article. The authors were fully responsible for all content and editorial decisions, were involved at all stages of development and have approved the final version.
Funding
The page processing charges for this article have been paid by Boehringer Ingelheim.
Availability of data and materials
Data sharing is not applicable to this article as no datasets were generated or analyzed.
Abbreviations
- CTD-ILDs
Connective tissue disease-related ILDs
- DLco
Diffusion capacity of the lungs for carbon monoxide
- EULAR
European League Against Rheumatism
- EUSTAR
EULAR Scleroderma Trials and Research group
- FVC
Forced vital capacity
- HP
Hypersensitivity pneumonitis
- ILD
Interstitial lung disease
- iNSIP
Idiopathic non-specific interstitial pneumonia
- IPAF
Interstitial pneumonia with autoimmune features
- IPF
Idiopathic pulmonary fibrosis
- MMP-7
Matrix metalloproteinase-7
- RA-ILD
Rheumatoid arthritis ILD
- SSc-ILD
Systemic sclerosis ILD
- UIP
Usual interstitial pneumonia
Authors’ contributions
Both authors contributed to writing and editing the manuscript and read and approved the final version.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
Martin Kolb reports receipt of grants and personal fees from Boehringer Ingelheim and Roche; personal fees from GlaxoSmithKline, Gilead, AstraZeneca, ProMetic and Genoa; and grants from Actelion, Respivert, the Canadian Institutes of Health Research (MOP-136950), and the Canadian Pulmonary Fibrosis Foundation. Martina Vašáková has received payment for consultancy, lectures and advisory board attendance from Roche and Boehringer Ingelheim and has received research grants from Roche.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Martin Kolb, Email: kolbm@mcmaster.ca.
Martina Vašáková, Email: martina.vasakova@ftn.cz.
References
- 1.Raghu G, Remy-Jardin M, Myers JL, Richeldi L, Ryerson CJ, Lederer DJ, et al. Diagnosis of idiopathic pulmonary fibrosis. An official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med. 2018;198(5):e44–e68. doi: 10.1164/rccm.201807-1255ST. [DOI] [PubMed] [Google Scholar]
- 2.Wells AU, Brown KK, Flaherty KR, Kolb M, Thannickal VJ; IPF Consensus Working Group. What's in a name? That which we call IPF, by any other name would act the same. Eur Respir J 2018;51(5) pii: 1800692. [DOI] [PubMed]
- 3.Lederer DJ, Martinez FJ. Idiopathic pulmonary fibrosis. N Engl J Med. 2018;378:1811–1823. doi: 10.1056/NEJMra1705751. [DOI] [PubMed] [Google Scholar]
- 4.Zamora-Legoff JA, Krause ML, Crowson CS, Ryu JH, Matteson EL. Progressive decline of lung function in rheumatoid arthritis-associated interstitial lung disease. Arthritis Rheumatol. 2017;69(3):542–549. doi: 10.1002/art.39971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Winstone TA, Assayag D, Wilcox PG, Dunne JV, Hague CJ, Leipsic J, et al. Predictors of mortality and progression in scleroderma-associated interstitial lung disease: a systematic review. Chest. 2014;146(2):422–436. doi: 10.1378/chest.13-2626. [DOI] [PubMed] [Google Scholar]
- 6.Marie I, Hatron PY, Dominique S, Cherin P, Mouthon L, Menard JF. Short-term and long-term outcomes of interstitial lung disease in polymyositis and dermatomyositis: a series of 107 patients. Arthritis Rheum. 2011;63(11):3439–3447. doi: 10.1002/art.30513. [DOI] [PubMed] [Google Scholar]
- 7.Spagnolo P, Rossi G, Trisolini R, Sverzellati N, Baughman RP, Wells AU. Pulmonary sarcoidosis. Lancet Respir Med. 2018;6(5):389–402. doi: 10.1016/S2213-2600(18)30064-X. [DOI] [PubMed] [Google Scholar]
- 8.Wang P, Jones KD, Urisman A, Elicker BM, Urbania T, Johannson KA, et al. Pathologic findings and prognosis in a large prospective cohort of chronic hypersensitivity pneumonitis. Chest. 2017;152(3):502–509. doi: 10.1016/j.chest.2017.02.011. [DOI] [PubMed] [Google Scholar]
- 9.Belloli EA, Beckford R, Hadley R, Flaherty KR. Idiopathic non-specific interstitial pneumonia. Respirology. 2016;21(2):259–268. doi: 10.1111/resp.12674. [DOI] [PubMed] [Google Scholar]
- 10.Hyldgaard C, Hilberg O, Pedersen AB, Ulrichsen SP, Løkke A, Bendstrup E, et al. A population-based cohort study of rheumatoid arthritis-associated interstitial lung disease: comorbidity and mortality. Ann Rheum Dis. 2017;76(10):1700–1706. doi: 10.1136/annrheumdis-2017-211138. [DOI] [PubMed] [Google Scholar]
- 11.Wijsenbeek M, Kreuter M, Fischer A, Mounir B, Zouad-Lejour L, Wells CD, et al. Non-IPF progressive fibrosing interstitial lung disease (PF-ILD): the patient journey. Am J Respir Crit Care Med. 2018;197:A167. [Google Scholar]
- 12.Olson AL, Gifford AH, Inase N, Fernández Pérez ER, Suda T. The epidemiology of idiopathic pulmonary fibrosis and interstitial lung diseases at risk of a progressive-fibrosing phenotype. Eur Respir Rev. 2018;27(150). pii: 180077. [DOI] [PMC free article] [PubMed]
- 13.O'Reilly S, Hügle T, van Laar JM. T cells in systemic sclerosis: a reappraisal. Rheumatology. 2012;51(9):1540–1549. doi: 10.1093/rheumatology/kes090. [DOI] [PubMed] [Google Scholar]
- 14.Vašáková M, Poletti V. Fibrosing interstitial lung diseases involve different pathogenic pathways with similar outcomes. Sarcoidosis Vasc Diffuse Lung Dis. 2015;32(3):246–250. [PubMed] [Google Scholar]
- 15.Strieter RM, Mehrad B. New mechanisms of pulmonary fibrosis. Chest. 2009;136(5):1364–1370. doi: 10.1378/chest.09-0510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maher TM, Wells AU, Laurent GJ. Idiopathic pulmonary fibrosis: multiple causes and multiple mechanisms? Eur Respir J. 2007;30(5):835–839. doi: 10.1183/09031936.00069307. [DOI] [PubMed] [Google Scholar]
- 17.Altorok N, Wang Y, Kahaleh B. Endothelial dysfunction in systemic sclerosis. Curr Opin Rheumatol. 2014;26(6):615–620. doi: 10.1097/BOR.0000000000000112. [DOI] [PubMed] [Google Scholar]
- 18.Bagnato G, Harari S. Cellular interactions in the pathogenesis of interstitial lung diseases. Eur Respir Rev. 2015;24(135):102–114. doi: 10.1183/09059180.00003214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Andersson-Sjöland A, de Alba CG, Nihlberg K, Becerril C, Ramírez R, Pardo A, et al. Fibrocytes are a potential source of lung fibroblasts in idiopathic pulmonary fibrosis. Int J Biochem Cell Biol. 2008;40(10):2129–2140. doi: 10.1016/j.biocel.2008.02.012. [DOI] [PubMed] [Google Scholar]
- 20.Willis BC, du Bois RM, Borok Z. Epithelial origin of myofibroblasts during fibrosis in the lung. Proc Am Thorac Soc. 2006;3(4):377–382. doi: 10.1513/pats.200601-004TK. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hung C, Linn G, Chow YH, Kobayashi A, Mittelsteadt K, Altemeier WA, et al. Role of lung pericytes and resident fibroblasts in the pathogenesis of pulmonary fibrosis. Am J Respir Crit Care Med. 2013;188(7):820–830. doi: 10.1164/rccm.201212-2297OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fernandez IE, Eickelberg O. New cellular and molecular mechanisms of lung injury and fibrosis in idiopathic pulmonary fibrosis. Lancet. 2012;380(9842):680–688. doi: 10.1016/S0140-6736(12)61144-1. [DOI] [PubMed] [Google Scholar]
- 23.Huang X, Yang N, Fiore VF, Barker TH, Sun Y, Morris SW, et al. Matrix stiffness-induced myofibroblast differentiation is mediated by intrinsic mechanotransduction. Am J Respir Cell Mol Biol. 2012;47(3):340–348. doi: 10.1165/rcmb.2012-0050OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Froese AR, Shimbori C, Bellaye PS, Inman M, Obex S, Fatima S, et al. Stretch-induced activation of transforming growth factor-β1 in pulmonary fibrosis. Am J Respir Crit Care Med. 2016;194(1):84–96. doi: 10.1164/rccm.201508-1638OC. [DOI] [PubMed] [Google Scholar]
- 25.Baron M, Sutton E, Hudson M, Thombs B, Markland J, Pope J, et al. The relationship of dyspnoea to function and quality of life in systemic sclerosis. Ann Rheum Dis. 2008;67(5):644–650. doi: 10.1136/ard.2007.075721. [DOI] [PubMed] [Google Scholar]
- 26.Kreuter M, Swigris J, Pittrow D, Geier S, Klotsche J, Prasse A, et al. Health related quality of life in patients with idiopathic pulmonary fibrosis in clinical practice: INSIGHTS-IPF registry. Respir Res. 2017;18(1):139. doi: 10.1186/s12931-017-0621-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kreuter M, Stansen W, Stowasser S, Schoof N. Impact of lung function decline on health-related quality of life in patients with idiopathic pulmonary fibrosis (IPF). Am J Respir Crit Care Med. 2018;197:A1604. Poster available at: http://ILDPosters2018.com/pdf/ATS_FVCandHRQL_Kreuter.pdf.
- 28.Tashkin DP, Elashoff R, Clements PJ, Goldin J, Roth MD, Furst DE, et al. Cyclophosphamide versus placebo in scleroderma lung disease. N Engl J Med. 2006;354(25):2655–2666. doi: 10.1056/NEJMoa055120. [DOI] [PubMed] [Google Scholar]
- 29.Walker UA, Tyndall A, Czirják L, Denton C, Farge-Bancel D, Kowal-Bielecka O, et al. Clinical risk assessment of organ manifestations in systemic sclerosis: a report from the EULAR scleroderma trials and research group database. Ann Rheum Dis. 2007;66(6):754–763. doi: 10.1136/ard.2006.062901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jaeger VK, Wirz EG, Allanore Y, Rossbach P, Riemekasten G, Hachulla E, et al. Incidences and risk factors of organ manifestations in the early course of systemic sclerosis: a longitudinal EUSTAR study. PLoS One. 2016;11(10):e0163894. doi: 10.1371/journal.pone.0163894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Guler SA, Winstone TA, Murphy D, Hague C, Soon J, Sulaiman N, et al. Does systemic sclerosis-associated interstitial lung disease burn out? Specific phenotypes of disease progression. Ann Am Thorac Soc. 2018;15(12):1427–1433. doi: 10.1513/AnnalsATS.201806-362OC. [DOI] [PubMed] [Google Scholar]
- 32.Vašáková M, Morell F, Walsh S, Leslie K, Raghu G. Hypersensitivity pneumonitis: perspectives in diagnosis and management. Am J Respir Crit Care Med. 2017;196(6):680–689. doi: 10.1164/rccm.201611-2201PP. [DOI] [PubMed] [Google Scholar]
- 33.Salisbury ML, Gu T, Murray S, Gross BH, Chughtai A, Sayyouh M, et al. Hypersensitivity pneumonitis: radiologic phenotypes are associated with distinct survival time and pulmonary function trajectory. Chest. 2018. 10.1016/j.chest.2018.08.1076 epub ahead of print. [DOI] [PMC free article] [PubMed]
- 34.Adegunsoye A, Oldham JM, Chung JH, Montner SM, Lee C, Witt LJ, et al. Phenotypic clusters predict outcomes in a longitudinal interstitial lung disease cohort. Chest. 2018;153(2):349–360. doi: 10.1016/j.chest.2017.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nardi A, Brillet PY, Letoumelin P, Girard F, Brauner M, Uzunhan Y, et al. Stage IV sarcoidosis: comparison of survival with the general population and causes of death. Eur Respir J. 2011;38(6):1368–1373. doi: 10.1183/09031936.00187410. [DOI] [PubMed] [Google Scholar]
- 36.Travis WD, Hunninghake G, King TE, Jr, Lynch DA, Colby TV, Galvin JR, et al. Idiopathic nonspecific interstitial pneumonia: report of an American Thoracic Society project. Am J Respir Crit Care Med. 2008;177(12):1338–1347. doi: 10.1164/rccm.200611-1685OC. [DOI] [PubMed] [Google Scholar]
- 37.Collard HR, Ryerson CJ, Corte TJ, Jenkins G, Kondoh Y, Lederer DJ, et al. Acute exacerbation of idiopathic pulmonary fibrosis. An international working group report. Am J Respir Crit Care Med. 2016;194(3):265–275. doi: 10.1164/rccm.201604-0801CI. [DOI] [PubMed] [Google Scholar]
- 38.Song JW, Lee HK, Lee CK, Chae EJ, Jang SJ, Colby TV, et al. Clinical course and outcome of rheumatoid arthritis-related usual interstitial pneumonia. Sarcoidosis Vasc Diffuse Lung Dis. 2013;30(2):103–112. [PubMed] [Google Scholar]
- 39.Hozumi H, Nakamura Y, Johkoh T, Sumikawa H, Colby TV, Kono M, et al. Acute exacerbation in rheumatoid arthritis-associated interstitial lung disease: a retrospective case control study. BMJ Open. 2013;3(9):e003132. doi: 10.1136/bmjopen-2013-003132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tomiyama F, Watanabe R, Ishii T, Kamogawa Y, Fujita Y, Shirota Y, et al. High prevalence of acute exacerbation of interstitial lung disease in Japanese patients with systemic sclerosis. Tohoku J Exp Med. 2016;239(4):297–230. doi: 10.1620/tjem.239.297. [DOI] [PubMed] [Google Scholar]
- 41.Okamoto M, Fujimoto K, Sadohara J, Furuya K, Kaieda S, Miyamura T, et al. A retrospective cohort study of outcome in systemic sclerosis-associated interstitial lung disease. Respir Investig. 2016;54(6):445–453. doi: 10.1016/j.resinv.2016.05.004. [DOI] [PubMed] [Google Scholar]
- 42.Miyazaki Y, Tateishi T, Akashi T, Ohtani Y, Inase N, Yoshizawa Y. Clinical predictors and histologic appearance of acute exacerbations in chronic hypersensitivity pneumonitis. Chest. 2008;134(6):1265–1270. doi: 10.1378/chest.08-0866. [DOI] [PubMed] [Google Scholar]
- 43.Raghu G, Chen SY, Yeh WS, Maroni B, Li Q, Lee YC, et al. Idiopathic pulmonary fibrosis in US Medicare beneficiaries aged 65 years and older: incidence, prevalence, and survival, 2001-11. Lancet Respir Med. 2014;2(7):566–572. doi: 10.1016/S2213-2600(14)70101-8. [DOI] [PubMed] [Google Scholar]
- 44.Strongman H, Kausar I, Maher TM. Incidence, prevalence, and survival of patients with idiopathic pulmonary fibrosis in the UK. Adv Ther. 2018;35(5):724–736. doi: 10.1007/s12325-018-0693-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ryerson CJ, Kolb M. The increasing mortality of idiopathic pulmonary fibrosis: fact or fallacy? Eur Respir J 2018;51(1). pii: 1702420. [DOI] [PubMed]
- 46.Elhai M, Meune C, Boubaya M, Avouac J, Hachulla E, Balbir-Gurman A, et al. Mapping and predicting mortality from systemic sclerosis. Ann Rheum Dis. 2017;76(11):1897–1905. doi: 10.1136/annrheumdis-2017-211448. [DOI] [PubMed] [Google Scholar]
- 47.Bongartz T, Nannini C, Medina-Velasquez YF, Achenbach SJ, Crowson CS, Ryu JH, et al. Incidence and mortality of interstitial lung disease in rheumatoid arthritis: a population-based study. Arthritis Rheum. 2010;62(6):1583–1591. doi: 10.1002/art.27405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kim EJ, Elicker BM, Maldonado F, Webb WR, Ryu JH, Van Uden JH, et al. Usual interstitial pneumonia in rheumatoid arthritis-associated interstitial lung disease. Eur Respir J. 2010;35(6):1322–1328. doi: 10.1183/09031936.00092309. [DOI] [PubMed] [Google Scholar]
- 49.Solomon JJ, Chung JH, Cosgrove GP, Demoruelle MK, Fernandez-Perez ER, Fischer A, et al. Predictors of mortality in rheumatoid arthritis-associated interstitial lung disease. Eur Respir J. 2016;47(2):588–596. doi: 10.1183/13993003.00357-2015. [DOI] [PubMed] [Google Scholar]
- 50.Fischer A, Antoniou KM, Brown KK, Cadranel J, Corte TJ, du Bois RM, et al. An official European Respiratory Society/American Thoracic Society research statement: interstitial pneumonia with autoimmune features. Eur Respir J. 2015;46(4):976–987. doi: 10.1183/13993003.00150-2015. [DOI] [PubMed] [Google Scholar]
- 51.Oldham JM, Adegunsoye A, Valenzi E, Lee C, Witt L, Chen L, et al. Characterisation of patients with interstitial pneumonia with autoimmune features. Eur Respir J. 2016;47(6):1767–1775. doi: 10.1183/13993003.01565-2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fernández Pérez ER, Kong AM, Raimundo K, Koelsch TL, Kulkarni R, Cole AL. Epidemiology of hypersensitivity pneumonitis among an insured population in the United States: a claims-based cohort analysis. Ann Am Thorac Soc. 2018;15(4):460–469. doi: 10.1513/AnnalsATS.201704-288OC. [DOI] [PubMed] [Google Scholar]
- 53.Park IN, Jegal Y, Kim DS, Do KH, Yoo B, Shim TS, et al. Clinical course and lung function change of idiopathic nonspecific interstitial pneumonia. Eur Respir J. 2009;33(1):68–76. doi: 10.1183/09031936.00158507. [DOI] [PubMed] [Google Scholar]
- 54.Guler SA, Ellison K, Algamdi M, Collard HR, Ryerson CJ. Heterogeneity in unclassifiable interstitial lung disease. A systematic review and meta-analysis. Ann Am Thorac Soc. 2018;15(7):854–863. doi: 10.1513/AnnalsATS.201801-067OC. [DOI] [PubMed] [Google Scholar]
- 55.Paterniti MO, Bi Y, Rekić D, Wang Y, Karimi-Shah BA, Chowdhury BA. Acute exacerbation and decline in forced vital capacity are associated with increased mortality in idiopathic pulmonary fibrosis. Ann Am Thorac Soc. 2017;14(9):1395–1402. doi: 10.1513/AnnalsATS.201606-458OC. [DOI] [PubMed] [Google Scholar]
- 56.Jo HE, Glaspole I, Grainge C, Goh N, Hopkins PMA, Moodley Y, et al. Baseline characteristics of idiopathic pulmonary fibrosis: analysis from the Australian Idiopathic Pulmonary Fibrosis Registry. Eur Respir J 2017;49. pii: 1601592. [DOI] [PubMed]
- 57.Snyder L, Neely ML, Hellkamp AS, O’Brien E, de Andrade J, Conoscenti CS, et al. Predictors of death or lung transplant after a diagnosis of idiopathic pulmonary fibrosis: insights from the IPF-PRO registry. Respir Res. in press. [DOI] [PMC free article] [PubMed]
- 58.Goh NS, Desai SR, Veeraraghavan S, Hansell DM, Copley SJ, Maher TM, et al. Interstitial lung disease in systemic sclerosis: a simple staging system. Am J Respir Crit Care Med. 2008;177(11):1248–1254. doi: 10.1164/rccm.200706-877OC. [DOI] [PubMed] [Google Scholar]
- 59.Sánchez-Cano D, Ortego-Centeno N, Callejas JL, Fonollosa Plá V, Ríos-Fernández R, Tolosa-Vilella C, et al. Interstitial lung disease in systemic sclerosis: data from the Spanish scleroderma study group. Rheumatol Int. 2018;38(3):363–374. doi: 10.1007/s00296-017-3916-x. [DOI] [PubMed] [Google Scholar]
- 60.Doubková M, Švancara J, Svoboda M, Šterclová M, Bartoš V, Plačková M, et al. EMPIRE registry, Czech part: impact of demographics, pulmonary function and HRCT on survival and clinical course in idiopathic pulmonary fibrosis. Clin Respir J. 2018;12(4):1526–1535. doi: 10.1111/crj.12700. [DOI] [PubMed] [Google Scholar]
- 61.Mooney JJ, Elicker BM, Urbania TH, Agarwal MR, Ryerson CJ, Nguyen MLT, et al. Radiographic fibrosis score predicts survival in hypersensitivity pneumonitis. Chest. 2013;144(2):586–592. doi: 10.1378/chest.12-2623. [DOI] [PubMed] [Google Scholar]
- 62.Gimenez A, Storrer K, Kuranishi L, Soares MR, Ferreira RG, Pereira CAC. Change in FVC and survival in chronic fibrotic hypersensitivity pneumonitis. Thorax. 2017;73(4):391–392. doi: 10.1136/thoraxjnl-2017-210035. [DOI] [PubMed] [Google Scholar]
- 63.Ryerson CJ, Urbania TH, Richeldi L, Mooney JJ, Lee JS, Jones KD, et al. Prevalence and prognosis of unclassifiable interstitial lung disease. Eur Respir J. 2013;42(3):750–757. doi: 10.1183/09031936.00131912. [DOI] [PubMed] [Google Scholar]
- 64.Tyndall AJ, Bannert B, Vonk M, Airò P, Cozzi F, Carreira PE, et al. Causes and risk factors for death in systemic sclerosis: a study from the EULAR scleroderma trials and research (EUSTAR) database. Ann Rheum Dis. 2010;69(10):1809–1815. doi: 10.1136/ard.2009.114264. [DOI] [PubMed] [Google Scholar]
- 65.Goh NS, Hoyles RK, Denton CP, Hansell DM, Renzoni EA, Maher TM, et al. Short-term pulmonary function trends are predictive of mortality in interstitial lung disease associated with systemic sclerosis. Arthritis Rheumatol. 2017;69(8):1670–1678. doi: 10.1002/art.40130. [DOI] [PubMed] [Google Scholar]
- 66.Zappala CJ, Latsi PI, Nicholson AG, Colby TV, Cramer D, Renzoni EA, et al. Marginal decline in forced vital capacity is associated with a poor outcome in idiopathic pulmonary fibrosis. Eur Respir J. 2010;35(4):830–836. doi: 10.1183/09031936.00155108. [DOI] [PubMed] [Google Scholar]
- 67.du Bois RM, Weycker D, Albera C, Bradford WZ, Costabel U, Kartashov A, et al. Forced vital capacity in patients with idiopathic pulmonary fibrosis: test properties and minimal clinically important difference. Am J Respir Crit Care Med. 2011;184(12):1382–1389. doi: 10.1164/rccm.201105-0840OC. [DOI] [PubMed] [Google Scholar]
- 68.Reichmann WM, Yu YF, Macaulay D, Wu EQ, Nathan SD. Change in forced vital capacity and associated subsequent outcomes in patients with newly diagnosed idiopathic pulmonary fibrosis. BMC Pulm Med. 2015;15:167. doi: 10.1186/s12890-015-0161-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ley B, Ryerson CJ, Vittinghoff E, Ryu JH, Tomassetti S, Lee JS, et al. A multidimensional index and staging system for idiopathic pulmonary fibrosis. Ann Intern Med. 2012;156(10):684–691. doi: 10.7326/0003-4819-156-10-201205150-00004. [DOI] [PubMed] [Google Scholar]
- 70.Morisset J, Vittinghoff E, Lee BY, Tonelli R, Hu X, Elicker BM, et al. The performance of the GAP model in patients with rheumatoid arthritis associated interstitial lung disease. Respir Med. 2017;127:51–56. doi: 10.1016/j.rmed.2017.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mango RL, Matteson EL, Crowson CS, Ryu JH, Makol A. Assessing mortality models in systemic sclerosis-related interstitial lung disease. Lung. 2018;196(4):409–416. doi: 10.1007/s00408-018-0126-6. [DOI] [PubMed] [Google Scholar]
- 72.Hyldgaard C, Bendstrup E, Wells AU, Hilberg O. Unclassifiable interstitial lung diseases: clinical characteristics and survival. Respirology. 2017;22(3):494–500. doi: 10.1111/resp.12931. [DOI] [PubMed] [Google Scholar]
- 73.Ryerson CJ, Vittinghoff E, Ley B, Lee JS, Mooney JJ, Jones KD, et al. Predicting survival across chronic interstitial lung disease: the ILD-GAP model. Chest. 2014;145(4):723–728. doi: 10.1378/chest.13-1474. [DOI] [PubMed] [Google Scholar]
- 74.Morisset J, Vittinghoff E, Elicker BM, Hu X, Le S, Ryu JH, et al. Mortality risk prediction in scleroderma-related interstitial lung disease: the SADL model. Chest. 2017;152(5):999–1007. doi: 10.1016/j.chest.2017.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wu W, Jordan S, Becker MO, Dobrota R, Maurer B, Fretheim H, et al. Prediction of progression of interstitial lung disease in patients with systemic sclerosis: the SPAR model. Ann Rheum Dis. 2018;77(9):1326–1332. doi: 10.1136/annrheumdis-2018-213201. [DOI] [PubMed] [Google Scholar]
- 76.Lynch DA, Godwin JD, Safrin S, Starko KM, Hormel P, Brown KK, et al. High-resolution computed tomography in idiopathic pulmonary fibrosis: diagnosis and prognosis. Am J Respir Crit Care Med. 2005;172(4):488–493. doi: 10.1164/rccm.200412-1756OC. [DOI] [PubMed] [Google Scholar]
- 77.Kelly CA, Saravanan V, Nisar M, Arthanari S, Woodhead FA, Price-Forbes AN, et al. Rheumatoid arthritis-related interstitial lung disease: associations, prognostic factors and physiological and radiological characteristics--a large multicentre UK study. Rheumatology (Oxford) 2014;53(9):1676–1682. doi: 10.1093/rheumatology/keu165. [DOI] [PubMed] [Google Scholar]
- 78.Walsh SL, Sverzellati N, Devaraj A, Wells AU, Hansell DM. Chronic hypersensitivity pneumonitis: high resolution computed tomography patterns and pulmonary function indices as prognostic determinants. Eur Radiol. 2012;22(8):1672–1679. doi: 10.1007/s00330-012-2427-0. [DOI] [PubMed] [Google Scholar]
- 79.Walsh SL, Wells AU, Sverzellati N, Keir GJ, Calandriello L, Antoniou KM, et al. An integrated clinicoradiological staging system for pulmonary sarcoidosis: a case-cohort study. Lancet Respir Med. 2014;2(2):123–130. doi: 10.1016/S2213-2600(13)70276-5. [DOI] [PubMed] [Google Scholar]
- 80.Lee SM, Seo JB, Oh SY, Kim TH, Song JW, Lee SM, et al. Prediction of survival by texture-based automated quantitative assessment of regional disease patterns on CT in idiopathic pulmonary fibrosis. Eur Radiol. 2018;28(3):1293–1300. doi: 10.1007/s00330-017-5028-0. [DOI] [PubMed] [Google Scholar]
- 81.Walsh SLF. Imaging biomarkers and staging in IPF. Curr Opin Pulm Med. 2018;24(5):445–452. doi: 10.1097/MCP.0000000000000507. [DOI] [PubMed] [Google Scholar]
- 82.Yokoyama A, Kondo K, Nakajima M, Matsushima T, Takahashi T, Nishimura M, et al. Prognostic value of circulating KL-6 in idiopathic pulmonary fibrosis. Respirology. 2006;11(2):164–168. doi: 10.1111/j.1440-1843.2006.00834.x. [DOI] [PubMed] [Google Scholar]
- 83.Lee YS, Kim HC, Lee BY, Lee CK, Kim MY, Jang SJ, et al. The value of biomarkers as predictors of outcome in patients with rheumatoid arthritis-associated usual interstitial pneumonia. Sarcoidosis Vasc Diffuse Lung Dis. 2016;33(3):216–223. [PubMed] [Google Scholar]
- 84.Yamakawa H, Hagiwara E, Kitamura H, Yamanaka Y, Ikeda S, Sekine A, et al. Serum KL-6 and surfactant protein-D as monitoring and predictive markers of interstitial lung disease in patients with systemic sclerosis and mixed connective tissue disease. J Thorac Dis. 2017;9(2):362–371. doi: 10.21037/jtd.2017.02.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Jiang Y, Luo Q, Han Q, Huang J, Ou Y, Chen M, et al. Sequential changes of serum KL-6 predict the progression of interstitial lung disease. J Thorac Dis. 2018;10(8):4705–4714. doi: 10.21037/jtd.2018.07.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kennedy B, Branagan P, Moloney F, Haroon M, O'Connell OJ, O'Connor TM, et al. Biomarkers to identify ILD and predict lung function decline in scleroderma lung disease or idiopathic pulmonary fibrosis. Sarcoidosis Vasc Diffuse Lung Dis. 2015;32(3):228–236. [PubMed] [Google Scholar]
- 87.Maher TM, Oballa E, Simpson JK, Porte J, Habgood A, Fahy WA, et al. An epithelial biomarker signature for idiopathic pulmonary fibrosis: an analysis from the multicentre PROFILE cohort study. Lancet Respir Med. 2017;5(12):946–955. doi: 10.1016/S2213-2600(17)30430-7. [DOI] [PubMed] [Google Scholar]
- 88.Rosas IO, Richards TJ, Konishi K, Zhang Y, Gibson K, Lokshin AE, et al. MMP1 and MMP7 as potential peripheral blood biomarkers in idiopathic pulmonary fibrosis. PLoS Med. 2008;5(4):e93. doi: 10.1371/journal.pmed.0050093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Richards TJ, Kaminski N, Baribaud F, Flavin S, Brodmerkel C, Horowitz D, et al. Peripheral blood proteins predict mortality in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2012;185(1):67–76. doi: 10.1164/rccm.201101-0058OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bauer Y, White ES, de Bernard S, Cornelisse P, Leconte I, Morganti A, et al. MMP-7 is a predictive biomarker of disease progression in patients with idiopathic pulmonary fibrosis. ERJ Open Res 2017;3(1). pii: 00074–2016. [DOI] [PMC free article] [PubMed]
- 91.Todd J, Vinisko R, Neely ML, Overton R, Flaherty KR, Noth I, et al. Peripheral blood matrix metalloproteinase profiling in the multicenter IPF-PRO Registry cohort. Poster presented at the 10th International Colloquium on Lung and Airway Fibrosis (ICLAF), September 2018. Available at http://uspubs-posters.com/iclaf2018/todd.
- 92.Jenkins RG, Simpson JK, Saini G, Bentley JH, Russell AM, Braybrooke R, et al. Longitudinal change in collagen degradation biomarkers in idiopathic pulmonary fibrosis: an analysis from the prospective, multicentre PROFILE study. Lancet Respir Med. 2015;3(6):462–472. doi: 10.1016/S2213-2600(15)00048-X. [DOI] [PubMed] [Google Scholar]
- 93.Maher TM, Stowasser S, Nishioka Y, White ES, Cottin V, Noth I, et al. Investigating the effects of nintedanib on biomarkers of extracellular matrix turnover in patients with IPF: design of the randomised placebo-controlled INMARK trial. BMJ Open Resp Res. 2018;5(1):e000325. doi: 10.1136/bmjresp-2018-000325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Peljto AL, Zhang Y, Fingerlin TE, Ma SF, Garcia JG, Richards TJ, et al. Association between the MUC5B promoter polymorphism and survival in patients with idiopathic pulmonary fibrosis. JAMA. 2013;309(21):2232–2239. doi: 10.1001/jama.2013.5827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Noth I, Zhang Y, Ma SF, Flores C, Barber M, Huang Y, et al. Genetic variants associated with idiopathic pulmonary fibrosis susceptibility and mortality: a genome-wide association study. Lancet Respir Med. 2013;1(4):309–317. doi: 10.1016/S2213-2600(13)70045-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ley B, Newton CA, Arnould I, Elicker BM, Henry TS, Vittinghoff E, et al. The MUC5B promoter polymorphism and telomere length in patients with chronic hypersensitivity pneumonitis: an observational cohort-control study. Lancet Respir Med. 2017;5(8):639–647. doi: 10.1016/S2213-2600(17)30216-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Newton CA, Oldham JM, Ley B, Anand V, Adegunsoye A, Liu G, et al. Telomere length and genetic variant associations with interstitial lung disease progression and survival. Eur Respir J 2019. pii: 1801641 doi: 10.1183/13993003.01641-2018 [Epub ahead of print]. [DOI] [PMC free article] [PubMed]
- 98.Juge PA, Lee JS, Ebstein E, Furukawa H, Dobrinskikh E, Gazal S, et al. MUC5B promoter variant and rheumatoid arthritis with interstitial lung disease. N Engl J Med. 2018;379(23):2209–2219. doi: 10.1056/NEJMoa1801562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Armanios MY, Chen JJ, Cogan JD, Alder JK, Ingersoll RG, Markin C, et al. Telomerase mutations in families with idiopathic pulmonary fibrosis. N Engl J Med. 2007;356(13):1317–1326. doi: 10.1056/NEJMoa066157. [DOI] [PubMed] [Google Scholar]
- 100.Stuart BD, Lee JS, Kozlitina J, Noth I, Devine MS, Glazer CS, et al. Effect of telomere length on survival in patients with idiopathic pulmonary fibrosis: an observational cohort study with independent validation. Lancet Respir Med. 2014;2(7):557–565. doi: 10.1016/S2213-2600(14)70124-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Dai J, Cai H, Li H, Zhuang Y, Min H, Wen Y, et al. Association between telomere length and survival in patients with idiopathic pulmonary fibrosis. Respirology. 2015;20(6):947–52. doi: 10.1111/resp.12566. [DOI] [PubMed] [Google Scholar]
- 102.Juge PA, Borie R, Kannengiesser C, Gazal S, Revy P, Wemeau-Stervinou L, et al. Shared genetic predisposition in rheumatoid arthritis-interstitial lung disease and familial pulmonary fibrosis. Eur Respir J. 2017:49(5). 10.1183/13993003.02314-2016. [DOI] [PubMed]
- 103.Mak AC, Tang PL, Cleveland C, Smith MH, Kari Connolly M, Katsumoto TR, et al. Brief report: whole-exome sequencing for identification of potential causal variants for diffuse cutaneous systemic sclerosis. Arthritis Rheumatol. 2016;68(9):2257–2262. doi: 10.1002/art.39721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Raghu G. Idiopathic pulmonary fibrosis: lessons from clinical trials over the past 25 years. Eur Respir J. 2017;50(4). 10.1183/13993003.01209-2017. [DOI] [PubMed]
- 105.Flaherty KR, Brown KK, Well AU, Clerisme-Beaty E, Collard HR, Cottin V, et al. Design of the PF-ILD trial: a double-blind, randomised, placebo-controlled phase III trial of nintedanib in patients with progressive fibrosing interstitial lung disease. BMJ Open Resp Res. 2017;4:e000212. doi: 10.1136/bmjresp-2017-000212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Distler O, Brown KK, Distler JHW, Assassi S, Maher TM, Cottin V, et al. Design of a randomised, placebo-controlled clinical trial of nintedanib in patients with systemic sclerosis-associated interstitial lung disease (SENSCIS). Clin Exp Rheumatol. 2017;35 Suppl 106(4):75–81. [PubMed]
- 107.Saunders P, Tsipouri V, Keir GJ, Ashby D, Flather MD, Parfrey H, et al. Rituximab versus cyclophosphamide for the treatment of connective tissue disease-associated interstitial lung disease (RECITAL): study protocol for a randomised controlled trial. Trials. 2017;18(1):275. doi: 10.1186/s13063-017-2016-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Maher TM, Corte TJ, Fischer A, Kreuter M, Lederer DJ, Molina-Molina M, et al. Pirfenidone in patients with unclassifiable progressive fibrosing interstitial lung disease: design of a double-blind, randomised, placebo-controlled phase II trial. BMJ Open Resp Res. 2018;5(1):e000289. doi: 10.1136/bmjresp-2018-000289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Pulmonary Fibrosis Foundation: PFF Patient Registry. https://www.pulmonaryfibrosis.org/medical-community/pff-patient-registry (2016). Accessed 24 October 2018.
- 110.O'Brien EC, Durheim MT, Gamerman V, Garfinkel S, Anstrom KJ, Palmer SM, et al. Rationale for and design of the Idiopathic Pulmonary Fibrosis-PRospective Outcomes (IPF-PRO) Registry. BMJ Open Resp Res. 2016;3(1):e000108. [DOI] [PMC free article] [PubMed]
- 111.Ryerson CJ, Tan B, Fell CD, Manganas H, Shapera S, Mittoo S, et al. The Canadian Registry for Pulmonary Fibrosis: design and rationale of a national pulmonary fibrosis registry. Can Respir J. 2016;2016:3562923. [DOI] [PMC free article] [PubMed]
- 112.Behr J, Hoeper MM, Kreuter M, Klotsche J, Wirtz H, Pittrow D. Investigating significant health trends in idiopathic pulmonary fibrosis (INSIGHTS-IPF): rationale, aims and design of a nationwide prospective registry. BMJ Open Respir Res. 2014;1(1):e000010. doi: 10.1136/bmjresp-2013-000010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kreuter M, Herth FJF, Wacker M, Leidl R, Hellmann A, Pfeifer M, et al. Exploring clinical and epidemiological characteristics of interstitial lung diseases: rationale, aims, and design of a nationwide prospective registry—the EXCITING-ILD registry. Biomed Res Int. 2015;2015:123876. doi: 10.1155/2015/123876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Schwarzkopf L, Witt S, Waelscher J, Polke M, Kreuter M. Associations between comorbidities, their treatment and survival in patients with interstitial lung diseases - a claims data analysis. Respir Res. 2018;19(1):73. doi: 10.1186/s12931-018-0769-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no datasets were generated or analyzed.